

Abstract
Ynamides consist of a polarized triple bond that is directly attached to a nitrogen atom carrying a sulfonyl, an alkoxycarbonyl, an acyl or another electron withdrawing group. The triple bond polarization renders ynamides broadly useful building blocks with synthetic opportunities that go far beyond traditional alkyne chemistry. The versatile reactivity of ynamides in cycloadditions, cycloisomerizations, regioselective additions with various nucleophiles or electrophiles, ring-closing metathesis, and many other reactions has been investigated in detail during the past decades. A common feature of these methods is that the triple bond is consumed and either cleaved or transformed to a new functionality. The wealth of reports on these ynamide reactions is in stark contrast to the dearth of carbon-carbon bond formations that leave the triple bond of terminal ynamides intact. The recent introduction of effective synthetic methods for the preparation of terminal ynamides has set the stage to fully explore the synthetic potential of this intriguing class of compounds. This digest letter summarizes the most effective routes to terminal ynamides and the current state of selective nucleophilic addition, substitution and coupling reactions, including the first examples of asymmetric synthesis.
Keywords: Terminal ynamides, nucleophilic additions, coupling reactions
1. Introduction
The distinctive chemical properties and synthetic versatility of ynamines and ynamides have attracted rapidly increasing attention among synthetic chemists. The reactivity of the electron-rich, strongly polarized triple bond in ynamines and analogues thereof varies significantly from that of simple alkynes. Ynamines are rather unstable and readily hydrolyze toward amides, which complicates the synthesis, storage and use of these intriguing building blocks. Because the presence of an electron withdrawing acyl or sulfonyl group effectively dimishes the triple bond polarization, ynamides have become practical alternatives that facilitate handling and improve reaction control (Figure 1).
Figure 1.
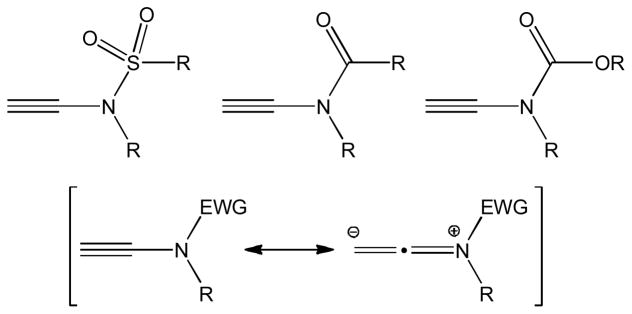
Structures of terminal ynamides.
The emergence of ynamides in the last 20 years has created new synthetic opportunities and challenges at the same time. Internal ynamides exhibiting a C-substituted triple bond have been used in many reactions and have been applied in the total synthesis of natural compounds.1 In stark contrast to alkynes, which have been employed extensively in Sonogashira couplings and nucleophilic addition and substitution reactions, the utilization of substrates carrying a terminal ynamide functionality typically trails behind the development of synthetic methods that exploit the more popular internal analogues. As a result, the majority of reactions of terminal ynamides reported to date do not conserve the triple bond,2 and cycloadditions,3 cycloisomerizations,4 Heck-Suzuki-Miyaura domino reactions,5 ring-closing metathesis,6 radical additions,7 and titanium-mediated carbon-carbon bond formations are among the most common synthetic transformations.8 Despite significant progress in the synthesis of terminal ynamides, carbon-carbon bond forming reactions that leave the triple bond intact are rare and have only recently been discovered (Scheme 1). This review discusses the current state of terminal ynamide synthesis and focusses on transformations that maintain the alkynyl motif.
Scheme 1.
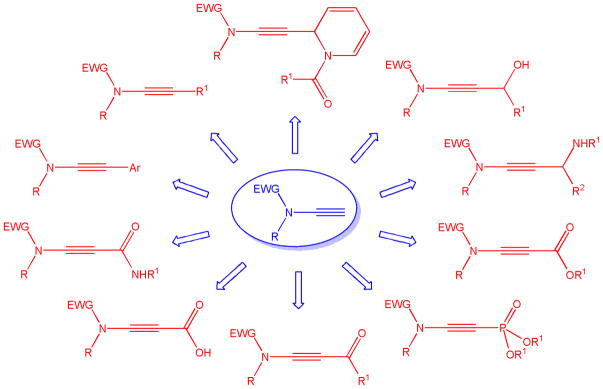
Overview of transformations of terminal ynamides leaving the triple bond intact.
2. Synthesis of terminal ynamides
To date, three major routes for the preparation of terminal ynamides have been developed.1,9 The first viable syntheses of terminal ynamides were based on elimination reactions of dichloro or trichloro enamides with n-butyllithium at low temperatures and subsequent quenching of the reaction with alcohol. More recently, the use of alkynyl iodonium salts and the extension of copper catalyzed C-N bond formation to ynamide synthesis have significantly improved the general scope and functional group compatibility. The reaction between lithiated amides and electrophilic alkynyl iodonium salts is believed to proceed via alkylidene carbene intermediates which preferentially rearrange to the corresponding ynamides, vide infra. Trimethylsilylated alkynyl iodonium salts were initially used to form silyl ynamides which were then subjected to deprotection with TBAF, but it was later found that terminal ynamides can be made directly from terminal alkynyl iodonium salts. The key step in the third main pathway to terminal ynamides is the copper catalyzed amidative cross-coupling of alkynyl halides, alkynyl trifluoroborates, alkynyl bismuthonium salts or terminal alkynes. In all cases, the alkyne moiety must be protected by a silyl group which is finally removed to yield the terminal ynamide (Scheme 2). Altogether, these three synthetic strategies provide convenient access to a variety of terminal ynamides that can easily be produced on the gram scale.
Scheme 2.
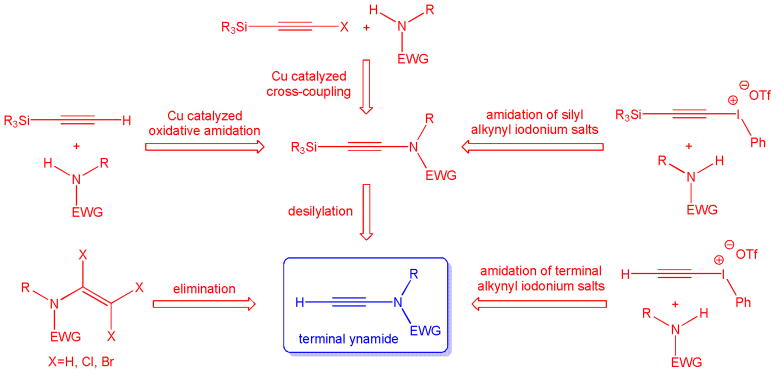
Synthetic routes to terminal ynamides.
2.1. Elimination method
In 1994, Zemlicka and coworkers showed successful conversion of purine and pyrimidine derived chloro enamines and enamides to terminal ynamines and ynamides, respectively.10 Deprotonation of thymine, 1, and cytosine, 2, with sodium hydride followed by addition of tetrachloroethylene, 3, and heating to 60 °C gave the trichloro enamides 4 and 5 in only up to 25% yield (Scheme 3). Treatment of 4 and 5 with n-butyllithium at −70 °C then furnished the terminal ynamides 6 and 7 in 34–51% yield. While this reaction sequence required the use of strong base and gave low overall yields, it represents the first access to terminal ynamides and it led to the development of quite successful elimination methods.
Scheme 3.
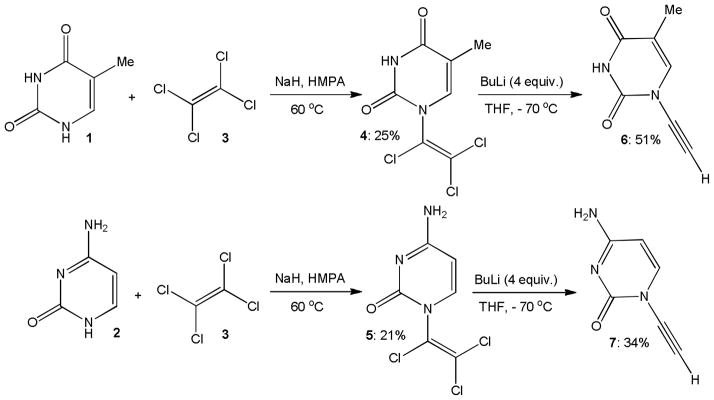
Thymine and cytosine derived ynamides.
Several years later, Brückner utilized dihalo enamides to develop a general synthetic entry to tosyl ynamides (Scheme 4).11 Inspired by the Corey-Fuchs transformation of aldehydes to alkynes,12 formamide 8 was transformed to the corresponding β, β-dibromo enamide 9 in 92% yield. Unfortunately, the conversion of 9 via 10 to ynesulfonamide 11 in the presence of n-butyllithium at −78 °C occurred with only 43% yield due to competing cleavage of the vinyl dibromide moiety and formation of considerable amounts of sulfonamide 12. This problem was solved by the replacement of carbon tetrabromide with carbon tetrachloride. Several β, β-dichloro enamide intermediates 14a–f were obtained in excellent yields and gave tosyl ynamides 15a–f under essentially the same conditions in 80–97% yield. Brückner’s method has been employed to make analogous ynamides 16 and 17 for use in coupling reactions and nucleophilic additions discussed below and ynamides 18–20 which were employed in Ru,13 Pt,4a and Au14 catalyzed isomerization reactions (Figure 2).
Scheme 4.

Brückner’s general synthesis of tosyl ynamides.
Figure 2.
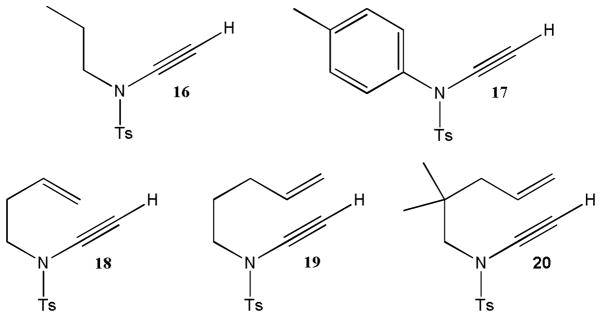
Selected ynamides synthesized with Brückner’s method.
The superior results observed with chloro enamides 14 compared to the low yield obtained with the brominated analogue 9 have been attributed to competing reaction pathways that may occur with the latter (Scheme 5).11b The dichloro enamides probably follow the Corey-Fuchs reaction course (pathway A). In this case, the dihalo enamide 21 preferentially undergoes deprotonation to 25 and subsequent lithium chloride elimination to chloro ynamide 26 which then reacts with another equivalent of butyllithium to the terminal ynesulfonamide 15. Alternatively, lithium-halide exchange of the diahalo enamide 21 can generate the lithiated enamide 22 which may eliminate tosylamide 23 (path B) or form an alkylidene carbene 27 that spontaneously rearranges to 15 (path C). The outcome of the reaction with 9 suggests that the lithium-bromide exchange predominates over the deprotonation path and thus leads to 12 as the major product.
Scheme 5.

Possible reaction pathways of dihalo enamides.
2.2. Amidation of alkynyl iodonium salts
Witulski and Stengel were first to realize that C-N bond formation with alkynyl iodonium salts, originally introduced by Stang et al. for the preparation of ynamines,15 provides new opportunities for terminal ynamide synthesis They employed trimethylsilylethynyl(phenyl)iodonium triflate, 29, which can be prepared from a stanna- or silaacetylene precursor, in the coupling reaction with amides and sulfonamides (Scheme 6).16 In this one-pot procedure, amides 28 are first deprotonated with butyllithium and then treated with 29 at room temperature.17 It is generally believed that the reaction proceeds through alkylidene carbene intermediates 30 which undergo spontaneous 1,2-migration of the silyl group to form silylated ynamides 31 in moderate to high yields. Interestingly, intramolecular CH-insertion to dihydropyrroles 33 was not observed. Desilylation of 31 with TBAF in wet THF at 0 °C gave the terminal ynamides 32 in high yields. Some of the ynamides prepared with this method were applied to inter- and intramolecular cycloadditions.16
Scheme 6.

Amidation with trimethylsilylethynyl(phenyl)iodonium triflate, 29.
The scope of alkynyl iodoinum salt amidation was further expanded to a variety of diynes which proved invaluable substrates for [2+2+2]cycloadditions producing an array of substituted indolines3j and carbazoles,3l and other ynamides used in [4+2]cycloadditions18 or Pauson-Khand reactions (Scheme 7).3i It is noteworthy that this method tolerates several functional groups, including alkenyl, alkynyl, carbamoyl, alkoxy, acetal, and alkoxycarbonyl moieties, albeit yields vary significantly. Interestingly, terminal ynamides can also be obtained directly from ethynyl(phenyl)iodonium triflate, 34, which eliminates the desilylation step. Comparison of the two methods shows that N-alkynylation with 34 gives consistently higher yields than the reaction with the silylated iodonium salt 29.
Scheme 7.

Amidations with free and TMS protected alkynyl iodonium salts 29 and 34.
The Witulski group also applied alkynyl iodonium triflate 34 in the synthesis of ynamides carrying a chiral substituent such as 36. Several N-sulfonyl protected allylglycine methyl esters 35 were found to undergo smooth amidation in the presence of cesium carbonate as base within 3 hours at room temperature and no sign of racemization was observed. Yields for 36a–d were generally high and ranged from 70% to 95%, but N-Boc and N-acetyl derivatives did not react and the corresponding ynamides 36e and 36f were not detected (Scheme 8).19
Scheme 8.
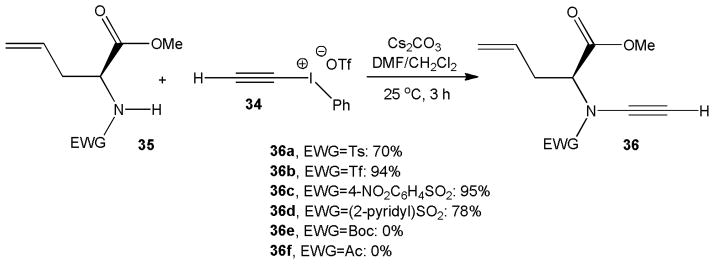
N-alkynylation of enantiopure allylglycine derivatives.
Other important ynamides synthesized by this method include the N-ethynylcarbamates 32c and 37 which have been employed in gold catalyzed cyclizations forming oxazolinones,20 and the oxazolidinone 38, another example of a chiral ynamide synthesized with Witulski’s method, which was used in Cadiot-Chodkiewicz reactions, vide infra (Figure 3).21
Figure 3.
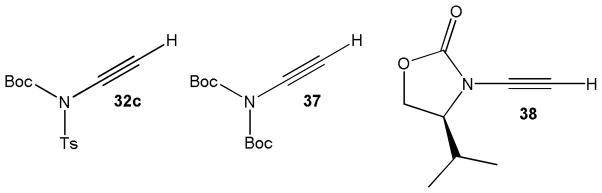
Other important ynamides prepared by the alkynyl iodonium method.
2.3. Copper catalyzed C-N bond formation
The coupling of silyl protected alkynyl halides and amides pioneered by the Hsung group has become the most versatile method for the synthesis of terminal ynamides (Scheme 9).22 Initial investigations with palladium catalysts, inspired by general progress with N-arylations of amides and amines, were unsuccessful due to predominant homocoupling toward 1,3-dialkynes. Copper-catalyzed coupling of amides 28 with alkynyl halides proved more promising, though competition with the homocoupling pathway persisted until alkynyl iodides were substituted with alkynyl bromides. In the presence of catalytic amounts of CuCN and N,N’-dimethyl ethylendiamine (DMEDA) and excess of potassium phosphate at 110–150 °C, the coupling of oxazolidinone nucleophiles with 1-bromo-2-triisopropylacetylene, 39, gave cyclic ynecarbamates 40a–c in 70–85% yield. The camphor-derived ynamide 40d and acylic ynecarbamates 40e and 40f, however, were obtained in poor yields with this method. The TIPS protected ynamides are readily converted to the corresponding terminal ynamides. For example, 40a was desilylated with TBAF in 71% yield within 5 minutes.
Scheme 9.
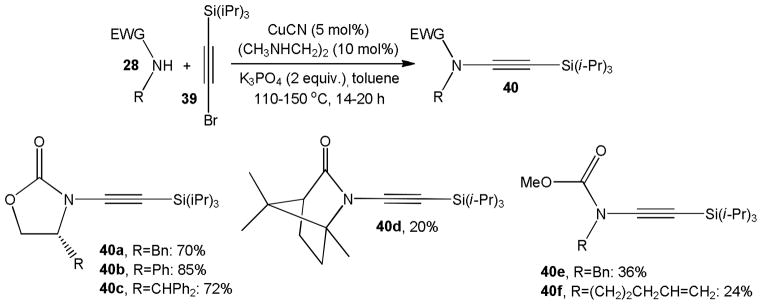
CuCN catalyzed N-alkynylation of oxazolidinones, amides and carbamates.
Under essentially the same conditions as shown in Scheme 9, Urabe et al. prepared several TMS protected ynesulfonamides 41a–e as well as 15e using 5 mol% of CuI as catalyst for the coupling of cyclic and acyclic substrates with 1-bromo-2-trimethylsilylacetylene, 42 (Figure 4).23 Although the free terminal ynamides were not isolated in this study, they are easily obtained via TBAF desilylation as has been shown by Witulski for ynesulfonamide 15e.16
Figure 4.

Ynesulfonamides synthesized with CuI as catalyst.
Danheiser introduced a stepwise approach to extend the scope of this approach and accomplished copper promoted ynamide formation under mild conditions.24 The amide substrate 43 was first converted to a copper complex with stoichiometric amounts of CuI and KHMDS and then treated with trialkylsilylethynyl halide 39 or 42 (Scheme 10). While this protocol is not catalytic, the reaction occurs at room temperature and several silyl protected ynecarbamates 44 were prepared in superior yields. As expected, 44b was successfully desilylated with TBAF to the corresponding terminal ynamide 44c in 81% yield.
Scheme 10.
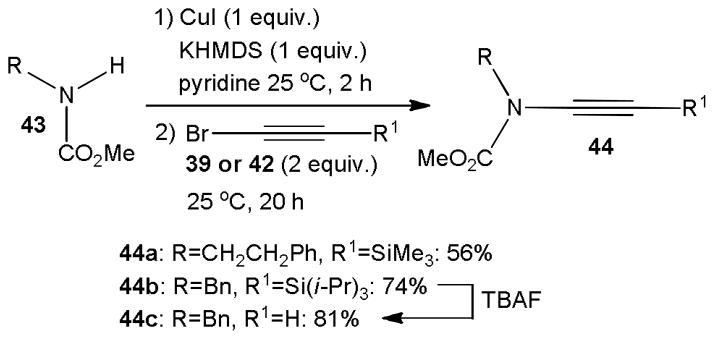
Copper mediated N-alkynylation of selected carbamates.
Hsung,25 Tam26 and others later increased the efficiency and scope of the catalytic ynamide formation using either copper sulfate, copper thiophene-2-carboxylate (CuTC) or cuprous iodide in combination with 1,10-phenanthroline or DMEDA as ligand.27 With these modifications, remarkable results have been obtained with several imidazolidinone, carbamate, 3-alkoxycarbonylindole, sulfonamide and phosphoramidate nucleophiles 28 and bromoalkyne 39, and the C-N bond formation was found to occur at lower temperatures than previously reported (Scheme 11). An impressive variety of terminal ynamides, including 40a–b and 40g–x, has been prepared by this method. For example, the cyclic ynecarbamate 40b was prepared in gram quantities on the 100 mmol scale with up to 97% yield which compares favorably with the CuCN method.25c It is noteworthy that Zhang et al. reported an iron catalyzed amidation protocol that uses FeCl3 under otherwise very similar conditions.28 The yields of the silylated ynamides prepared by this method were generally lower than those obtained by the copper catalyzed processes shown in Scheme 11 and the amidation reaction may have been partly affected by the presence of small copper impurities in the iron salt used.29 As discussed above, the silyl compounds 40 are readily cleaved with TBAF or potassium carbonate to furnish the corresponding terminal ynamides.24b
Scheme 11.

Scope of the ynamide synthesis using catalytic amounts of copper and phenanthroline.
Evano’s group introduced potassium trifluoroborates, which can be prepared from terminal alkynes by subsequent lithiation, addition of trimethyl borate and treatment with potassium hydrogen difluoride, as alkynyl transfer agents to copper catalyzed ynamide synthesis and obtained 31d from 45 in 51% yield.30 Remarkable features of this approach are that the reaction occurs under air at room temperature and in the absence of strong base (Scheme 12). The sparse solubility of the alkynyl trifluoroborate 46 in dichloromethane is critical to the success of the catalytic C-N coupling. The use of DMSO as solvent, in which 46 is readily soluble, led to exclusive formation of the undesired alkyne homodimer. The heterogeneous reaction conditions, however, seem to be the major reason for the long reaction times. The overall scope of this method for the synthesis of terminal ynamides remains to be fully explored.
Scheme 12.
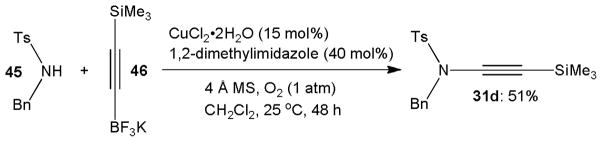
Catalytic C-N bond formation with an alkynyl trifluoroborate.
A significant contribution to this field came from the Stahl group.31 They developed an aerobic oxidative procedure that directly couples amides 28 and terminal alkynes 47, thus avoiding the requirement for a halogenated alkyne precursor (Scheme 13). This method provides convenient access to a variety of silylated ynamides 48a–i that were obtained with remarkable yields. The oxidative C-N coupling of carbamate, urea and sulfonamide nucleophiles occurs in the presence of catalytic amounts of copper chloride and mild base at 70 °C. The use of indole derived nucleophiles, however, typically requires stoichiometric copper amounts. A remaining drawback is that large excess (5 equivalents) of the N-nucleophile is necessary to limit the formation of alkynyl chloride and homocoupled alkyne byproducts. Ynamides obtained by this method have been desilylated to provide terminal alkynes.32 For example, the silyl group of 48i was quantitatively removed with TBAF within 5 minutes at room temperature.
Scheme 13.
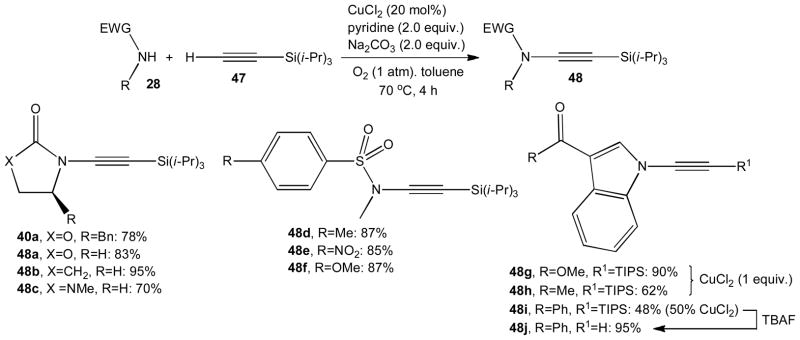
Oxidative alkynylation of various N-nucleophiles.
A mechanism explaining the reaction course was proposed by the same group (Scheme 14).31 During formation of 48d the copper catalyst 49 probably first reacts with trimethylsilylacetylene, 47a, to form the acetylide complex 50 in the presence of base. Complex 50 can then either accept a second acetylide to yield 51 and subsequently dialkyne 52 or react with the sulfonamide 53 to the copper(II) species 54. The latter direction should be favored and outperform the homocoupling pathway toward 52 when 53 is present in excess. Finally, reductive elimination and reoxidation of the copper catalyst produces ynamide 48d and regenerates 49.
Scheme 14.
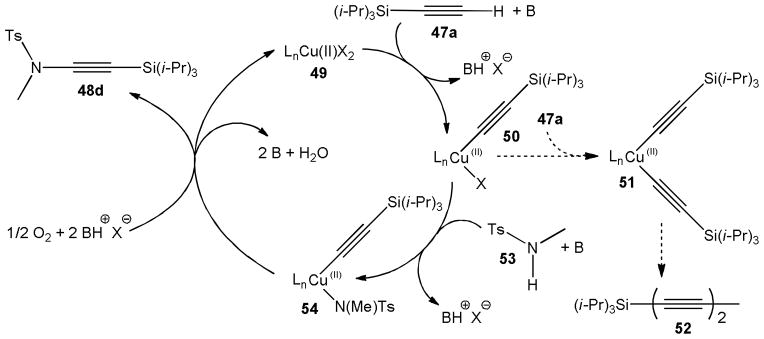
Mechanism of the copper catalyzed oxidative C-N bond formation with terminal alkynes as postulated by Stahl.
Few TMS protected ynimides have been synthesized by coupling of succinimide or phthalimide 55 with alkynyl bismuthonium salts 56 which first have to be prepared from the corresponding potassium alkynyltrifluoroborate precursors (Scheme 15).33 The yields of the silylated ynimides were initially poor due to significant side reactions forming the homocoupled dialkyne derivatives and N-aryl imide byproducts. Following Stahl’s work, the homocoupling pathway was suppressed by using the imide in excess and 57a and 57b were produced with 52 to 57% yield. Terminal ynimides cannot be directly synthesized with ethynyl bismuthonium salts, but 57b was desilylated to the corresponding free terminal ynimide with TBAF at 0 °C in 88% yield.
Scheme 15.

Ynimide synthesis with alkynyl bismuthonium salts.
3. Nucleophilic addition and substitution reactions
Despite the general success and utility of carbon-carbon bond formation with terminal alkynes, few sporadic reports of nucleophilic 1,2-additions of terminal ynamides and ynamines to carbonyl compounds and other electrophiles have appeared in the literature.34 Saá and coworkers were first to show the broad potential of nucleophilic ynamide additions. Because deprotonation of tosyl ynamides with LDA and other strong bases followed by trapping with trimethylsilyl chloride or benzaldehyde gave unsatisfactory results, they resorted to in situ formation of the lithiated ynamide 58 from its β, β-dichloro enamide precursor 14.35 Treatment of 14 with two equivalents of butyllithium at −78 °C and subsequent reaction of the lithiated ynamide with various electrophiles gave the expected products 59 in good to high yields (Table 1). For example, addition of TMSCl and dimethyl sulfate gave 59a and 59b in 78% and 74% yield, respectively (entries 1 and 2). With the exception of tert-butyl isocyanate, which gave 59f in only 53% yield, benzaldehyde and other carbonyl nucleophiles were converted to the corresponding 1,2-addition products 59c–g in 88% to 96% yield and the procedure was also successfully applied to diethyl chlorophosphate, producing 59h in 73% yield (entries 3–8). As expected, similar results were obtained by acetylation of other tosyl ynamides to 59i–l (entries 9–12).
Table 1.
Ynamide additions to common nucleophiles.
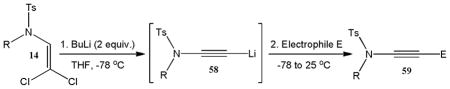
| ||||
|---|---|---|---|---|
| Entry | R | Electrophile | Product | Yield (%) |
| 1 | Ph | TMSCl |
 59a |
78 |
| 2 | Ph | Me2SO4 |
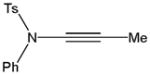 59b |
74 |
| 3 | Ph | PhCHO |
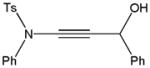 59c |
88 |
| 4 | Ph | Ac2O |
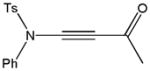 59d |
90 |
| 5 | Ph | ClCO2Et |
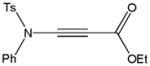 59e |
96 |
| 6 | Ph | t-BuNCO |
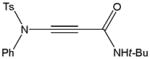 59f |
53 |
| 7 | Ph | CO2 |
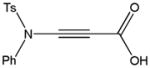 59g |
90 |
| 8 | Ph | ClP(O)(OEt)2 |
 59h |
73 |
| 9 | 4-MeC6H4 | Ac2O |
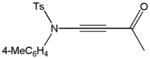 59i |
86 |
| 10 | Bn | Ac2O |
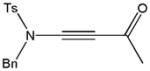 59j |
84 |
| 11 | Pr | Ac2O |
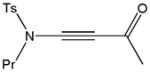 59k |
84 |
| 12 | CH2=CHCH2 | Ac2O |
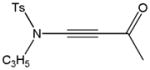 59l |
77 |
Careful deprotonation of terminal ynamides and ynehydrazides such as 60 and 62 with lithium hexamethyldisilazide (LHMDS) or another strong base has remained an attractive alternative to Saá’s in situ generation of lithium ynamides shown above.4b,22,36 Additions of the alkylnyllithium intermediates to ethyl chloroformate gave 61 and 63 in 90% and 47% yield, respectively. A particularly interesting example is the SN2 methylation of the phosphoramidate-derived ynamide 64 to the farnesol derivative 65 which produced a mixture of bicyclic enamine 66 and tricyclic imine 67 upon heating (Scheme 16).37 In analogy to the addition to carbonyl electrophiles, the deprotonation of terminal ynamides 68 with LHMDS at −50 °C followed by addition of activated aromatic imines 69 was reported to give a variety of N-carbamoyl- and N-sulfonyl-γ-amino ynamides 70a–h in good yields.38
Scheme 16.

Deprotonation of terminal ynamides for nucleophilic addition and substitution reactions.
We recently demonstrated that copper iodide catalyzes the addition of ynesulfonamide 14f to acyl chlorides 71 and several N-heterocycles 73 activated in situ with ethyl chloroformate at room temperature.39 The reaction with aromatic acyl chlorides 71 gave a series of 3-aminoynones 72a–f in high yields ranging from 79 to 99% (Scheme 17). Steric hindrance does not seem to affect the reaction and the ynamide addition to pivaloyl chloride gave 72g in 90% yield. Aliphatic acyl chlorides that can undergo ketene formation and related side reactions remain suitable substrates for 3-aminoynone synthesis at slightly reduced temperatures. For example, 72h was obtained in 70% yield when the reaction was performed at 15 °C.
Scheme 17.

Copper catalyzed nucleophilic ynamide addition to acyl chlorides and activated N-heterocycles.
We found that the copper catalyzed addition of 14f to alkyl chloroformates does not occur under the same conditions. This observation prompted us to investigate if this ynamide can be added to pyridine and other N-heterocycles activated in situ with ethyl chloroformate. Again, copper iodide catalysis proved successful and we were able to produce 1,2-dihydropyridines 74a–e in 71–96% yield (Scheme 17). The addition product obtained from 4-methoxypyridine was unstable and hydrolyzed during chromatographic purification to ketone 74f which was isolated in 71% yield. Several quinolines and phenanthridine were then employed in the same protocol to give 74g–j in 82–95% yield. This reaction generally occurs with high conversion but yields can be compromised if 1,4-addition is possible. Significant amounts of the 1,4-regioisomer were obtained with pyridine and other substrates unless the para position was blocked. A proposed catalytic cycle of the ynamide addition to the activated N-heterocyclic substrate 77 formed in situ from quinoline, 73g, is shown in Scheme 18. It is assumed that 14f reacts with CuI via 75 to a copper acetylide complex 76. Nucleophilic attack at 77 then produces the 1,2-dihydroquinoline derivative 74g and regenerates the copper catalyst.
Scheme 18.

Catalytic cycle of the copper catalyzed ynamide addition to activated quinoline.
The progress with nucleophilic ynamide additions to various electrophiles discussed above set the stage for stereoselective variants. Hsung and coworkers were first to show that ynamides can be practical prenucleophiles in asymmetric additions.40 They observed that lithiation of N-sulfonyl ynamides 78 with LHMDS followed by addition of chiral N-tert-butanesulfinyl imines 79 at −40 °C affords the corresponding 1,2-addition products 80a–g in good to high yields and with impressive diastereoselectivity (Scheme 19). The reactions between the N-benzyl-N-tosyl ynamide 78 with 4 different activated aldimines carrying a tert-butanesulfinyl moiety gave the propargylic amines 80a–d in 63-77% yield and with at least 20:1 dr. For example, 80a was obtained from the benzaldehyde derived N-tert-butanesulfinyl imine in 69% yield and 25:1 dr. The para-substituent in the arylsulfonyl group exerts a strong influence on the yield and diastereoselectivity of this reaction. Replacement of the para-methyl group in 78 by a methoxy group increases the yield to 95% while the diastereomeric ratio remains unchanged. The para-nitro analogue, however, gives 80f in 79% yield and only 4:1 dr. In all cases, the presence of an (S)-tert-butanesulfinyl auxiliary was found to favor a Re-face attack leading to the (S)-propargylic amines. This was attributed to a Zimmermann-Traxler chair-like transition state.
Scheme 19.
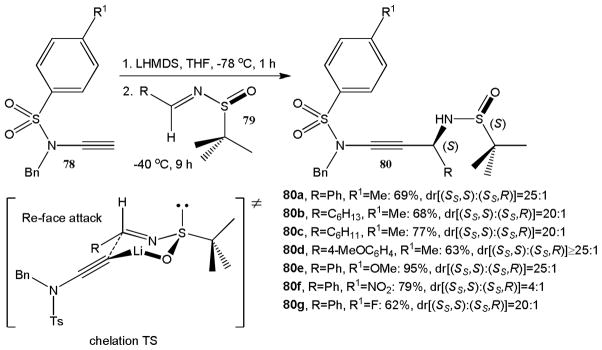
Diastereoselective synthesis of propargylic amines 80 from tert-butanesulfinyl derived imines 79.
Interestingly, addition of selected Lewis acids can completely reverse the sense of asymmetric induction (Scheme 20). When stoichiometric amounts of boron trifluoride diethyl etherate were added to the lithiated ynamide, the reaction with 79 gave the (R)-configured addition products 80a–g with high diastereoselectivity and yields were generally higher than under Lewis acid free conditions. The reversal of the chiral induction was rationalized with two possible open chain transition states, both favoring a Si-face attack on the (S)-tert-butanesulfinyl imine.
Scheme 20.

Boron promoted diastereoselective addition of sulfonyl ynamides 78 to N-tert-butanesulfinyl imines 79.
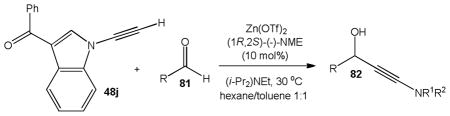
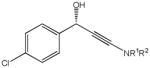
The first catalytic enantioselective nucleophilic 1,2-addition with ynamides was accomplished in our laboratory.32 We realized that terminal ynamides undergo zinc catalyzed addition with aldehydes under mild conditions and started with the development of an asymmetric procedure. Screening of a wide range of chiral ligands, solvents, base and variation of the prenucleophile revealed that indole derived ynamides are particularly useful and give the corresponding propargylic alcohols in superior yields and ee compared to ynesulfonamides and ynecarbamates. The initial reaction between 3-benzoylindolyl ynamide 48j and 4-bromobenzaldehyde, 81a, in the presence of catalytic amounts of zinc triflate and N-methyl ephedrine in toluene gave 82a in 87% yield and 77% ee. An interesting feature of this reaction is that it is reversible. We observed considerable racemization of the product leading to reduced ee’s unless apolar solvents in which the addition products precipitate are used. We therefore switched to toluene/hexane mixtures to impede racemization and isolated 82a in 97% yield and 93% ee (Table 2, entry 1). The procedure is operationally simple and has a wide substrate scope. Very high yields and ee’s were obtained with other electron-rich and electron-deficient aromatic aldehydes 81b–l and the 3-aminopropargylic alcohols 82b–l were isolated in 81–93% yield and up to 96% ee (entries 2–12). Similar results were also obtained with aliphatic aldehydes 81m and 81n (entries 13 and 14).
Table 2.
Catalytic asymmetric ynamide addition to aldehydes.
| |||||
|---|---|---|---|---|---|
| Entry | Aldehyde | Product | Time (h) | Yield (%) | Ee (%) |
| 1 |
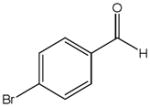 81a |
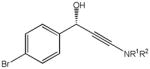 82a |
18 | 97 | 93 |
| 2 |
 81b |
 82b |
16 | 92 | 95 |
| 3 |
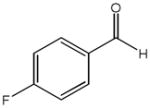 81c |
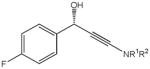 82c |
18 | 85 | 93 |
| 4 |
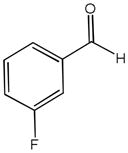 81d |
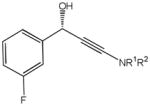 82d |
18.5 | 87 | 88 |
| 5 |
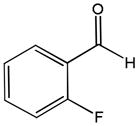 81e |
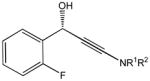 82e |
13 | 92 | 71 |
| 6b |
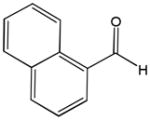 81f |
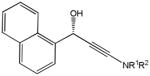 82f |
18 | 92 | 96 |
| 7 |
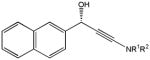
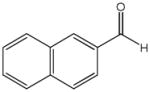 81g |
 82g |
17 | 92 | 90 |
| 8 |
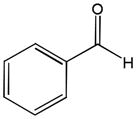 81h |
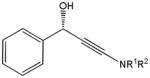 82h |
18 | 87 | 84 |
| 9 |
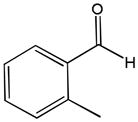 81i |
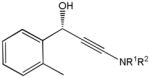 82i |
15 | 93 | 95 |
| 10 |
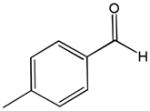 81j |
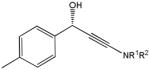 82j |
20 | 80 | 88 |
| 11 |
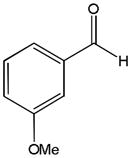 81k |
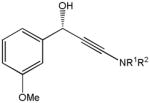 82k |
26 | 92 | 87 |
| 12 |
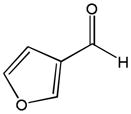 81l |
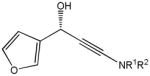 82l |
21 | 81 | 87 |
| 13b |
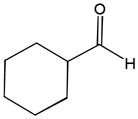 81m |
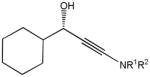 82m |
15 | 89 | 90 |
| 14 |
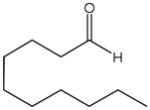 81n |
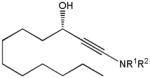 82n |
13 | 87 | 95 |
4. Coupling reactions
The Sonogashira reaction and oxidative dimerizations of terminal alkynes have become very popular methods for practical carbon-carbon bond formation. Surprisingly, the possibility of catalytic cross- and homocoupling with ynamides has rarely been explored.41 Saá and coworkers successfully applied a Glaser-Hay coupling protocol to a few ynesulfonamides 15 and obtained diynes 83a–e in excellent yields. The oxidative coupling is catalyzed by CuI in the presence of TMEDA and atmospheric dioxygen, and it is generally complete within 3 hours at room temperature (Scheme 21).42 Alternatively, copper catalyzed Cadiot-Chodkiewicz cross-coupling of sulfonyl and carbamoyl ynamides 32 with the electron-deficient bromoalkyne 84 has been reported to proceed at ambient temperatures. The push-pull diynes 85 were isolated in up to 93% yield.21
Scheme 21.

Glaser-Hay coupling and Cadiot-Chodkiewicz cross-coupling of ynamides.
Saá and coworkers also reported palladium catalyzed cross coupling of zinc ynamides with aryl iodides. This coupling procedure requires lithiation of dichlorovinyl amides 14 and subsequent treatment with zinc dibromide. The in situ formed zinc ynamides 86 were then employed in palladium catalyzed coupling reactions to afford a variety of C-substituted ynamides 87 (Scheme 22).43 The coupling with simple aryl iodides furnished 87a–c in moderate 63–69% yield. The reaction is of limited use when electron-rich aryl iodides are employed and the methoxy-derived products 87d–f and 87j were obtained in only 24–48% yield. Not unexpectedly, the reaction with electron-deficient aryl halides such as 4-nitro-iodobenzene and pyridine or pyrimidines gave 87g–i, 87k, and 87l in up to 92% yield.
Scheme 22.

Palladium catalyzed cross coupling of zinc ynamides with aryl iodides.
Hsung demonstrated that terminal ynecarbamates 88 are suitable substrates for typical Sonogashira reaction protocols.44 They used Pd(PPh3)4 as catalyst and CuI or CuCN as cocatalyst to affect smooth transmetallation with aryl iodides at room temperature (Scheme 23). The coupling products 89a–d were obtained with varying yields and the screening of the copper source is apparently important. Unfortunately, bromobenzene gave poor results and effective ynamide coupling with aryl bromides and chlorides is still elusive to date. A noteworthy variation of the Sonogashira reaction with ynamides is the coupling with 2-iodoanilines. In this case, the C-arylation of the terminal ynamide is followed by spontaneous 5-endo-dig cyclization toward 2-amidoindoles.45
Scheme 23.

Sonogashira coupling with ynamides.
5. Iodination
2-Haloynamides such as 26 may not only serve as intermediate precursors in the synthesis of terminal ynamides but also as highly versatile, isolable reagents (Scheme 5). Danheiser’s group recognized the synthetic potential of 2-iodoynamides and prepared 90a–c by deprotonation of terminal ynamides with either n-butyllithium or KHMDS and subsequent addition of iodine or 1,2-diiodoethane as the iodination reagent (Scheme 24).46 The use of 2-iodoynamides in [2+2] cycloadditions with ketenes was found to provide invaluable access to multifunctional iodocyclobutenones.
Scheme 24.

Iodination of terminal ynamides.
6. Outlook
The unique properties and the distinct reactivity of terminal ynamides continue to draw increasing attention to this synthetically very useful class of compounds. While most ynamides can be stored conveniently at room temperature and under air, which greatly facilitates preparation and use, they often do not react like simple alkynes. In stark contrast to terminal alkynes, reactions with the highly polarized ynamide analogues that leave the triple bond intact have rarely been investigated. It is evident that the progress with the development of orthogonal methods for the synthesis of a wide variety of terminal ynamides has greatly facilitated and inspired recent studies exploring efficient functionalization of these versatile building blocks. The general utility of terminal ynamides in catalytic cross-couplings, nucleophilic additions, asymmetric carbon-carbon bond formations and other reactions that do not consume the N-substituted alkyne moiety, however, remains to be fully explored. It is expected that the number of publications on both synthetic and mechanistic aspects of this emerging area will continue to rapidly increase and that terminal ynamides will soon find broad use in many applications including total synthesis.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM106260).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References and notes
- 1.For excellent reviews, see: Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei LL. Tetrahedron. 2001;57:7575–7606.Evano G, Coste A, Jouvin K. Angew Chem Int Ed. 2010;49:2840–2859. doi: 10.1002/anie.200905817.DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem Rev. 2010;110:5064–5106. doi: 10.1021/cr100003s.Wang XN, Yeom HS, Fang LC, He S, Ma ZX, Kedrowski BL, Hsung RP. Acc Chem Res. 2014;47:560–578. doi: 10.1021/ar400193g.
- 2.Selected examples: Hydroboration: Witulski B, Buschmann N, Bergsträsser U. Tetrahedron. 2000;56:8473–8480.Oxoarylations and cyclizations: Bhunia S, Chang CJ, Liu RS. Org Lett. 2012;14:5522–5525. doi: 10.1021/ol302621z.Yang LQ, Wang KB, Li CY. Eur J Org Chem. 2013:2775–2779.Couty S, Meyer C, Cossy J. Synlett. 2007:2819–282.Al-Rashid ZF, Hsung RP. Org Lett. 2008;10:661–663. doi: 10.1021/ol703083k.Homologation reactions: You L, Al-Rashid ZF, Figueroa R, Ghosh SK, Ti G, Lu T, Hsung RP. Synlett. 2007:1656–1662.
- 3.[3+2] Cycloadditions: IJsselstijn M, Cintrat JC. Tetrahedron. 2006;62:3837–3842.Li H, You L, Zhang X, Johnson WL, Figueroa R, Hsung RP. Heterocycles. 2007;74:553–568.Li H, Hsung RP. Org Lett. 2009;11:4462–4465. doi: 10.1021/ol901860b.Zhang X, Li H, You L, Tang Y, Hsung RP. Adv Synth Catal. 2006;348:2437–2442.Zhang X, Hsung RP, Li H. Chem Commun. 2007:2420–2422. doi: 10.1039/b701040k.Kim JY, Kim SH, Chang S. Tetrahedron Lett. 2008;49:1745–1749.Zhang X, Hsung RP, Li H, Zhang Y, Johnson WL, Figueroa R. Org Lett. 2008;10:3477–3479. doi: 10.1021/ol801257j. [4+2] Cycloadditions: Martinez-Esperon MF, Rodriguez D, Castedo L, Saá C. Org Lett. 2005;7:2213–2216. doi: 10.1021/ol050609a.Dunetz JR, Danheiser RL. J Am Chem Soc. 2005;127:5776–5777. doi: 10.1021/ja051180l.[2+2+1] Cycloadditions: Witulski B, Goessmann M. Synlett. 2000:1793–1797.[2+2+2] Cycloadditions: Witulski B, Stengel T. Angew Chem Int Ed. 1999;38:2426–2430.Witulski B, Stengel T, Fernandez-Hernandez J. Chem Commun. 2000:1965–1966.Witulski B, Alayrac C. Angew Chem Int Ed. 2002;41:3281–3284. doi: 10.1002/1521-3773(20020902)41:17<3281::AID-ANIE3281>3.0.CO;2-G.Dateer RB, Shaibu BS, Liu RS. Angew Chem Int Ed. 2012;51:113–117. doi: 10.1002/anie.201105921.[2+2] Cycloadditions: Kohnen AL, Mak XY, Lam TY, Dunetz JR, Danheiser RL. Tetrahedron. 2006;62:3815–3822. doi: 10.1016/j.tet.2005.11.088.
- 4.(a) Marion F, Coulomb J, Courillon C, Fensterbank L, Malacria M. Org Lett. 2004;6:1509–1511. doi: 10.1021/ol049530g. [DOI] [PubMed] [Google Scholar]; (b) Marion F, Coulomb J, Servais A, Courillon C, Fensterbank L, Malacria M. Tetrahedron. 2006;62:3856–3871. [Google Scholar]; (c) Hashmi AS, Rudolph M, Bats J, Frey W, Rominger F, Oeser T. Chem Eur J. 2008;14:6672–6678. doi: 10.1002/chem.200800210. [DOI] [PubMed] [Google Scholar]; (d) Couty S, Meyer C, Cossy J. Tetrahedron. 2009;65:1809–1832. [Google Scholar]
- 5.(a) Couty S, Liegault B, Meyer C, Cossy J. Org Lett. 2004;6:2511–2514. doi: 10.1021/ol049302m. [DOI] [PubMed] [Google Scholar]; (b) Couty S, Liegault B, Meyer C, Cossy J. Tetrahedron. 2006;62:3882–3895. [Google Scholar]
- 6.Saito N, Sato Y, Mori M. Org Lett. 2002;4:803–805. doi: 10.1021/ol017298y. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee B, Litvinov DN, Kang J, Bettale JD, Castle SL. Org Lett. 2010;12:2650–2652. doi: 10.1021/ol1008679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Tanaka R, Hirano S, Urabe H, Sato F. Org Lett. 2003;5:67–70. doi: 10.1021/ol027209x. [DOI] [PubMed] [Google Scholar]; (b) Tanaka R, Yuza A, Watai Y, Suzuki D, Takayama Y, Sato F, Urabe M. J Am Chem Soc. 2005;127:7774–7780. doi: 10.1021/ja050261e. [DOI] [PubMed] [Google Scholar]; (c) Tanaka D, Sato Y, Mori M. J Am Chem Soc. 2007;129:7730–7731. doi: 10.1021/ja071954t. [DOI] [PubMed] [Google Scholar]
- 9.For additional literature that discusses ynamide synthesis: Lu T, Hsung RP. Arch Org Chem. 2014:127–141.Evano G, Gaumont AC, Alayrac C, Wrona IE, Giguere JR, Delacroix O, Bayle A, Jouvin K, Theunissen C, Gatignol J, Silvanus AC. Tetrahedron. 2014;70:1529–1616.Evano G, Jouvin K, Coste A. Synthesis. 2013;45:17–26.Beletskaya IP, Cheprakov AV. Organometallics. 2012;31:7753–7808.Brand JP, Waser J. Chem Soc Rev. 2012;41:4165–4179. doi: 10.1039/c2cs35034c.Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379–1390.
- 10.Ramachandra VJ, Xu ZQ, Ksebati MB, Kessel D, Corbett TH, Drach JC, Zemlicka J. J Chem Soc Perkin, Trans. 1994;1:1089–1098. [Google Scholar]
- 11.(a) Brückner D. Synlett. 2000:1402–1404. [Google Scholar]; (b) Brückner D. Tetrahedron. 2006;62:3809–3814. [Google Scholar]
- 12.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769–3772. [Google Scholar]
- 13.(a) Saito N, Sato Y, Mori M. Org Lett. 2002;4:803–805. doi: 10.1021/ol017298y. [DOI] [PubMed] [Google Scholar]; (b) Mori M, Wakamatsu H, Saito N, Sato Y, Narita R, Sato Y, Fujita R. Tetrahedron. 2006;62:3872–3881. [Google Scholar]
- 14.Couty S, Meyer C, Cossy J. Angew Chem Int Ed. 2006;45:6726–6730. doi: 10.1002/anie.200602270. [DOI] [PubMed] [Google Scholar]
- 15.Murch P, Williamson BL, Stang PJ. Synthesis. 1994:1255–1256. [Google Scholar]
- 16.Witulski B, Stengel T. Angew Chem Int Ed. 1998;37:489–492. doi: 10.1002/(SICI)1521-3773(19980302)37:4<489::AID-ANIE489>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 17.For other examples of TMS protected ynamides prepared by the alkynyl iodonium method, see: Rainier JD, Imbriglio JE. Org Lett. 1999;1:2037–2039.Rainier JD, Imbriglio JE. J Org Chem. 2000;65:7272–7226. doi: 10.1021/jo001044t.For N-ethynylimidazoles: Kerwin SM, Nadipuram A. Synlett. 2004:1404–1408.Kerwin SM, Nadipuram A. Tetrahedron Lett. 2006;47:353–356.
- 18.Witulski B, Lumtscher J, Bergsträsser U. Synlett. 2003:708–710. [Google Scholar]
- 19.Witulski B, Göβmann M. Chem Commun. 1999:1879–1880. [Google Scholar]
- 20.Hashmi ASK, Salathe R, Frey W. Synlett. 2007:1763–1766. [Google Scholar]
- 21.Witulski B, Schweikert T, Schollmeyer D, Nemkovich NA. Chem Commun. 2010;46:2953–2955. doi: 10.1039/b919275a. [DOI] [PubMed] [Google Scholar]
- 22.Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J Am Chem Soc. 2003;125:2368–2369. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]
- 23.(a) Hirano S, Tanaka R, Urabe H, Sato F. Org Lett. 2004;6:727–729. doi: 10.1021/ol036396b. [DOI] [PubMed] [Google Scholar]; (b) Hirano S, Fukudome Y, Tanaka R, Sato F, Urabe H. Tetrahedron. 2006;62:3896–3916. [Google Scholar]
- 24.(a) Dunetz JR, Danheiser RL. Org Lett. 2003;5:4011–4014. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohnen AL, Dunetz JR, Danheiser RL. Org Synth. 2007;84:88–101. [PMC free article] [PubMed] [Google Scholar]; (c) Wang YP, Danheiser RL. Tetrahedron Lett. 2011;52:2111–2114. doi: 10.1016/j.tetlet.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org Lett. 2004;6:1151–1154. doi: 10.1021/ol049827e. [DOI] [PubMed] [Google Scholar]; (b) Sagamanova IK, Kurtz KCM, Hsung RP. Org Synth. 2007;84:359–367. [Google Scholar]; (c) Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Shen L, Tracey MR. J Org Chem. 2006;71:4170–4177. doi: 10.1021/jo060230h. [DOI] [PubMed] [Google Scholar]; (d) DeKorver KA, Walton MC, North TD, Hsung RP. Org Lett. 2011;13:4862–4865. doi: 10.1021/ol201947b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riddell N, Villeneuve K, Tam W. Org Lett. 2005;7:3681–3684. doi: 10.1021/ol0512841. [DOI] [PubMed] [Google Scholar]
- 27.Dooleweerdt K, Birkedal H, Ruhland T, Skrydstrup T. J Org Chem. 2008;73:9447–9450. doi: 10.1021/jo801935b. [DOI] [PubMed] [Google Scholar]
- 28.Yao B, Liang Z, Niu T, Zhang Y. J Org Chem. 2009;74:4630–4633. doi: 10.1021/jo900595c. [DOI] [PubMed] [Google Scholar]
- 29.Buchwald SL, Bolm C. Angew Chem Int Ed. 2009;48:5586–5587. doi: 10.1002/anie.200902237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jouvin K, Couty F, Evano G. Org Lett. 2010;12:3272–3275. doi: 10.1021/ol101322k. [DOI] [PubMed] [Google Scholar]
- 31.Hamada T, Ye X, Stahl SS. J Am Chem Soc. 2008;130:833–835. doi: 10.1021/ja077406x. [DOI] [PubMed] [Google Scholar]
- 32.Cook AM, Wolf C. Chem Commun. 2014;50:3151–3154. doi: 10.1039/c4cc00394b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sueda T, Oshima A, Teno N. Org Lett. 2011;13:3996–3999. doi: 10.1021/ol2014973. [DOI] [PubMed] [Google Scholar]
- 34.(a) Joshi RV, Xu Z-Q, Ksebati MB, Kessel D, Corbett TH, Drach JC, Zemlicka JJ. Chem Soc Perkin Trans I. 1994:1089–1098. [Google Scholar]; (b) Egi M, Yamaguchi Y, Fujiwara N, Akai S. Org Lett. 2008;10:1867–1870. doi: 10.1021/ol800596c. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez D, Martinez-Esperon MF, Castedo L, Saá C. Synlett. 2007:1963–1965. [Google Scholar]
- 36.Beveridge RE, Batey RA. Org Lett. 2012;14:540–543. doi: 10.1021/ol2031608. [DOI] [PubMed] [Google Scholar]
- 37.(a) DeKorver KA, Wang XN, Walton MC, Hsung RP. Org Lett. 2012;14:1768–1771. doi: 10.1021/ol300366e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X-N, Winston-McPherson GN, Walton MC, Zhang Y, Hsung RP, DeKorver KA. J Org Chem. 2013;78:6233–6244. doi: 10.1021/jo400960e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi R, Wang XN, DeKorver KA, Tang Y, Wang CC, Li Q, Li H, Lv MC, Yu Q, Hsung RP. Synthesis. 2013;45:1749–1758. doi: 10.1055/s-0033-1338476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang P, Cook AM, Liu Y, Wolf C. J Org Chem. 2014;79:4167–4173. doi: 10.1021/jo500365h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Wang XN, Hsung RP, Fox SK, Lv M-C, Qi R. Heterocycles. 2014;88:1233–1254. doi: 10.3987/COM-13-S(S)88. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang XN, Hsung RP, Qi R, Fox SK, Lv MC. Org Lett. 2013;15:2514–2517. doi: 10.1021/ol400989x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.One example of a palladium catalyzed dimerization of an ynesulfonamide to a 2,4-disulfonamido-1-penten-3-yne has been reported: Saito S, Uchiyama N, Gevorgyan V, Yamamoto Y. J Org Chem. 2000;65:4338–4341. doi: 10.1021/jo000171m.
- 42.Rodriguez D, Castedo L, Saá C. Synlett. 2004;10:377–379. [Google Scholar]
- 43.(a) Rodriguez D, Castedo L, Saá C. Synlett. 2004;10:783–786. [Google Scholar]; (b) Martinez-Esperon M, Rodriguez D, Castedo L, Saá C. Tetrahedron. 2006;62:3843–3855. [Google Scholar]
- 44.Tracey MR, Zhang Y, Frederick MO, Mulder JA, Hsung RP. Org Lett. 2004;6:2209–2212. doi: 10.1021/ol0493251. [DOI] [PubMed] [Google Scholar]
- 45.Dooleweerdt K, Ruhland T, Skrydstrup T. Org Lett. 2009;11:221–224. doi: 10.1021/ol802477d. [DOI] [PubMed] [Google Scholar]
- 46.Wang YP, Danheiser RL. Tetrahedron Lett. 2011;52:2111–2114. doi: 10.1016/j.tetlet.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]