

Introduction
Pulmonary arterial hypertension (PAH) is defined foremost by a distinct pulmonary vascular pathophenotype that occurs as a result of dysregulated vascular cell proliferation, intimal and medial hypertrophy, inflammation, and fibrosis. This plexogenic arteriopathy with subtotal luminal obliteration increases pulmonary vascular resistance and imposes a hemodynamic stress on the right ventricle (RV). The chronically increased afterload leads to RV hypertrophy and failure that contributes to premature death. Owing to the functional interrelatedness of the pulmonary vasculature and the RV, these two compartments are increasingly considered as a collective unit and assessed by examining RV-pulmonary arterial coupling in studies that focus on the pathophysiology of PAH.1 Ideal RV-pulmonary arterial coupling is present when contractility of the RV is sufficient to match the afterload imposed by the pulmonary artery, which is determined by its distensibility or compliance, and is recognized by minimal pressure fluctuation during systole.2 Optimal RV-pulmonary arterial coupling may be defined further based on the ratio of ventricular to arterial elastance, which typically achieves a ratio of 1.5–2.0 and reflects a balance between RV work and oxygen consumption.3 RV-pulmonary arterial uncoupling, therefore, occurs when there is a mismatch between RV contractility and pulmonary arterial compliance. Based on this premise, it is now acknowledged that factors that decrease normal pulmonary vascular compliance to increase resistance adversely affect RV-pulmonary arterial coupling kinetics. This results in a decline in ventricular efficiency and an increased probability of developing right-sided heart failure.4 Similarly, a decrease in RV contractility owing to RV cardiomyocyte dysfunction or impaired RV myocardial performance decreases RV tolerance to increased afterload and lowers the threshold at which pulmonary vascular disease may become clinically evident.5
While PAH is a disease of pulmonary vascular origin with subsequent effects on the RV, the pathobiology and PAH disease severity may be subject to variation by circulating neurohormonal mediators (e.g., sympathetic nervous system and renin-angiotensin-aldosterone system) that act as disease modifiers. Activation of the sympathetic nervous system and the renin-angiotensin-aldosterone system have been demonstrated in PAH and initially suggested to occur as a consequence of right heart failure and end-stage disease. Recent evidence, however, has led to a paradigm shift in our understanding of the role of the sympathetic nervous system and the renin-angiotensin-aldosterone system in PAH and implicated these factors as candidate pathogenic contributors to the pulmonary vascular remodeling process and RV dysfunction.6, 7 Moreover, the cellular and molecular mechanisms by which these effectors mediate cardiopulmonary structural and functional changes in PAH cannot always be extrapolated from what is known about their actions in the systemic vasculature and left ventricle (LV). Differences that exist between systemic and pulmonary vascular cells as well as RV and LV cardiomyocytes underlie the response of the RV-pulmonary circuit to neurohormonal activation in the development of PAH.8, 9 This is consistent with the prevailing observation that pulmonary vascular and RV remodeling occurs in PAH without evidence of substantial systemic vascular or LV dysfunction. This review will focus on the roles of the sympathetic nervous system and the renin-angiotensin-aldosterone system in PAH, with an emphasis on the cellular and molecular mechanisms important for the pathobiology of the disease.
The sympathetic nervous system in the cardiopulmonary circuit
The cardiopulmonary circuit is a principle target of the sympathetic nervous system. Preganglionic neurons arise from the spinal cord (T1-L2), synapse with postganglionic neurons that innervate the lung, pulmonary arterioles and pulmonary veins or innervate the adrenal gland directly.10,11 Activation of sympathetic nerves leads to local or adrenal release of catecholamines to initiate pulmonary vasoconstriction via stimulation of pulmonary vascular adrenergic receptors (AR). For example, α1-AR, which are stimulated by (nor)epinephrine to induce blood vessel constriction, are expressed in pulmonary vascular tissue (24.0 ± 5.2 fmol/mg protein) at density levels akin to resistance blood vessels (23.0 ± 5.9 fmol/mg protein), and at levels greater than aorta (9.8 ± 1.8 fmol/mg protein) or epicardial coronary arteries (2.1 ± 0.7 fmol/mg protein).12 Human pulmonary vessels also express β1- and β2-AR in a 1:3 distribution in the endothelium that can oppose the effects of α1-receptors and promote vasodilation to regulate vascular tone.11
The sympathetic nervous system also regulates RV function by reducing venous capacitance to affect preload as well as increasing heart rate and contractility. The sympathetic nerve fibers are located subepicardially and there is a gradient of innervation from the base to the apex of the ventricle, suggesting that RV hypertrophic remodeling may lead to changes in relative nerve density and responsiveness to adrenergic stimulation.13 The RV expresses β1- and β2-AR at a ratio of 70:30 as well as β3- receptors, which oppose the positive inotropic effects of the β1- and β2-receptors.14, 15 The RV also expresses dopamine receptors and α1A- and α1B-receptors, albeit at lower levels than β-receptors.16 Under physiologic conditions, they are not believed to play a major role in regulating cardiac function.17
Sympathetic nervous system activation in PAH
There are several lines of evidence to indicate that the systemic sympathetic nervous system is activated in PAH patients, although it is important to note that these findings do not necessarily indicate organ-specific sympathetic nervous system activity. In one study involving a cohort of 60 PAH [Group 1] patients, plasma venous norepinephrine levels correlated inversely with cardiac output (r= −0.29, p<0.05) and were ~3-fold higher in patients with end-stage heart failure compared to patients with minimally impaired functional capacity.18 These findings were supported by another small study that included 15 PAH patients who were found to have increased plasma levels and arterial-venous gradient of norepinephrine, similar to what was observed in patients with LV failure.19 Despite these observations, elevated levels of plasma catecholamines have not been reported consistently in PAH patients and there was concern that the absolute catecholamine concentrations measured in these studies did not achieve pathophysiological levels.18, 20, 21 Other data in support of sympathetic activation in PAH are provided by a study of 17 patients with PAH that assessed postganglionic muscle sympathetic nerve activity, which measures muscle-directed sympathetic nerve traffic and is a validated predictor of LV heart failure severity.21 Muscle sympathetic nerve activity was increased significantly in PAH patients compared to controls (76 ± 4 vs. 57 ± 6 bursts/min, p<0.01) and correlated positively with worsening New York Heart Association functional class (r=0.52, p=0.046) (Figure 1).21, 22 There is also evidence of decreased heart rate variability in PAH patients with several studies confirming that the spectral power of heart rate variability was reduced in all frequency domain indices (i.e., high-frequency, low-frequency, and very low-frequency bands). These findings correlated with an increase in muscle sympathetic nerve burst frequency, longer QTc intervals on the electrocardiogram, and a reduction in peak oxygen uptake during cardiopulmonary exercise testing.3, 23–25 Other reports of selective downregulation of RV β1-AR and decreased RV uptake of the radiotracer 123I-metaiodobenzylguanidine (MIBG), which is a norepinephrine analog that competes with norepinephrine for uptake, support the concept of sympathetic nervous system activation in PAH, in some cases in advance of RV failure.22, 25, 26 In fact, 123I-MIBG imaging may be beneficial clinically as a metric of RV and lung sympathetic nervous system activation (Figure 1). In small series of patients with pulmonary hypertension, the myocardial uptake ratio of 123I-MIBG (interventricular septum-to-LV) was shown to correlate negatively with mean pulmonary artery pressure.22 The heart-to-mediastinum 123I-MIBG activity ratio was found to associate with survival; patients with a ratio of ≥ 2.0 had better cumulative survival as compared to patients with a ratio of < 2.0.27 The lung-to-heart 123I-MIBG activity ratio has also been examined as an indicator of pulmonary sympathetic nervous system activity. In patients with dilated cardiomyopathy, a delay in activity was found to correlate significantly with pulmonary vascular resistance, duration of disease, and heart failure episodes, suggesting that this measure may also be applicable to patients with PAH.28
Figure 1.

Sympathetic nervous system activation is increased in pulmonary arterial hypertension (PAH). (A) Muscle sympathetic nerve activity neurograms demonstrate increased sympathetic nerve traffic in skeletal muscle of PAH patients compared to controls. (B) This correlates with more severe heart failure symptoms as assessed by the New York Heart Association (NYHA) functional class. (C) Cardiac imaging with 123I- metaiodobenzylguanidine (MIBG), a norepinephrine analog that competes with norepinephrine for uptake, showing a reduction in 123I-MIBG uptake (i.e., higher norepinephrine uptake) in the RV of a patient with severe pulmonary hypertension. HR, heart rate; BP, blood pressure; s, seconds, PH, pulmonary hypertension. Reproduced with permission from references 21, 22.
Adrenergic signaling in the pulmonary vasculature in PAH
The sympathetic nervous system maintains low levels of basal tone in normal pulmonary vessels via the actions of both α- and β-AR that are expressed by the pulmonary endothelium and pulmonary vascular smooth muscle cells. Typically, β-AR signaling is responsible for ambient pulmonary vascular tone as antagonism of β-AR leads to vasoconstriction while antagonism of α-receptors results in vasodilation.29 In human pulmonary arteries, the G protein-coupled β1- and β2- receptors mediate vasodilation via Gαs-stimulated activation of adenylyl cyclase/cAMP/protein kinase A. Protein kinase A, in turn, phosphorylates I-1 protein and phospholamban to decrease intracellular Ca2+ levels through sarcoplasmic reticulum Ca2+ uptake. At present, the physiological role of β3- receptors in pulmonary vessels has not been well characterized 29, 30 In human pulmonary artery endothelium, stimulation of β1- and β2-receptors also modulates pulmonary vascular tone, in part, by inducing synthesis of the vasodilator nitric oxide (NO•), which also possesses antimitogenic properties.11
In PAH, pulmonary vascular adrenergic signaling results in AR-mediated vasoconstriction, dysregulated proliferation, and fibrosis through signaling mechanisms that involve cross-talk with the renin-angiotensin-aldosterone system (discussed below), loss of NO•, and increases in intracellular Ca2+ (Figure 2). Elevated levels of reactive oxygen and nitrogen species (e.g., superoxide, hydrogen peroxide, and peroxynitrite) disrupt Gi-subunit function to deactivate the β-receptors as well as interrupt normal β-receptor-NO• signaling by scavenging NO• and uncoupling eNOS.31, 32 The relevance of changes in α1-AR signaling for pulmonary vascular function is less well established. Stimulation of α1-receptors in pulmonary artery smooth muscle cells and adventitial fibroblasts increases cellular DNA and protein content to facilitate blood vessel hypertrophy 33 Stimulation of α1- and α2-receptors expressed by pulmonary arterial smooth muscle cells also leads to vasoconstriction and proliferation through activation of Gαq to activate phospholipase C and increase intracellular Ca2+ levels or by activation of Gαi to inhibit adenylyl cyclase.34 When there is prolonged AR activation, a feedback loop involving protein kinase A-induced phosphorylation of the β-receptors exists; this leads to β-receptor desensitization.30 The factors underpinning the transition from physiological to pathogenic α1-AR signaling is largely unknown, but may involve cross-talk between α- and β-receptors.35
Figure 2.

Adrenergic receptor signaling and cross-talk with the renin-angiotensin aldosterone system. Activation of β-adrenergic receptor signaling increases cAMP levels and activates protein kinase A (PKA) to promote vasodilation and an increase in cardiac contractility. These effects are opposed by α2-AR signaling. PKA also activates the MAP kinase signaling pathway, which is downstream from the angiotensin type 1 receptor (AT1R). AT1R also activates protein tyrosine kinases (PTK), which have been implicated in pulmonary hypertension, and similar to α1-AR, activates phospholipase C (PLC). Both AT1R and mineralocorticoid receptor (MR) activate NADPH oxidase to increase reactive oxygen species formation (ROS), which decrease bioavailable nitric oxide (NO•).
Adrenergic signaling in PAH: focus on the RV
Maladaptive RV remodeling, particularly RV hypertrophy, is an early indicator of impending RV failure in patients with PAH, and is therefore considered a key risk factor for PAH-associated morbidity and mortality.5 Although targeting AR stimulation by catecholamines is a longstanding therapeutic cornerstone in LV heart failure, comparatively less is known regarding the differential effects of this axis on RV performance, particularly in the setting of increased RV afterload as occurs in PAH.
The RV expresses subtypes of both α- and β-AR. Under physiological conditions, β1- and β2- AR increase cardiac contractility, frequency, and rate of relaxation through the activation of Gs protein-coupled receptors or both Gi and Gs protein-coupled receptors, respectively. This, in turn, activates adenylyl cyclase/cAMP/protein kinase A signaling to regulate calcium handling via phosphorylation of the L-type calcium channels and ryanodine receptors, phospholamban, troponin I as well as phospholemman, which mediates activity of the Na+/K+-ATPase pump. Protein kinase A also phosphorylates β-AR’s leading to desensitization and uncoupling of the receptors. The RV expresses β3- ARs that initiate a negative inotropic effect when stimulated and α1- ARs that activate phospholipase C, which also regulates intracellular Ca2+ levels (reviewed in 30).
Studies have shown that in PAH the hypertrophied RV undergoes molecular remodeling with downregulation of α1- and β1-receptor mRNA and protein levels as well as receptor desensitization in RV cardiomyocytes isolated from Sugen-5416/hypoxia-, monocrotaline-, or pulmonary artery banding rodent models of PAH and in patients with idiopathic PAH (iPAH) (Figure 3).16 Changes in receptor expression levels were more severe in models associated with decreased cardiac output, which confirms earlier observations from patients with PAH and cor pulmonale.26 Interestingly, diminished D1 dopamine receptor expression in RV cardiomyocytes implicates decreased cAMP formation (or increased degradation) in RV hypertrophy, which may underlie the abnormal cardiopulmonary hemodynamic response patterns to dopamine in experimental PAH in vivo and in patients with right-sided heart failure.16, 36 In support of this, in the RV, increased activity of G protein-coupled receptor kinase-2, which regulates G protein-coupled receptor function, and AR-dependent cAMP formation in RV cardiomyocytes, results in desensitization of β1-AR in the RV in experimental PAH while inhibition of G protein-coupled receptor kinase-2 restores RV contractility.16
Figure 3.
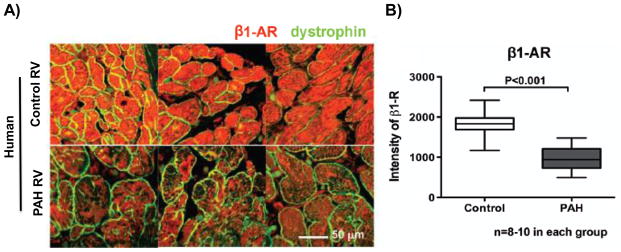
β1-adrenergic receptor expression is down regulated in the right ventricle (RV) of patients with pulmonary arterial hypertension (PAH). (A) A representative cross-sectional image of RV cardiomyocytes and (B) mean values from human tissue microarrays showing down regulation of the β1-adrenergic receptor in hypertrophied RVs from patients with PAH compared to age- and sex-matched controls. Reproduced with permission from reference 16.
These and other similar observations suggest that strategies to modulate abnormal adrenergic signaling patterns in PAH may, in turn, offset RV hypertrophy.37 Several investigators have explored this and shown that pharmacological inhibition of α1-, β1-, and β2-receptor signaling improves RV structure and performance in experimental PAH. In Sugen-5416/hypoxia-PAH rats treated with carvedilol, although no significant change was noted in the pulmonary arterioles, drug treatment promoted reverse RV remodeling, improved tricuspid plane annular excursion, and increased cardiac output. This effect was associated with decreased RV thickness and intramyocardial fibrosis as well as alterations to the molecular signature of the hypertrophied RV vis-á-vis cardiomyocyte fetal gene activation, including decreased α-myosin heavy chain and increased fetal β-myosin heavy chain gene expression levels.38 Similar findings were observed for the selective β1-receptor antagonist bisoprolol, which delayed the time to RV failure by decreasing RV hypertrophy, fibrosis, and inflammation to improve RV contractility, partially restore RV-arterial coupling, and increase cardiac output in monocrotaline-PAH. At a molecular level, there was evidence of increased phosphorylation of the β-receptor targets myosin binding protein C and troponin I.8 Other studies performed with the non-selective β-receptor antagonist propranolol also demonstrated improved RV function under alternative experimental conditions such as high altitude and hypobaric hypoxia.39
Interventions that target the sympathetic nervous system in PAH
Pharmacologic
Although the balance of available data points to sympathetic nervous system activation and dysregulated AR signaling in the pulmonary vasculature and RV in PAH, translating the beneficial findings from preclinical studies of β-blockers in PAH to patients has evoked controversy. This has occurred as a consensus is pending regarding the risk-benefit balance of using AR-modulating pharmacotherapeutics in patients with PAH. While attempts have been made to recapitulate the (substantial) benefits of β-blockers observed in left heart disease in PAH patients with RV dysfunction, this strategy has been evaluated empirically only in small observational studies, limited largely to patients with selected forms of pulmonary vascular disease (e.g., portopulmonary hypertension) and in whom treatment was initiated with early generation drugs (e.g., propranolol or atenolol) at high doses.40 In a frequently cited study of 10 patients with moderate to severe pulmonary hypertension, ~2 months after withdrawal of β-blockers, 90% of patients demonstrated significant improvement in their 6-minute walk distance, a 28% increase in cardiac output, and a 19% decrease in pulmonary vascular resistance.40 Another observational study that followed a cohort of PAH patients treated with β-blockers for up to 20 months reported no difference between treated and untreated individuals with respect to exercise tolerance or RV remodeling.41 This led to the suggestion that these agents may be tolerated well in PAH patients; however, concerns were raised pertaining to the atypical PAH population studied, noting that there was a high prevalence of systemic hypertension, atrial fibrillation, and coronary artery disease as well as the absence of information about the number of patients that were intolerant of the drug.7, 41 Some experts in the PAH field maintain reservations regarding the safety profile of β-blockers, citing decreased exercise tolerance due to chronotropic and RV inotropic insufficiency in patients with (exercise-induced) pulmonary hypertension.42 Furthermore, recently published expert consensus guidelines referenced insufficient clinical data to outline formal recommendations for the use of β-blockers in practice. 43 To bridge this knowledge gap, several randomized, prospective clinical trials have been announced assessing the effect of β-blockers, including carvedilol, on outcome in PAH (NCT00964678; NCT01723371; NCT01586156 at clinicaltrials.gov) (Table 1). Nonetheless, further efforts are still required to clarify the optimal timing, duration, and dosing of β-blocker therapy in patients with PAH.
Table 1.
Registered clinical trials investigating therapies that target neurohormonal activation in pulmonary arterial hypertension (PAH).
| Study Name | Identifier/Duration | Treatment | Inclusion Criteria | Endpoint
fc |
|
|---|---|---|---|---|---|
| Primary | Secondary | ||||
| Beta-blockers for the treatment of PAH in children |
NCT01723371 6 mo End date: 9/2014 |
Carvedilol | Age: ≥8 and ≤17.5 yr mPAP >25 mmHg PCWP <15 mmHg PVR >3 Wood units Clinically stable (3 mo) |
Adverse Event Incidence | Δ 6MWD, VO2 Δ TAPSE Δ RVEF |
| Beta-blockers in PAH |
NCT01246037 6 mo End date: 4/2014 |
Bisoprolol | Age: ≥18 yr Stable iPAH WHO Class II/III |
ΔRVEF by CMR Safety measures |
RV diastolic function Sympathetic activity levels |
| PAH Treatment with Carvedilol for Heart Failure (PAHTCH) |
NCT01586156 6 months End date: 7/2018 |
Carvedilol | Age: 18–65 yr PAH WHO Class I–III |
ΔHIF/NO/AR Recovery ΔRV function |
|
| Pilot Study of the Safety and Efficacy of Carvedilol in PAH |
NCT00964678 6 mo End date: 5/2014 |
Carvedilol | Age: ≥18 yr WHO Group 1 PH NHYA Class II/III mPAP >25 mm Hg 6MWD >100m |
ΔRVEF by CMR | ΔRVESV Δ6MWD ΔTAPSE |
| Spironolactone for PAH |
NCT01712620 6 mo End date: 11/2015 |
Spironolactone | Age: ≥18 yr WHO Group 1 PH Stable therapy (4 wk) mPAP >25 mmHg PCWP<15 mmHg PVR >3.0 Wood units NYHA Class I–III |
Δ6MWD Clinical worsening |
ΔVO2 ΔRV Function Inflammation biomarkers Drug Safety |
| Effects of Spironolactone on Collagen Metabolism in Patients with PAH |
NCT01468571 16 wk End date: 12/2015 |
Spironolactone | Age: ≥18 yr Body weight >40 kg WHO Group 1 PH Stable therapy (4 wk) |
Δ Fibrosis markers | Adverse events Δ6MWD Δ Functional class Clinical worsening |
| Modulating Effects of Lisinopril on Sildenafil Activity in PAH (MELISSA) |
NCT01181284 32 wk End date: 7/2011 |
Lisinopril added to sildenafil | Age: 18–75 yr WHO Group I PAH PVR >3 Wood units PCWP ≤16 6MWD 150–575 m PDE-Vi (3 mo) |
ACE-I tolerability | ΔN-BNP levels Δ Gas exchange measures Δ6MWD |
| Hormonal, Metabolic, and Signaling Interactions in PAH |
NCT01884051 5 yr End date: 9/2017 |
ACE-2 Metformin | Sex hormone metabolite levels Δ6MWD Glucose metabolism |
Hemodynamics PET scan results |
|
NCT, national clinical trial (clinicaltrials.gov); mPAP, mean pulmonary artery pressure; PVR, pulmonary vascular resistance; PCWP, pulmonary capillary wedge pressure; 6MWD, 6-minute walk distance; VO2, peak volume of oxygen consumption; TAPSE, tricuspid annual plane excursion; RVEF, right ventricular ejection fraction; iPAH, idiopathic pulmonary arterial hypertension; WHO, World Health Organization; CMR, cardiac magnetic resonance; PH, pulmonary hypertension; NYHA, New York Heart Association; PDE-Vi, phosphodiesterase-type 5 inhibitor; ACE-I, angiotensin converting enzyme inhibitor; N-BNP, N-terminal brain natriuretic peptide; PET, positron emission tomography; mo, month; wk, week; yr, year.
Non-pharmacologic
Another therapeutic strategy is to target key signaling molecules that are play a direct or indirect role in adrenergic signaling, are downregulated in PAH, and serve a compensatory function to overcome the deleterious consequences of dysregulated catecholamine-AR signaling. Among candidate molecules, the sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) is one that regulates intracellular Ca2+ levels, and, thereby, pulmonary smooth muscle cell proliferation and vasoconstriction, and is known to be downregulated in remodeled pulmonary blood vessels isolated from patients with PAH as compared to patients with other forms of lung disease. 44 As activation of α-adrenergic signaling increases intracellular Ca2+ levels in PAH, restoring SERCA2a expression and activity is one method to decrease intracellular Ca2+ levels and modify pulmonary vascular remodeling (Figure 4). In monocrotaline-PAH rats with documented downregulation of SERCA2a in the pulmonary arterioles and RV, intratracheal aerosolized gene transfer of aerosolized adeno-associated virus serotype 1 (AAV1) carrying SERCA2a resulted in efficient transduction of the pulmonary vasculature leading to a reduction in pulmonary artery pressures, vascular remodeling, and RV hypertrophy.45 Recent results from the phase I/II Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) trial demonstrated safety and possible benefit of intracoronary SERCA2a gene transfer in symptomatic patients with LV dysfunction, suggesting that a combination intratracheal and intracoronary delivery strategy may also benefit patients with PAH and concomitant severe RV failure.46
Figure 4.

Adrenergic receptor signaling, intracellular calcium, and pulmonary artery smooth muscle cell proliferation. In pulmonary artery smooth muscle cells (PASMC), β-adrenergic receptor (AR) activates adenylyl cyclase (AC) to increase cAMP and, thereby, activate protein kinase A (PKA). PKA phosphorylates phospholamban (PLN) and increase activity of the sarcoplasmic reticulum Ca2+-ATPase (SERCA2), which decreases intracellular Ca2+ levels. In pulmonary hypertension, SERCA2 expression is downregulated. This results in high intracellular Ca2+ levels through the actions of the ryanodine receptors (RYR) and α-AR signaling. The elevated levels of intracellular Ca2+ stimulate proliferation through protein phosphatase 2B (PP2B), translocation of nuclear factor of activated T cells (NFAT) to the nucleus to promote transcription of cyclin D1. PLC, phospholipase C, inositol triphosphate.
As pulmonary nerve stimulation is associated with frequency-dependent increases in pulmonary artery perfusion pressure, investigators have also attempted sympathetic ganglion block and pulmonary artery denervation as therapeutic interventions in PAH.11 In a preclinical study of monocrotaline-PAH rats, daily injections of ropivicaine into the left superior cervical ganglion for 14 days decreased pulmonary arteriole remodeling, RV systolic filling pressures, and RV hypertrophy. This was also associated with a decrease in oxidant stress and improved indices of bioavailable NO•, including plasma nitrite and cGMP in lung tissue.47 Preclinical studies of direct pulmonary artery denervation suggested that catheter ablation <2 mm proximal to the main artery bifurcation improved pulmonary vascular and RV filling pressures; however, histology demonstrating adequate nerve damage was not provided and the short follow-up precluded an assessment of the long term durability of the procedure.48, 49 Results from a pilot first-in-man pulmonary artery denervation trial that enrolled 13 patients and 8 controls with idiopathic PAH nonresponsive to optimal medial therapy have been reported. In these patients, pulmonary artery denervation at 3 sites (ostium of the right and left pulmonary arteries and bifurcation of the main pulmonary artery) was performed. In 12 of 13 patients procedural success, defined as a decrease in the pulmonary artery pressure of ≥10 mmHg and no complication, was achieved. At 3 month follow-up, an increase from baseline in the 6-minute walk distance (324 ± 21 vs. 491 ± 38 m, p<0.01), Tei index (a measure of RV systolic function) (0.7 ± 0.04 vs. 0.5 ± 0.04, p<0.01), and improvements in the World Health Organization class and Borg score were seen in patients that underwent the denervation procedure.50 Although these results are intriguing, the nonrandomized study design and enrollment of patients with normal right atrial pressures elicited concern leading to a call for more rigorous testing in a more representative PAH patient population with longer-term follow-up. This latter point is indeed necessary owing to the fact that sympathetic reinnervation has been shown to occur in a previously denervated lung in experimental models, possibly as a result of increased nerve growth factor secretion by proliferating pulmonary arterial smooth muscle cells.51–53 Furthermore, the negative findings from the randomized, sham-controlled, Renal Denervation in Patients with Uncontrolled Hypertension (SYMPICITY HTN-3) trial of renal denervation have stalled catheter development suggesting that there will be a significant delay in obtaining additional data using this technique.54
The renin-angiotensin-aldosterone system in the pathophysiology of PAH
There has also been a resurgence of interest in the renin-angiotensin system as a modifiable contributor to PAH.55, 56 The integral role of the lungs in regulating angiotensin II levels has long been recognized as the pulmonary endothelium are a rich source of angiotensin converting enzyme (ACE), and generation of angiotensin II from angiotensin I takes place predominantly in the pulmonary vascular bed (Figure 5).57 In the systemic circulation, angiotensin II has been linked to proliferation, hypertrophy, and dysfunction of vascular cells. It is, therefore, not surprising that activation of the renin-angiotensin system has also been associated with pulmonary vascular diseases characterized by hyperproliferative vessel remodeling, such as PAH (Figure 2).
Figure 5.

Angiotensin II and related metabolites. Angiotensin II is generated from angiotensin I via the actions of angiotensin converting enzyme (ACE). Angiotensin I and II may also be metabolized to the vasoactive heptapeptides angiotensin-(1-9) and angiotensin-(1-7) through the actions of angiotensin converting enzyme 2 (ACE2) and neutral endopeptidsase (NEP) and prolylendopeptidase (PEP). Angiotensin II may stimulate angiotensin type-1 (AT-1) receptors to increase pulmonary artery smooth muscle cell (PASMC) proliferation, vasoconstriction, and disrupt right ventricular (RV)-arterial coupling. By contrast, stimulation of angiotensin II receptor type 2 (AT-2) or the Mas receptor by angiotensin II or angiotensin-(1-7) is associated with vasculoprotective effects, including increased nitric oxide generation and a decrease in cell proliferation and vasodilation. PRCP, prolylcarboxypeptidase. Adapted with modifications from reference 55.
Findings from preclinical studies in rodent models of PAH identified renin-angiotensin activation in the cardiopulmonary circuit and suggested that it may play a causal role in pulmonary vascular and RV remodeling processes. Studies performed in a rat hypobaric hypoxia model revealed increased pulmonary vascular ACE expression and activity, upregulation of the angiotensin receptor type 1 (AT1R), and a 3.4-fold increase in RV ACE activity.58 Subsequent analysis of human lung tissue from PAH patients confirmed upregulation of ACE and AT1R in hypertrophic remodeled pulmonary vessels.59, 60 Pulmonary artery endothelial cells isolated from PAH patients were found to have increased ACE activity and angiotensin II production, which, in turn, augmented pulmonary smooth muscle cell proliferation through AT1R signaling.60 Recently, definitive evidence of increased circulating levels of components of the renin-angiotensin system in PAH was provided by a study of 58 patients with symptomatic Group 1 PAH. In this study, patients with progressive PAH, defined as a >10% decrease in 6-minute walk distance over a mean of 39 months of follow-up, had elevated levels of plasma renin activity, angiotensin I, and angiotensin II. Moreover, increased levels of angiotensin II were associated with an increased risk of lung transplantation or death (HR=3.02, 95% CI: 1.40–6.48, p=0.005).60
Activation of the renin-angiotensin system in the cardiopulmonary system promotes pulmonary vascular and RV remodeling by stimulating cell proliferation, hypertrophy, and migration; vasoconstriction; and fibrosis. Angiotensin II initiates these cellular processes through AT1R-mediated activation of mitogen-activated protein kinases, receptor tyrosine kinases, and non-receptor tyrosine kinases that have been linked to PAH.56 It has also been proposed that angiotensin II contributes to adverse vascular remodeling by facilitating hypoxia inducible factor-1α (HIF-1α) accumulation under normoxic conditions, activating the cyclin-dependent kinase p27(Kip1) to promote cell hypertrophy, and increasing oxidant stress.61, 62 In fact, angiotensin II is known to stimulate NADPH oxidase to generate reactive oxygen species, which leads to vasoconstriction and vessel inflammation that contribute to disease progression.56
Angiotensin II has also been implicated in pulmonary vascular and RV fibrosis. Infusion of angiotensin II in rats increases pulmonary artery perivascular collagen deposition and fibrosis that occurs mainly in areas of increased ACE and AT1R activity.63 Angiotensin II has also been related to RV fibrosis. Studies performed in a rabbit pulmonary artery banding model attributed the ~3-fold increase in RV collagen volume to angiotensin II. In the RV, fibrosis was associated with increased expression of the profibrotic mediators transforming growth factor-β1, connective tissue growth factor, and endothelin-1.64
Seminal observations reported over 2 decades ago determined that ACE inhibitors attenuated hypoxic pulmonary vasoconstriction and RV dysfunction by decreasing levels of angiotensin II directly rather than altering levels of angiotensin-II-associated vasoactive factors, such as bradykinin metabolism.65 Theoretically, use of these agents should, therefore, limit vascular remodeling and improve RV-pulmonary arterial coupling in PAH; however, the use of ACE inhibitors and angiotensin receptor blockers in the treatment of PAH remains controversial. Preclinical studies have demonstrated that ACE inhibition decreased pulmonary artery smooth muscle cell proliferation and concentric thickening of pulmonary arterioles in experimental models of PAH. 66 Similarly, the selective AT1R inhibitor losartan was shown to delay progression of pulmonary hypertension and improve RV-pulmonary arterial hemodynamics in PAH rats in vivo (Figure 6).60 Nonetheless, findings from small clinical trials of ACE inhibitors performed in the era prior to the availability of PAH-specific therapy failed to demonstrate a consistent effect on cardiopulmonary hemodynamics and reported systemic hypotension as a limiting side-effect in treated patients.67–69 Of note, the one study that did find an improvement in pulmonary artery pressures and RV function linked this to a decrease in plasma angiotensin levels.69 Overall, the observed hypotension associated with ACE inhibitor use has led to the speculation that selective AT1R antagonists may be more appropriate in this patient population based on the comparatively lower risk of systemic hypotension.7
Figure 6.

Angiotensin II type-1 receptor (AT1R) inhibition improves hemodynamics in pulmonary arterial hypertension (PAH). (A) Expression levels of AT1R are increased in pulmonary arterioles harvested from patients with idiopathic PAH compared to controls. (B) Inhibition of AT1R with losartan (20 mg/kg) improves RV systolic pressure (left) and pulmonary vascular resistance (right) in monocrotaline-PAH. Reprinted from Ref. 60 with permission of the American Thoracic Society. Copyright ©2014 American Thoracic Society
Additional angiotensin peptides and PAH pathobiology
The deleterious effects of angiotensin II in the cardiopulmonary system are counterbalanced by its catabolism via angiotensin converting enzyme-2 (ACE2)-mediated cleavage of angiotensin I and angiotensin II to yield angiotensin-(1-7) (Ang-(1-7)), angiotensin-(1-9), and angiotensin-(1-5).70, 71 Angiotensin converting enzyme-2 is a homologue of ACE that is insensitive to ACE inhibitors, indicating that the adverse effects observed with in patients treated with ACE inhibitors could not be attributed to inhibition of ACE2 and Ang-(1-7).72 These vasculo- and cardio-protective peptides activate the Mas and angiotensin type 2 receptors to limit pulmonary vascular and RV remodeling in PAH. Although decreased ACE2 levels and ACE2 autoantibodies have been detected in serum from patients with PAH, increased ACE2 activity was identified in the failing RV from PAH patients at the time of heart-lung transplantation.73–75 These seemingly contradictory findings are reconciled by the observation that ACE2 activity and Ang-(1-7) increase, likely as a compensatory mechanism, when local angiotensin levels are elevated. Thus, factors that downregulate ACE2 or failure to increase ACE2 activity sufficiently are permissive for angiotensin II-induced cardiopulmonary injury.
In the RV-pulmonary vascular unit, ACE2 is expressed by the pulmonary endothelium, smooth muscle cells, and the RV myocardium.76 In rodent models of experimental PAH, ACE2 expression is elevated with disease manifestation but is inadequate to prohibit pulmonary vascular and RV hypertrophy and fibrosis. Strategies to increase ACE2 expression or activity as a therapeutic intervention in PAH models of disease using recombinant human ACE2; pharmacological ACE2 activators; the antitrypanosomal agent diminazene aceturate, which increases ACE2 activity as an off-target effect; and gene transfer of ACE2 or Ang-(1-7) have all been shown to lower pulmonary pressures, improve RV function, and limit or reverse pulmonary vascular remodeling, RV hypertrophy, and fibrosis. 6, 70
Mechanistically, ACE2/Ang-(1-7)/Mas receptor activation inhibits cell proliferation, hypertrophy, and pro-fibrotic signaling pathways that contribute to cardiopulmonary remodeling in PAH.77 ACE2 inhibits ERK 1/2 and JAK2-STAT3 cell survival signaling to prevent pulmonary artery smooth muscle cell proliferation and migration.78 ACE2/Ang-(1-7) has also been shown to influence vascular remodeling by decreasing cellular oxidant stress through downregulation of NADPH oxidase to limit hydrogen peroxide levels. This, in turn, decreases pulmonary smooth muscle cell proliferation and hypertrophy and improves pulmonary NO• synthesis and NO•-dependent vascular reactivity.77 The antifibrotic effects of ACE2/Ang-(1-7) are related to a reduction in oxidant stress, transforming growth factor-β levels and collagen production.77 Within the RV, ACE2/Ang-(1-7) limits functional and electrical remodeling, in part, by maintaining NO• levels, enhancing cardiomyocyte calcium handling, increasing expression of SERCA2a, and normalizing expression of the cell-cell communication gap protein connexin 37, to improve myocardial contractility.70 Thus, targeting ACE2/Ang-(1-7)/Mas signaling in the pulmonary vasculature and RV likely holds therapeutic promise in PAH despite the limited data currently available with respect to the clinical relevance of this signaling pathway in PAH.
Aldosterone is a PAH disease modifier
Elevated levels of the mineralocorticoid hormone aldosterone are present systemically and within the pulmonary vascular compartment in PAH. Aldosterone levels are increased in plasma and lung tissue isolated from monocrotaline- and Sugen-5416/hypoxia-rat models of PAH as well as in patients with PAH where pulmonary arterial aldosterone levels are increased by 4.9-fold compared with controls.9, 79 In PAH patients, aldosterone levels correlated inversely with cardiac output and positively with key measures of pulmonary vascular remodeling, including pulmonary vascular resistance and the transpulmonary pressure gradient.79 Adrenal stimulation by upregulation of the renin-angiotensin axis is likely to be the chief source of aldosterone production in PAH; however, recent evidence indicates that the pulmonary vascular endothelium is an extra-adrenal source of aldosterone production.9, 80 In fact, endocrine functionality of the lung was suggested originally five decades previously by several groups based on observations demonstrating that isolated synthesis of angiotensin I occurs in lung tissue.81 In support of this, a transpulmonary increase in plasma levels of the aldosterone secretagogues angiotensin II and the endothelin-1 precursor big endothelin-1 has been observed in selected subtypes of PAH, although whether these trends are generalizable to the larger pulmonary vascular disease population remains unknown.82–84
Nevertheless, it is noteworthy that key proteins required for extra-adrenal de novo aldosterone synthesis, including steroidogenic acute regulatory protein (StAR), 11-β-hydroxylase (CYP11B1), and aldosterone synthase (CYP11B2) are constitutively expressed or inducible in cardiomyocytes, pulmonary artery endothelial cells, and pulmonary vascular smooth muscle cells by factors linked to the pathogenesis of PAH.85, 86 For example, treatment of human pulmonary artery endothelial cells with endothelin-1 at levels similar to those observed in PAH patients induces association of the steroidogenic transcription factors steroidogenic factor-1 (SF-1) and peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α to the CYP11B2 promoter, which increases pulmonary endothelial CYP11B2 expression and aldosterone levels by 2-fold in vitro.9 Similarly, hypoxia increases pulmonary artery endothelial cell StAR expression and de novo aldosterone synthesis measured by mass spectrometry by 2.1-fold.80 Pharmacological antagonism of the mineralocorticoid receptor with spironolactone attenuates angiotensin-II-mediated pulmonary artery smooth muscle cell hypertrophy, providing some evidence in support of functionally active aldosterone biosynthesized in pulmonary vascular tissue.85 In fact, the bioactivity of pulmonary artery endothelial cell de novo aldosterone synthesis was confirmed by demonstrating upregulation of the profibrotic mediator connective tissue growth factor in pulmonary artery smooth muscle cells exposed to conditioned medium from hypoxia-treated pulmonary artery endothelial cells.80 Thus, localized de novo aldosterone synthesis is likely to function as a disease modifier in PAH, similar to adrenal-derived aldosterone. Moreover, it is plausible that this mechanism may serve to increase aldosterone levels locally and the levels achieved or duration of exposure may be important factors that determine the contribution of extra-adrenal aldosterone to vascular and ventricular dysfunction in PAH, although this remains to be proven definitively.
Vasculopathy and RV dysfunction associated with hyperaldosteronism
Elevated levels of aldosterone promote a structural and functional vasculopathy in systemic blood vessels that overlaps, in part, with the pulmonary vascular pathophenotype of PAH.87 In patients with primary hyperaldosteronism (i.e., unstimulated adrenal aldosterone synthesis), for example, vascular smooth muscle cell proliferation, increased arterial stiffness, and vascular fibrosis define the remodeling pattern of resistance blood vessels.88 Within the pulmonary vasculature, pathophysiologically relevant levels of aldosterone have been shown to increase human pulmonary artery endothelial cell NADPH oxidase type-4 (NOX4)-derived hydrogen peroxide generation. This, in turn, oxidatively modifies the endothelin-B (ETB) receptor at Cys405 from the reduced (R-SH) to the sulfenic acid (R-SOH) form, interrupts ETB signal transduction and, thus, ETB-dependent NO• synthesis is impaired. The consequence of both Cys405 oxidation and decreased bioavailable NO• is concentric hypertrophy and pulmonary vascular fibrosis in monocrotaline- and Sugen-5416/hypoxia-PAH (Figure 7). 9 Aldosterone is also a mitogenic trigger in pulmonary vascular cells. Treatment of pulmonary artery smooth muscle cells harvested from idiopathic PAH patients with aldosterone stimulates cellular proliferation and mitosis, in part, through upregulation of mitogen activated protein kinase signaling.89 This effect was augmented by co-incubation with bone morphogenetic protein-2 and -7, thus identifying aldosterone as a contributor to the effects of PAH-associated proteins involved in pulmonary vascular remodeling.89
Figure 7.

A proposed mechanism by which hyperaldosteronism decreases pulmonary endothelial eNOS activation and nitric oxide (NO•) generation in PAH. Hyperaldosteronism (ALDO) in pulmonary arterial hypertension (PAH) may occur via i) endothelin-1 (ET-1)-mediated activation of PPARγ coactivator-1α (PGC-1α)/steroidogenesis factor-1 (SF) to increase CYP11B2 (aldosterone synthase) gene transcription in human pulmonary artery endothelial cells (HPAECs); ii) hypoxia-stimulated upregulation of steroidogenic acute regulatory protein (StAR) by increasing binding of c-fos and c-jun to the promoter; and, iii) adrenal ALDO synthesis via ET-1 and/or overactivation of the renin-angiotensin pathway. Stimulation of the mineralocorticoid receptor (MR) in HPAECs by ALDO activates NADPH oxidase type 4 (NOX4) to increase levels of hydrogen peroxide (H2O2), which, in turn, oxidatively modifies redox sensitive, functional cysteinyl thiol(s) in the ETB receptor (Cys405) to impair ETB-dependent activation of eNOS and decrease synthesis of nitric oxide (NO•). eNOS, endothelial nitric oxide synthase; R-SOXH, higher oxidative intermediaries of cysteine. Adapted from ref 9.
While RV remodeling occurs as a consequence of the PAH vasculopathy, evidence indicates that elevated levels of aldosterone contribute to RV hypertrophy and fibrosis. Preclinical studies in the rat monocrotaline model of PAH have shown that hyperaldosteronism is associated with RV cardiomyocyte hypertrophy and antagonism with spironolactone was found to decrease RV cardiomyocyte size without influencing RV total mass.90 Similarly, in the pig pulmonary vein banding model of pulmonary hypertension, hyperaldosteronism was linked to maladaptive RV remodeling with RV cardiomyocyte hypertrophy and dysfunction as demonstrated by downregulation of SERCA2a.91 This is not surprising as in other experimental models of hyperaldosteronism, RV and LV cardiomyocytes exhibit heterogeneity in size. This may be attributable to increased cardiomyocyte free Ca2+ and oxidant stress as well as activation of transforming growth factor-β, tumor necrosis factor-α and insulin signaling, which are all involved in the remodeling process.92
The fibrosis pathophenotype of PAH and aldosterone
Pulmonary vascular and RV fibrosis is a critical component of the cardiopulmonary remodeling pattern in PAH and, when present, is an end-stage finding associated with increased mortality.93 Activation of transforming growth factor-β signaling by aldosterone promotes collagen deposition in pulmonary vascular, cardiovascular, and various non-vascular tissue beds.94 Likewise, transforming growth factor -β is a master regulator of lung and pulmonary vascular fibrosis, and is implicated in the development of pulmonary hypertension in PAH patients. 95 Aldosterone also increases levels of the profibrotic connective tissue growth factor, collagen, and the matrix remodeling proteins matrix metalloproteinase-2 and matrix metalloproteinase -9 in cultured human pulmonary artery endothelial cells.96 Similarly, in other experimental models, aldosterone has been implicated in increased synthesis and deposition of the extracellular matrix proteins collagen I and III, fibronectin, and matrix metalloproteinases-3, -7, 12, -13 as well as the matricellular proteins thrombospondin 1, osteonectin, periostin, and tenascin C.97 In turn, these changes are associated with fibrillar collagen deposition in pulmonary arterioles and frank RV replacement fibrosis in PAH in vivo.9, 80, 90 Aldosterone may also exert these pathogenic effects on cardiopulmonary tissue through alternative fibrotic signaling pathways linked to mineralocorticoid receptor activation, including reactive oxygen species generation, NF-κB signaling, and other pro-inflammatory pathways.98, 99
Clinical relevance of aldosterone inhibition in PAH
Accumulating data from several experimental animal models of PAH indicates that mineralocorticoid receptor antagonism with spironolactone or eplerenone is able to reverse or prevent pulmonary vascular remodeling, improve pulmonary hypertension, and decrease RV size.9, 80, 90 Nevertheless, the role of mineralocorticoid receptor antagonism in the treatment of PAH patients is limited to case reports or small observational studies.100 The first effort to systematically characterize the effect of spironolactone on outcome in PAH was from a retrospective analysis of the Pulmonary Arterial Hypertension, Randomized Double-Blind, Placebo-Controlled, Multicenter, Efficacy Study 1 (ARIES-1) and Study 2 (ARIES-2) trial.101 In this hypothesis-generating study, the clinical relevance of biological data showing that hyperaldosteronism in PAH impairs ETB-dependent vasodilatory signaling was assessed by examining the effects of spironolactone as an added therapy to ambrisentan to inhibit ETA-dependent pulmonary vasoconstriction and remodeling.79 In that study, concurrent spironolactone use (mean dose 31.2 mg/d) was identified in 10 patients randomized to ambrisentan (10 mg/d) (N=67). Compared to ambrisentan alone (N=57), therapy with ambrisentan plus spironolactone improved the 6-minute walk distance (change from baseline at 12 weeks:+38.2 ± 8.1 vs. +74.2 ± 27.4 m, p = 0.11) and was associated with a 90% relative increase in the number of patients improving at least one World Health Organization functional class (p=0.08).101 Forthcoming prospective clinical data on the effect of mineralocorticoid receptor antagonism in PAH is anticipated with the announcement of several clinical trials (NCT01468571; NCT01712620) (Table 1).
Conclusions
Evidence from numerous lines of research implicates involvement of the sympathetic nervous system and the renin-angiotensin-aldosterone system in the pathobiology of pulmonary vascular disease and RV dysfunction in PAH. In particular, mechanistic data are available associating AR stimulation, angiotensin II, and aldosterone to the development of pulmonary vascular and RV histopathophenotypic changes observed in experimental models of PAH. It is recognized that there is cross-talk between the sympathetic nervous system and the renin-angiotensin-aldosterone system with although, in PAH, it remains unclear which of the two systems is activated first. Importantly, the preponderance of data suggests that activation of these systems functions primarily as a PAH disease modifier rather than a causal role in the disease per se. Although preclinical studies have offered a wealth of data to support pharmacological intervention of these signaling pathways, controversial findings from early clinical studies performed in the era prior to PAH-specific therapies and newer selective agents has limited adoption of this strategy in PAH. Patients with PAH have clinical markers indicating activation of the sympathetic nervous system (i.e., elevated circulating catecholamine levels, muscle sympathetic nerve activity, heart rate variability, decreased MIBG uptake) and the renin-angiotensin-aldosterone system (i.e., increased renin activity, elevated levels of angiotensin I and aldosterone, decreased ACE2 activity and related peptides). Although activation of these systems has been linked experimentally to the end pathophenotype of PAH, clinical therapy with AR blockers, ACE inhibitors/AT1 receptor blockers, and mineralocorticoid receptor antagonists remains empirical. Enhanced insight into the potential therapeutic benefit(s) of targeting these factors in PAH is anticipated through the completion of several ongoing clinical trials.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants 1K08HL111207-01A1 (B.A.M.) and HL105301 (J.A.L.), in addition to The Thomas W. Smith, M.D. Foundation (J.A.L.), Pulmonary Hypertension Association, Klarman Foundation and CIMIT award (B.A.M.).
Footnotes
Conflict of Interest Disclosures: B.A.M. receives funding to research pulmonary hypertension from Gilead Sciences Inc.
References
- 1.Forfia PR, Vaidya A, Wiegers SE. Pulmonary heart disease: The heart-lung interaction and its impact on patient phenotypes. Pulm Circ. 2013;3:5–19. doi: 10.4103/2045-8932.109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Rourke MF, Yaginuma T, Avolio AP. Physiological and pathophysiological implications of ventricular/vascular coupling. Ann Biomed Eng. 1984;12:119–134. doi: 10.1007/BF02584226. [DOI] [PubMed] [Google Scholar]
- 3.Vonk-Noordegraaf A, Westerhof N. Describing right ventricular function. Eur Respir J. 2013;41:1419–1423. doi: 10.1183/09031936.00160712. [DOI] [PubMed] [Google Scholar]
- 4.Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, Chamera E, Corretti MC, Champion HC, Abraham TP, Girgis RE, Hassoun PM. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174:1034–1041. doi: 10.1164/rccm.200604-547OC. [DOI] [PubMed] [Google Scholar]
- 5.Mehra MR, Park MH, Landzberg MJ, Lala A, Waxman AB. Right heart failure: toward a common language. J Heart Lung Transplant. 2014;33:123–126. doi: 10.1016/j.healun.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 6.Maron BA, Leopold JA. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series) Pulm Circ. 2014;4:200–210. doi: 10.1086/675984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Man FS, Handoko ML, Guignabert C, Bogaard HJ, Vonk-Noordegraaf A. Neurohormonal axis in patients with pulmonary arterial hypertension: friend or foe? Am J Respir Crit Care Med. 2013;187:14–19. doi: 10.1164/rccm.201209-1663PP. [DOI] [PubMed] [Google Scholar]
- 8.de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5:97–105. doi: 10.1161/CIRCHEARTFAILURE.111.964494. [DOI] [PubMed] [Google Scholar]
- 9.Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold JA. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation. 2012;126:963–974. doi: 10.1161/CIRCULATIONAHA.112.094722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan AY, Chen PS, Chen LS, Fishbein MC. Autonomic nerves in pulmonary veins. Heart Rhythm. 2007;4:S57–60. doi: 10.1016/j.hrthm.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes PJ, Liu SF. Regulation of pulmonary vascular tone. Pharmacol Rev. 1995;47:87–131. [PubMed] [Google Scholar]
- 12.Rudner XL, Berkowitz DE, Booth JV, Funk BL, Cozart KL, D’Amico EB, El-Moalem H, Page SO, Richardson CD, Winters B, Marucci L, Schwinn DA. Subtype specific regulation of human vascular alpha(1)-adrenergic receptors by vessel bed and age. Circulation. 1999;100:2336–2343. doi: 10.1161/01.cir.100.23.2336. [DOI] [PubMed] [Google Scholar]
- 13.Pierpont GL, DeMaster EG, Reynolds S, Pederson J, Cohn JN. Ventricular myocardial catecholamines in primates. J Lab Clin Med. 1985;106:205–210. [PubMed] [Google Scholar]
- 14.Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Jr, Trendelenburg U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- 15.Rozec B, Erfanian M, Laurent K, Trochu JN, Gauthier C. Nebivolol, a vasodilating selective beta(1)-blocker, is a beta(3)-adrenoceptor agonist in the nonfailing transplanted human heart. J Am Coll Cardiol. 2009;53:1532–1538. doi: 10.1016/j.jacc.2008.11.057. [DOI] [PubMed] [Google Scholar]
- 16.Piao L, Fang YH, Parikh KS, Ryan JJ, D’Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012;126:2859–2869. doi: 10.1161/CIRCULATIONAHA.112.109868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shannon R, Chaudhry M. Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am Heart J. 2006;152:842–850. doi: 10.1016/j.ahj.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Nagaya N, Nishikimi T, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Kakishita M, Fukushima K, Okano Y, Nakanishi N, Miyatake K, Kangawa K. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102:865–870. doi: 10.1161/01.cir.102.8.865. [DOI] [PubMed] [Google Scholar]
- 19.Mak S, Witte KK, Al-Hesayen A, Granton JJ, Parker JD. Cardiac sympathetic activation in patients with pulmonary arterial hypertension. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1153–1157. doi: 10.1152/ajpregu.00652.2011. [DOI] [PubMed] [Google Scholar]
- 20.Nootens M, Kaufmann E, Rector T, Toher C, Judd D, Francis GS, Rich S. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol. 1995;26:1581–1585. doi: 10.1016/0735-1097(95)00399-1. [DOI] [PubMed] [Google Scholar]
- 21.Velez-Roa S, Ciarka A, Najem B, Vachiery JL, Naeije R, van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation. 2004;110:1308–1312. doi: 10.1161/01.CIR.0000140724.90898.D3. [DOI] [PubMed] [Google Scholar]
- 22.Morimitsu T, Miyahara Y, Sinboku H, Ikeda S, Naito T, Nishijima K, Takao M. Iodine-123-metaiodobenzylguanidine myocardial imaging in patients with right ventricular pressure overload. J Nucl Med. 1996;37:1343–1346. [PubMed] [Google Scholar]
- 23.McGowan CL, Swiston JS, Notarius CF, Mak S, Morris BL, Picton PE, Granton JT, Floras JS. Discordance between microneurographic and heart-rate spectral indices of sympathetic activity in pulmonary arterial hypertension. Heart. 2009;95:754–758. doi: 10.1136/hrt.2008.157115. [DOI] [PubMed] [Google Scholar]
- 24.Yi HT, Hsieh YC, Wu TJ, Huang JL, Lin WW, Liang KW, Su CS, Tsai WJ, Wang KY. Heart rate variability parameters and ventricular arrhythmia correlate with pulmonary arterial pressure in adult patients with idiopathic pulmonary arterial hypertension. Heart Lung. 2014;43:534–540. doi: 10.1016/j.hrtlng.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 25.Wensel R, Jilek C, Dorr M, Francis DP, Stadler H, Lange T, Blumberg F, Opitz C, Pfeifer M, Ewert R. Impaired cardiac autonomic control relates to disease severity in pulmonary hypertension. Eur Respir J. 2009;34:895–901. doi: 10.1183/09031936.00145708. [DOI] [PubMed] [Google Scholar]
- 26.Bristow MR, Minobe W, Rasmussen R, Larrabee P, Skerl L, Klein JW, Anderson FL, Murray J, Mestroni L, Karwande SV, et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89:803–815. doi: 10.1172/JCI115659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakamaki F, Satoh T, Nagaya N, Kyotani S, Oya H, Nakanishi N, Kuribayashi S, Ishida Y. Correlation between severity of pulmonary arterial hypertension and 123I-metaiodobenzylguanidine left ventricular imaging. J Nucl Med. 2000;41:1127–1133. [PubMed] [Google Scholar]
- 28.Kamiyoshi Y, Yazaki Y, Urushibata K, Koizumu T, Kasai H, Izawa A, Kinoshita O, Hongo M, Ikeda U. Risk stratification assessed by combined lung and heart iodine-123 metaiodobenzylguanidine uptake in patients with idiopathic dilated cardiomyopathy. Am J Cardiol. 2008;101:1482–1486. doi: 10.1016/j.amjcard.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 29.Boe J, Simonsson BG. Adrenergic receptors and sympathetic agents in isolated human pulmonary arteries. Eur J Respir Dis. 1980;61:195–202. [PubMed] [Google Scholar]
- 30.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–1762. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Kim GH, Ryan JJ, Archer SL. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid Redox Signal. 2013;18:1920–1936. doi: 10.1089/ars.2012.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chakraborti S, Roy S, Chowdhury A, Mandal A, Chakraborti T. Role of PKCalpha-p38 MAPK-Gialpha axis in peroxynitrite-mediated inhibition of beta-adrenergic response in pulmonary artery smooth muscle cells. Cell Signal. 2013;25:512–526. doi: 10.1016/j.cellsig.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Faber JE, Szymeczek CL, Salvi SS, Zhang H. Enhanced alpha1-adrenergic trophic activity in pulmonary artery of hypoxic pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol. 2006;291:H2272–2281. doi: 10.1152/ajpheart.00404.2006. [DOI] [PubMed] [Google Scholar]
- 34.Minneman KP. Alpha 1-adrenergic receptor subtypes, inositol phosphates, and sources of cell Ca2+ Pharmacol Rev. 1988;40:87–119. [PubMed] [Google Scholar]
- 35.Hool LC, Oleksa LM, Harvey RD. Role of G proteins in alpha1-adrenergic inhibition of the beta-adrenergically activated chloride current in cardiac myocytes. Mol Pharmacol. 1997;51:853–860. doi: 10.1124/mol.51.5.853. [DOI] [PubMed] [Google Scholar]
- 36.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 37.Leineweber K, Brandt K, Wludyka B, Beilfuss A, Ponicke K, Heinroth-Hoffmann I, Brodde OE. Ventricular hypertrophy plus neurohumoral activation is necessary to alter the cardiac beta-adrenoceptor system in experimental heart failure. Circ Res. 2002;91:1056–1062. doi: 10.1161/01.res.0000045088.59360.b7. [DOI] [PubMed] [Google Scholar]
- 38.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. doi: 10.1164/rccm.201003-0335OC. [DOI] [PubMed] [Google Scholar]
- 39.Voelkel NF, McMurtry IF, Reeves JT. Chronic propranolol treatment blunts right ventricular hypertrophy in rats at high altitude. J Appl Physiol Respir Environ Exerc Physiol. 1980;48:473–478. doi: 10.1152/jappl.1980.48.3.473. [DOI] [PubMed] [Google Scholar]
- 40.Provencher S, Herve P, Jais X, Lebrec D, Humbert M, Simonneau G, Sitbon O. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology. 2006;130:120–126. doi: 10.1053/j.gastro.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 41.So PP, Davies RA, Chandy G, Stewart D, Beanlands RS, Haddad H, Pugliese C, Mielniczuk LM. Usefulness of beta-blocker therapy and outcomes in patients with pulmonary arterial hypertension. Am J Cardiol. 2012;109:1504–1509. doi: 10.1016/j.amjcard.2012.01.368. [DOI] [PubMed] [Google Scholar]
- 42.Peacock A, Ross K. Pulmonary hypertension: a contraindication to the use of {beta}-adrenoceptor blocking agents. Thorax. 2010;65:454–455. doi: 10.1136/thx.2008.111955. [DOI] [PubMed] [Google Scholar]
- 43.Gomberg-Maitland M, Bull TM, Saggar R, Barst RJ, Elgazayerly A, Fleming TR, Grimminger F, Rainisio M, Stewart DJ, Stockbridge N, Ventura C, Ghofrani AH, Rubin LJ. New trial designs and potential therapies for pulmonary artery hypertension. J Am Coll Cardiol. 2013;62:D82–91. doi: 10.1016/j.jacc.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maack C. Dissecting the role of g-protein-coupled receptor kinase 2 for excitation-contraction coupling. Circulation. 2012;125:2054–2056. doi: 10.1161/CIRCULATIONAHA.112.109389. [DOI] [PubMed] [Google Scholar]
- 45.Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmuller P, Ladage D, Guignabert C, Ishikawa K, Aguero J, Ibanez B, Turnbull IC, Kohlbrenner E, Liang L, Zsebo K, Humbert M, Hulot JS, Kawase Y, Hajjar RJ, Leopold JA. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation. 2013;128:512–523. doi: 10.1161/CIRCULATIONAHA.113.001585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Na S, Kim OS, Ryoo S, Kweon TD, Choi YS, Shim HS, Oh YJ. Cervical ganglion block attenuates the progression of pulmonary hypertension via nitric oxide and arginase pathways. Hypertension. 2014;63:309–315. doi: 10.1161/HYPERTENSIONAHA.113.01979. [DOI] [PubMed] [Google Scholar]
- 48.Chen SL, Zhang YJ, Zhou L, Xie DJ, Zhang FF, Jia HB, Wong SS, Kwan TW. Percutaneous pulmonary artery denervation completely abolishes experimental pulmonary arterial hypertension in vivo. EuroIntervention. 2013;9:269–276. doi: 10.4244/EIJV9I2A43. [DOI] [PubMed] [Google Scholar]
- 49.Bhamra-Ariza P, Keogh AM, Muller DW. Percutaneous interventional therapies for the treatment of patients with severe pulmonary hypertension. J Am Coll Cardiol. 2013;63:611–618. doi: 10.1016/j.jacc.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 50.Chen SL, Zhang FF, Xu J, Xie DJ, Zhou L, Nguyen T, Stone GW. Pulmonary artery denervation to treat pulmonary arterial hypertension: the single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension) J Am Coll Cardiol. 2013;62:1092–1100. doi: 10.1016/j.jacc.2013.05.075. [DOI] [PubMed] [Google Scholar]
- 51.Takachi T, Maeda M, Shirakusa T, Hayashida Y. Sympathetic reinnervation of unilaterally denervated rat lung. Acta Physiol Scand. 1995;154:43–50. doi: 10.1111/j.1748-1716.1995.tb09884.x. [DOI] [PubMed] [Google Scholar]
- 52.Nam J, Onitsuka I, Hatch J, Uchida Y, Ray S, Huang S, Li W, Zang H, Ruiz-Lozano P, Mukouyama YS. Coronary veins determine the pattern of sympathetic innervation in the developing heart. Development. 2013;140:1475–1485. doi: 10.1242/dev.087601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kimura K, Ieda M, Kanazawa H, Yagi T, Tsunoda M, Ninomiya S, Kurosawa H, Yoshimi K, Mochizuki H, Yamazaki K, Ogawa S, Fukuda K. Cardiac sympathetic rejuvenation: a link between nerve function and cardiac hypertrophy. Circ Res. 2007;100:1755–1764. doi: 10.1161/01.RES.0000269828.62250.ab. [DOI] [PubMed] [Google Scholar]
- 54.Bhatt DL, Kandzari DE, O’Neill WW, D’Agostino R, Flack JM, Katzen BT, Leon MB, Liu M, Mauri L, Negoita M, Cohen SA, Oparil S, Rocha-Singh K, Townsend RR, Bakris GL. A controlled trial of renal denervation for resistant hypertension. N Engl J Med. 2014;370:1393–1401. doi: 10.1056/NEJMoa1402670. [DOI] [PubMed] [Google Scholar]
- 55.Bader M. ACE2, angiotensin-(1-7), and Mas: the other side of the coin. Pflugers Arch. 2013;465:79–85. doi: 10.1007/s00424-012-1120-0. [DOI] [PubMed] [Google Scholar]
- 56.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 57.Hall JE, Guyton AC, Mizelle HL. Role of the renin-angiotensin system in control of sodium excretion and arterial pressure. Acta Physiol Scand Suppl. 1990;591:48–62. [PubMed] [Google Scholar]
- 58.Morrell NW, Danilov SM, Satyan KB, Morris KG, Stenmark KR. Right ventricular angiotensin converting enzyme activity and expression is increased during hypoxic pulmonary hypertension. Cardiovasc Res. 1997;34:393–403. doi: 10.1016/s0008-6363(97)00049-7. [DOI] [PubMed] [Google Scholar]
- 59.Orte C, Polak JM, Haworth SG, Yacoub MH, Morrell NW. Expression of pulmonary vascular angiotensin-converting enzyme in primary and secondary plexiform pulmonary hypertension. J Pathol. 2000;192:379–384. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH715>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 60.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, Francois C, Schalij I, Dorfmuller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, van der Velden J, Vonk-Noordegraaf A, Humbert M, Eddahibi S, Guignabert C. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:780–789. doi: 10.1164/rccm.201203-0411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patten DA, Lafleur VN, Robitaille GA, Chan DA, Giaccia AJ, Richard DE. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: the essential role of mitochondrial-derived reactive oxygen species. Mol Biol Cell. 2010;21:3247–3257. doi: 10.1091/mbc.E10-01-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolf G. Role of reactive oxygen species in angiotensin II-mediated renal growth, differentiation, and apoptosis. Antioxid Redox Signal. 2005;7:1337–1345. doi: 10.1089/ars.2005.7.1337. [DOI] [PubMed] [Google Scholar]
- 63.Sun Y, Ramires FJ, Weber KT. Fibrosis of atria and great vessels in response to angiotensin II or aldosterone infusion. Cardiovasc Res. 1997;35:138–147. doi: 10.1016/s0008-6363(97)00097-7. [DOI] [PubMed] [Google Scholar]
- 64.Friedberg MK, Cho MY, Li J, Assad RS, Sun M, Rohailla S, Honjo O, Apitz C, Redington AN. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased transforming growth factor-beta1 signaling and is abrogated by angiotensin receptor blockade. Am J Respir Cell Mol Biol. 2013;49:1019–1028. doi: 10.1165/rcmb.2013-0149OC. [DOI] [PubMed] [Google Scholar]
- 65.Morrell NW, Morris KG, Stenmark KR. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol. 1995;269:H1186–1194. doi: 10.1152/ajpheart.1995.269.4.H1186. [DOI] [PubMed] [Google Scholar]
- 66.Morrell NW, Atochina EN, Morris KG, Danilov SM, Stenmark KR. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96:1823–1833. doi: 10.1172/JCI118228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bertoli L, Fusco M, Lo Cicero S, Micallef E, Busnardo I. Influence of ACE inhibition on pulmonary haemodynamics and function in patients in whom beta-blockers are contraindicated. Postgrad Med J. 1986;62 (Suppl 1):47–51. [PubMed] [Google Scholar]
- 68.Leier CV, Bambach D, Nelson S, Hermiller JB, Huss P, Magorien RD, Unverferth DV. Captopril in primary pulmonary hypertension. Circulation. 1983;67:155–161. doi: 10.1161/01.cir.67.1.155. [DOI] [PubMed] [Google Scholar]
- 69.Ikram H, Maslowski AH, Nicholls MG, Espiner EA, Hull FT. Haemodynamic and hormonal effects of captopril in primary pulmonary hypertension. Br Heart J. 1982;48:541–545. doi: 10.1136/hrt.48.6.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shenoy V, Qi Y, Katovich MJ, Raizada MK. ACE2, a promising therapeutic target for pulmonary hypertension. Curr Opin Pharmacol. 2011;11:150–155. doi: 10.1016/j.coph.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S, Liao S, Yang K, Li Q, Wan H. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L631–640. doi: 10.1152/ajplung.90415.2008. [DOI] [PubMed] [Google Scholar]
- 72.Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Ann Tallant E, Smith RD, Chappell MC. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005;68:2189–2196. doi: 10.1111/j.1523-1755.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- 73.Dai HL, Guo Y, Guang XF, Xiao ZC, Zhang M, Yin XL. The changes of serum angiotensin-converting enzyme 2 in patients with pulmonary arterial hypertension due to congenital heart disease. Cardiology. 2013;124:208–212. doi: 10.1159/000346884. [DOI] [PubMed] [Google Scholar]
- 74.Takahashi Y, Haga S, Ishizaka Y, Mimori A. Autoantibodies to angiotensin-converting enzyme 2 in patients with connective tissue diseases. Arthritis Res Ther. 2010;12:R85. doi: 10.1186/ar3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, Canver CC. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108:1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]
- 76.Johnson JA, West J, Maynard KB, Hemnes AR. ACE2 improves right ventricular function in a pressure overload model. PLoS One. 2011;6:e20828. doi: 10.1371/journal.pone.0020828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D, Venugopal CS, Francis J, Reudelhuber T, Santos RA, Patel JM, Raizada MK, Katovich MJ. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010;182:1065–1072. doi: 10.1164/rccm.200912-1840OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKinney CA, Fattah C, Loughrey CM, Milligan G, Nicklin SA. Angiotensin-(1-7) and angiotensin-(1-9): function in cardiac and vascular remodelling. Clin Sci (Lond) 2014;126:815–827. doi: 10.1042/CS20130436. [DOI] [PubMed] [Google Scholar]
- 79.Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail. 2013;15:277–283. doi: 10.1093/eurjhf/hfs173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, Loscalzo J, Leopold JA. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation. 2014;130:168–179. doi: 10.1161/CIRCULATIONAHA.113.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heinemann HO, Ryan JW, Ryan US. Is the lung a para-endocrine organ? Am J Med. 1977;63:595–603. doi: 10.1016/0002-9343(77)90205-4. [DOI] [PubMed] [Google Scholar]
- 82.Wenz M, Steinau R, Gerlach H, Lange M, Kaczmarczyk G. Inhaled nitric oxide does not change transpulmonary angiotensin II formation in patients with acute respiratory distress syndrome. Chest. 1997;112:478–483. doi: 10.1378/chest.112.2.478. [DOI] [PubMed] [Google Scholar]
- 83.Wilkens H, Bauer M, Forestier N, Konig J, Eichler A, Schneider S, Schafers HJ, Sybrecht GW. Influence of inhaled iloprost on transpulmonary gradient of big endothelin in patients with pulmonary hypertension. Circulation. 2003;107:1509–1513. doi: 10.1161/01.cir.0000056104.49686.4b. [DOI] [PubMed] [Google Scholar]
- 84.Peacock AJ, Dawes KE, Shock A, Gray AJ, Reeves JT, Laurent GJ. Endothelin-1 and endothelin-3 induce chemotaxis and replication of pulmonary artery fibroblasts. Am J Respir Cell Mol Biol. 1992;7:492–499. doi: 10.1165/ajrcmb/7.5.492. [DOI] [PubMed] [Google Scholar]
- 85.Takeda Y, Miyamori I, Yoneda T, Hatakeyama H, Inaba S, Furukawa K, Mabuchi H, Takeda R. Regulation of aldosterone synthase in human vascular endothelial cells by angiotensin II and adrenocorticotropin. J Clin Endocrinol Metab. 1996;81:2797–2800. doi: 10.1210/jcem.81.8.8768832. [DOI] [PubMed] [Google Scholar]
- 86.Young MJ, Clyne CD, Cole TJ, Funder JW. Cardiac steroidogenesis in the normal and failing heart. J Clin Endocrinol Metab. 2001;86:5121–5126. doi: 10.1210/jcem.86.11.7925. [DOI] [PubMed] [Google Scholar]
- 87.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bernini G, Galetta F, Franzoni F, Bardini M, Taurino C, Bernardini M, Ghiadoni L, Bernini M, Santoro G, Salvetti A. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J Hypertens. 2008;26:2399–2405. doi: 10.1097/HJH.0b013e32831286fd. [DOI] [PubMed] [Google Scholar]
- 89.Yamanaka R, Otsuka F, Nakamura K, Yamashita M, Otani H, Takeda M, Matsumoto Y, Kusano KF, Ito H, Makino H. Involvement of the bone morphogenetic protein system in endothelin- and aldosterone-induced cell proliferation of pulmonary arterial smooth muscle cells isolated from human patients with pulmonary arterial hypertension. Hypertens Res. 2010;33:435–445. doi: 10.1038/hr.2010.16. [DOI] [PubMed] [Google Scholar]
- 90.Preston IR, Sagliani KD, Warburton RR, Hill NS, Fanburg BL, Jaffe IZ. Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2013;304:L678–688. doi: 10.1152/ajplung.00300.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aguero J, Ishikawa K, Hadri L, Santos-Gallego CG, Fish K, Hammoudi N, Chaanine AH, Torquato S, Naim C, Ibanez B, Pereda D, Garcia-Alvarez A, Fuster V, Sengupta PP, Leopold JA, Hajjar RJ. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am J Physiol Heart Circ Physiol. 2014;307:H1204–1215. doi: 10.1152/ajpheart.00246.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheema Y, Zhao W, Zhao T, Khan MU, Green KD, Ahokas RA, Gerling IC, Bhattacharya SK, Weber KT. Reverse remodeling and recovery from cachexia in rats with aldosteronism. Am J Physiol Heart Circ Physiol. 2012;303:H486–495. doi: 10.1152/ajpheart.00192.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carlsen J, Hasseriis Andersen K, Boesgaard S, Iversen M, Steinbruchel D, Bogelund Andersen C. Pulmonary arterial lesions in explanted lungs after transplantation correlate with severity of pulmonary hypertension in chronic obstructive pulmonary disease. J Heart Lung Transplant. 2013;32:347–354. doi: 10.1016/j.healun.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 94.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 95.Graham BB, Chabon J, Gebreab L, Poole J, Debella E, Davis L, Tanaka T, Sanders L, Dropcho N, Bandeira A, Vandivier RW, Champion HC, Butrous G, Wang XJ, Wynn TA, Tuder RM. Transforming growth factor-beta signaling promotes pulmonary hypertension caused by Schistosoma mansoni. Circulation. 2013;128:1354–1364. doi: 10.1161/CIRCULATIONAHA.113.003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, Loscalzo J, Leopold JA. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation. 2014;130:168–179. doi: 10.1161/CIRCULATIONAHA.113.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Munoz-Pacheco P, Ortega-Hernandez A, Caro-Vadillo A, Casanueva-Eliceiry S, Aragoncillo P, Egido J, Fernandez-Cruz A, Gomez-Garre D. Eplerenone enhances cardioprotective effects of standard heart failure therapy through matricellular proteins in hypertensive heart failure. J Hypertens. 2013;31:2309–2318. doi: 10.1097/HJH.0b013e328364abd6. [DOI] [PubMed] [Google Scholar]
- 98.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. 2013;9:459–469. doi: 10.1038/nrneph.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, Jaffe IZ. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. doi: 10.1161/CIRCRESAHA.108.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kokubu T, Kazatani Y, Hamada M, Matsuzaki K, Ito T, Nishimura K, Ochi T, Daimon F, Joh T. Is captopril effective in primary pulmonary hypertension? Jpn Circ J. 1982;46:1095–1097. doi: 10.1253/jcj.46.1095. [DOI] [PubMed] [Google Scholar]
- 101.Maron BA, Waxman AB, Opotowsky AR, Gillies H, Blair C, Aghamohammadzadeh R, Loscalzo J, Leopold JA. Effectiveness of spironolactone plus ambrisentan for treatment of pulmonary arterial hypertension (from the [ARIES] study 1 and 2 trials) Am J Cardiol. 2013;112:720–725. doi: 10.1016/j.amjcard.2013.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.