

Abstract
The protozoans Trypanosoma cruzi, Trypanosoma brucei and Leishmania major (Tritryps), are evolutionarily ancient eukaryotes which cause worldwide human parasitosis. They present unique biological features. Indeed, canonical DNA/RNA cis-acting elements remain mostly elusive. Repetitive sequences, originally considered as selfish DNA, have been lately recognized as potentially important functional sequence elements in cell biology. In particular, the dinucleotide patterns have been related to genome compartmentalization, gene evolution and gene expression regulation. Thus, we perform a comparative analysis of the occurrence, length and location of dinucleotide repeats (DRs) in the Tritryp genomes and their putative associations with known biological processes. We observe that most types of DRs are more abundant than would be expected by chance. Complementary DRs usually display asymmetrical strand distribution, favoring TT and GT repeats in the coding strands. In addition, we find that GT repeats are among the longest DRs in the three genomes. We also show that specific DRs are non-uniformly distributed along the polycistronic unit, decreasing toward its boundaries. Distinctive non-uniform density patterns were also found in the intergenic regions, with predominance at the vicinity of the ORFs. These findings further support that DRs may control genome structure and gene expression.
Keywords: Trypanosoma, DNA, Gene, Regulation, Microsatellite, Polycistron
1. Introduction
The protozoans Trypanosoma cruzi, Trypanosoma brucei and Leishmania major, the so called Tritryps (El-Sayed et al., 2005b), are the human pathogens responsible of American trypanosomiasis, African trypanosomiasis and old-world cutaneous leishmaniasis, respectively. They belong to the Family Trypanosomatidae, Order Kinetoplastida, constituting evolutionarily ancient eukaryotes that display exceptional gene expression hallmarks. For instance, protein coding genes are mainly arranged in unidirectional clusters that are transcribed as long polycistronic RNAs (Clayton, 2002). The strand-switch regions (SSRs) have been implicated in transcription and also in replication initiation and chromosome segregation (Martinez-Calvillo et al., 2004; Martinez-Calvillo et al., 2003; Obado et al., 2005; Respuela et al., 2008). Nevertheless, the DNA sequence elements that mediate these processes have been largely elusive so far. The absence of canonical eukaryotic promoters, together with the lack of classical regulatory DNA signals for the transcription of protein coding genes, has led to the idea that transcription is not a major control level of gene expression in trypanosomatids. In this context, gene expression regulation is considered to be fundamentally dependent on post-transcriptional events (Palenchar and Bellofatto, 2006).
Although repetitive sequences were initially perceived as “junk” DNA, their functional importance in cell biology has been widely documented (Plohl et al., 2008; Sharma et al., 2007). Nevertheless, broader questions regarding the origin, evolution and functional significance of microsatellites remain essentially unsolved (Buschiazzo and Gemmell, 2006).
Dinucleotide repeats (DRs) constitute one type of microsatellite. The pattern of dinucleotide occurrence has been associated with genome compartmentalization, gene evolution and gene expression level (Sharma et al., 2007). Moreover, DRs have been reported as target sequences for specific protein recognition (Epplen et al., 1996; Zhang et al., 2009). We have previously analyzed DRs in the genome of T. cruzi using the sequences deposited in the GenBank up to the year 2001 (Duhagon et al., 2001). Our results suggested active roles of these sequences in gene expression. In further support to the latter hypothesis, some DRs were recently found at the SSRs of head to head transcriptional units (Cribb et al., 2010; Respuela et al., 2008; Siegel et al., 2009; Thomas et al., 2009), which have been involved in polymerase II transcription initiation.
Here we extend the analysis of the presence, length (nt) and location of DRs to the whole genome of T. cruzi and also T. brucei and L. major. The use of the three species would allow the finding of conserved patterns. The analysis of the completely assembled genomes permits the study of DRs in the context of the overall genomic architecture, which could shed light on their use in position dependent molecular mechanisms.
2. Materials and Methods
Occurrence of DRs was examined for each of the 10 possible combinations of nucleotides (AA, TT, GG, CC, AT/TA, AG/GA, AC/CA, CT/TC, CG/GC, GT/TG) in: a) L. major genome, consisting of 33 Mb distributed in 36 chromosomes available in GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) (base frequency composition: A: 0.20, C: 0.30, G: 0.30, T: 0.20); b) T. brucei genome, consisting of 26 Mb distributed in 11 chromosomes available in GenBank (base frequency A: 0.27, C: 0.23, G: 0.23, T: 0.27) and c) T. cruzi genome, consisting of 58 Mb distributed in 638 scaffolds available in TcruziDB (http://tcruzidb.org/tcruzidb/) (base frequency A: 0.25, C: 0.25, G: 0.25, T: 0.25). For each genome, strand-switch localization was determined using PERL programming language (Wall et al., 2000) scripts developed for this purpose. We defined a polycistronic unit as any region consisting of at least two annotated coding sequences (CDSs) oriented in the same direction, and intergenic region as the sequence between two consecutive CDSs. In addition, the occurrence (O), length in nucleotides (l) and mismatch (a) (as a measure of the deviation from the perfect dinucleotide repetitive sequence) were determined by the Tandem Repeat Finder program (TRF) (Benson, 1999). The TRF parameters were set to Match.Mismatch.Delta.PM.PI.Minscore.MaxPeriod = 2.7.7.80.10.7.2 to enable the detection of imperfect DRs of l≥8nt and perfect DRs of l≥6nt. Relational databases were built and tables combining strand-switch and repeat content information were created. In addition, similar tables containing the location of the DRs with respect to CDSs were constructed. The data were analyzed using R software environment for statistical computing. The randomly expected frequencies for each class of perfect DR of a defined length was set as the product of each nucleotide frequency: ((X/N)*(Y/N))n * ((X/N)l-2n+ (Y/N)l-2n) for n≥3, where n is the copy number of the dinucleotide (XY)n and N the genome size (nt). This formula takes into account both even (l=2n) and odd (l=2n+1) DR lengths. The resulting frequencies were transformed to expected occurrences (E) multiplying by the corresponding N. Only integral values of E were considered. The difference between the expected frequency for a given length (l) and the expected frequency of the subsequently longer DR (l+1) renders the non-cumulative expected occurrence. The significance of the deviation of observed (O) from expected (E) values was measured using the χ2 test. For each dinucleotide class, we determined the previously defined variable D=Σ l.a (Duhagon et al., 2001), that combines the occurrence, length and mismatch factor from the TRF program (Benson, 1999).
3. Results and Discussion
3.1. DRs display specific pattern of occurrence in the coding strands
In an attempt to uncover a putative functionality of the DRs, we analyzed their frequency in the genomes of L. major, T. brucei and T. cruzi (Berriman et al., 2005). Strand-switches were defined as indicated in Material and Methods, and only the coding strands were used for all the analyses. Perfect and imperfect DRs were detected with the TRF program (Benson, 1999) as detailed in Materials and Methods. The occurrence of each type of DR with l≥8 in the genome of the Tritryps is presented in Figure 1A. The global pattern of dinucleotide abundance conforms to early reports from partial genome sequences of T. cruzi (Aguero et al., 2000), and interestingly, of other organisms too (Toth et al., 2000). Overall, T. brucei and T. cruzi patterns of DR occurrence are more similar than L. major's.
Figure 1. Abundance of DRs in the Tritryps.

A. Occurrence of the ten classes of DRs in the coding strands. Imperfect repeats of length l≥8 were used in the analysis. B. D value (D=Σ l.a) normalized by the genome size for the ten classes of DRs with l≥8nt in the coding strands. Genome size (N) and G+C genomic content (G+C (%)) of each parasite are indicated in the upper table. Tb: T. brucei, Tc: T. cruzi, Lm: L. major.
Additionally, (TT)n and (AA)n are the most abundant DRs in T. brucei and T. cruzi, but only (TT)n is prominent in L. major. This difference can be due to the higher G+C content of L. major genome (60%) in comparison to the 46% for T. brucei and 51% for T. cruzi. In contrast, L. major presents a higher proportion of DRs containing C. In an attempt to evaluate the influence of the genomic compositional bias in the occurrence of the different DRs, we determine the deviation of the observed repeat frequencies from those expected by chance, based on the nucleotide composition of the genomes (Table 1). In accordance with our earlier report (Duhagon et al., 2001), the occurrence of the majority of DRs is strongly and significantly deviated from serendipity (χ2 test p<0.001). Above l=8nt, the majority of non-cumulative observed DRs (O) are higher than the E, and this tendency becomes gradually more noticeable as the repeat length increases. Some studies have interpreted this overrepresentation as a result of polymerase mediated strand slippage above a specific repeat length threshold (reviewed in Kelkar et al., 2010). The biggest difference in O vs. E values is observed for (TT)n in the three parasites, which is 6-23 times (O/E) overrepresented (i.e. O>E) for l=8nt to more than a thousand for l=14nt. Following TT, (AA)n presents the highest overrepresentation. Although (TT)n and (AA)n repeats are the most abundant in T. cruzi and T. brucei but not in L. major, they still present the highest O/E ratios in the three parasites. This finding suggests the existence of a conserved force that overcomes compositional bias and leads to the overrepresentation of these two types of repeats in the three genomes. The high frequency of (AA)n and (TT)n has been attributed to their involvement in putative DNA conformations and chromatin structure (Sinden, 1994). It is worth mentioning that polyA tails from retroposons, or even processed pseudogenes, could also contribute to their abundance (Toth et al., 2000). In addition, A and T rich post-transcriptional regulatory DNA elements have been described in intergenic regions of the Tritryps and other organisms (Clayton and Shapira, 2007; Di Noia et al., 2000; D'Orso et al., 2003; Haile et al., 2008; McKee and Silver, 2007). Additionally, (GT)n is conspicuous in the genomes of all Tritryp, ranking at the second highest position in L major and third in T. cruzi and T. brucei (Figure 1A). In L major, the occurrence of this DR is even higher than the occurrences of the (TT)n. Particularly, (GT)8 repeat is 2-4 times more frequent than is expected by chance (χ2 test, p<0.0001) in the three parasites (Table 1) reaching 744-6478 O/E ratio at l=14nt. The abundance of poly-(GT), that we have previously communicated for T. cruzi (Duhagon et al., 2001), has also been reported in human (Sharma et al., 2005) and in ten other different taxa (Toth et al., 2000) pointing out to a strongly conserved mutational or selective mechanism maintaining this repeat.
Table 1. Observed (O) and expected (E) occurrences of perfect poly-dinucleotide repeats of lenght (l) 6≤l≤14 in the coding strands of the Tritryp genomes.
| T. brucei | T. cruzi | L. major | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| l=6nt | E | O | E | O | E | O |
| AA | 7367 | 2011 | 10166 | 3666 | 1736 | 540 |
| AC | 9943 | 692 | 22588 | 1401 | 11249 | 1313 |
| AG | 9816 | 744 | 22357 | 1740 | 10979 | 899 |
| AT | 15127 | 1019 | 20176 | 1238 | 3642 | 276 |
| CC | 3357 | 522 | 12548 | 884 | 18353 | 1559 |
| CG | 6629 | 293 | 24839 | 859 | 35818 | 2247 |
| CT | 10240 | 829 | 22466 | 2701 | 11817 | 2441 |
| GG | 3272 | 586 | 12292 | 1836 | 17476 | 843 |
| GT | 10110 | 1148 | 22235 | 3063 | 11533 | 2046 |
| TT | 7816 | 2735 | 10056 | 7751 | 1915 | 1432 |
|
| ||||||
| Total | 83678 | 10579 | 179723 | 25139 | 124517 | 13596 |
| l=7nt | E | O | E | O | E | O |
|
| ||||||
| AA | 1963 | 3766 | 2499 | 6847 | 347 | 1196 |
| AC | 2485 | 290 | 5655 | 991 | 2847 | 837 |
| AG | 2449 | 274 | 5587 | 736 | 2763 | 438 |
| AT | 4114 | 457 | 5012 | 831 | 738 | 221 |
| CC | 781 | 423 | 3198 | 605 | 5503 | 908 |
| CG | 1539 | 111 | 6320 | 222 | 10695 | 1042 |
| CT | 2574 | 338 | 5620 | 1194 | 3009 | 1444 |
| GG | 758 | 526 | 3122 | 1371 | 5196 | 673 |
| GT | 2537 | 582 | 5552 | 2086 | 2920 | 1324 |
| TT | 2104 | 4927 | 2467 | 13817 | 389 | 3050 |
|
| ||||||
| Total | 21305 | 11694 | 45032 | 28700 | 34408 | 11133 |
| l=8nt | E | O | E | O | E | O |
|
| ||||||
| AA | 523 | 2194 | 614 | 2671 | 69 | 677 |
| AC | 616 | 754 | 1415 | 4175 | 674 | 1591 |
| AG | 606 | 642 | 1396 | 2117 | 652 | 1045 |
| AT | 1039 | 884 | 1174 | 1276 | 144 | 274 |
| CC | 182 | 159 | 815 | 161 | 1650 | 819 |
| CG | 357 | 268 | 1608 | 389 | 3193 | 2974 |
| CT | 641 | 605 | 1405 | 1868 | 720 | 3768 |
| GG | 176 | 230 | 793 | 355 | 1545 | 580 |
| TG | 630 | 1488 | 1385 | 4680 | 696 | 2617 |
| TT | 566 | 3133 | 605 | 5992 | 79 | 1803 |
|
| ||||||
| Total | 5337 | 10357 | 11210 | 23684 | 9422 | 16148 |
| l=9nt | E | O | E | O | E | O |
|
| ||||||
| AA | 139 | 1868 | 151 | 1687 | 14 | 767 |
| AC | 154 | 272 | 354 | 2580 | 171 | 861 |
| AG | 151 | 203 | 349 | 694 | 164 | 509 |
| AT | 341 | 313 | 345 | 600 | 33 | 119 |
| CC | 42 | 161 | 208 | 74 | 495 | 1464 |
| CG | 83 | 81 | 409 | 275 | 954 | 1249 |
| CT | 161 | 214 | 351 | 1374 | 183 | 1695 |
| GG | 41 | 259 | 201 | 184 | 459 | 712 |
| GT | 158 | 624 | 346 | 4237 | 176 | 1238 |
| TT | 152 | 3035 | 149 | 4286 | 16 | 2156 |
|
| ||||||
| Total | 1424 | 7030 | 2864 | 15991 | 2666 | 10770 |
| l=10nt | E | O | E | O | E | O |
|
| ||||||
| AA | 37 | 1264 | 37 | 1172 | 3 | 477 |
| AC | 38 | 109 | 89 | 1206 | 40 | 335 |
| AG | 37 | 71 | 87 | 324 | 39 | 227 |
| AT | 29 | 224 | 28 | 521 | 2 | 81 |
| CC | 10 | 184 | 53 | 61 | 148 | 1356 |
| CG | 19 | 27 | 104 | 27 | 285 | 552 |
| CT | 40 | 69 | 88 | 295 | 44 | 768 |
| GG | 9 | 285 | 51 | 147 | 136 | 640 |
| GT | 39 | 255 | 86 | 1526 | 42 | 525 |
| TT | 41 | 2247 | 36 | 3129 | 3 | 1440 |
|
| ||||||
| Total | 300 | 4735 | 659 | 8408 | 742 | 6401 |
| T. brucei | T. cruzi | L. major | ||||
|
| ||||||
| l=11nt | E | O | E | O | E | O |
|
| ||||||
| AA | 10 | 768 | 9 | 862 | 1 | 176 |
| AC | 10 | 107 | 22 | 615 | 10 | 313 |
| AG | 9 | 50 | 22 | 203 | 10 | 133 |
| AT | 21 | 119 | 18 | 375 | 1 | 81 |
| CC | 2 | 86 | 14 | 48 | 45 | 731 |
| CG | 4 | 5 | 27 | 9 | 85 | 226 |
| CT | 10 | 52 | 22 | 453 | 11 | 508 |
| GG | 2 | 159 | 13 | 109 | 41 | 324 |
| GT | 10 | 165 | 22 | 2283 | 11 | 405 |
| TT | 11 | 1417 | 9 | 2287 | 1 | 641 |
|
| ||||||
| Total | 90 | 2928 | 177 | 7244 | 215 | 3538 |
| l=12nt | E | O | E | O | E | O |
|
| ||||||
| AA | 3 | 427 | 2 | 674 | 0 | 76 |
| AC | 2 | 42 | 6 | 478 | 2 | 203 |
| AG | 2 | 30 | 5 | 147 | 2 | 112 |
| AT | 6 | 127 | 4 | 331 | 0 | 55 |
| CC | 1 | 62 | 3 | 37 | 13 | 288 |
| CG | 1 | 0 | 7 | 0 | 25 | 85 |
| CT | 3 | 33 | 5 | 121 | 3 | 359 |
| GG | 1 | 76 | 3 | 109 | 12 | 136 |
| GT | 2 | 102 | 5 | 619 | 2 | 308 |
| TT | 3 | 948 | 2 | 1965 | 0 | 286 |
|
| ||||||
| Total | 23 | 1847 | 44 | 4481 | 61 | 1908 |
| l=13nt | E | O | E | O | E | O |
|
| ||||||
| AA | 1 | 227 | 1 | 644 | 0 | 15 |
| AC | 1 | 54 | 1 | 242 | 1 | 281 |
| AG | 1 | 28 | 1 | 132 | 1 | 122 |
| AT | 2 | 91 | 1 | 295 | 0 | 110 |
| CC | 0 | 43 | 1 | 41 | 4 | 136 |
| CG | 0 | 0 | 2 | 2 | 8 | 51 |
| CT | 1 | 37 | 1 | 376 | 1 | 386 |
| GG | 0 | 41 | 1 | 92 | 4 | 69 |
| GT | 1 | 118 | 2 | 866 | 1 | 319 |
| TT | 1 | 496 | 1 | 1635 | 0 | 96 |
|
| ||||||
| Total | 6 | 1847 | 11 | 4325 | 18 | 1585 |
| l=14nt | E | O | E | O | E | O |
|
| ||||||
| AA | 0 | 102 | 0 | 66 | 0 | 12 |
| AC | 0 | 27 | 0 | 27 | 0 | 169 |
| AG | 0 | 28 | 0 | 26 | 0 | 85 |
| AT | 0 | 121 | 0 | 89 | 0 | 103 |
| CC | 0 | 20 | 0 | 1 | 1 | 88 |
| CG | 0 | 0 | 0 | 0 | 2 | 8 |
| CT | 0 | 34 | 0 | 16 | 0 | 369 |
| GG | 0 | 16 | 0 | 0 | 1 | 34 |
| GT | 0 | 81 | 0 | 78 | 0 | 311 |
| TT | 0 | 209 | 0 | 217 | 0 | 38 |
|
| ||||||
| Total | 1 | 429 | 2 | 303 | 5 | 1179 |
The values are not significant deviated from serendipity (X2 test p<0.001) are highlighted.
Interestingly, the (CT)n is the most frequent DR in L. major (Figure 1A). However, (CT)n overrepresentation is seen at l=8nt and over, whereas (AA)n and (TT)n deviation is already detected at l=7nt, suggesting that (AA)n and (TT)n still show an important bias in this organism. Indeed, the relative occurrences of (CT)n within the global occurrences of DRs in each Tritryp seems to accompany the A+T genomic content. (CT)n proportion could also be favored by the reported role of polypyrimidine sequence in the intergenic regions of coding strands in Tritryps (Stern et al., 2009).
Finally, (CG)n is the least frequent DR, with the exception of L. major (Figure 1A). Table 1 shows that (CG)8 and (CC)8 are the only underrepresented (i.e. O<E) classes in all the Trityps (O/E =0.2-0.9). In fact, (CG)n is the only repeat that is less abundant than expected by chance at 10≤l≤14nt (only in trypanosomes), suggesting a selective pressure against these sequences that overcomes the compositional bias of the genome. In fact, the low frequency of CG repeats has been well recognized (Jurka and Pethiyagoda, 1995; Lowenhaupt et al., 1989; Stallings, 1992; Tautz et al., 1986; Toth et al., 2000) and it has been attributed to the high C→T mutational rate in CpG dinucleotides. This phenomenon could support an increase in the TpG content, thus providing an explanation of the high occurrence of (GT)n (reviewed in Frank and Lobry, 1999). It has also been proposed that the (CG)n propensity to adopt particular conformational structures could be implicated in the negative selection of these sequences (Stallings, 1992).
In addition, we analyzed the pattern of dinucleotides using the variable “D” that combines occurrence, length and mismatch (Duhagon et al., 2001) (Figure 1B). The result observed for D is similar to that for occurrences (Figure 1A), further supporting the significance of the findings. Nevertheless, there are some differences between O (Figure 1A) and D (Figure 1B) outcomes. Indeed, considering the D value, the (AT)n becomes the fourth most represented class (instead of the fifth) in T. cruzi and, it rises above (AA)n and (AG)n in L major. In addition, differences among the three organisms were observed within a class. Note that while the occurrence of (GT)n is higher in T. cruzi than in the other two Tritryps, the corresponding D value turns out to be the highest in L. major. The difference in O and D patterns could be due to the preferential weight on length assumed by variable D. This observation prompted us to perform a more detailed analysis of the DR length distribution.
Although Tritryp multigenic families are known to be rich in diverse repetitive amino acid tracts (CITA), the low conservation of their nucleotide sequences together with the limited coding possibilities of the ten DRs and the degeneration of the genetic code, prevent these repetitive domains to skew the global DR frequency (data not shown).
3.2. The lengths of DRs are class specific and display commonalities among the parasite genomes
To further understand the putative roles of DR sequences, we studied their length distribution. A box-plot representation of each class of DR in the non-coding regions of sense strands is shown in Figure 2. On the whole, the medians of the lengths are similar both inter and intra-genomes. Strikingly, the AT repeat presents the longest median and the broader spread of the outliers in the three parasites, perhaps contributing to the differences between O and D discussed above. The biological significance of this signal in the three parasite genomes should be defined. In addition, the (CG)n shows the narrowest spread of outliers followed by (GG)n and (CC)n, except in L. major where (TT)n and (AA)n are comparable. Interestingly, the longest DRs, both in T. brucei and L. major, are the (GT)n. For T. cruzi, longer DRs are formed by the (AA)n and (TT)n stretches. In addition, (GT)n medians rank second after (AT)n in the three genomes. This finding is in agreement with previous reports on four mammalian organisms (H. sapiens, M. musculus, C. elegans and S. cerevisiae) by Dokholyan et al. (Dokholyan et al., 2000), who found that (GT/AC)n and (AG/CT)n are the longest DRs in these organisms and suggested the existence of a specific role for lengths between 20 and 60nt.
Figure 2. Length dispersion of DRs.
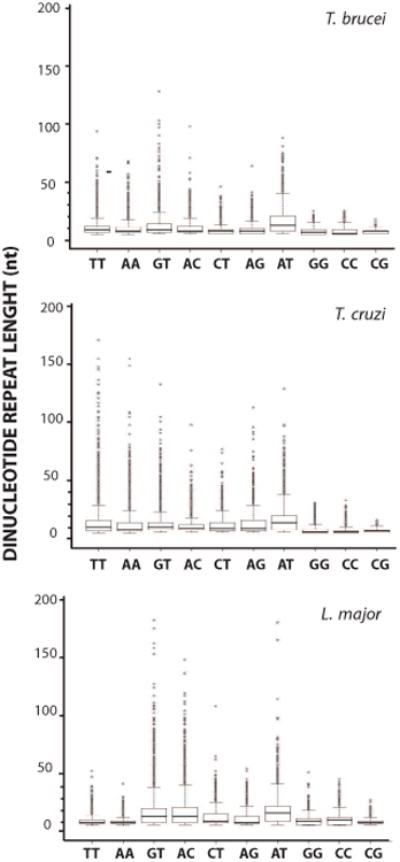
Length boxplots of each class of DRs of length l≥8nt in coding strands. A maximum length of 200 nt was arbitrarily graphed for comparison among the organisms. Outliers are set as 98th percentile. Species name corresponding to the data is indicated above each panel.
In an attempt to further study the potential importance of specific repeat lengths we looked at the length density distribution (Figure 3). The density of DRs represents the relative frequency of DRs of a given length. Overall, DRs present a monophasic decay as expected by randomness. This is evident for (AA)n and (TT)n. Nevertheless, most of the curves also display local deviations. For instance, the (AT)n shows a significant contribution of sequences ranging from 15 to 20nt that is evidenced by a secondary peak in the distribution. This is in agreement with its longest median in Figure 2. A conspicuous secondary peak is also observed for (GG)n and (CC)n in L. major, together with other minor peaks. Likewise, (CG)n repeats show at least two major peaks in a narrow length range that is similar for the three genomes. Multi-peak distributions composed by several minor peaks are observed for the (GT)n, (AC)n, (CT)n and (AG)n. Altogether, the finding of specific DR length profiles, which are sometimes conserved among the species, suggests they hold distinctive roles.
Figure 3. Length density of DRs.

The abundance of DRs in coding strands is shown as a distribution (relative frequency of a dinucleotide for a particular length range). The abscissa plots the length of the repeat (nt) with an arbitrary maximum of 35 nt and the ordinate the relative frequency of repeats for a given length. Axis labels are only shown for the left most panels, being identical in the rest of them within a single species. Species name corresponding to the data is indicated above each panel.
3.3. Asymmetrical strand distribution of DRs is widespread in Tritryp genomes
It is well documented that both complementary DNA strands bear base compositional differences that may be due to either selective or mutational differences between them (Frank and Lobry, 1999). The strand asymmetry is proposed to arise from asymmetric DNA related processes such as replication, repair, transcription and chromatin packing. We have previously detected an asymmetrical strand distribution between coding and non-coding strand, for the complementary pair of DRs (GT)n and (AC)n (Duhagon et al., 2001). Here, a similar analysis of strand distribution was carried out for the complete Tritryp genomes. The analysis of perfect DRs with lengths l≥8nt shows that all the complementary pairs, except for CT/AG in T. brucei, are asymmetrically distributed among the strands (Figure 4). As previously shown (Duhagon et al., 2001), the (GT)n is more frequent in the coding strand than the (AC)n. In addition, (TT)n is also more frequent in the coding strand in the three genomes and (CT)n in T. cruzi and L. major. Interestingly, the (GG)n is less abundant in the coding strand of L. major, representing the unique reverse strand distribution of DRs among the three parasites. When imperfect DRs are included in the analysis, a similar pattern of asymmetric strand distribution between complementary repeated sequences is also observed (data not shown). In conclusion, disproportionate distribution of complementary DRs in the coding/non-coding strands is a common feature in Tritryp genomes. In addition, three out of the four pairs display the same strand orientation bias in the three genomes, pointing out to the existence of a directional mechanism for the generation or maintenance of the asymmetry. Local variations in base content (known as AT skew: (A-T)/(A+T) and GC skew: (G-C)/(G+C)) in the coding/non-coding DNA strands have been described in most organisms (Rocha and Danchin, 2001; Mugal et al., 2010; Green et al., 2003). They have been related to the direction of replication and transcription. The G and T are usually more abundant than the C and A in the coding strands. In trypanosomatid parasites, the pattern of GC and AT skews has been previously studied by McDonagh and Nilsson (El-Sayed et al., 2005b; McDonagh et al., 2000; Nilsson and Andersson, 2005). The authors found that trypanosome base skew patterns were similar to those reported for eubacteria but different from L major. They proposed that the gene density and inherent synonymous codon usage could explain, at least in part, the GC and AT skews in these organisms. However, the intergenic regions could be greatly influencing the base skews through the existence of strand asymmetrical specific regulatory DNA elements or even simple repeats. The GC and AT skews observed in eubacteria and trypanosomes could lead to an increase of the (GG)n, (GT)n and (TT)n in the leader strand. In fact, these DRs are more frequent in the coding strands of the Tritryps (with the exception of GG in L. major). In the particular case of L. major, there is an increase in C content in the coding strand (Nilsson and Andersson, 2005), thus (CC)n, (CT)n and (TT)n would be expected to be augmented. These are in fact the DRs more represented in the coding strands in L. major. Therefore, the abundance of DRs in the coding strands described here, accompanies the strand base compositional bias. Although strand dissymmetry has been proposed to neutrally arise from genome mutational skews (Eckert and Hile, 2009), it might also be selectively maintained because of its putative functionality (e.g. in transcription or DNA replication).
Figure 4. Relative proportion of complementary DRs.

The percentages of complementary dinucleotides repeats of length l≥8nt in the coding strands of the four parasite genomes are shown. The percentage of each DR (■) and its complementary (■) is presented (the occurrence of one member of the pair/the addition of the occurrence of each member of the pair*100). Significance of the deviation between complementary dinucleotides repeats (p<0.0001) yielded by the two proportions hypothesis test (Z) is indicated by *. Species name corresponding to the data is indicated above each panel.
3.4. DRs are not uniformly distributed along the co-directional clusters
The Tritryp polycistronic gene arrangements are necessarily controlled by sequence elements that still remain poorly characterized. In order to study a possible role of DRs in the polycistron dynamics we determine the occurrence of DRs along the co-directional clusters ordered from head to tail (as defined in Materials and Methods). When we analyzed absolute polycistronic distances, we observed a DR distribution that mainly resembles polycistron length distribution in all the Tritryps (data not shown). To eliminate the influence in the analysis of length variation among polycistrons, we used the relative distances (Figure 5). Strikingly, non-parametric Smirnov-Kolmogorov goodness of fit test indicates that most of the DRs are non-uniformly distributed along the polycistrons (24 out of the 30, p≤0.05). The uniformity displayed by (CT)n, (AG)n, (GG)n, (CC)n, (CG)n in T. brucei and (AC)n in T. cruzi, suggests that these sequences are not involved in global polycistron structuring or functionality. Among the non-uniform distributions, 16 out of 30 present a decrease of repeat density at both polycistron boundaries (e.g., (TT)n and (AT)n in T. brucei, (GT)n in the three parasites). This trend towards a decrease of DR density at the boundary of the polycistronic unit might be linked to the maintenance of its structural integrity. In fact, in spite of the important sequence divergence in trypanosomatid genomes, they display a strong synteny that might be sustained by the polycistronic architecture. DRs may enhance recombinational processes or polymerase inadequacy resulting in increased local mutational rates (Majewski and Ott, 2000; Jeffreys et al., 1998). Alternatively, position dependent genomic stability might arise from local variations in the efficiency of repair mechanism. Finally, the recent finding of a histone code at the SSRs of T. cruzi and T. brucei (Respuela et al., 2008; Siegel et al., 2009) strengthens the importance of chromatin configuration in polycistron dynamics (Li et al., 2002). Thus, since microsatellites have been shown to affect chromatin structure, we cannot rule out an involvement of these sequences in the establishment of polycistronic epigenetic signatures. A remarkable exception is represented by (AA)n in L. major, which is conversely more abundant at the ends of the polycistron. Regardless of the direction, the symmetry of these distributions (i.e., similar change at both edges of the polycistron) argues against DR involvement in DNA directional processes. Lastly, some DRs (e.g. (AC)n, or (GG)n) display a more complex distribution. It is worth noting that short and long polycistrons may be governed by different mechanisms; therefore, a unified analysis comprising all lengths might be masking specific roles for DRs in particular classes of polycistrons. In conclusion, although the biological basis of the patterns described above is still unclear, they support an active role of DRs in polycistron dynamics. To our knowledge this is the first report of DR distribution along polycistronic units.
Figure 5. DRs distribution along polycistrons.
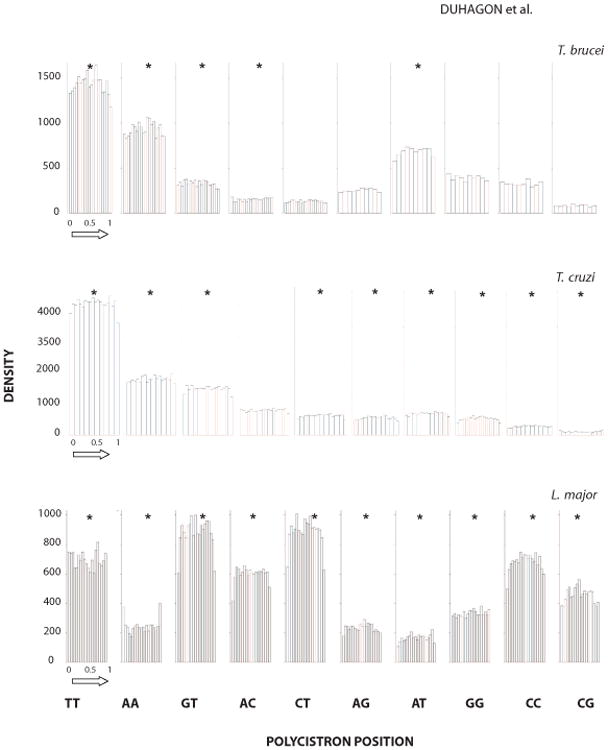
The histogram of each class of DR in the sense strand of polycistrons is shown. The relative distance from the strand-switch from head to tail (as indicated by the arrow) is depicted in the abscissa. Rejection of the null hypothesis of uniform distribution by Smirnov-Kolmogorov goodness of fit test (p≤0.0001) is indicated by *. Species name corresponding to the data is indicated above each panel.
3.5. Tritryps exhibit differences in location of DRs within the intergenic region
Previous research has shown that DRs are abundant in the intergenic regions of trypanosomatid genes (Andersson et al., 1998; Duhagon et al., 2001; Nilsson and Andersson, 2005). Considering the hypothesis of these sequences bearing roles in post-transcriptional stages of gene expression, we asked about their distribution along the intergenic regions. Since, the analysis using absolute intergenic distance was strongly biased by the interCDS length distribution (data not shown), we used the relative distance of the repeat from the end of the CDS. In Figure 6, the occurrence of DRs along the region between the 3′ end of one CDS (5′ end of the interCDS region) to the 5′ beginning of the following one (3′ end of the interCDS region) is presented. The boundaries of the interCDS are therefore enriched in UTRs. The Smirnov-Kolmogorov goodness of fit test showed that most DRs are not uniformly distributed along the intergenic regions (P≤0.05). Exceptions are (CG)n in L. major and T. brucei, and (CT)n (AG)n, (AT)n, (CC)n in T. brucei. Some plots show a predominance of repeats towards one end of the intergenic region; e.g., in T. brucei and L. major (GT)n are prevalent towards the 3′UTRs and conversely, (AC)n towards the 5′UTRs in L. major. Interestingly, most of T. cruzi plots show increased DR abundance at both intergenic ends simultaneously, producing “U” shaped curves. Meanwhile, edge single peak curves are predominant in L. major whereas smoother patterns are detected in the genome of T. brucei. Finally, few DRs are under-represented near the CDSs. One example is (AA)n in L. major. In particular, all polypyrimidine DRs (TT; CC and TC) are under-represented at the 3′ end of the intergenic region, i.e. the beginning of the downstream coding region. It is known that the conserved pyrimidine-rich tract that precedes the SL addition site is located at an average of 53 nucleotides to the CDS (Campos et al., 2008) and that the average intergenic region length has been calculated to be 1024 nt in T. cruzi (El-Sayed et al., 2005a). Thus, in agreement with Campos et al, it is possible to speculate that the five percent 3′ most region of the intergenic region is devoid of pyrimidine-rich DRs as a strategy to achieve the positional conservation of the functional polypyrimidine tract. Similar conclusions can be withdrawn for L. major and T. brucei considering their average intergenic and UTR regions size. Besides, a reduction of polypyrimidine containing DRs is also observed at the 5′end of the intergenic region in L. major (for TT; CC and TC), and T. brucei (only for CT). This could be analogously explained, as driven by the conservation of the polypyrimidine tract that modulates the poly-adenylation process. However, the comparatively less conservation of the 5′end versus the 3′end of the intergenic region patterns could be attributed to the broader size distribution of 5′UTR lengths compared to 3′UTR (Campos et al., 2008). In addition; since there are at least two polypyrimidine motifs at the vicinity of the polyA addition site (Campos et al., 2008), the interpretation of the polypyrimidine distribution at this region becomes more complex.
Figure 6. DR distribution along intergenic regions.
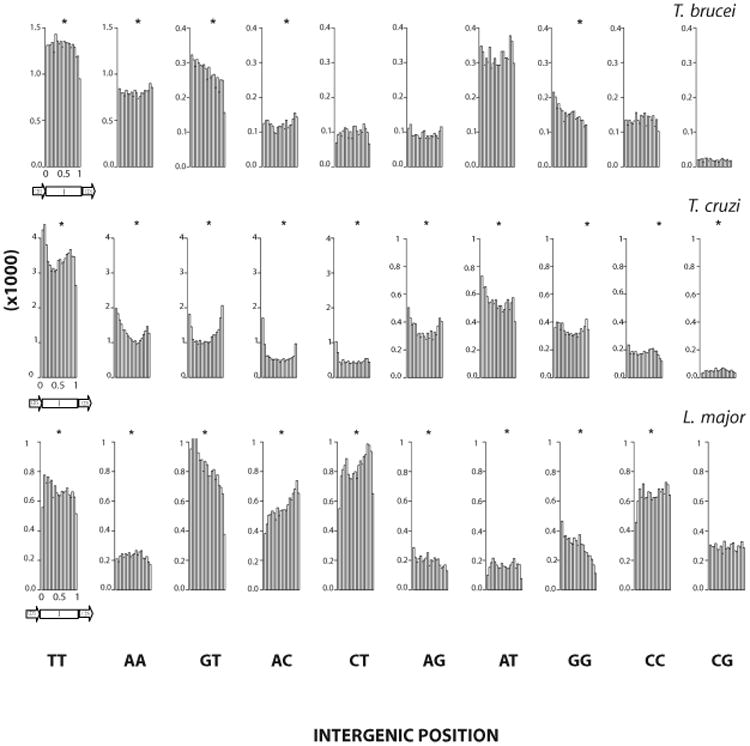
The histogram of each class of DRs in the inter-CDS regions (I) is shown. The relative distance from the 3′ end of one CDS to the 5′ beginning of the following CDS is depicted (and is indicated by the arrow). Rejection of the null hypothesis of uniform distribution by Smirnov-Kolmogorov goodness of fit test (p≤0.0001) is indicated by *. Species name corresponding to the data is indicated above each panel.
Inter-specific differences in repeat distribution along the intercistronic regions might reflect a divergent use of DRs in the post-translational regulation of each parasite. Nevertheless, the general enrichment of DRs in the proximity of the CDSs seems to be a common feature in the three genomes, thus favoring their putative function as UTR sequence elements.
3.6. Conclusion
The origin, evolution and functionality of microsatellites in the eukaryotic genome are still scarcely understood. Various reports have uncovered the non-stochastic patterns of abundance and distribution of DRs in the genomes. Although many roles have been ascribed to them, the relative extent of neutrality and selectivity of these sequences is not yet understood. Trypanosomes are well known for the lack of canonical higher eukaryote sequence elements; thus, their molecular biology may rely on signals from non-traditional sequences. We and others have proposed a functional role for DRs in the gene expression of these parasites. Here we address this hypothesis through the comparative bioinformatic analysis of their distribution in the coding strands of the complete genomes of the Tritryps. We found that DRs are present at frequencies that greatly differ from the stochastic expectations. In addition, they exhibit common and specific patterns of occurrence, length, strand distribution, and location both along the polycistron and intergenic regions. These associations with genomic regions actively involved in molecular processes suggest that DRs may constitute evolutionary selected sequence elements.
Acknowledgments
This work was financially supported by FIRCA n° R03 TW05665-01, PEDECIBA and CSIC, UdelaR. MAD received a PEDECIBA fellowship. We thank Dr. F. Alvarez-Valin and Dr. H. Musto for helpful discussions.
Abbreviations
- DR
dinucleotide repeat
- (XY)n
oligodi(deoxynucleotide) of n nucleotides in length
- SSR
strand-switch region
- ORF
open reading frame
- CDS
coding sequence
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguero F, Verdun RE, Frasch AC, Sanchez DO. A random sequencing approach for the analysis of the Trypanosoma cruzi genome: general structure, large gene and repetitive DNA families, and gene discovery. Genome Res. 2000;10:1996–2005. doi: 10.1101/gr.gr-1463r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson B, Aslund L, Tammi M, Tran AN, Hoheisel JD, Pettersson U. Complete sequence of a 93.4-kb contig from chromosome 3 of Trypanosoma cruzi containing a strand-switch region. Genome Res. 1998;8:809–816. doi: 10.1101/gr.8.8.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Bohme U, Hannick L, Aslett MA, Shallom J, Marcello L, Hou L, Wickstead B, Alsmark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K, Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN, Corton CH, Cronin A, Davies RM, Doggett J, Djikeng A, Feldblyum T, Field MC, Fraser A, Goodhead I, Hance Z, Harper D, Harris BR, Hauser H, Hostetler J, Ivens A, Jagels K, Johnson D, Johnson J, Jones K, Kerhornou AX, Koo H, Larke N, Landfear S, Larkin C, Leech V, Line A, Lord A, Macleod A, Mooney PJ, Moule S, Martin DM, Morgan GW, Mungall K, Norbertczak H, Ormond D, Pai G, Peacock CS, Peterson J, Quail MA, Rabbinowitsch E, Rajandream MA, Reitter C, Salzberg SL, Sanders M, Schobel S, Sharp S, Simmonds M, Simpson AJ, Tallon L, Turner CM, Tait A, Tivey AR, Van Aken S, Walker D, Wanless D, Wang S, White B, White O, Whitehead S, Woodward J, Wortman J, Adams MD, Embley TM, Gull K, Ullu E, Barry JD, Fairlamb AH, Opperdoes F, Barrell BG, Donelson JE, Hall N, Fraser CM, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Buschiazzo E, Gemmell NJ. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays. 2006;28:1040–1050. doi: 10.1002/bies.20470. [DOI] [PubMed] [Google Scholar]
- Campos PC, Bartholomeu DC, DaRocha WD, Cerqueira GC, Teixeira SM. Sequences involved in mRNA processing in Trypanosoma cruzi. Int J Parasitol. 2008;38:1383–1389. doi: 10.1016/j.ijpara.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Clayton C, Shapira M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol Biochem Parasitol. 2007;156:93–101. doi: 10.1016/j.molbiopara.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Clayton CE. Life without transcriptional control? From fly to man and back again. Embo J. 2002;21:1881–1888. doi: 10.1093/emboj/21.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribb P, Esteban L, Trochine A, Girardini J, Serra E. Trypanosoma cruzi TBP shows preference for C/G-rich DNA sequences in vitro. Exp Parasitol. 2010;124:346–349. doi: 10.1016/j.exppara.2009.11.003. [DOI] [PubMed] [Google Scholar]
- D'Orso I, De Gaudenzi JG, Frasch AC. RNA-binding proteins and mRNA turnover in trypanosomes. Trends Parasitol. 2003;19:151–155. doi: 10.1016/s1471-4922(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, D'Orso I, Sanchez DO, Frasch AC. AU-rich elements in the 3′-untranslated region of a new mucin-type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. J Biol Chem. 2000;275:10218–10227. doi: 10.1074/jbc.275.14.10218. [DOI] [PubMed] [Google Scholar]
- Dokholyan NV, Buldyrev SV, Havlin S, Stanley HE. Distributions of dimeric tandem repeats in non-coding and coding DNA sequences. J Theor Biol. 2000;202:273–282. doi: 10.1006/jtbi.1999.1052. [DOI] [PubMed] [Google Scholar]
- Duhagon MA, Dallagiovanna B, Garat B. Unusual features of poly[dT-dG].[dC-dA] stretches in CDS-flanking regions of Trypanosoma cruzi genome. Biochem Biophys Res Commun. 2001;287:98–103. doi: 10.1006/bbrc.2001.5545. [DOI] [PubMed] [Google Scholar]
- Eckert KA, Hile SE. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol Carcinog. 2009;48:379–388. doi: 10.1002/mc.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005a;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Peacock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005b;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- Epplen JT, Kyas A, Maueler W. Genomic simple repetitive DNAs are targets for differential binding of nuclear proteins. FEBS Lett. 1996;389:92–95. doi: 10.1016/0014-5793(96)00526-1. [DOI] [PubMed] [Google Scholar]
- Frank AC, Lobry JR. Asymmetric substitution patterns: a review of possible underlying mutational or selective mechanisms. Gene. 1999;238:65–77. doi: 10.1016/s0378-1119(99)00297-8. [DOI] [PubMed] [Google Scholar]
- Green P, Ewing B, Miller W, Thomas PJ, Green ED. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514–517. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- Haile S, Dupe A, Papadopoulou B. Deadenylation-independent stage-specific mRNA degradation in Leishmania. Nucleic Acids Res. 2008;36:1634–1644. doi: 10.1093/nar/gkn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Murray J, Neumann R. High-resolution mapping of crossovers in human sperm defines a minisatellite-associated recombination hotspot. Mol Cell. 1998;2:267–273. doi: 10.1016/s1097-2765(00)80138-0. [DOI] [PubMed] [Google Scholar]
- Jurka J, Pethiyagoda C. Simple repetitive DNA sequences from primates: compilation and analysis. J Mol Evol. 1995;40:120–126. doi: 10.1007/BF00167107. [DOI] [PubMed] [Google Scholar]
- Kelkar YD, Strubczewski N, Hile SE, Chiaromonte F, Eckert KA, Makova KD. What Is a Microsatellite: A Computational and Experimental Definition Based upon Repeat Mutational Behavior at A/T and GT/AC Repeats. Genome Biology and Evolution. 2010;2:620–635. doi: 10.1093/gbe/evq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11:2453–2465. doi: 10.1046/j.1365-294x.2002.01643.x. [DOI] [PubMed] [Google Scholar]
- Lowenhaupt K, Rich A, Pardue ML. Nonrandom distribution of long mono- and dinucleotide repeats in Drosophila chromosomes: correlations with dosage compensation, heterochromatin, and recombination. Mol Cell Biol. 1989;9:1173–1182. doi: 10.1128/mcb.9.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Ott J. GT repeats are associated with recombination on human chromosome 22. Genome Res. 2000;10:1108–1114. doi: 10.1101/gr.10.8.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Nguyen D, Stuart K, Myler PJ. Transcription initiation and termination on Leishmania major chromosome 3. Eukaryot Cell. 2004;3:506–517. doi: 10.1128/EC.3.2.506-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Yan S, Nguyen D, Fox M, Stuart K, Myler PJ. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol Cell. 2003;11:1291–1299. doi: 10.1016/s1097-2765(03)00143-6. [DOI] [PubMed] [Google Scholar]
- McDonagh PD, Myler PJ, Stuart K. The unusual gene organization of Leishmania major chromosome 1 may reflect novel transcription processes. Nucleic Acids Res. 2000;28:2800–2803. doi: 10.1093/nar/28.14.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AE, Silver PA. Systems perspectives on mRNA processing. Cell Res. 2007;17:581–590. doi: 10.1038/cr.2007.54. [DOI] [PubMed] [Google Scholar]
- Mugal CF, Wolf JB, von Grunberg HH, Ellegren H. Conservation of neutral substitution rate and substitutional asymmetries in mammalian genes. Genome Biol Evol. 2010;2:19–28. doi: 10.1093/gbe/evp056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson D, Andersson B. Strand asymmetry patterns in trypanosomatid parasites. Exp Parasitol. 2005;109:143–149. doi: 10.1016/j.exppara.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Obado SO, Taylor MC, Wilkinson SR, Bromley EV, Kelly JM. Functional mapping of a trypanosome centromere by chromosome fragmentation identifies a 16-kb GC-rich transcriptional “strand-switch” domain as a major feature. 2005:36–43. doi: 10.1101/gr.2895105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palenchar JB, Bellofatto V. Gene transcription in trypanosomes. Mol Biochem Parasitol. 2006;146:135–141. doi: 10.1016/j.molbiopara.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Plohl M, Luchetti A, Mestrovic N, Mantovani B. Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene. 2008;409:72–82. doi: 10.1016/j.gene.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Respuela P, Ferella M, Rada-Iglesias A, Aslund L. Histone acetylation and methylation at sites initiating divergent polycistronic transcription in Trypanosoma cruzi. J Biol Chem. 2008;283:15884–15892. doi: 10.1074/jbc.M802081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Ongoing evolution of strand composition in bacterial genomes. Mol Biol Evol. 2001;18:1789–1799. doi: 10.1093/oxfordjournals.molbev.a003966. [DOI] [PubMed] [Google Scholar]
- Sharma PC, Grover A, Kahl G. Mining microsatellites in eukaryotic genomes. Trends Biotechnol. 2007;25:490–498. doi: 10.1016/j.tibtech.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Sharma VK, Brahmachari SK, Ramachandran S. (TG/CA)n repeats in human gene families: abundance and selective patterns of distribution according to function and gene length. BMC Genomics. 2005;6:83. doi: 10.1186/1471-2164-6-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel TN, Hekstra DR, Kemp LE, Figueiredo LM, Lowell JE, Fenyo D, Wang X, Dewell S, Cross GA. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 2009;23:1063–1076. doi: 10.1101/gad.1790409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden RR. DNA structure and function. Academic Press; San Diego: 1994. [Google Scholar]
- Stallings RL. CpG suppression in vertebrate genomes does not account for the rarity of (CpG)n microsatellite repeats. Genomics. 1992;13:890–891. doi: 10.1016/0888-7543(92)90178-u. [DOI] [PubMed] [Google Scholar]
- Stern MZ, Gupta SK, Salmon-Divon M, Haham T, Barda O, Levi S, Wachtel C, Nilsen TW, Michaeli S. Multiple roles for polypyrimidine tract binding (PTB) proteins in trypanosome RNA metabolism. RNA. 2009;15:648–665. doi: 10.1261/rna.1230209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Trick M, Dover GA. Cryptic simplicity in DNA is a major source of genetic variation. Nature. 1986;322:652–656. doi: 10.1038/322652a0. [DOI] [PubMed] [Google Scholar]
- Thomas S, Green A, Sturm NR, Campbell DA, Myler PJ. Histone acetylations mark origins of polycistronic transcription in Leishmania major. BMC Genomics. 2009;10:152. doi: 10.1186/1471-2164-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth G, Gaspari Z, Jurka J. Microsatellites in Different Eukaryotic Genomes: Survey and Analysis. Genome Res. 2000;10:967–981. doi: 10.1101/gr.10.7.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall L, Christiansen T, Orwant J. Programming Perl. 3rd. O'Reilly; Beijing; Cambridge, Mass: 2000. [Google Scholar]
- Zhang W, He L, Liu W, Sun C, Ratain MJ. Exploring the relationship between polymorphic (TG/CA)n repeats in intron 1 regions and gene expression. Hum Genomics. 2009;3:236–245. doi: 10.1186/1479-7364-3-3-236. [DOI] [PMC free article] [PubMed] [Google Scholar]