

Abstract
Thymine DNA glycosylase (TDG) is an essential multifunctional enzyme involved in DNA base excision repair, DNA demethylation, and transcription regulation. TDG is the predominant enzyme to remove thymine from T/G mispair, which arises due to deamination of 5-methylcytosine at the CpG dinucleotide, thereby preventing C to T mutations. SIRT1 is a member of class III NAD+-dependent histone/protein deacetylases. In this study, we demonstrate that SIRT1 interacts with the residues 67–110 of human TDG (hTDG). In addition, SIRT1 enhances TDG glycosylase activity and deacetylates acetylated TDG. TDG acetylation weakens its interaction with SIRT1. Although acetylated TDG has reduced glycosylase activity toward T/G, 5-formylcytosine/G, and 5-carboxylcytosine/G, it has a stronger activity toward 5-fluorouracil/G substrate as compared to unmodified TDG. SIRT1 weakly stimulates acetylated hTDG activity toward T/G, 5-formylcytosine/G, and 5-carboxylcytosine/G as compared to control hTDG. Sirt1 knockout mouse embryonic fibroblast cells have higher levels of TDG expression and acetylation. The physical and functional interactions between SIRT1 and TDG may mediate DNA repair, gene expression, and FU-mediated cytotoxicity.
Keywords: acetylation, deacetylase, SIRT1, DNA repair, base excision repair, TDG DNA glycosylase, 5-fluorouracil
INTRODUCTION
The human genome is vulnerable to DNA damaging agents of both endogenous and environmental origin. Alkylation, deamination, and oxidation produce thousands of cytotoxic and mutagenic base lesions per cell per day [1]. In the absence of DNA repair, DNA damage leads to genomic instability. Thymine DNA glycosylase (TDG) is a key multifunctional enzyme involved in DNA repair, gene expression, and DNA demethylation (reviewed in [2]). TDG recognizes a broad range of DNA lesions and initiates base excision repair (BER) [2–6]. The major substrates of TDG are U/G and T/G which are generated by deamination. T/O6-methylguanine (MeG), 5-fluorouracil (FU, a chemotherapeutic), 5-chlorouracil (a marker of DNA damage), 3, N4-ethenocytosine/G (εC/G), and oxidized bases (thymine glycol, 5-formyl-U, 5-hydroxy-U, and 5-hydroxy-methyl-U) are also the substrates of TDG, suggesting a diverse role of TDG on drug sensitivity and against oxidative damage. The resulting DNA containing apurinic/apyrimidinic (AP) site is transferred to and cleaved by apurinic/apyrimidinic (AP) endonuclease I (APE1). T/G mispairs can arise through spontaneous deamination of 5-methylcytosine (mC) at the CpG dinucleotide, hence their removal by TDG prevents C:G to T:A mutations. In humans, mutations at CpG sequences account for about 30% of all germline mutations [7]. Importantly, mutations at CpG sites within promoter regions can alter gene expression [8].
Another biological function of TDG is to regulate gene expression. TDG interacts with and modulates many transcription factors and transcription coactivators including the retinoic acid receptor, estrogen receptors and p300/CBP histone/protein acetyltransferases (reviewed in [2]). The glycosylase activity of TDG has recently been implicated in epigenetic regulation through the process of active demethylation [9,10]. Active DNA demethylation (reviewed by [11]) may involve enzyme-induced mC deamination to thymine and followed by TDG initiated BER. Recent studies show that an alternative mechanism of DNA demethylation involves oxidation of mC to 5-hydroxymethylcytosine (hmC), then to 5-formylcytosine (fC), and further to 5-carboxylcytosine (caC) by Tet enzymes (ten-eleven translocation protein, Tet1-3) [12–15]. fC and caC can subsequently be excised by TDG, which leads to DNA demethylation [13,16]. Consist with this role, TDG is indispensible for embryonic development and TDG defective cells contain higher levels of CpG methylation and repressive histone methylation at the promoters of developmental genes [9,10].
In eukaryotes, DNA repair is coordinated with other cellular pathways [17]. The N-terminal domain of TDG is the nexus of a variety of partner proteins including p300/CBP [18], APE1 [18], and Hus1 (a subunit of the cell cycle sensor Rad9-Rad1-Hus1 (9–1–1) complex) [19]. The N-terminal domain of TDG binds DNA non-specifically [20], affects T/G (but not U/G) processing [19]. It can be modified by acetylation [18] and phosphorylation [21]. Tini et al. [18] have reported that acetylation of TDG triggers the release of p300 from DNA-TDG-p300 ternary complex and weakens its interaction with APE1. Acetylated TDG (Ac-TDG) has reduced T/G glycosylase activity but has normal U/G glycosylase activity [21]. TDG acetylation-deacetylation has been proposed as a molecular switch to coordinate its DNA repair and transcription functions [21]. Although TDG is known to be acetylated by p300/CBP [18], the enzyme(s) responsible for its deacetylation has not been identified thus far.
Histone deacetylases (HDACs) deacetylate histones and non-histone proteins [22]. Sirtuins (SIRTs) are class III NAD+-dependent HDACs that play critical role in cellular physiology, calorie restriction, and aging (reviewed in [23,24]). There are seven sirtuin members in human, SIRT1-7, of which SIRT1, SIRT6, and SIRT7 are localized in the nucleus. Increasing evidence indicates that sirtuins are involved in DNA repair. SIRT1 is involved in stress response and deacetylates different proteins including many DNA repair enzymes [25–28]. In response to oxidative stress and ionizing irradiation, SIRT1 dissociates from repetitive DNA and relocalizes to DNA breaks to promote DNA repair [29]. Sirt1 knockout mice die in early postnatal stages due to altered histone modification, impaired DNA damage response, and genomic instability [30,31].
The precise mechanism by which SIRT1 regulates genomic stability is currently under intensive investigation. The role of SIRT1 in cancer has been shown to be cell type dependent and complex. This may be due to its diverse functions and interaction with many proteins. SIRT1 can serve as a tumor promoter or a tumor suppressor in different tissue contexts [32,33]. Particularly, SIRT1 reduction promotes tumor formation but sanitizes cells to apoptosis by FU [34]. To study the role of SIRT1 in DNA repair, we investigated whether it has a role in TDG-mediated BER pathway. In this study, we establish that SIRT1 affects DNA repair through binding to TDG, stimulating TDG glycosylase activity, maintaining TDG in hypoacetylated state, and regulating TDG expression. Although acetylated TDG has reduced glycosylase activities toward T/G, fC/G and caC/G, it has a stronger activity toward FU/G substrate as compared to unmodified TDG. SIRT1 weakly stimulates acetylated hTDG activity toward T/G, fC/G, and caC/G as compared to control hTDG. Our results provide new insights into the direct role of SIRT1 in the maintenance of genomic stability.
EXPERIMENTAL
Reagents and antibodies
All reagents were obtained from Sigma-Aldrich unless otherwise stated. The antibodies used for Western blotting were: SIRT1 (Millipore), acetyl-lysine (Calbiochem), APE1 (Abcam), FLAG-tag (Sigma-Aldrich), β-actin (Sigma-Aldrich), GFP (Invitrogen), horseradish peroxidase-conjugated anti-mouse/anti-rabbit antibodies (BioRad), and anti-rabbit IgG DyLight 800 (Thermo Scientific). TDG antibody for immunoprecipitation was from ABNOVA and for Western blotting was from Primo Schar (University of Basel, Switzerland) or generated in our own laboratory.
Polyclonal TDG antibodies against a TDG domain (residues 58–305 of hTDG) were raised in rabbits. The IgG antibodies were purified from antisera by affinity chromatography. CNBr sepharose matrices (GE Health) were activated with 1 mM HCl, washed with 1 mM HCl and 0.125 M phosphate coupling buffer and then coupled with the TDG (residue 67–308) at 4°C overnight. The coupled matrices were washed with coupling buffer and incubated with 1 M ethanolamine, pH 8.0 (blocking buffer) for 5 h at 4°C. The matrices were washed with phosphate buffered saline (PBS) and packed into a 10 ml column. An equal volume of PBS was added to 6 ml of antisera, filtered and loaded onto the column containing sepharose-TDG. After washing with 40 ml of PBS, the antibodies against the TDG were eluted with elution buffer containing 63 mM glycine, pH 2.3 and 0.2 M NaCl. Samples (0.75 ml) were collected into tubes containing 0.25 ml of neutralizing buffer (0.5 M potassium phosphate, pH 7.5). Antibody titer was performed by ELISA.
Polyclonal Ac-TDG antibodies against a peptide (residues 67–93 of hTDG with cysteine at the N-terminus) containing three acetylated K83, K84, and K87 residues were raised in rabbits. The Ac-TDG peptide antibodies were purified similarly as described above for the TDG antibodies. The antisera were first passed through a column containing sepharose-TDG and the flow-thru factions were then loaded to the column containing sepharose-Ac-TDG-peptide. Antibody titer was performed by ELISA The specificity of the Ac-TDG antibody was verified with unmodified (lane 1) and in vitro p300 acetylated hTDG (lane 2) (Figure S3 in supplementary materials).
Purification of SIRT1
Full length human SIRT1 (SIRT1-FL) cloned in pECE vector (clone 1791) and N-terminus truncated SIRT1 (SIRT1-NT with residues 7–83 deleted) cloned in pCDNA3.1 vector (clone 13812) were obtained from Addgene. To express the FLAG-tagged hSIRT1 proteins, the hSIRT1 constructs were transfected into the HEK293T cells. The cells from ten of 100 × 20 mm tissue culture dishes were lysed with FLAG M2 lysis buffer (Sigma-Aldrich) and centrifuged at 10,000 × g for 10 min. The FLAG-tagged hSIRT1 proteins were purified by FLAG M2 affinity chromatography according to the manufacturer’s protocol. FLAG-SIRT1 was confirmed by SDS-PAGE and Western blotting with FLAG antibody. The pooled fractions were then divided into small aliquots and stored at −80°C. The FLAG-hSIRT1-FL and FLAG-hSIRT1-NT proteins were approximately 90% pure (Figure S1 in supplementary materials, lanes 2 and 3) and their concentrations were determined by the Bradford method (BioRad).
Cell culture
Human HEK 293T cells were obtained from ATCC. Cells were maintained at 37°C in 5% CO2 in MEM (Invitrogen) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin. Sirt1+/+ (Wild type, WT) and sirt1−/− (knockout, KO) mouse embryonic fibroblast (MEF) cells (obtained from Dr. Toren Finkel at NIH) were maintained on in DMEM (Invitrogen) supplemented with 15% fetal bovine serum and 1% Penicillin-Streptomycin. Cell transfection and cell extract preparation can be found in supplementary materials.
GST pull-down assay
The pull-down assay was performed similarly as described previously [19]. GST fusion proteins (500 ng) were incubated with purified proteins (100 ng) in 0.2 ml volume of buffer G (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 2 mM EDTA) or buffer S (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 4 mM MgCl2) containing 0.1% (v/v) NP40 at 4°C with shaking overnight. After centrifugation at 1000g for 2 min, the pellets were washed five times with 1 ml of buffer G containing 0.1% (v/v) NP40. Bound proteins were eluted by boiling in SDS loading buffer (30 mM Tris–HCl, pH 6.8, 5% (v/v) glycerol, 1% SDS, 0.5 mg/ml bromophenol blue and 1% β-mercapoethanol) and resolved on a 10% SDS–polyacrylamide gel. The proteins were subsequently analyzed by western blot using the corresponding antibodies as described above. The membranes were blocked with phosphate-buffered saline (PBS) with 0.1% Tween-20 and 10% nonfat dry milk, reacted with primary antibodies, and then incubated with secondary antibodies with wash between each step. Western blotting was detected either by the Enhanced Chemiluminescence (ECL) analysis system (USB Corporation, 72552) or ECL Plex (GE Healthcare) according to the manufacturer’s protocols.
Co-immunoprecipitation
Cell extracts (2 mg) were precleared by adding 30 μl Protein G agarose (Invitrogen) for 2 hours at 4°C. After centrifugation at 1000 × g, the supernatant was incubated with 4 μg of first antibody overnight at 4°C. Protein G agarose (30 μl) was added and incubated for 4 hours at 4°C. After centrifugation at 1000 × g, the supernatant was saved and the pellet was washed. The pellet fractions were resolved on a 10% or 12% SDS-PAGE. Western blot analysis was performed with antibody against second protein, probed with anti-rabbit or anti-mouse HRP-conjugated secondary antibody, and detected by the Enhanced Chemiluminescence (ECL) analysis system (USB Corporation) according to the manufacturer’s protocol. Because the heavy chain of IgG runs very close to endogenous TDG on SDS-PAGE, the Western blot analysis described above cannot be applied to examine the co-immunoprecipitation of TDG with SIRT1 antibody. Purified TDG antibody (generated in our laboratory) was labeled with Cy5 (GE Healthcare, PA25001) in the coupling solution (0.1M sodium carbonate/bicarbonate pH 9.26) for according to the manufacturer’s instructions. This Cy5-labeled TDG antibody was then subjected to Western blot directly to detect endogenous TDG in the immuno-precipitant. The fluorescence signal was detected by Typhoon FLA9500 (GE HealthCare).
TDG glycosylase activity assay
The DNA substrates of hTDG are a 40-mer duplex containing a T/G mismatch and 28-mer duplexes containing FU/G, fC/G or caC/G mismatch (Table S1 in the supplementary materials). The strand containing the mispaired T, FU, fC or caC was labeled at the 5′ end with [γ-32P]ATP by polynucleotide kinase, annealed with the mispaired G strand as described by Lu et al [35]. The hTDG reaction was carried out with 1.8 fmol (0.18 nM) or 50 fmol (5 nM) DNA substrate in 10 μl reaction containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 50 μg/ml BSA, 1 mM EDTA at 37°C for 30 min as described [19]. To exam the effect of NAD+ and SIRT1 on Ac-TDG glycosylase, the reactions used higher concentrations of TDG and were carried out in buffer containing 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 50 μg/ml BSA, 37.5 mM NaCl, 0.75 mM KCl, 1 mM MgCl2, and various amount of SIRT1 in the absence or presence of 3 mM NAD+. For both T/G- and FU/G-DNA, the reaction products were heated at 90°C for 30 min in the presence of 0.1 N NaOH. For both fC/G- and caC/G-DNA, the reaction products were heated at 80°C for 12 min in the presence of 0.1 N NaOH to avoid non-enzyme catalyzed strand cleavage. The reaction samples were supplemented with 5 μl of formamide dye (90% formamide, 10 mM EDTA, 0.1% xylene cyanol and 0.1% bromophenol blue) and 7 μl of the mixture was loaded onto a 14% polyacrylamide sequencing gel containing 7 M urea. The gel images were detected with Image screen and viewed on Typhoon FLA 9500 imager and quantified using the ImageQuant software (GE Healthcare). The hTDG cleavage activity was calculated by the percentage of product over total DNA (product plus substrate bands).
TDG acetylation and deacetylation
Purified recombinant hTDG protein (200 ng) (gift from Alex C. Drohat, University of Maryland) was incubated with 50 ng of p300 acetyltransferase (gift from Phillip A. Cole, Johns Hopkins University) in acetylation buffer (50 mM Tris–HCl (pH 8.0), 0.1 mM EDTA, 10% (v/v) glycerol, 1 mM DTT, 50 mM Acetyl CoA) for 1 h at 30°C. Following incubation, the mixture was supplemented with 10 units of SIRT1 (Biomol), 3 mM NAD+, 150 mM NaCl, 3 mM KCl, 4 mM MgCl2 to initiate the deacetylation reaction. The deacetylation reaction was performed with or without 10 mM NAM. For SDS-PAGE analyses, after the incubation for 90 min at 37°C, the reaction was terminated by adding 3× SDS sample buffer and samples were subjected to analysis on 12% SDS–PAGE and immunoblotted with antibody against acetyl-lysine (Ac-Lys) or TDG to evaluate TDG acetylation. To check glycosylase activity of Ac-TDG, the reaction solution following acetylation with p300 was passed through an Amicon Ultra 0.5 ml 10K filter (Millipore) and the buffer was exchanged to acetylation buffer without Acetyl CoA. The mock reaction was performed without Acetyl CoA. The amounts of mock TDG and Ac-TDG were compared by SDS-PAGE and silver strain. TDG was diluted with TDG reaction buffer and assayed as described above.
RESULTS
SIRT1 interacts with TDG
Recent studies elucidating the involvement of SIRT1 in DNA repair pathways including BER [26,36], prompted us to investigate the link between SIRT1 and TDG. First, we examined the physical interaction between SIRT1 and TDG by glutathione S-transferase (GST) pull-down method (Figure 1A). GST-TDG or 3 fold molar excess of GST was immobilized on glutathione-sepharose and incubated with purified FLAG-hSIRT1-FL. The pellets were fractionated on a SDS-polyacrylamide gel followed by Western blot analysis with the FLAG antibody. hSIRT1 interacted with GST-hTDG but not with GST (Figure 1A, upper panel). We also showed that FLAG-hSIRT1-NT (residues 7–83 deleted) could be pulled down by GST-hTDG (Figure 1A, lower panel). Conversely, GST-hSIRT1-NT or GST in 3 fold molar excess immobilized on glutathione-sepharose was incubated with purified His-hTDG in the pull-down assay. TDG was pulled down by immobilized GST-hSIRT1-NT protein but not by GST (Figure 1B).
Figure 1. Physical Interaction of TDG with SIRT1.
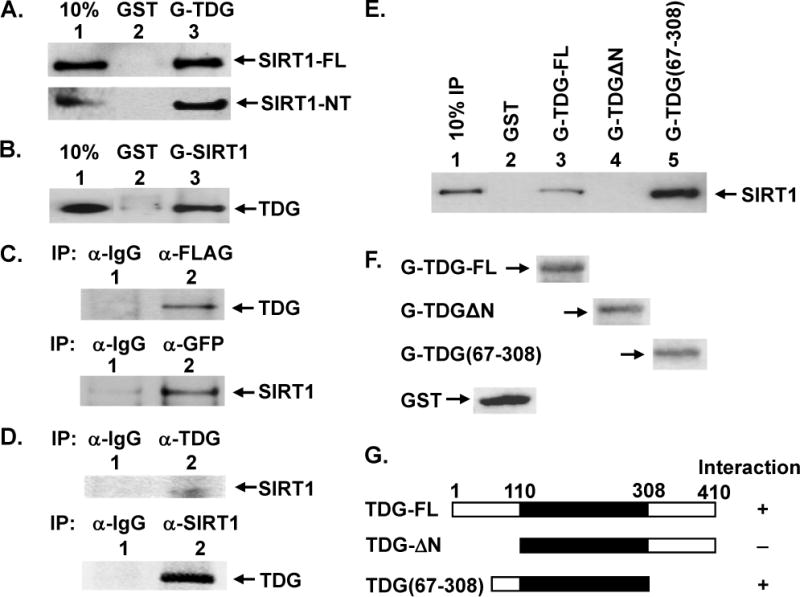
(A) Direct interaction between TDG and SIRT1 was demonstrated by GST pull-down method. GST alone (lane 2) and GST-hTDG (lane 3) were immobilized on glutathione-sepharose and incubated with 100 ng purified FLAG-tagged hSIRT1-FL (upper panel) or 100 ng purified FLAG-tagged hSIRT1-NT (with residues 7–83 deleted) (lower panel) in buffer G (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 2 mM EDTA). GST is about 3 fold molar excess of GST-hTDG. The pellets were fractionated on a 10% SDS-polyacrylamide gel followed by Western blot analysis with the FLAG antibody. Lane 1 contains 10 ng (10% of the total input) of FLAG-hSIRT1-FL. (B) Immobilized GST alone (lane 2, in 3–4 fold molar excess) and GST-hSIRT1-NT (lane 3) were incubated with purified His-hTDG. Western blot analysis was performed with the His antibody. Lane 1 contains 10% of the total input of His-hTDG. (C) hTDG and hSIRT1 co-immunoprecipitated in extracts prepared from HEK293T cells expressing FLAG-SIRT1 and GFP-hTDG. Upper panel, immunoprecipation were performed with antibody against FLAG and the Western blot was detected by hTDG antibody (lane 2). Lane 1 is a negative control in which the immunoprecipation was performed with antibody against IgG. Lower panel, immunoprecipation were performed with antibody against GFP and the Western blot was detected by FLAG antibody (lane 2). Lane 1 is a negative control. (D) hTDG and hSIRT1 co-immunoprecipitated in HEK293T extracts. Upper panel, immunoprecipation was performed with antibody against TDG and the Western blot was detected by SIRT1 antibody (lane 2). Lane 1 is a negative control. Lower panel, immunoprecipation was performed with SIRT1 antibody. The Western blot was detected by Cy5-labeled TDG antibody which was prepared according to the manufacturer’s instructions and detected by Typhoon FLA9500 (GE HealthCare). Lane 1 is a negative control. (E) SIRT1 binds to residues 67–110 of TDG. Two TDG deletion constructs were used to determine the region within hTDG involved in binding to SIRT1. FLAG-hSIRT1-FL (100 ng) was incubated with GST-TDG constructs (as labeled) or GST alone immobilized on beads as in (A). (F) The amounts of GST and GST-TDG constructs were determined by Ponceau S stain. The same membrane of (E) was stained by Ponceau S. Due to the large size difference, only the bands of GST-tagged proteins were shown on each lane. As indicated, GST is about 3 fold molar excess of GST-hTDG-FL which is in similar molar amount of GST-TDGΔN and GST-TDG(67–308). (G) Graphic depiction of hTDG constructs and the summary of physical interaction of these constructs with SIRT1. The intact hTDG (TDG-FL) contains 410 amino acid residues and the core catalytic domain is within residues 111–308 (shown in black box). The physical interactions of hTDG and SIRT1 are derived from results of Figure 1E. “+” represents for positive and “−” represents for negative interactions.
Interaction between SIRT1 and TDG was also demonstrated by co-immunoprecipitation. FLAG-SIRT1 and GFP-TDG were co-expressed in HEK293T cells; the cell extract was subjected to immunoprecipitation with FLAG antibody followed by Western analysis with TDG antibody. As shown in Figure 1C (upper panel), hTDG was found in the pellet with FLAG antibody but not with control IgG. Thus, TDG co-precipitates with hSIRT1. Conversely, the cell extract was subjected to immunoprecipitation with GFP antibody followed by Western analysis with FLAG antibody. hSIRT1 was co-immunoprecipitated with GFP antibody (Figure 1C, lower panel). To show that endogenous SIRT1 and TDG also interact, HEK293T cell extract was subjected to immunoprecipitation with TDG antibody followed by Western analysis with SIRT1 antibody. As shown in Figure 1D (upper panel), SIRT1 was found in the pellet with TDG antibody but not with control IgG. Conversely, the cell extract was subjected to immunoprecipitation with SIRT1 antibody followed by Western analysis with TDG antibody. TDG1 was co-immunoprecipitated with SIRT1 antibody (Figure 1D, lower panel). These results suggest that the association of SIRT1 and TDG in human cells.
To map the region of TDG that interacts with SIRT1, we constructed two GST-TDG deletion mutants. Analyses of these GST-TDG deletion constructs indicated that residues 67–110 of TDG were important for SIRT1 interaction (Figure 1E). Interestingly, TDG(67–308) had a stronger interaction with SIRT1 than TDG-full (Figure 1E, compare lane 5 with lane 3). It is also interesting to note that many regulatory factors including CBP/p300 [18], APE1 [18], and Hus1 [19] also interact within the same region of the N-terminal domain of TDG.
SIRT1 enhances TDG glycosylase activity
Since hTDG physically interacts with hSIRT1, we tested whether they interact functionally. First, we used purified recombinant hTDG expressed in E. coli and purified hSIRT1-FL and hSIRT1-NT expressed in HEK293T cells to examine the effect of hSIRT1 on the hTDG glycosylase activity. When increasing amounts of hSIRT1-FL or hSIRT1-NT were added to the hTDG glycosylase reactions with DNA substrate containing a T/G mismatch, the hTDG activity was enhanced (Figures 2A and 2B, compare lane 2 with lanes 3–5). The quantification results (Figure 2C) showed that hSIRT1-FL and hSIRT1-NT had similar extent of stimulation on TDG glycosylase activity on T/G-DNA substrate. At a SIRT1/TDG ratio of 4, both hSIRT1-FL and hSIRT1-NT could enhance TDG activity by 6 folds. hSIRT1-FL and hSIRT1-NT alone at 2 nM did not have glycosylase or nicking activity on the T/G-DNA substrate (Figures 2A and 2B, lane 6).
Figure 2. Human TDG glycosylase activity can be stimulated by hSIRT1.
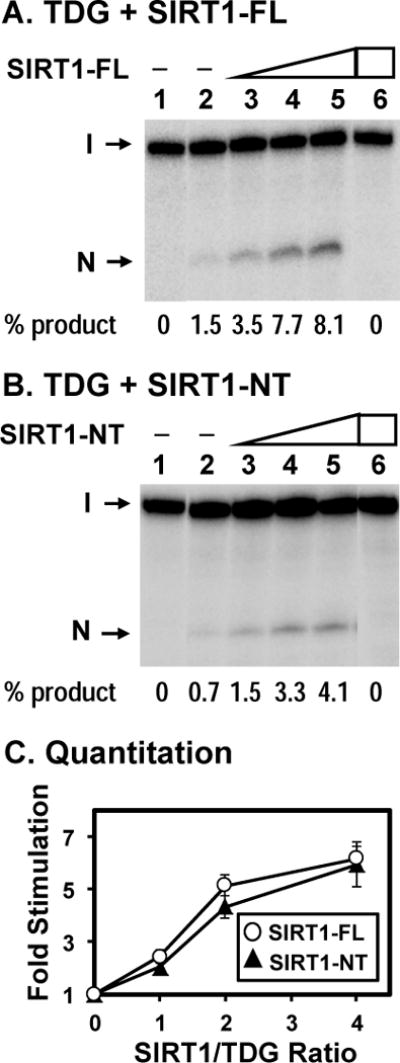
Human SIRT1-FL (A) and SIRT1-NT (B) purified from HEK293T cells enhance the activities of TDG. Lane 1, T/G-containing DNA substrate. Lane 2, 0.18 nM T/G-containing DNA substrate was incubated with 0.5 nM untreated hTDG Lanes 3–5 are similar to lane 2 but with added 0.5, 1, and 2 nM hSIRT1-FL (or hSIRT1-NT), respectively. Lane 6, T/G-containing DNA substrate was incubated with 2 nM hSIRT1-FL (or hSIRT1-NT) without TDG. The products were then treated with 0.1 M NaOH at 90°C for 30 min and separated on a 14% DNA sequencing gel. The images are pasted together from the same gel. Arrows mark the intact DNA substrate (I) and the cleavage product (N). Percentage (%) of product generated is shown below each lane. (C) Quantitative analyses of the fold of stimulation of hSIRT1-FL (open circles) and hSIRT1-NT (filled triangles) on the TDG glycosylase activity on T/G-containing DNA substrate from three experiments. The error bars reported are the standard deviations of the averages.
To investigate the nature of SIRT1 enhancement on TDG activity, we performed the TDG glycosylase assays under substrate-limiting and enzyme-limiting conditions. In the substrate-limiting conditions, the reactions contained 0.18 nM T/G-DNA, 0.5 nM TDG, and 2 nM SIRT1-NT. At SIRT1/TDG ratio of 4, time course studies indicated that SIRT1 could stimulate TDG glycosylase on T/G-DNA by maximum 6-fold (Fig. 3A–3C). Thus, SIRT1 stimulates the excision rate of TDG on T/G-DNA. In the enzyme-limiting conditions, the reactions contained 5 nM T/G-DNA, 0.5 nM TDG, and 2 nM SIRT1-NT. As shown in Fig. 3D–3F, at 64 min incubation, the product amount of TDG was increased by 5-fold in the presence of SIRT1 as compared to reactions without SIRT1. The maximum amount of DNA cleaved by TDG alone was 0.2 nM which was 40% of 0.5 nM of measured TDG concentration. Under this condition, TDG excises the mismatched T and remains tightly bound to the AP-DNA [20,37,38]. Due to this slow turnover, the TDG preparation may be only 40% active on T/G substrate. The loss of activity in this TDG preparation may be caused by freeze-thaw. In the presence of SIRT1, the maximum amount of DNA cleaved by TDG became higher than 1 nM which is more than 2-fold of 0.5 nM of measured TDG concentration and more than 5-fold of 0.2 nM of active TDG concentration. Thus, SIRT1 can stimulate AP-product release from TDG. From these data, we interpret that SIRT1 stimulates both the excision and turnover rates of TDG.
Figure 3. Time course of TDG glycosylase reaction under single-turnover and multiple-turnover conditions.
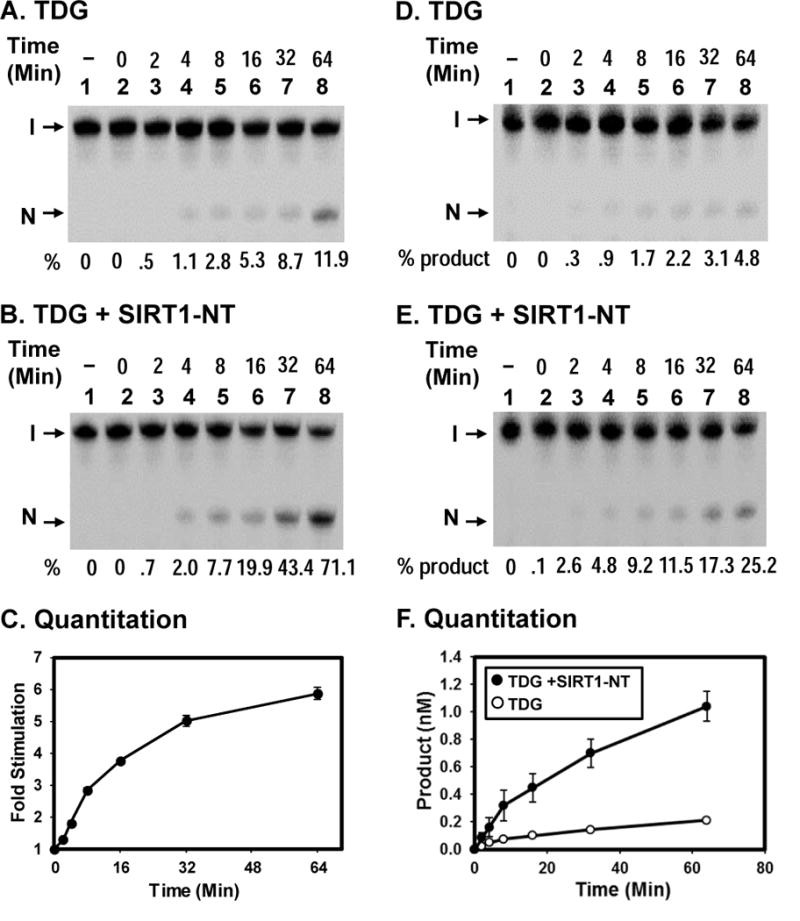
(A) Reactions are similar to Fig. 2B (lane 2) with 0.18 nM T/G-DNA and 0.5 nM TDG at various time points as indicated. (B) Reactions are similar to (A) except with 2 nM of SIRT1-NT. Percentage (%) of products generated is shown below each lane. (C) Quantitative analyses of the fold of stimulation of hSIRT1-NT on the TDG glycosylase activity on T/G-containing DNA substrate from three experiments. The error bars reported are the standard deviations of the averages. (D) and (E) Time course of TDG reaction in the absence or presence of hSIRT1-NT, respectively, under multiple-turnover condition. Reactions contained 5 nM T/G-DNA and 0.5 nM TDG in the absence or presence of 2 nM SIRT1-NT. Percentage (%) of products generated is shown below each lane. (F) Plots of product formation vs. time from three experiments as in (D) (open circles) and in (E) (closed circles). The error bars reported are the standard deviations of the averages.
Next, we tested whether TDG has any effect on SIRT1 deacetylase activity. An in vitro fluorescence based assay of SIRT1 deacetylase (see supplementary materials) was performed in the presence of increasing amounts of recombinant TDG. As shown in Figure S3 in supplementary materials, SIRT1 deacetylation activity was not affected by TDG.
SIRT1 deacetylates TDG in vitro and in vivo
TDG can be acetylated by p300/CBP [18], however the enzyme(s) responsible for its deacetylation has not been identified thus far. SIRT1 has been shown to have robust deacetylation activity towards several non-histone proteins [39], including APE1 and PARP1 [25,26], two of the enzymes involved in BER pathway, hence it is plausible that SIRT1 may deacetylate TDG. His-tagged TDG was in vivo acetylated in HEK293T cells, by overexpression of TDG and p300. Acetylated TDG (Ac-TDG) was purified and subjected to deacetylation assay with SIRT1 and SIRT6. SIRT1 deacetylated TDG in the presence of NAD+ (Figure 4A, lane 3). In the absence of NAD+, SIRT1 had no deacetylase activity as expected (Figure 4A, lane 2). Addition of nicotinamide (NAM, a SIRT1 inhibitor) abolished SIRT1 deacetylase activity on Ac-TDG (Figure 4A, lane 4). SIRT6, another member of class III HDACs, also has a role in DNA repair [40]. However, SIRT6 was unable to deacetylate TDG under similar conditions (Figure S4 in supplementary materials). In addition, recombinant TDG isolated from E. coli was acetylated in vitro with p300 in the presence of acetyl-coA, followed by deacetylation with SIRT1. Data in Figure 4A and 4B indicate that SIRT1 can deacetylate Ac-TDG.
Figure 4. SIRT1 can deacetylate TDG.
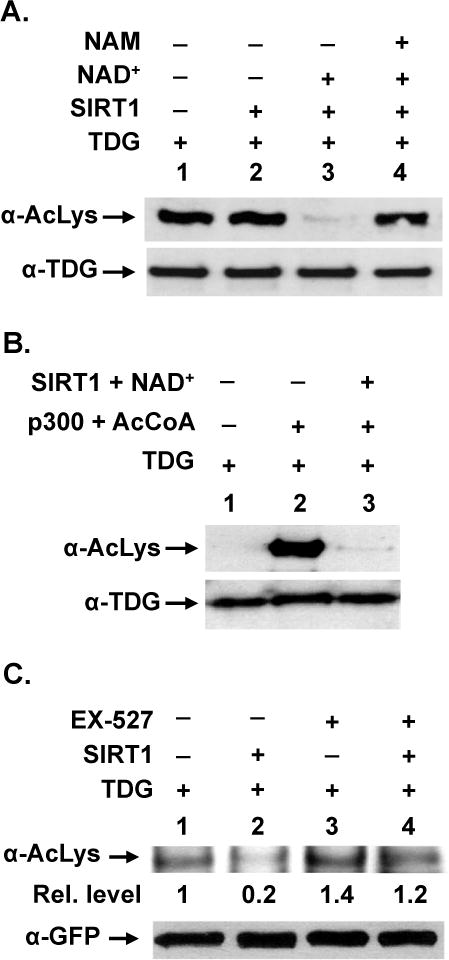
(A) In vivo acetylated hTDG is a substrate for SIRT1. His-hTDG and p300 plasmids were transiently transfected into HEK293T cells. Acetylated TDG was purified and incubated with SIRT1-FL in the absence or presence of its cofactor NAD+ or its inhibitor nicotinamide (NAM). Western blotting was performed with antibody against acetyl-lysine (α-AcLys) or TDG (α-TDG) to evaluate TDG acetylation. (B) In vitro acetylated hTDG is a substrate for SIRT1. Purified recombinant hTDG protein was incubated with p300 acetyltransferase and acetyl CoA (AcCoA) and then treated with SIRT1-FL in the presence of NAD+. Western blotting was performed with antibody against acetyl-lysine or TDG to evaluate TDG acetylation. (C) SIRT1 controls the in vivo acetylation level of TDG. GFP-hTDG was transiently transfected into HEK293T cells in the absence or presence of SIRT1-FL plasmid. SIRT1 specific inhibitor EX-527 was added 6 hrs before cell harvest. Equal amounts of cell extracts were subjected to immunoprecipitation by anti-GFP. Western blotting was performed with α-acetyl lysine antibody (upper panel) or GFP antibody (lower panel). Relative acetylation level is shown below each lane on upper panel.
To test whether TDG is a target substrate of SIRT1 in human cells, we expressed GFP-TDG in HEK293T cells with or without the co-expression of FLAG-SIRT1 or the presence of SIRT1 specific inhibitor EX-527 [41]. TDG was immunoprecipitated by anti-GFP antibody and analyzed by Western blotting with antibody against acetylated lysine (Ac-Lys). When SIRT1 was co-expressed with GFP-TDG, the TDG acetylation level was reduced by about 5-fold (Figure 4C, upper panel, compare lanes 1 and 2). When TDG transformed cells were treated with 10 μM of EX-527 for 48 hrs, TDG acetylation level was increased by only about 1.5-fold (Figure 4C, upper panel, compare lanes 1 and 3). EX-527 increased the TDG acetylation level by about 6-fold in TDG and SIRT1 co-expressed cells (Figure 4C, upper panel, compare lanes 2 and 4). The levels of GFP-TDG in the immunoprecipitants were similar (Figure 4C, lower panel). Thus, SIRT1 deacetylates TDG both in vitro and in human cells.
TDG acetylation weakens its interaction with SIRT1
It has been shown that mouse TDG can be acetylated by p300/CBP at four lysines and that acetylation of TDG weakens its interactions with p300 and APE1 [18]. The four corresponding lysine residues in hTDG are K59, K83, K84, and K87. Three of these acetylated sites of hTDG (K83, K84, and K87) are within the SIRT1 binding domain. Therefore, we examined the effects of TDG acetylation and NAD+ on its interaction with SIRT1. We tested the pull-down reaction with GST-SIRT1 with unacetylated TDG and Ac-TDG in the SIRT1 reaction buffer (50 mM Tris-HCl, pH 8.0, 3 mM NAD+, 150 mM NaCl, 3 mM KCl, and 4 mM MgCl2). We found that control TDG had a stronger binding with GST-SIRT1 under this condition (compare Figure 5A, lane 3 with Figure 1B, lane 3). Acetylated TDG had about 3.7-fold reduced binding to GST-SIRT1 immobilized on beads (Figure 5A, compare lanes 3 and 6). In the presence of NAD+ in SIRT1 reaction buffer, binding of Ac-TDG to GST-SIRT1 was 2.3-fold better than that without NAD+ (Figure 5A, compare lanes 6 and 9), but is 1.6-fold weaker than the interaction between control TDG and GST-SIRT1 (Figure 5A, compare lanes 3 and 9). We reasoned that in presence of NAD+, Ac-TDG was deacetylated by SIRT1 but the deacetylation reaction was not complete.
Figure 5. Effect of acetylation on TDG interaction with SIRT1.
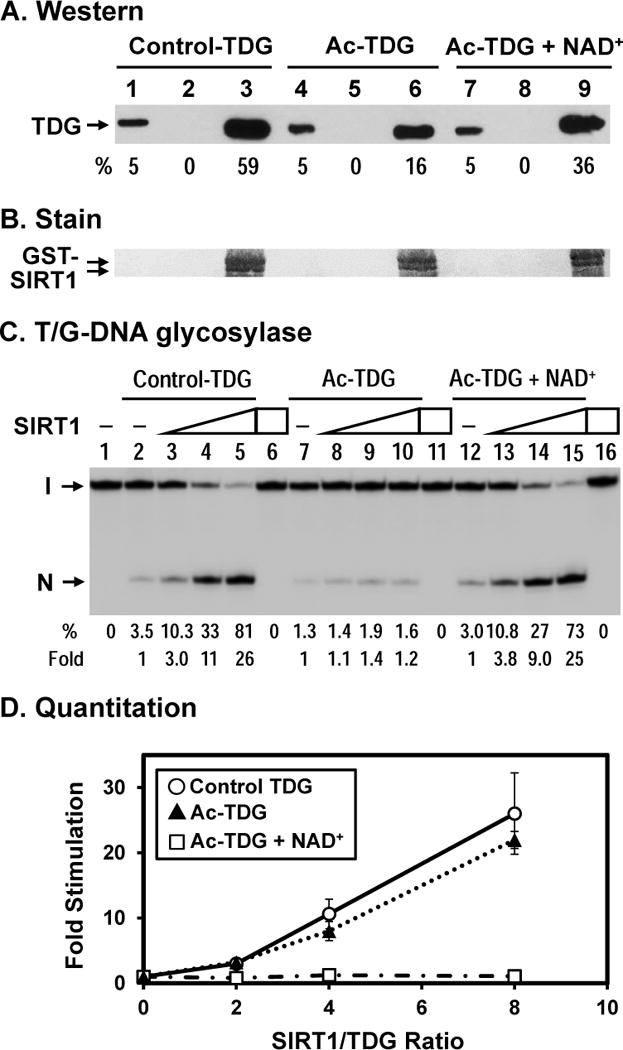
(A) Acetylated TDG has a reduced physical interaction with hSIRT1. hTDG was acetylated in vitro by p300 (lanes 4–9) and control TDG was treated similarly but without acetyl-CoA (lanes 1–3). GST alone (lanes 2, 5, and 8) and GST-hSIRT1 (lanes 3, 6, and 9) were immobilized on glutathione-sepharose and incubated with 100 ng TDG in buffer S (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 4 mM MgCl2). Lane 1, 10 ng His-tagged TDG (5% of input), lane 2, unacetylated control TDG bound to GST alone, and lane 3, control TDG bound to GST-SIRT1-FL immobilized on beads. Lanes 4–6 are similar to lanes 1–3 except using in vitro acetylated TDG. Lanes 7–9 are similar to lanes 4–6 except adding 3 mM NAD+ in the incubation buffer. Western blot was detected by TDG primary antibody made in our laboratory and anti-rabbit IgG DyLight 800 (Thermo Scientific) secondary antibody. The image was detected by Typhoon FLA9500 and quantitated by ImageQuant software (GE Healthcare). Percentage (%) of input TDG (lanes 1, 4, and 7) and precipitated TDG as compared to input TDG and adjusted with GST-SIRT1 amounts on membrane in (B) are shown below each lane. (B) The GST-SIRT1 immobilized on membrane of (A) was stained by Ponceau S. As indicated, the amounts of GST-SIRT1 are about the same. (C) SIRT1 has a reduced stimulation on acetylated TDG with T/G-DNA. Control hTDG and acetylated TDG were the same used in (A). 0.18 nM T/G-DNA was incubated with 10 nM TDG in reaction buffer (50 mM Tris-HCl, pH 8.0, 1 mM DTT, 50 μg/ml BSA, 37.5 mM NaCl, 0.75 mM KCl, 1 mM MgCl2) in the absence or presence of SIRT1 or 3 mM NAD+. Lanes 1, DNA alone, Lane 2, 0.18 nM T/G-containing DNA substrate was incubated with 10 nM control hTDG Lanes 3–5 are similar to lane 2 but with added 20, 40, and 80 nM hSIRT1-FL, respectively. Lane 6, T/G-DNA was incubated with 80 nM hSIRT1-FL without TDG. Lanes 7–10 are similar to lanes 2–5 except using Ac-TDG. Lanes 12–15 are similar to lanes 7–10 except adding 3 mM NAD+. Percentage (%) of product generated and fold of stimulation of hSIRT1-FL on the hTDG glycosylase activity from three experiments are shown below each lane. (C) Quantitative analyses of the fold of stimulation of hSIRT1-FL on the TDG glycosylase activity on T/G-DNA substrate from three experiments. Open circles, control TDG; closed triangles, Ac-TDG in the absence of NAD+; and open rectangulars, Ac-TDG in the presence of NAD+. The error bars reported are the standard deviations of the averages.
Acetylation alters DNA glycosylase activity and subtract specificity of TDG
It has been shown that acetylated TDG has reduced T/G glycosylase activity but has normal U/G glycosylase activity [21]. We confirmed that acetylated hTDG had 2.5-fold decreased T/G glycosylase activity (Figure 6A, compare lanes 2 with 6). Acetylated hTDG could still be enhanced by SIRT1 but at reduced levels as compared to control TDG (Figure 6A, compare lanes 2–5 with lanes 6–9).
Figure 6. Effect of acetylation on TDG subtract specificity.
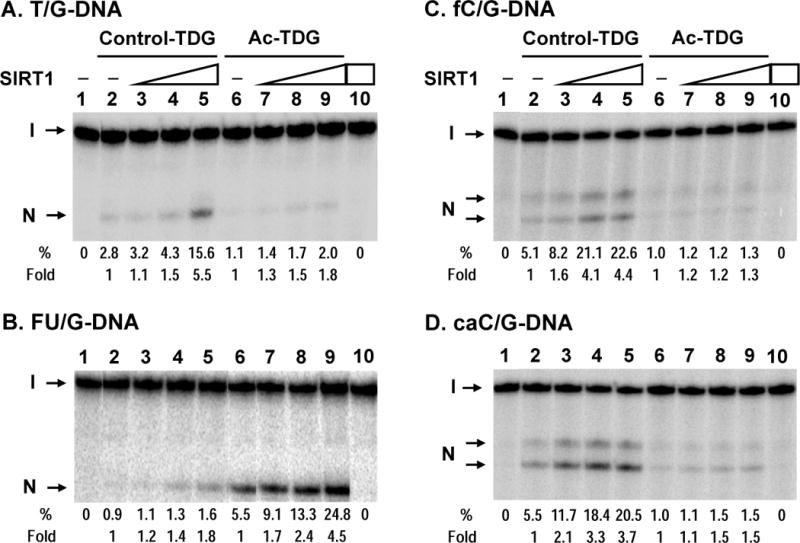
(A) Human SIRT1-FL enhances the T/G glycosylase activities of TDG and acetylated TDG. hTDG was acetylated in vitro by p300 (lanes 6–9) and control TDG was treated similarly but without acetyl-CoA (lanes 2–5). Lanes 1–5 and 6–9 are similar to lanes 1–5 in Fig. 2A. Lanes 3–5 and 7–9, 0.18 nM T/G-containing DNA substrate was incubated with 0.5 nM hTDG with added 0.5, 1, and 2 nM hSIRT1-FL, respectively. Lane 10, T/G-containing DNA substrate was incubated with 2 nM hSIRT1-FL without TDG. Percentage (%) of product generated and fold of stimulation of hSIRT1-FL on the hTDG glycosylase activity are shown below each lane. (B) Acetylated hTDG has a stronger activity on FU/G mismatches and can be better stimulated by SIRT1 as compared to unmodified TDG. hTDG glycosylase activity was assayed similarly as in (A) except using 0.2 nM TDG with 0.18 nM FU/G substrate in the presence of 0.2, 0.4, and 0.8 nM of SIRT1-FL. (C) and (D), Acetylated hTDG on fC/G and caC/G mismatches, respectively, can be stimulated by SIRT1. hTDG glycosylase activity was assayed similarly as in (A) except using fC/G or caC/G substrate in the presence of 0.5, 1, and 2 nM of SIRT1-NT and the reaction products were heated at 80°C for 12 min to avoid non-enzyme catalyzed strand cleavage at fC and caC. Under this condition, the AP site is not completely processed by β/δ-elimination, thus two cleavage products (labeled as N) were observed: the upper one is derived by β-elimination only and the lower one is produced by β/δ-elimination.
The TDG deacetylation by SIRT1 requires NAD+ cofactor. Thus, we examined whether NAD+ has any effect on SIRT1 stimulation of TDG activity. We observed that TDG glycosylase activity was much weaker in SIRT1 reaction buffer (50 mM Tris–HCl, pH 8.0, 3 mM NAD+, 150 mM NaCl, 3 mM KCl, and 4 mM MgCl2) (data not shown). Thus, to examine the effect of NAD+ and SIRT1 on Ac-TDG glycosylase, TDG glycosylase reactions were supplemented with 3 mM NAD+, 37.5 mM NaCl, 0.75 mM KCl, 1 mM MgCl2, and various amount of SIRT1. Under this condition, at SIRT1/TDG ratios of 4 and 8, hSIRT1-FL could enhance TDG activity by 11 and 26 folds, respectively (Fig. 5B, compare lanes 4 and 5 with lane 2). Similar to the results in Figure 6A, acetylated hTDG had 2.5-fold decreased T/G glycosylase activity (Figure 5C, compare lanes 2 with 7) and was hardly enhanced by SIRT1 (Figure 6B, compare lanes 2–5 with lanes 7–10). In the presence of NAD+, Ac-TDG had 2-fold increased T/G glycosylase activity (Figure 5C, compare lanes 7 with 12) and was enhanced by SIRT1 (Fig. 5C, lanes 12–15) at similar levels as control TDG and at much higher level than in the absence of NAD+ (Figure 5D). At SIRT1/TDG ratios of 4 and 8, hSIRT1-FL could enhance TDG activity by 9 and 25 folds, respectively (Fig. 5C, compare lanes 14 and 15 with lane 12). We suggest that in the presence of NAD+, Ac-TDG is deacetylated by SIRT1 and has activity like control TDG. With unacetylated TDG, NAD+ did not have effect (neither stimulation nor inhibition) on SIRT1 enhancement of T/G excision activity (Figure S2 in supplementary materials).
FU, a chemotherapeutic drug commonly used in cancer treatment, inhibits thymidylate synthase, imbalances nucleotide pools, thereby favoring misincorporation of uracil and FU into genomic DNA. It has been shown that FU/G is a substrate of TDG and that TDG mediates DNA-directed cytotoxic effect of FU [3,5,10]. We thus tested whether Ac-TDG had an altered activity to FU/G-containing DNA. Surprisingly, Ac-TDG had a 6-fold stronger activity toward FU/G substrate as compared to unmodified TDG (Figure 6B, compare lanes 2 and 6). Acetylated hTDG could be enhanced by SIRT1 at increased levels as compared to control TDG (Figure 6B, compare lanes 2–5 with lanes 6–9). At a SIRT1/TDG ratio of 4, hSIRT1-FL could enhance the activities of control TDG and acetylated TDG on FU/G-DNA substrate by 2 and 4.5 folds, respectively. SIRT1 alone at 0.8 nM did not have glycosylase or nicking activity on the FU/G-DNA substrate (Figure 6B, lane 10).
Recent studies show that the role of TDG in DNA demethylation may involve removing 5-formylcytosine (fC) and 5-carboxylcytosine (caC) [13,16]. Thus we checked the effects of TDG acetylation on its glycosylase activity on fC/G- and caC/G-substates. Because fC and caC are labile in the presence of 0.1 N NaOH at 90°C, the glycosylase reactions were heated at 80°C for 12 min to observe AP products but to avoid non-enzyme catalyzed strand cleavage at fC and caC. Under this condition, the AP site is not completely processed by β/δ-elimination, thus two cleavage products were observed: the upper one is derived by β-elimination only and the lower one is produced by β/δ-elimination. Unmodified TDG had activity on fC/G- and caC/G-substrates (Figure 6C and 6D, lane 2) as reported [13,16]. Ac-TDG had about 5 fold weaker activities toward these substrates as compared to unmodified TDG (Figure 6C and 6D, compare lanes 2 and 6). SIRT1 stimulated the glycosylase activity toward fC/G of unmodified TDG but not of Ac-TDG (Figure 6C). On caC/G-substate, SIRT1 exerted 2.5 fold higher enhancements on unmodified TDG than on Ac-TDG (Figure 6D, compare lanes 3–5 with lane 2 and compare lanes 7–9 with lane 6). SIRT1 alone did not have glycosylase or nicking activity on these two DNA substrates (Figure 6C and 6D, lane 10). These results suggest that deacetylation and stimulation of the TDG activity by SIRT1 will enhance DNA demethylation.
SIRT1 influences TDG expression
SIRTs act as gene silencers by deacetylating histones (reviewed in [23,24]). To determine whether the TDG expression level is regulated by SIRT1, we monitored TDG protein levels in total cell extracts prepared from wild-type and sirt1 knockout MEFs by Western blotting (Figure 7A). The level of TDG protein increased by approximately 3-folds in sirt1 knockout cells (Figure 7A, 2nd panel) as normalized to the amounts of β-actin (Figure 7A, 5th panel). In contrast, the APE1 protein level was nearly unchanged (Figure 7A, 4th panel). To determine the acetylation level of TDG in sirt1 knockout MEF, we develop Ac-TDG specific antibodies against a peptide (residues 67–93 of hTDG) containing three acetylated K83, K84, and K87 residues. The specificity of the Ac-TDG antibody was verified with unmodified (lane 1) and in vitro p300 acetylated hTDG (lane 2) (Figure S5 in supplementary materials). Consistent with the finding of Figure 4 that TDG is deacetylated by SIRT1, a Western blot with Ac-TDG specific antibodies showed a preferentially reactive band in sirt1 knockout cell extracts as compared to control extracts (Figure 7A, 3rd panel). To determine whether the increased TDG protein level in sirt1 knockout cells is controlled at the transcription level, we examined the Tdg mRNA level. As shown in Figure 6B, analysis by reverse transcription quantitative PCR (RT-qPCR) indicated that the Tdg mRNA level in sirt1 knockout cells was 1.9 ± 0.2 fold of that of the wild-type cells using mRNA of β-actin as an internal control. Thus, the level of Tdg mRNA is up-regulated in sirt1 knockout cells.
Figure 7. TDG is overexpressed in sirt1 knockout cells.
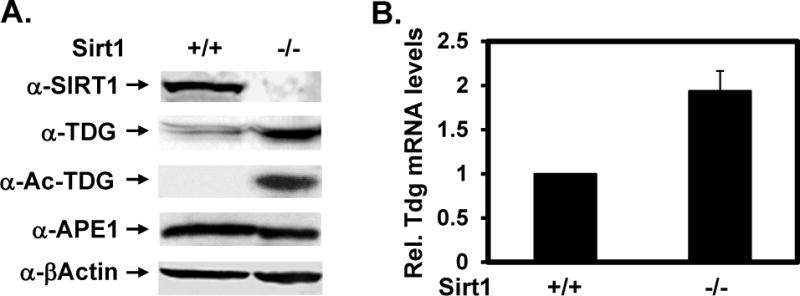
(A) Protein expression levels in wild-type (+/+) and sirt1 knockout (−/−) MEFs. Western blotting was performed with antibodies against SIRT1, TDG, Ac-TDG, APE1, and β-actin in total cell extracts. The TDG protein levels were normalized to the amounts of β-actin. (B) The Tdg mRNA levels in wild-type (+/+) and sirt1 knockout (−/−) MEFs. The mRNA levels were measured by RT-qPCR analysis. The mRNA level of Tdg was calculated relative to that of β-actin as described in Supplementary Materials. The relative Tdg mRNA levels of WT cells over sirt1 KO cells are presented in X-axis. The reactions were carried out in duplicate and data are averaged from three independent experiments.
DISCUSSION
Mammalian sirtuins play vital role in many biological processes, such as insulin secretion, fat mobilization, response to stress, and lifespan regulation (reviewed in [23,24]). SIRT1 exerts multiple functions through regulation of different targets including histones and many non-histone proteins [32]. It has been shown that SIRT1 is involved in DNA repair [28,31,36,42]. SIRT1-defective or knockdown cells have been shown to be more sensitive to ultraviolet light (UV), methyl methanesulfonate (MMS), H2O2, ionizing irradiation (IR), and FU [26,28,31,34,42–44].
SIRT1 can deacetylate three enzymes involved in BER pathway: TDG (Figure 4), APE1 [26], and PARP1 [25]. In the case of PARP, SIRT1-dependent deacetylation blocks its activity and protects cells from PARP1-mediated cell death [25]. In addition, SIRT1 suppresses PARP1 gene expression. Interestingly, both PARP1 and SIRT1 activities are dependent on NAD+. In contrast, SIRT1 does not regulate APE1 gene expression (Figure 7A) and deacetylation of APE1 by SIRT1 does not affect APE1 activity in vitro [26]. However, deacetylation of APE1 promotes its binding to X-ray cross-complementing-1 (XRCC1), thus stimulating DNA repair activity in vivo [26]. In this study, we demonstrate that SIRT1 modulates BER pathway through enhancing TDG glycosylase activity, deacetylating TDG, and suppressing TDG gene expression. Hence, SIRT1 is a key regulator of the BER.
The N-terminal domain of TDG is required for SIRT1 interaction. This region also interacts with many other protein partners including p300/CBP [18], APE1 [18], and the 9–1–1 complex [19]. It will be valuable to know how TDG uses the same domain to select its partners and how to regulate these interactions. The interaction of TDG with SIRT1 results in modest stimulation of hTDG glycosylase activity toward T/G, FU/G, fC/G and caC/G mismatches. It will be interesting to test the SIRT1 effects on TDG activity with other DNA substrates [2–6]. Because TDG has a protective role against oxidative damage, the SIRT1-mediated stimulation may play an important role in the balance between accumulation of oxidative DNA damage and its repair.
The physical and functional interactions between SIRT1 and TDG are altered by TDG acetylation. TDG acetylation weakens its physical interaction with SIRT1. The T/G glycosylase activity of unacetylated TDG was increased by 6 fold in TDG reaction buffer (Figure 2) and by 11 fold in the presence of 4-fold molar excess of SIRT1 in quarter strength of SIRT1 reaction buffer (Figure 5C and 5D). Acetylated TDG has reduced glycosylase activity toward T/G, fC/G, and caC/G, but it has a stronger activity toward FU/G substrate as compared to unmodified TDG (Figure 6). Such a regulatory network of protein interaction and protein acetylation/deacetylation allows TDG to function in DNA repair and transcription [21]. Thus, unacetylated TDG is the preferred form for DNA repair and DNA demethylation while acetylated TDG enhances FU sensitivity (Figure 8). We propose that SIRT1 has two interactions with TDG: its deacetylase activity acts on Ac-TDG and it stimulates the T/G glycosylase activity of the resulting unacetylated TDG. Given that SIRT1 can both bind and deacetylated TDG, TDG deacetylation by SIRT1 may suffer from minor product inhibition. Although the mechanism for these interactions is unclear, the product inhibition has been observed in many DNA glycosylases including TDG [38].
Figure 8. A model depicting the role of SIRT1 in mediating TDG function in DNA repair, gene expression, and drug cytotoxicity.
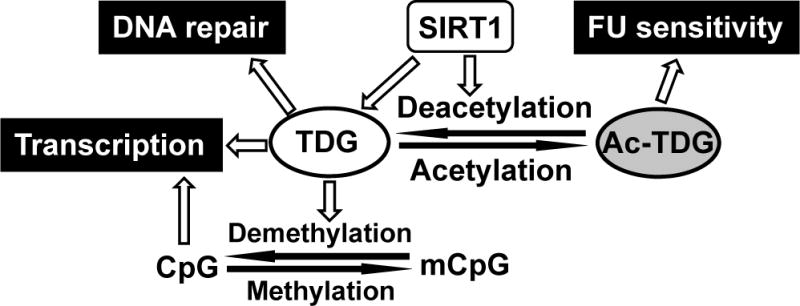
SIRT1 enhances TDG glycosylase activity, deacetylates TDG, and suppresses TDG gene expression. Acetylated TDG has reduced glycosylase activities toward T/G, fC/G, and caC/G mismatches, but has a stronger activity toward FU/G substrate as compared to unmodified TDG. Thus, we propose that unmodified TDG is responsible for DNA repair of T/G mismatches and DNA demethylation while acetylated TDG promotes FU removal. If the resulting AP sites are not efficiently repaired by BER after FU removal, spontaneous breakage at AP sites leads to the persistence of single-strand DNA lesions. During DNA replication, these DNA nicks can be converted to double-strand breaks. Alternatively, spontaneous breakage at nearby AP sites generates double-strand breaks and thus causes FU cytotoxicity and cell death.
The anticancer effect of FU, one of the widely used anticancer agents, is thought to involve multiple pathways [45]. This includes the incorporation of U and FU into DNA followed by their excision by a DNA glycosylase, leading to DNA fragmentation and cell death [45]. Consistent with a role for TDG in this process, Kunz et al. [46] have shown that inactivation of TDG significantly increases cellular resistance towards FU. This likely reflects TDG activity against FU/G lesions because its FU/A activity is relatively weak [6]. It has been shown that downregulation of SIRT1 increase cellular sensitivity to FU [34,43]. However, the molecular mechanism for the SIRT1’s effect on protecting cell from apoptosis upon FU treatment has not been investigated. We hypothesize that the physical and functional interactions between SIRT1 and TDG may mediate FU cytotoxicity (Figure 8). Because Ac-TDG has 6-fold higher activity toward FU/G substrate as compared to unmodified TDG (Figure 6B, compare lanes 2 with 6), TDG glycosylase activity on FU/G mismatches may increase in sirt1 knockout cells in which TDG is overproduced and hyperacetylated.
TDG also functions in DNA demethylation and gene regulation [9,10,13,14,16,47–50]. Interaction between TDG and SIRT1 may coordinate DNA methylation with histone acetylation. It has been shown that CpG methylation is associated with histone hypo-acetylation and epigenetic silencing [51]. SIRT1 maintains TDG in the hypoacetylated form which has higher glycosylase activity toward T/G, fC/G, and caC/G mismatches. Moreover, SIRT1 exerted higher enhancement on unmodified TDG than on Ac-TDG on these DNA substrates. Both of these SIRT1 actions are crucial for TDG’s function in active DNA demethylation [9,10]. This suggests that SIRT1 may act as a gene activator, contradicting its usual role as a gene silencer. Thus, we hypothesize that SIRT1 has dual roles and may be dependent on its interaction with TDG: SIRT1 deacetylates histones and induces compact chromatin at silenced genes; however, it deacetylates and enhances TDG at active genes. The dual roles of SIRT1 are in synchronization with the observations that SIRT1 has oncogenic properties but, paradoxically, can act as a tumor suppressor in different cell types [32,33]. These apparent pleiotropic effects of SIRT1 may due to its diverse functions and interactions with many proteins, one of which would be TDG.
Supplementary Material
Acknowledgments
The authors thank Dr. Phillip A. Cole (Johns Hopkins University) for kindly providing p300 protein, Alex C. Drohat (University of Maryland) for hTDG protein as well as FU/G-, fC/G-, and caC/G-containing DNA, and Dr. Toren Finkel (NIH) for SIRT1+/+ and SIRT1−/− mouse embryonic fibroblast cells. We thank Drs. Primo Schar (University of Basel, Switzerland) and Jianyuan Luo (University of Maryland) for providing TDG antibody and p300 expression plasmid, respectively. This work was supported by the National Cancer Institute of the National Institutes of Health under award number R01-CA78391 and a pilot grant from University of Maryland Greenebaum Cancer Center (to A-L. L.).
Abbreviations used
- Ac
acetylated
- AP
apurinic/apyrimidinic
- APE1
AP endonuclease I
- BER
base excision repair
- caC
5-carboxylcytosine
- fC
5-formylcytosine
- FL
Full length
- FU
5-fluorouracil
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
- HDACs
histone deacetylases
- hmC
5-hydroxymethylcytosine
- IR
ionizing irradiation
- KO
knockout
- MBD4
methyl binding domain IV
- mC
5-methylcytosine
- MEF
mouse embryonic fibroblast
- MeG
O6-methyl-guanine
- MMS
methyl methanesulfonate
- NAD+
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NT
N-terminal deletion
- PCR
polymerase chain reaction
- q-PCR
quantitative PCR
- RT
reverse transcription
- SIRT1
Sir2 homolog 1
- SIRTs
Sir2 homologs
- TDG
thymine DNA glycosylase
- UV
ultraviolet light
- WT
wild type
- XRCC1
X-ray cross-complementing-1
Footnotes
AUTHOR CONTRIBUTION
Amrita Madabushi performed protein purification, most GST pull-down analyses, immunoprecipitation, SIRT1 stimulation of TDG activity, SIRT1 deacetylation reaction, TDG expression in sirt1 kockout cells, data analysis, experimental design, and prepared the paper. Bor-Jang Hwang performed immunoprecipitation, GST pull-down of GST-SIRT1 with Ac-TDG, TDG acetylation in sirt1 knockout cells, and TDG deacetylation by SIRT1. Jin Jin performed TDG glycosylase assays with FU-, fC- and caC-DNA. A-Lien Lu conceived the work and assisted in experimental design and preparation of the paper. All authors read and approved the final paper.
Supporting Information Available: Supplementary materials related to this article can be found online.
References
- 1.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 2.Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J Am Chem Soc. 2006;128:12510–12519. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardeland U, Bentele M, Lettieri T, Steinacher R, Jiricny J, Schar P. Thymine DNA glycosylase. Prog Nucleic Acid Res Mol Biol. 2001;68:235–253. doi: 10.1016/s0079-6603(01)68103-0. [DOI] [PubMed] [Google Scholar]
- 5.Hardeland U, Bentele M, Jiricny J, Schar P. The versatile thymine DNA-glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan MT, Bennett MT, Drohat AC. Excision of 5-halogenated uracils by human thymine DNA glycosylase. Robust activity for DNA contexts other than CpG. J Biol Chem. 2007;282:27578–27586. doi: 10.1074/jbc.M704253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 8.Patra SK, Patra A, Rizzi F, Ghosh TC, Bettuzzi S. Demethylation of (Cytosine-5-C-methyl) DNA and regulation of transcription in the epigenetic pathways of cancer development. Cancer Metastasis Rev. 2008;27:315–334. doi: 10.1007/s10555-008-9118-y. [DOI] [PubMed] [Google Scholar]
- 9.Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 10.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rai K, Sarkar S, Broadbent TJ, Voas M, Grossmann KF, Nadauld LD, Dehghanizadeh S, Hagos FT, Li Y, Toth RK, et al. DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell. 2010;142:930–942. doi: 10.1016/j.cell.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8:1076–1092. doi: 10.1038/sj.cdd.4400948. [DOI] [PubMed] [Google Scholar]
- 18.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 19.Guan X, Madabushi A, Chang DY, Fitzgerald ME, Shi G, Drohat AC, Lu A-L. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates DNA repair enzyme TDG glycosylase. Nucleic Acids Res. 2007;35:6207–6218. doi: 10.1093/nar/gkm678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinacher R, Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol. 2005;15:616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 21.Mohan RD, Litchfield DW, Torchia J, Tini M. Opposing regulatory roles of phosphorylation and acetylation in DNA mispair processing by thymine DNA glycosylase. Nucleic Acids Res. 2010;38:1135–1148. doi: 10.1093/nar/gkp1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- 23.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 24.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–4129. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, Jung SB, Kim CS, Irani K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010;38:832–845. doi: 10.1093/nar/gkp1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2010;62:35–41. doi: 10.1016/j.phrs.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149–162. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, Kaushal D, Cheng HL, Fischer MR, Stokes N, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5:147–152. doi: 10.7150/ijbs.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang Y, Nicholl MB. Sirtuin 1 in malignant transformation: friend or foe? Cancer Lett. 2011;306:10–14. doi: 10.1016/j.canlet.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 34.Kabra N, Li Z, Chen L, Li B, Zhang X, Wang C, Yeatman T, Coppola D, Chen J. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J Biol Chem. 2009;284:18210–18217. doi: 10.1074/jbc.M109.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu A-L, Tsai-Wu J-J, Cillo J. DNA determinants and substrate specificities of Escherichia coli MutY. J Biol Chem. 1995;270:23582–23588. doi: 10.1074/jbc.270.40.23582. [DOI] [PubMed] [Google Scholar]
- 36.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzgerald ME, Drohat AC. Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J Biol Chem. 2008;283:32680–32690. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 39.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 41.Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, Huber LJ. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. 2010;39:247–258. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Akao Y, Noguchi S, Iio A, Kojima K, Takagi T, Naoe T. Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett. 2011;300:197–204. doi: 10.1016/j.canlet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 44.Jeong J, Juhn K, Lee H, Kim SH, Min BH, Lee KM, Cho MH, Park GH, Lee KH. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007;39:8–13. doi: 10.1038/emm.2007.2. [DOI] [PubMed] [Google Scholar]
- 45.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 46.Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, Schar P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009;7:e91. doi: 10.1371/journal.pbio.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 49.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 50.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 51.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.