

Abstract
ERBB2 is an oncogenic receptor tyrosine kinase overexpressed in a subset of human breast cancer and other cancers. We recently found that human prolidase (PEPD), a dipeptidase, is a high affinity ERBB2 ligand and cross-links two ERBB2 monomers. Here, we show that recombinant human PEPD (rhPEPD) strongly inhibits ERBB2-overexpressing tumors in mice, whereas it does not impact tumors without ERBB2 overexpression. rhPEPD causes ERBB2 depletion, disrupts oncogenic signaling orchestrated by ERBB2 homodimers and heterodimers, and induces apoptosis. The impact of enzymatically-inactive mutant rhPEPDG278D on ERBB2 is indistinguishable from that of rhPEPD, but rhPEPDG278D is superior to rhPEPD for tumor inhibition. The enzymatic function of rhPEPD stimulates HIF-1α and other pro-survival factors in tumors, which likely attenuates its antitumor activity. rhPEPDG278D is also attractive in that it may not interfere with the physiologic function of endogenous PEPD in normal cells. Collectively, we have identified a human protein as an inhibitory ERBB2 ligand that inhibits ERBB2-overexpressing tumors in vivo. Several anti-ERBB2 agents are on the market but are hampered by drug resistance and high drug cost. rhPEPDG278D may synergize with these agents and may also be highly cost-effective, since it targets ERBB2 with a different mechanism and can be produced in bacteria.
Keywords: ERBB2, ERBB2 ligand, Prolidase, PEPD, Anti-ERBB2 agent
Highlights
-
•
Human prolidase (rhPEPD) inhibits the growth of ErbB2-overexpressing tumors.
-
•
rhPEPD disrupts the signaling complexes assembled by ErbB2 homo- and hetero-dimers.
-
•
Enzymatically-inactive rhPEPDG278D is superior to rhPEPD for tumor inhibition.
1. Introduction
ERBB2, also known as HER2 or v-erb-B2 erythroblastic leukemia viral oncogene homolog 2 among other names, is an oncogenic cell surface receptor tyrosine kinase and an important cancer therapeutic target. ERBB2 amplification or overexpression occurs in 20–30% of breast cancer and is a strong predictor of poor disease prognosis (Ross et al., 2009, Slamon et al., 1987). ERBB2 overexpression also occurs in several other types of cancer (Junttila et al., 2003, Lassus et al., 2004, Saffari et al., 1995, Tanner et al., 2005). A number of ERBB2-targeting agents have been developed, including monoclonal antibodies, small molecule tyrosine kinase inhibitors, and antibody-cytotoxic agent conjugates (Incorvati et al., 2013), but they are limited by drug resistance and high drug cost. For example, trastuzumab, a humanized monoclonal antibody, has been the leading agent for treatment of ERBB2-positive breast cancer, but about half of patients do not respond to trastuzumab-based therapy (Pohlmann et al., 2009, Romond et al., 2005, Vogel et al., 2002). Moreover, trastuzumab, which is produced in mammalian cells, costs over $50,000 for a full course of treatment. On the other hand, it is now widely recognized that combination of agents with different targeting mechanisms improves treatment outcome.
Prolidase (PEPD) is a cytosolic enzyme that splits dipeptides with proline or hydroxyproline at the carboxy terminus and is believed to be important for collagen homeostasis, as high levels of the imino acids are present in collagen (Kitchener and Grunden, 2012). Interestingly, we recently found that recombinant human PEPD (rhPEPD) binds as a homodimer, with high affinity (Kd = 7.3 nM, based on ELISA), to subdomain 3 in the extracellular domain (ECD) of ERBB2, with each PEPD subunit (molecular mass of 54 kD) binding to an ECD, thereby cross-linking two ERBB2 monomers (Yang et al., 2014). This was unexpected, as it had long been believed that ERBB2 exists in a closed conformation and cannot be liganded. Indeed, no ERBB2 ligand had been previously identified. The ability of rhPEPD to cross-link two ERBB2 monomers represents a new ERBB2-binding mechanism, as neither trastuzumab nor pertuzumab (another therapeutic anti-ERBB2 monoclonal antibody) cross-links ERBB2 monomers (Cho et al., 2003, Franklin et al., 2004). Also, no ligands of ERBB2 family members cross-link their respective receptors (Hynes and Lane, 2005, Leahy, 2004). In cells overexpressing ERBB2, rhPEPD binds to preformed ERBB2 dimers and blocks ERBB2-SRC signaling by causing SRC disassociation from ERBB2, apparently resulting from alteration of ERBB2 conformation (Yang et al., 2014). SRC plays a key role in ERBB2 oncogenesis (Muthuswamy et al., 1994, Sheffield, 1998, Zhang et al., 2011). rhPEPD also binds to ERBB2 monomers, causing ERBB2 dimerization and activation (Yang et al., 2014). However, such ERBB2 activation is transient and may be functionally insignificant, as rhPEPD-bound ERBB2 is efficiently internalized and degraded, causing ERBB2 depletion, and rhPEPD strongly inhibits the malignant phenotype of cells overexpressing ERBB2, while showing little effect on cells without ERBB2 overexpression (Yang et al., 2014). These results not only reveal a new function of human PEPD, but also show that the protein is primarily an inhibitory ligand of ERBB2 and suggest that rhPEPD may be a potential antitumor agent. Among the ERBB2 family members, rhPEPD does not bind to ERBB3 and ERBB4 (Yang et al., 2014), but also binds to ERBB1 ECD (Yang et al., 2013). Interestingly, despite relatively low binding affinity of rhPEPD (Kd = 5.3 μM, based on ELISA) towards ERBB1, it modulates the receptor at low nM concentrations, causing transient activation and then depletion of the latter (Yang et al., 2013). Notably, intracellular human PEPD does not modulate ERBB1 and ERBB2, and the dipeptidase activity of rhPEPD is not required for modulation of the receptors (Yang et al., 2013, Yang et al., 2014).
In this study, we investigate the ability of rhPEPD to inhibit tumor growth in vivo, whether ERBB2 overexpression is a critical indicator of rhPEPD efficacy, and whether rhPEPD is able to silence ERBB2 signaling in the tumor tissues. Notably, trastuzumab, which binds to subdomain 4 in the ERBB2 ECD (Cho et al., 2003), shows similar effect on ERBB2 to rhPEPD in cultured cells (Cuello et al., 2001, Nagata et al., 2004). However, the ability of trastuzumab to downregulate ERBB2 expression or to inhibit ERBB2 tyrosine phosphorylation in tumor tissues in vivo seems to be limited (Gennari et al., 2004, Gijsen et al., 2010), and its Fc domain may play a significant role in tumor inhibition by engaging Fc receptors on immune effector cells and eliciting antibody-dependent cell-mediated cytotoxicity (ADCC) (Barok et al., 2007, Clynes et al., 2000, Spiridon et al., 2004). In contrast, rhPEPD does not have an Fc domain. We also compare the antitumor efficacy of rhPEPD with that of rhPEPDG278D, a mutant which lacks the dipeptidase activity, in order to assess the relevance of the dipeptidase function for rhPEPD to target ERBB2 and to inhibit tumor growth in vivo.
2. Materials and Methods
2.1. Biochemicals, Cell Lines and Reagents
rhPEPD and rhPEPDG278D with 6xhistidine tagged to their carboxy terminus were generated as previously described (Yang et al., 2013). Briefly, full-length human PEPD cDNA and its variant coding PEPDG278D were cloned to the bacterial pBAD/TOPO expression vector, and the His-tagged recombinant proteins were generated in Escherichia coli and purified by Ni-NTA agarose chromatography. We obtained enoxaparin (EP) from Sanofi-Aventis via Roswell Park Cancer Institute (RPCI) Pharmacy. Recombinant human epidermal growth factor (EGF) and human neuregulin 1 (NRG-1) were obtained from R&D Systems and Cell Signaling, respectively. All cell lines and their culture conditions were described previously (Yang et al., 2013, Yang et al., 2014). The following antibodies were used: anti-PEPD (Abcam, ab86507), anti-ERBB1 (Cell Signaling, 2232), anti-p-ERBB1 (Y1173) (Cell Signaling, 4407), anti-ERBB2 (Cell Signaling, 2165), anti-p-ERBB2 (Y1221/1222) (Cell Signaling, 2243), anti-ERBB3 (Santa Cruz, sc-285), anti-p-ERBB3 (Y1328) (Santa Cruz, sc-135654), anti-AKT (Cell Signaling, 4691), anti-p-AKT (Cell Signaling, 4060), anti-ERK (Cell Signaling, 9102), anti-p-ERK (Cell Signaling, 9101), anti-PI3K p85 (Cell Signaling, 4257), anti-SRC (Cell Signaling, 2123), anti-p-SRC (Cell Signaling, 6943), anti-STAT3 (Cell Signaling, 4904), anti-p-STAT3 (Cell Signaling, 9145), anti-caspase-3 (Cell Signaling, 9662), anti-cleaved caspase-8 (Cell Signaling, 9496), anti-cleaved caspase-9 (Cell Signaling, 9501), anti-BCL-2 (Cell Signaling, 2870), anti-BAX (Cell Signaling, 2772), anti-VEGF (Santa Cruz, sc-152), anti-GLUT-1 (Santa Cruz, sc-7903), anti-HIF-1α (Santa Cruz, sc-53546), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Millipore, MAB374), and biotin-conjugated anti-His (Bethyl, A190-113B). HRP-conjugated Streptavidin (N100) was purchased from Thermo Scientific. Matrigel was purchased from BD Biosciences. A goat anti-rabbit IgG-HRP was purchased from Jackson ImmunoResearch (111-035-003).
2.2. Tumor Xenograft Study in Mice
Athymic nude mice (female, 6–7 weeks of age) from Harlan were used. The experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at RPCI. rhPEPD and rhPEPDG278D were evaluated in combination with EP which serves as a dose reducer for the PEPDs. We established subcutaneous tumors by inoculating CHO-K1/ERBB2 cells or CHO-K1 cells to the flanks of the mice at 1 × 106 cells per site in 100 μl of PBS-Matrigel mixture (1:1 ratio). Four days after cell inoculation, EP (2.5 mg/kg) or vehicle was administered to the mice via intraperitoneal injection (i.p.) daily. Three days later, tumor size reached about 40 mm3 (CHO-K1/ERBB2 tumors) or 30 mm3 (CHO-K1 tumors), and the EP-treated mice also began treatment with rhPEPD (0.02 or 0.2 mg/kg) or vehicle i.p. thrice weekly (Monday, Wednesday, Friday). Blood samples were collected from the mice when they were killed 24 h after the final treatment for measurement of plasma levels of PEPD and sERBB2.
To establish orthotopic mammary tumors, we implanted the mice with 1.7 mg 60-day release 17β-estradiol pellets (Innovative Research of America) subcutaneously and 2 days later inoculated BT-474 cells to the mammary fat pads at 2 × 106 per site in 100 μl of PBS-Matrigel mixture (1:1). The mice were used in two experiments as described below. In experiment 1, the mice were either untreated (control) or treated with EP (0.5 mg/kg) i.p. daily, starting 23 days after cell inoculation. Four days later, tumor size reached about 60 mm3, and the EP-treated mice also began treatment with vehicle, rhPEPD or rhPEPDG278D (each at 2 mg/kg) i.p. thrice weekly (Monday, Wednesday, Friday), while daily EP treatment continued. All treatments were stopped 30 days later, and the mice were kept under observation. One day after treatment stop, blood samples were collected from the mice via retro-orbital bleeding. Blood samples were also collected from the untreated mice at the same time, but these mice were killed following blood draw. Each mouse kept under observation was given another 17β-estradiol pellet 2 days later (day 61 after cell inoculation). Approximately 4 weeks post treatment, the mice that were initially treated with EP alone were retreated with EP or EP plus rhPEPDG278D, and the mice that were previously treated with EP plus rhPEPD or EP plus rhPEPDG278D but showed tumor relapse were retreated with EP plus rhPEPDG278D. Again, EP (0.5 mg/kg) was given i.p. daily, and rhPEPDG278D treatment (2 mg/kg) began 4 days later, which was given i.p. every 2–3 days (a total of 4–5 doses). The mice were killed 48 h after the final treatment. In experiment 2, tumors were allowed to reach approximately 220 mm3. The mice were then treated with vehicle, EP, EP plus rhPEPD, and EP plus rhPEPDG278D. We gave EP (0.5 mg/kg) i.p. once daily for 7 days, and on the fifth and seventh days of EP treatment, we also gave rhPEPD or rhPEPDG278D i.p. at 2 mg/kg. The mice were killed 24 h after the final treatment.
In all experiments, on the days when both EP and rhPEPD or rhPEPDG278D were given, EP was always given 1 h earlier than the other agent. EP, rhPEPD and rhPEPDG278D were given in PBS. We calculated tumor size using length × width2 / 2. Tumor images were captured using a Canon EOS Digital Rebel Xsi camera.
2.3. Measurement of Plasma PEPD and Plasma sERBB2
Plasma PEPD concentrations were determined by ELISA as previously described (Yang et al., 2013). Plasma sERBB2 concentrations were measured using the Human Soluble Her2 ELISA Kit (Aviscera Bioscience), following manufacturer's instruction.
2.4. Measurement of SRC Activity and PI3K Activity
SRC and PI3K activities in cell and tissue samples were measured as previously described (Yang et al., 2014).
2.5. Plasmids and Gene Transfection
To construct pCMV6-XL5-ERBB1 which expresses human ERBB1, the full length human ERBB1 coding sequence was amplified by PCR from LNCaP cDNA using Kpn1-forward primer (5′-GGTACCCGGCCCCCTGACTCCGTCCAG-3′) and HindIII-reverse primer (5′-AAGCTTTCATGCTCCAATAAATTCACTGCTTTGTGGC-3′). The amplified PCR product was digested by KpnI and HindIII (New England BioLabs), followed by ligation into pCMV6-XL5 (Origene) which was pre-digested with the same restriction enzymes. The construct was sequenced to ensure the integrity of the entire coding sequence and correct orientation of the gene. We obtained human ERBB3-expressing plasmid (pCMV6-XL4-ERBB3) and human SRC-expressing plasmid (pCMV6-XL4-SRC) from Origene. Transient gene transfection was carried out using cells grown in 6-well plates and FuGENE HD (Promega).
2.6. Immunoprecipitation and Immunoblotting
Sample preparation, immuoprecipitation and immunoblotting as well as cross-linking reactions using bis(sulfosuccinimidyl) suberate (BS3, Pierce) were performed as previously described (Yang et al., 2013, Yang et al., 2014).
2.7. Statistical Analysis
Analysis of variance and t-test were used for comparison of multiple groups and two groups, respectively. A mixed model (fixed effects plus random effects including time effect) was used for two-group comparison of longitudinal tumor data, using SAS 9.3. We considered a difference with P < 0.05 (two-sided) statistically significant.
3. Results
3.1. rhPEPD Inhibits Tumor Growth, but ERBB2 Overexpression Is Critical for rhPEPD Efficacy
Therapeutically relevant plasma concentrations of rhPEPD could be achieved in mice by given rhPEPD at 10 mg/kg i.p. (Supplementary Fig. 1A). However, pretreatment with enoxaparin (EP) at 2.5 mg/kg i.p. allowed rhPEPD dose to be reduced by at least 50 fold without decreasing its plasma concentration (Supplementary Fig. 1B). EP is a clinically used low molecular weight heparin (LMWH). EP and other LMWHs were shown to significantly elevate plasma level of endogenous PEPD in rats (Caliskan et al., 2014). EP also significantly increased plasma level of endogenous PEPD in mice (Supplementary Fig. 1B). LMWHs including EP are known to inhibit several blood coagulation proteases, including factors Xa and IIa, via binding and activating antithrombin, a serine protease inhibitor (Hirsh and Raschke, 2004). Our preliminary observation suggests that EP inhibits PEPD proteolysis in the plasma, but neither factor Xa nor factor IIa degrades PEPD. EP was used as a dose reducer for rhPEPD and its mutant in animal studies described below.
We first evaluated rhPEPD in tumors derived from Chinese hamster ovary cells (CHO-K1), which express a low level of ERBB2 but no other ERBBs, and CHO-K1 cells with stable overexpression of human ERBB2 (CHO-K1/ERBB2) (Yang et al., 2014). Subcutaneous CHO-K1/ERBB2 tumors grew rapidly; EP (2.5 mg/kg i.p. daily) had no impact on tumor growth, but rhPEPD at 0.02 and 0.2 mg/kg i.p. (thrice weekly for 2.3 weeks) in the presence of EP (2.5 mg/kg i.p. daily) inhibited tumor growth by 26.7% (373.5/1401.2) and 65.1% (911.9/1401.2; P < 0.0001) respectively (Fig. 1A). Although tumor inhibition by rhPEPD at the low dose was not statistically significant, other effects of rhPEPD at this dose level were highly significant, as shown below. Plasma rhPEPD concentrations, measured at 24 h after the last dose (Fig. 1B), correlated with rhPEPD dose levels and with tumor inhibition; rhPEPD at similar concentrations targets ERBB2-overexpressing cells in vitro (Yang et al., 2014). Subcutaneous tumors of CHO-K1 cells grew slower, compared with CHO-K1/ERBB2 tumors, reflecting the oncogenic activity of ERBB2, but rhPEPD at 0.2 mg/kg i.p. (thrice weekly for 2 weeks) in the presence of EP (2.5 mg/kg i.p. daily) had no effect on tumor growth (Fig. 1C), even though plasma concentrations of rhPEPD were high (Fig. 1D). The rhPEPD-treated mice showed no sign of toxicity and no change in body weight gain (Supplementary Fig. 2A and B). As expected, ERBB2 was overexpressed in CHO-K1/ERBB2 tumors along with tyrosine phosphorylation (auto-phosphorylation) as well as activation/phosphorylation of all evaluated downstream signals, including SRC (Bjorge et al., 2000), AKT (Gao et al., 2005), extracellular signal-regulated kinase (ERK) (Spencer et al., 2000) and signal transducer and activator of transcription 3 (STAT3) (Ren and Schaefer, 2002), none of which was impacted by EP but uniformly suppressed by rhPEPD in a dose-dependent manner (Fig. 1E). As expected, CHO-K1 tumors showed low ERBB2 expression and no ERBB2 tyrosine phosphorylation, although significant phosphorylation of SRC, AKT, ERK and STAT3 existed in the tumors, apparently independent of ERBB2 (Fig. 1E). None of these proteins in CHO-K1 tumors was modulated by EP, and rhPEPD only slightly reduced ERBB2 level and SRC phosphorylation but had little effect on the other proteins (Fig. 1E). ERBB2 sheds its ECD (sERBB2) via proteolytic cleavage (Codony-Servat et al., 1999). Plasma sERBB2 concentration reached ~ 3 ng/ml in mice bearing CHO-K1/ERBB2 tumors but was undetectable (< 5 pg/ml) in mice bearing CHO-K1 tumors (Fig. 1F). EP did not significantly alter plasma sERBB2 level, but rhPEPD reduced plasma sERBB2 by up to 68.4% (2092.0/3060.7; P = 0.005, Fig. 1F), consistent with ERBB2 depletion induced by rhPEPD in the tumor tissues. Notably, rhPEPD does not directly modulate ERBB2 ECD shedding (Yang et al., 2014). rhPEPD, but not EP, also strongly and dose-dependently reduced SRC kinase activity in CHO-K1/ERBB2 tumors, but such effect was minimal in CHO-K1 tumors (Fig. 1G). Collectively, these data show that rhPEPD potently targets ERBB2 oncogenenesis in vivo.
Fig. 1.

rhPEPD inhibits tumor growth by targeting ERBB2. [A] Sizes of subcutaneous CHO-K1/ERBB2 tumors upon treatment with vehicle, EP, EP plus rhPEPD. EP was administered to the mice i.p. at 2.5 mg per kg body weight daily, starting 3 days before rhPEPD. rhPEPD was administered to the mice i.p. at 0.02 mg (L) or 0.2 mg (H) per kg body weight thrice weekly (Monday, Wednesday and Friday), starting on day 7 post cell inoculation and lasted for 2.3 weeks. [B] Plasma levels of PEPD (endogenous mouse PEPD and/or rhPEPD) at 24 h after the final dose of each treatment as indicated in A. [C] Sizes of subcutaneous CHO-K1 tumors upon treatment of the mice with EP or EP plus PEPD as indicated in A, except that rhPEPD treatment lasted for 2 weeks and was evaluated only at 0.2 mg/kg. [D] Plasma levels of PEPD (endogenous PEPD and/or rhPEPD) at 24 h after the final dose of each treatment as indicated in C. [E] Immunoblots comparing major cell signaling changes in tumor specimens obtained 24 h after the last dose of each treatment as indicated in A and C, as well as in untreated CHO-K1 tumor specimens. Each sample represents one tumor. [F] Plasma levels of sERBB2 at 24 h after the final dose of each treatment as indicated in A and C. [G] SRC kinase activities in tumor specimens at 24 h after the final dose of each treatment as indicated in A and C. Error bars for the tumor sizes are SEM (n = 8–12). All other error bars are SD (n = 3). Scale bars in A and C: 5 cm.
3.2. A Dipeptidase-deficient rhPEPD Mutant Is Superior to Wild-type rhPEPD for Tumor Inhibition
We next turned to orthotopic BT-474 tumors. Human breast cancer BT-474 cells constitutively overexpress ERBB2 but also express a low level of ERBB1 and ERBB3 (Yang et al., 2014). rhPEPD dose was escalated to 2 mg/kg thrice weekly, in view of its performance in CHO-K1/ERBB2 tumors. EP dose was lowered to 0.5 mg/kg daily, which was adequate for sustaining blood PEPD level (Supplementary Fig. 3). rhPEPDG278D, an enzymatically inactive mutant (Ledoux et al., 1996), was also evaluated. EP had no effect on BT-474 tumor, but tumor began to shrink after the first dose of rhPEPD or rhPEPDG278D, and the remaining tumors were consistently smaller in the rhPEPDG278D-treated mice than in rhPEPD-treated mice during the treatment period (P = 0.0003, Fig. 2A). Plasma rhPEPDG278D concentration was 16.8% (25.9/154.5) lower than that of rhPEPD at 24 h after the final treatment, albeit not statistically significant (Fig. 2B). Plasma sERBB2 concentrations decreased greatly after treatment with rhPEPD or rhPEPDG278D and were undetectable in 50% (3/6) rhPEPD-treated mice and 67% (4/6) rhPEPDG278D-treated mice (Fig. 2C). Initial treatments lasted 30 days, with 27.3% (3/11) of rhPEPD-treated tumors and 41.7% (5/12) of rhPEPDG278D-treated tumors achieving complete remission (CR) (Fig. 2A; Supplementary Fig. 4A and B). Nearly all of the tumors in partial remission relapsed by approximately 25 days after treatment termination; however, these tumors were still exquisitely sensitive to rhPEPDG278D (Fig. 2A), indicating that tumor relapsed due to incomplete initial treatment but not due to the presence of rhPEPD/rhPEPDG278D-resistant cells. Moreover, very large tumors (~ 500 mm3) also responded exquisitely to rhPEPDG278D (Fig. 2A). The experiment was stopped due to heavy tumor burden in the mice treated by EP alone. Molecular changes in the tumor tissues could not be determined, because tumors in some treatment groups were extremely small or no longer present. There were no signs of adverse effects in the mice treated by rhPEPD or rhPEPDG278D. Neither rhPEPD nor rhPEPDG278D impacted animal body weight gain, the weights of major organs or heart histology (Supplementary Fig. 4C–E). Notably, cardiotoxicity occurs in some patients receiving anti-ERBB2 therapies (Sendur et al., 2013).
Fig. 2.

rhPEPDG278D is superior to rhPEPD for combating ErbB2-overexpressing tumors. [A] Sizes of orthotopic BT-474 tumors with no treatment, upon treatment with EP, EP plus rhPEPD, or EP plus rhPEPDG278D, during treatment pause, and upon retreatment with EP, or EP plus rhPEPDG278D. All treatment days are relative to the day of cancer cell inoculation. EP was administered to the mice i.p. at 0.5 mg/kg daily, starting 4 days before rhPEPD or rhPEPDG278D. rhPEPD or rhPEPDG278D was administered to the mice i.p. at 2 mg/kg; during the initial phase of treatment, each agent was administered thrice weekly (Monday, Wednesday and Friday); during the retreatment phase, rhPEPDG278D was administered every 2–3 days. The tumor growth curves in mice treated initially with EP plus rhPEPD or EP plus rhPEPDG278D were redrawn on tumor growth day 83 into those with relapse or with CR. Error bars are SEM (n = 6 in no treatment group; n = 10–12 in all other groups during the initial phase of treatment; n = 4–8 during the second phase of treatment). *tumors were removed and photographed 2 days before the other tumors. Scale bars: 3 cm. [B] Plasma levels of PEPD (endogenous PEPD, rhPEPD and/or rhPEPDG278D) at 24 h after the last dose during the initial phase of treatment. Error bars are SD (n = 3). [C] Scatter plots of plasma levels of sERBB2 at 24 h after the last dose during the initial phase of treatment; 3–6 samples per group.
3.3. The Molecular Changes in the Tumor Tissues after Treatment with rhPEPD or rhPEPDG278D
After treatment with only two doses of rhPEPD or rhPEPDG278D at 2 mg/kg, separated by 2 days, in the presence of EP (0.5 mg/kg daily), BT-474 tumors shrank to 42.2% (136.6/323.8) or 37.3% (120.8/323.8) of the control respectively (Fig. 3A). EP impacted neither tumor growth nor ERBB2 signaling in the tumor tissues, while tumor inhibition by rhPEPD or rhPEPDG278D was associated with marked depletion and dephosphorylation of ERBB2, and dephosphorylation of key downstream signals, including SRC, AKT, ERK and STAT3 (Fig. 3B). Low levels of ERBB1 and ERBB3 were present in BT-474 tumors; both agents strongly reduced ERBB1 expression and its tyrosine phosphorylation, and strongly reduced ERBB3 tyrosine phosphorylation but not its expression (Fig. 3B). While SRC is activated upon binding to activated ERBB1 or ERBB2, phosphoinositide 3-kinase (PI3K) activation and signaling in ERBB2-overexpressing breast cancer cells depend on its recruitment to activated ERBB3 (Holbro et al., 2003). Accordingly, both agents strongly inhibited SRC kinase activity and PI3K activity in the tumor tissues (Fig. 3C and D). Each agent also downregulated B-cell lymphoma 2 (BCL-2), up regulated BCL-2-associated X protein (BAX), and activated caspases-3/-8/-9 in the tumor tissues (Fig. 3B). However, rhPEPD, but not rhPEPDG278D, stimulated hypoxia-inducible factor 1α (HIF-1α) and its downstream targets, including vascular endothelial growth factor (VEGF) and glucose transporter 1 (GLUT-1) in the tumor tissues (Fig. 3E). The effects of rhPEPD on HIF-1α, VEGF and GLUT-1 likely stem from the metabolism of imidodipeptides by rhPEPD upon its internalization and the inhibition of HIF-1α degradation by the metabolites (Surazynski et al., 2008). Both rhPEPD and rhPEPDG278D accumulated in the tumor tissues (Fig. 3E) via ERBB2-mediated internalization (Supplementary Fig. 5). Again, we detected no adverse effects in mice treated by the test agents. Neither rhPEPD or rhPEPDG278D nor EP impacted animal body weight gain, the weight and histology of heart, kidney and liver (Supplementary Fig. 6).
Fig. 3.

Molecular changes in tumors after treatment with rhPEPD or rhPEPDG278D. [A] Sizes of orthotopic BT-474 tumors upon treatment with vehicle, EP, EP plus rhPEPD or EP plus rhPEPDG278D. EP was administered to the mice i.p. at 0.5 mg/kg daily, starting 4 days before rhPEPD or rhPEPDG278D. rhPEPD or rhPEPDG278D was administered to the mice i.p. at 2 mg/kg twice, separated by 2 days. Error bars are SEM (n = 4–6). Scale bars: 3 cm. [B] Immunoblots comparing major cell signaling changes in tumor specimens obtained 24 h after the final dose of each treatment as indicated in A. Each sample represents one tumor. [C & D] SRC kinase activity and PI3K activity in tumor specimens obtained 24 h after the final dose of each treatment as indicated in A. Error bars are SD (n = 3). [E] Immunoblots comparing HIF-1α signaling changes in tumor specimens obtained 24 h after the final dose of each treatment as indicated in A. Each sample represents one tumor.
3.4. The Effects of rhPEPD and rhPEPDG278D on ERBB1 and ERBB1-ERBB2 Heterodimer Interaction
rhPEPD and rhPEPDG278D downregulate both ERBB1 and ERBB2 in BT-474 tumors (Fig. 3B). Downregulation of ERBB2 by the agents results from its internalization and degradation and is independent of ERBB1 (Yang et al., 2014) (see also Fig. 1E). We investigated whether downregulation of ERBB1 by the agents may involve ERBB2, since ERBB2 is a well-known preferred heterodimerization partner for other ERBBs. In cells overexpressing both ERBB1 and ERBB2 (CHO-K1/ERBB1 + ERBB2), with no expression of other ERBBs, rhPEPD (5 nM) caused rapid tyrosine phosphorylation of both ERBB1 and ERBB2 but rapid tyrosine dephosphorylation of SRC, followed by downregulation of both ERBB1 and ERBB2, with downregulation of ERBB2 occurring much faster than that of ERBB1 (Fig. 4A). Similar results were shown previously in other cell lines (Yang et al., 2013, Yang et al., 2014). In ERBB1-overexpressing murine myeloid 32D cells (32D/ERBB1), with no expression of ERBB2 or other ERBBs (Fan et al., 2004), the ERBB1 changes induced by both rhPEPD and rhPEPDG278D are almost identical to that in CHO-K1/ERBB1 + ERBB2 cells (Fig. 4B). Clearly, ERBB1 phosphorylation and subsequent downregulation induced by the agents are not related to ERBB2. The relatively slow rate of ERBB1 downregulation in response to the PEPDs, compared to that of ERBB2, is likely related to a difference in their internalization and degradation (Yang et al., 2014). In 32D/ERBB1 cells in which binding of rhPEPD and rhPEPDG278D to ERBB1 was cross-linked by BS3, we were able to show that both agents bind to ERBB1 (monomer MW of 175 kD) as a homodimer (dimer MW of 108 kD), first forming heterotrimer (1 ERBB1 monomer linked to 1 dimer of rhPEPD or rhPEPDG278D) and then heterotetramer (1 ERBB1 dimer linked to 1 dimer of rhPEPD or rhPEPDG278D) (Fig. 4C). Increased level of ERBB1 dimer free of the PEPDs was also detected (Fig. 4C), but this likely results from incomplete cross-linking of the proteins by BS3. ERBB1 closely resembles ERRB2 (Yang et al., 2014) in PEPD binding. As mentioned before, SRC is activated upon binding to activated ERBB1 or ERBB2. Both rhPEPD and rhPEPDG278D caused rapid SRC dissociation from ERBB2 and SRC dephosphorylation (Fig. 4D), consistent with previous finding (Yang et al., 2014). 32D/ERBB1 cells do not express SRC, but after transient transfection of human SRC, rhPEPD and rhPEPDG278D promote SRC association with ERBB1 and SRC phosphorylation in these cells (Fig. 4E and F). Thus, both agents promote ERBB1 and SRC interaction, in contrast to their inhibitory effect on ERBB2 and SRC interaction.
Fig. 4.

The effect of rhPEPD and rhPEPDG278D on ERBB1 and ERBB1-ERBB2 heterodimer interaction. [A] CHO-K1/ERBB2 cells were transfected with human ERBB1 for 24 h and then treated with rhPEPD (5 nM), followed by immunoblotting. [B] 32D/ERBB1 cells were treated with rhPEPD or rhPEPDG278D, followed by immunoblotting. [C] 32D/ERBB1 cells were treated with vehicle, rhPEPD or rhPEPDG278D (5 nM), and then treated with cross-linking agent BS3 (2 mM, 30 min); cell lysates were immunoblotted for ERBB1. [D] CHO-K1/ERBB2 cells were treated with solvent, rhPEPD or rhPEPDG278D (each at 5 nM) for 1 h. Cell lysates were pulled down with an ERBB2 antibody, followed by immunoblotting. Sample loading was adjusted to contain an equal amount of ERRB2. An example of immunoblot without adjustment of ERBB2 content is shown in Supplementary Fig. 7. [E] 32D/ERBB1 cells were transfected with human SRC for 48 h and then treated with rhPEPD or rhPEPDG278D (5 nM), followed by immunoblotting. [F] 32D/ERBB1 cells were transfected with human SRC for 48 h and then treated with solvent, rhPEPD or rhPEPDG278D (each at 5 nM) for 1 h. Cell lysates were immunoprecipitated using an ERBB1 antibody, followed by immunoblotting. [G & H] CHO-K1/ERBB2 cells were transfected with human ERBB1 for 24 h and then treated with solvent, rhPEPD or rhPEPDG278D (each at 5 nM) for 1 h, with or without pretreatment with EGF (20 nM, 15 min). Cell lysates were immunoprecipitated using an ERBB2 antibody or an ERBB1 antibody, followed by immunoblotting. For the immunoblots shown in G, sample loading was adjusted to contain an equal amount of ERBB2.
Even though rhPEPD and rhPEPDG278D bind to both ERBB1 and ERBB2, both agents disrupt ERBB1-ERBB2 association in CHO-K1/ERBB1 + ERBB2 cells, whether the heterodimer forms spontaneously or is stimulated by EGF (an ERBB1 ligand) (Fig. 4G). Interestingly, even though EGF significantly simulated ERBB2-ERBB1 association, it did not stimulate SRC association with the heterodimer or SRC phosphorylation by the heterodimer (Fig. 4G), indicating that SRC interacts only with ERBB2 in the heterodimer and that such interaction is not modulated by EGF. Remarkably, both agents caused EGF-bound ERBB1 to separate from ERBB2 (Fig. 4H). It is possible that the agents bind to ERBB2 in the heterodimer to force homodimerization at the expense of heterodimerization and/or disrupt the heterodimer by causing ERBB2 depletion.
3.5. rhPEPD and rhPEPDG278D Disrupt ERBB2-ERBB3 Heterodimer Interaction
In BT-474 tumors (Fig. 3B), neither rhPEPD nor rhPEPDG278D downregulated ERBB3, consistent with our recent finding that the agents do not bind to ERBB3, but both agents caused ERBB3 to dephosphorylate along with loss of PI3K activity. In cells overexpressing both ERBB2 and ERBB3 (CHO-K1/ERBB2 + ERBB3), with no expression of other ERBBs, rhPEPD (5 nM) caused ERBB3 dephosphorylation without protein downregulation, along with transient increase in ERBB2 phosphorylation, rapid ERBB2 protein downregulation and rapid SRC dephosphorylation, whereas PI3K expression remained unchanged (Fig. 5A). As mentioned before, SRC is activated upon binding to activated ERBB2, whereas PI3K is activated by binding to activated ERBB3. In CHO-K1/ERBB2 + ERBB3 cells, both rhPEPD and rhPEPDG278D disassembled the ErbB2–ErbB3 signaling unit, causing dissociation of ERBB3 from ERBB2 along with dissociation of SRC and PI3K from the ERBBs (Fig. 5B and C), whether the signaling complex formed spontaneously or was stimulated by NRG-1 (an ERBB3 ligand). Moreover, both agents caused NRG-1-bound ERBB3 to dissociate from ERBB2 (Fig. 5C). It is possible that the agents may bind to ERBB2 in the heterodimer to force ERBB2 homodimerization at the expense of heterodimerization and/or disrupt the heterodimer by causing ERBB2 depletion.
Fig. 5.
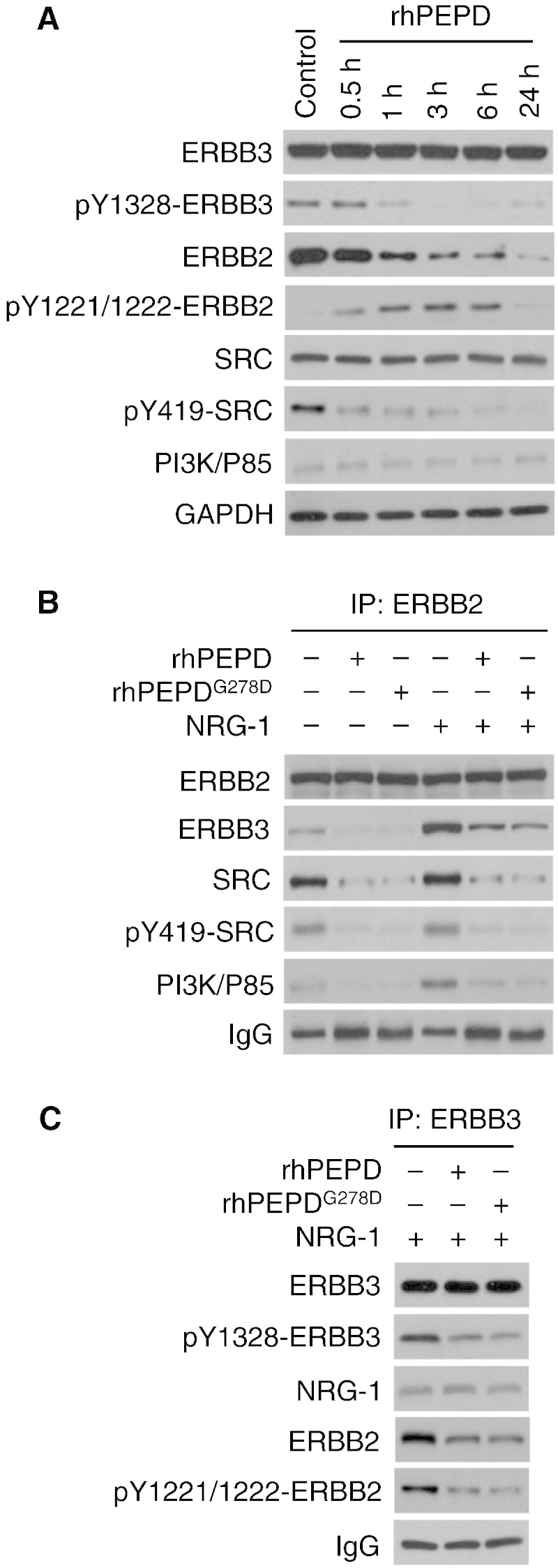
The effect of rhPEPD and rhPEPDG278D on ERBB2-ERBB3 heterodimer interaction. [A] CHO-K1/ERBB2 cells were transfected with human ERBB3 for 24 h and then treated with rhPEPD (5 nM), followed by immunoblotting. [B & C] CHO-K1/ERBB2 cells were transfected with human ERBB3 for 24 h and then treated with solvent, rhPEPD or rhPEPDG278D (each at 5 nM) for 1 h, with or without pretreatment with NRG-1 (20 nM, 15 min). Cell lysates were immunoprecipitated using an ERBB2 antibody or an ERBB3 antibody, followed by immunoblotting. For the immunoblots shown in B, sample loading was adjusted to contain an equal amount of ERBB2.
4. Discussion
The tumors in the three mouse models used herein either expressed minimal ERBB2 (CHO-K1) or overexpressed it (CHO-K1/ERBB2 and BT-474), and either grew subcutaneously (CHO-K1 and CHO-K1/ERBB2) or orthotopically in the mammary fat pad (BT-474). We show that rhPEPD strongly inhibits the growth of ERBB2-overexpressing tumors, regardless of tumor location, type or size, but it does not inhibit tumors without ERBB2 overexpression. The response of the tumors to rhPEPD in vivo mimics the response of the same cell lines to rhPEPD in vitro (Yang et al., 2014). However, rhPEPDG278D is more attractive than rhPEPD as a potential antitumor agent for several reasons. First, the antitumor efficacy of rhPEPDG278D is superior to that of rhPEPD, as judged by both partial and complete tumor remission. Second, rhPEPD, rather than rhPEPDG278D, stimulates HIF-1α and its downstream targets (VEGF, GLUT-1) in the tumor tissues via its dipeptidase activity. PEPD was also shown to stimulate TGFβ and its receptor via its dipeptidase function (Surazynski et al., 2010). The stimulating effect of rhPEPD on the prosurvival factors mentioned above likely attenuates its antitumor activity. Third, rhPEPDG278D may not interfere with the physiologic function of endogenous PEPD in normal cells and tissues. In the present study, rhPEPDG278D achieved durable CR in 41.7% (5/12) BT-474 tumors, but it is possible that a higher CR rate may be achieved with dose escalation. Other dipeptidase-defective mutants of human PEPD may also possess strong antitumor activity. However, we previously showed that deletion mutants of human PEPD, which are incapable of forming homodimers, have no ability to bind and modulate ERBB2 (Yang et al., 2014).
rhPEPD and rhPEPDG278D are indistinguishable in terms of modulating ERBB2 and its family members in tumor cells; both agents cause the depletion of ERBB1 and ERBB2 via internalization and degradation, and both disrupt ERBB2–ERBB1 and ERBB2–ERBB3 association and signaling (Fig. 6). We speculate that they may also disrupt ERBB2–ERBB4 heterodimerization and signaling. Both agents also cause transient phosphorylation/activation of ERBB1 and ERBB2 in cultured cells; however, these changes were not detected in tumor tissues in vivo and therefore may not significantly impede their antitumor activity. The inhibitory impact of both rhPEPD and rhPEPDG278D on ERBB2 and its family members is further shown by decrease in plasma sERBB2 level and dephosphorylation/inactivation of all analyzed downstream signaling proteins (SRC, AKT, ERK and STAT3) in tumor tissues. Moreover, stimulation of apoptosis in the tumor tissues by the agents, including downregulation of antiapoptotic BCL-2, upregulation of proapoptotic BAX, and stimulation of caspases-3/-8/-9, which is indicative of activation of both mitochondria-dependent and -independent apoptosis, is consistent with ERBB2 suppression (Kumar et al., 1996, Martin-Perez et al., 2014, Petry et al., 2010). These findings together with those shown recently in cultured cells (Yang et al., 2014) indicate conclusively that human PEPD is an inhibitory ligand of ERBB2 and that the dipeptidase activity of PEPD is not involved in suppressing ERBB2 oncogenesis.
Fig. 6.

Paradigm of molecular changes induced by rhPEPD and rhPEPDG278D in cancer cells. rhPEPD and rhPEPDG278D share the same antitumor mechanism, except that the dipeptidase function of rhPEPD stimulates HIF-1α and other prosurvival factors, which are not shown in the paradigm. The paradigm is constructed based on the present data and those published recently (Yang et al., 2013, Yang et al., 2014). SRC and PI3K are shown as representative downstream signaling molecules. rhPEPD and rhPEPDG278D exist as homodimers and cross-link preformed ERBB2 homodimers, silencing ERBB2-SRC signaling by causing SRC dissociation from ERBB2. They also cross-link ERBB2 monomers, causing dimerization and transient phosphorylation. Upon binding to the PEPDs, ERBB2 is internalized and degraded, leading to profound and persistent ERBB2 depletion. The PEPDs do not bind to ERBB3 but disrupt ligand-independent and NRG-1-induced ERBB2–ERBB3 dimerization and signaling (inactivation of both SRC and PI3K). The PEPDs also cross-link ERBB1, causing dimerization and transient phosphorylation, followed by ERBB1 degradation. Despite binding to both ERBB1 and ERBB2, the PEPDs do not cross-link ERBB1 and ERBB2; rather, they disrupt ligand-independent and EGF-induced ERBB2–ERBB1 dimerization. The PEPDs promote SRC association with ERBB1 homodimer and ERBB1-mediated SRC phosphorylation, but such effect is transient as both agents subsequently cause ERBB1 depletion. Overall, the impact of the PEPDs on ERBB2 and its family members in cancer cells is inhibitory, leading to shutdown of ERBB signaling, induction of apoptosis and inhibition of cancer growth.
Compared to trastuzumab and related agents, rhPEPDG278D is exciting for several reasons. First, rhPEPDG278D is produced in bacteria, likely lowering manufacturing cost considerably, compared to trastuzumab and other agents. Studies to demonstrate the relative activity of rhPEPDG278D in comparison with trastuzumab and related agents will become important, once the antitumor efficacy of rhPEPDG278D has been more thoroughly assessed. Second, rhPEPDG278D directly attacks the ERBB2 signaling system in the tumor, whereas trastuzumab seems to rely significantly on ADCC for tumor inhibition in vivo. Therefore, studies to investigate whether PEPDG278D may synergize with trastuzumab and may circumvent trastuzumab resistance are warranted. These studies may be pursued, once the antitumor activity of rhPEPDG278D has been more thoroughly evaluated and better understood. Third, because PEPDG278D lacks an Fc domain, a PEPDG278D-Fc hybrid may allow it to engage ADCC, thereby enhancing its antitumor activity. Fourth, a conjugate of PEPDG278D with a cancer chemotherapeutic agent, similar to ado-trastuzumab emtansine (a conjugate of trastuzumab with a microtubule inhibitor) (Amiri-Kordestani et al., 2014), may boost its antitumor activity. In this connection, we show that rhPEPDG278D as well as rhPEPD are internalized by tumor cells in an ERBB2-dependent manner. Finally, unlike trastuzumab which only disrupts ligand-independent ERBB2–ERBB3 signaling (Junttila et al., 2009) and has no effect on ERBB1, rhPEPDG278D disrupts both ligand-independent and ligand-stimulated ERBB2–ERBB3 and ERBB1–ERBB2 interaction and signaling, and downregulates both ERBB1 and ERBB2. By targeting both ERBB1 and ERBB2, PEPDG278D may also overcome resistance to ERBB1-directed therapies, resulting from activation of ERBB2 signaling and/or insensitivity to tyrosine kinase inhibitors (Lin and Bivona, 2012, Yonesaka et al., 2011).
In summary, we have shown the in vivo evidence of human PEPD as an inhibitory ligand of ERBB2, and have demonstrated that rhPEPDG278D is a highly promising agent for combating ERBB2-driven cancer. rhPEPDG278D may be a strong candidate for combination with trastuzumab or other clinically used anti-ERBB2 agents. Given its ability to attack ERBB2-overexpressing tumors with multiple mechanisms, studies to investigate whether rhPEPDG278D circumvent drug resistance to current anti-ERBB2 therapies are warranted. Studies to investigate whether rhPEPDG278D may inhibit ERBB1-driven tumors and tumors resistant to current ERBB1-directed therapies are also warranted. The strong antitumor activity of rhPEPD also raises the question of whether endogenous PEPD may impede the growth of ERBB2-overexpressing tumors. Our present data show that PEPD at low nM plasma concentrations can inhibit ERBB2-overexpressing tumors. PEPD is present in normal blood, and blood PEPD level increases significantly in patients of breast cancer (Kir et al., 2003). However, PEPD was measured via its dipeptidase activity in human blood, and actual blood PEPD concentration or the source of blood PEPD is not known. PEPD is expressed at significantly higher level in human breast cancer tissues than in normal breast tissues (Cechowska-Pasko et al., 2006), which may account for the increase in blood PEPD level in patients.
Author Contributions
LY, YL, AB, and YZ designed experiments and analyzed data. LY, YL, and AB carried out experiments. LY and YZ wrote the manuscript. All authors edited and approved the final manuscript.
Conflict of Interest
The authors have no conflicting financial interests.
Acknowledgments
We thank Leslie Curtin in the Department of Laboratory Animal Resources, RPCI for assistance for animal experiments. We also thank Zhengyu Yang in the Department of Biostatistics & Bioinformatics, RPCI for assistance with statistical analysis. This work was supported in part by the US National Institutes of Health grants R01-CA164574 and P30-CA016056, and a Roswell Park Cancer Institute Support Fund.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2015.03.016.
Appendix A. Supplementary data
Supplementary material.
References
- Amiri-Kordestani L., Blumenthal G.M., Xu Q.C., Zhang L., Tang S.W., Ha L., Weinberg W.C., Chi B., Candau-Chacon R., Hughes P. FDA approval: ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin. Cancer Res. 2014;20:4436–4441. doi: 10.1158/1078-0432.CCR-14-0012. [DOI] [PubMed] [Google Scholar]
- Barok M., Isola J., Palyi-Krekk Z., Nagy P., Juhasz I., Vereb G., Kauraniemi P., Kapanen A., Tanner M., Szollosi J. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol. Cancer Ther. 2007;6:2065–2072. doi: 10.1158/1535-7163.MCT-06-0766. [DOI] [PubMed] [Google Scholar]
- Bjorge J.D., Jakymiw A., Fujita D.J. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000;19:5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- Caliskan A., Yavuz C., Karahan O., Yazici S., Guclu O., Demirtas S., Mavitas B. Factor-Xa inhibitors protect against systemic oxidant damage induced by peripheral-ischemia reperfusion. J. Thromb. Thrombolysis. 2014;37:464–468. doi: 10.1007/s11239-013-1019-4. [DOI] [PubMed] [Google Scholar]
- Cechowska-Pasko M., Palka J., Wojtukiewicz M.Z. Enhanced prolidase activity and decreased collagen content in breast cancer tissue. Int. J. Exp. Pathol. 2006;87:289–296. doi: 10.1111/j.1365-2613.2006.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.S., Mason K., Ramyar K.X., Stanley A.M., Gabelli S.B., Denney D.W., Jr., Leahy D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- Clynes R.A., Towers T.L., Presta L.G., Ravetch J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- Codony-Servat J., Albanell J., Lopez-Talavera J.C., Arribas J., Baselga J. Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Res. 1999;59:1196–1201. [PubMed] [Google Scholar]
- Cuello M., Ettenberg S.A., Clark A.S., Keane M.M., Posner R.H., Nau M.M., Dennis P.A., Lipkowitz S. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001;61:4892–4900. [PubMed] [Google Scholar]
- Fan Y.X., Wong L., Deb T.B., Johnson G.R. Ligand regulates epidermal growth factor receptor kinase specificity: activation increases preference for GAB1 and SHC versus autophosphorylation sites. J. Biol. Chem. 2004;279:38143–38150. doi: 10.1074/jbc.M405760200. [DOI] [PubMed] [Google Scholar]
- Franklin M.C., Carey K.D., Vajdos F.F., Leahy D.J., de Vos A.M., Sliwkowski M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–328. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- Gao T., Furnari F., Newton A.C. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Gennari R., Menard S., Fagnoni F., Ponchio L., Scelsi M., Tagliabue E., Castiglioni F., Villani L., Magalotti C., Gibelli N. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin. Cancer Res. 2004;10:5650–5655. doi: 10.1158/1078-0432.CCR-04-0225. [DOI] [PubMed] [Google Scholar]
- Gijsen M., King P., Perera T., Parker P.J., Harris A.L., Larijani B., Kong A. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol. 2010;8:e1000563. doi: 10.1371/journal.pbio.1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh J., Raschke R. Heparin and low-molecular-weight heparin: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest. 2004;126:188S–203S. doi: 10.1378/chest.126.3_suppl.188S. [DOI] [PubMed] [Google Scholar]
- Holbro T., Beerli R.R., Maurer F., Koziczak M., Barbas C.F., 3rd, Hynes N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. U. S. A. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes N.E., Lane H.A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Incorvati J.A., Shah S., Mu Y., Lu J. Targeted therapy for HER2 positive breast cancer. J. Hematol. Oncol. 2013;6:38. doi: 10.1186/1756-8722-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila T.T., Laato M., Vahlberg T., Soderstrom K.O., Visakorpi T., Isola J., Elenius K. Identification of patients with transitional cell carcinoma of the bladder overexpressing ErbB2, ErbB3, or specific ErbB4 isoforms: real-time reverse transcription-PCR analysis in estimation of ErbB receptor status from cancer patients. Clin. Cancer Res. 2003;9:5346–5357. [PubMed] [Google Scholar]
- Junttila T.T., Akita R.W., Parsons K., Fields C., Lewis Phillips G.D., Friedman L.S., Sampath D., Sliwkowski M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- Kir Z.O., Oner P., Iyidogan Y.O., Turkmen S., Kocak H., Koser M., Kucucuk S.O. Serum prolidase I activity and some bone metabolic markers in patients with breast cancer: in relation to menopausal status. Clin. Biochem. 2003;36:289–294. doi: 10.1016/s0009-9120(03)00028-6. [DOI] [PubMed] [Google Scholar]
- Kitchener R.L., Grunden A.M. Prolidase function in proline metabolism and its medical and biotechnological applications. J. Appl. Microbiol. 2012;113:233–247. doi: 10.1111/j.1365-2672.2012.05310.x. [DOI] [PubMed] [Google Scholar]
- Kumar R., Mandal M., Lipton A., Harvey H., Thompson C.B. Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin. Cancer Res. 1996;2:1215–1219. [PubMed] [Google Scholar]
- Lassus H., Leminen A., Vayrynen A., Cheng G., Gustafsson J.A., Isola J., Butzow R. ERBB2 amplification is superior to protein expression status in predicting patient outcome in serous ovarian carcinoma. Gynecol. Oncol. 2004;92:31–39. doi: 10.1016/j.ygyno.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Leahy D.J. Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Adv. Protein Chem. 2004;68:1–27. doi: 10.1016/S0065-3233(04)68001-6. [DOI] [PubMed] [Google Scholar]
- Ledoux P., Scriver C.R., Hechtman P. Expression and molecular analysis of mutations in prolidase deficiency. Am. J. Hum. Genet. 1996;59:1035–1039. [PMC free article] [PubMed] [Google Scholar]
- Lin L., Bivona T.G. Mechanisms of resistance to epidermal growth factor receptor inhibitors and novel therapeutic strategies to overcome resistance in NSCLC patients. Chemother. Res. Pract. 2012;2012:817297. doi: 10.1155/2012/817297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Perez R., Palacios C., Yerbes R., Cano-Gonzalez A., Iglesias-Serret D., Gil J., Reginato M.J., Lopez-Rivas A. Activated ERBB2/HER2 licenses sensitivity to apoptosis upon endoplasmic reticulum stress through a PERK-dependent pathway. Cancer Res. 2014;74:1766–1777. doi: 10.1158/0008-5472.CAN-13-1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy S.K., Siegel P.M., Dankort D.L., Webster M.A., Muller W.J. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol. Cell. Biol. 1994;14:735–743. doi: 10.1128/mcb.14.1.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata Y., Lan K.H., Zhou X., Tan M., Esteva F.J., Sahin A.A., Klos K.S., Li P., Monia B.P., Nguyen N.T. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Petry I.B., Fieber E., Schmidt M., Gehrmann M., Gebhard S., Hermes M., Schormann W., Selinski S., Freis E., Schwender H. ERBB2 induces an antiapoptotic expression pattern of Bcl-2 family members in node-negative breast cancer. Clin. Cancer Res. 2010;16:451–460. doi: 10.1158/1078-0432.CCR-09-1617. [DOI] [PubMed] [Google Scholar]
- Pohlmann P.R., Mayer I.A., Mernaugh R. Resistance to trastuzumab in breast cancer. Clin. Cancer Res. 2009;15:7479–7491. doi: 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z., Schaefer T.S. ErbB-2 activates Stat3 alpha in a Src- and JAK2-dependent manner. J. Biol. Chem. 2002;277:38486–38493. doi: 10.1074/jbc.M112438200. [DOI] [PubMed] [Google Scholar]
- Romond E.H., Perez E.A., Bryant J., Suman V.J., Geyer C.E., Jr., Davidson N.E., Tan-Chiu E., Martino S., Paik S., Kaufman P.A. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005;353:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- Ross J.S., Slodkowska E.A., Symmans W.F., Pusztai L., Ravdin P.M., Hortobagyi G.N. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- Saffari B., Jones L.A., el-Naggar A., Felix J.C., George J., Press M.F. Amplification and overexpression of HER-2/neu (c-erbB2) in endometrial cancers: correlation with overall survival. Cancer Res. 1995;55:5693–5698. [PubMed] [Google Scholar]
- Sendur M.A., Aksoy S., Altundag K. Cardiotoxicity of novel HER2-targeted therapies. Curr. Med. Res. Opin. 2013;29:1015–1024. doi: 10.1185/03007995.2013.807232. [DOI] [PubMed] [Google Scholar]
- Sheffield L.G. C-Src activation by ErbB2 leads to attachment-independent growth of human breast epithelial cells. Biochem. Biophys. Res. Commun. 1998;250:27–31. doi: 10.1006/bbrc.1998.9214. [DOI] [PubMed] [Google Scholar]
- Slamon D.J., Clark G.M., Wong S.G., Levin W.J., Ullrich A., McGuire W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Spencer K.S., Graus-Porta D., Leng J., Hynes N.E., Klemke R.L. ErbB2 is necessary for induction of carcinoma cell invasion by ErbB family receptor tyrosine kinases. J. Cell Biol. 2000;148:385–397. doi: 10.1083/jcb.148.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiridon C.I., Guinn S., Vitetta E.S. A comparison of the in vitro and in vivo activities of IgG and F(ab′)2 fragments of a mixture of three monoclonal anti-Her-2 antibodies. Clin. Cancer Res. 2004;10:3542–3551. doi: 10.1158/1078-0432.CCR-03-0549. [DOI] [PubMed] [Google Scholar]
- Surazynski A., Donald S.P., Cooper S.K., Whiteside M.A., Salnikow K., Liu Y., Phang J.M. Extracellular matrix and HIF-1 signaling: the role of prolidase. Int. J. Cancer. 2008;122:1435–1440. doi: 10.1002/ijc.23263. [DOI] [PubMed] [Google Scholar]
- Surazynski A., Miltyk W., Prokop I., Palka J. Prolidase-dependent regulation of TGF beta (corrected) and TGF beta receptor expressions in human skin fibroblasts. Eur. J. Pharmacol. 2010;649:115–119. doi: 10.1016/j.ejphar.2010.09.034. [DOI] [PubMed] [Google Scholar]
- Tanner M., Hollmen M., Junttila T.T., Kapanen A.I., Tommola S., Soini Y., Helin H., Salo J., Joensuu H., Sihvo E. Amplification of HER-2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann. Oncol. 2005;16:273–278. doi: 10.1093/annonc/mdi064. [DOI] [PubMed] [Google Scholar]
- Vogel C.L., Cobleigh M.A., Tripathy D., Gutheil J.C., Harris L.N., Fehrenbacher L., Slamon D.J., Murphy M., Novotny W.F., Burchmore M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- Yang L., Li Y., Ding Y., Choi K.S., Kazim A.L., Zhang Y. Prolidase directly binds and activates epidermal growth factor receptor and stimulates downstream signaling. J. Biol. Chem. 2013;288:2365–2375. doi: 10.1074/jbc.M112.429159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Li Y., Zhang Y. Identification of prolidase as a high affinity ligand of the ErbB2 receptor and its regulation of ErbB2 signaling and cell growth. Cell Death Dis. 2014;5:e1211. doi: 10.1038/cddis.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonesaka K., Zejnullahu K., Okamoto I., Satoh T., Cappuzzo F., Souglakos J., Ercan D., Rogers A., Roncalli M., Takeda M. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Huang W.C., Li P., Guo H., Poh S.B., Brady S.W., Xiong Y., Tseng L.M., Li S.H., Ding Z. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011;17:461–469. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.