

Summary
Neurovascular coupling (NVC) allows increased blood flow to metabolically active neurons and involves the Ca2+-dependent release of vasodilator influences by astrocyte endfeet encasing parenchymal arterioles. We have previously reported inversion of NVC from dilation to constriction in brain slices from subarachnoid hemorrhage (SAH) model rats. Corresponding to NVC inversion, there was a marked increase in the amplitude of spontaneous Ca2+ oscillations in astrocyte endfeet. Calcium-permeable transient receptor potential vanilloid 4 (TRPV4) channels have been reported in astrocyte endfeet and activators of these channels enhance Ca2+ oscillations in healthy animals. Here, we examined the role of TRPV4 channels in the development of high amplitude spontaneous Ca2+ oscillations in astrocyte endfeet and the inversion of neurovascular coupling after SAH. Treatment of brain slices with the TRPV4 channel antagonist, HC-067047 (10 μM), did not alter the amplitude of spontaneous Ca2+ oscillations after SAH. In addition, HC-067047 did not inhibit or change SAH-induced inversion of neurovascular coupling. In summary, TRPV4 channels do not appear to be involved in the inversion of neurovascular coupling after SAH. Further studies examining the impact of SAH on additional Ca2+ signaling pathways in astrocytes are likely to reveal valuable insights into new therapeutic strategies to advance SAH treatments.
Key words and/or reference phrases: subarachnoid hemorrhage, neurovascular coupling, astrocytes, Ca2+ oscillation, transient receptor potential channels, TRPV4
Introduction
Neurovascular coupling forms the basis of functional hyperemia and ensures adequate delivery of oxygen and nutrients to active neurons. This neurally-evoked vasodilation matches blood flow to task-dependent increases in regional brain function and involves the coordinated activity of neurons, astrocytes and intracerebral (parenchymal) arterioles. Under physiological conditions, neurovascular coupling involves 1) neuronal activation and release of the neurotransmitter, glutamate; 2) activation of metabotropic glutamate receptors (mGluRs) on astrocyte processes leading to a wave of elevated Ca2+ in astrocytes due to activation of inositol triphosphate receptor (IP3Rs); and 3) Ca2+-dependent release of vasodilatory signals by astrocyte endfeet that encase parenchymal arterioles [8, 23, 25]. A number of pathologies, such as Alzheimer’s disease, ischemic stroke and hypertension, have been reported to impair neurovascular coupling [11]. We have recently demonstrated a fundamental change in the polarity of the neurovascular response in brain slices from SAH animals [12, 13, 14]. We have found that neuronal activation of similar intensity has the opposite effect in brain slices from SAH animals causing vasoconstriction rather than the vasodilation observed in control and sham-operated animals. Local vasoconstriction in response to neuronal activity after SAH could potentially restrict blood flow, compromise neuronal viability and contribute to the development of delayed cerebral ischemic injury that manifests in humans several days after cerebral aneurysm rupture [24].
Our previous findings also indicate that increased amplitude of spontaneous Ca2+ oscillations in astrocyte endfeet after SAH is a key determinant in the inversion of neurovascular coupling [13]. However, the molecular mechanism leading to enhancement of these endfoot Ca2+ events is presently unclear. Increased activity of TRPV4 channels, a subtype of Ca2+ permeable ion channels within the transient receptor potential (TRP) channel family [18], is one potential contributor to enhanced endfoot Ca2+ signaling after SAH. TRPV4 channels are located on the plasma membrane of cortical astrocytes [3] and evidence indicates that a synthetic activator of these channels can increase the amplitude of spontaneous Ca2+ oscillations in astrocyte endfeet encompassing parenchymal arterioles of healthy mice [6]. Therefore, the goal of the present study was to examine the role of TRPV4 channels in the SAH-induced increased amplitude of spontaneous Ca2+ events and the inversion of neurovascular coupling observed in brain slices obtained from SAH model rats. Our present findings indicate that TRPV4 channel activity does not contribute to altered astrocyte Ca2+ signaling or the inversion of neurovascular coupling that occurs after SAH.
Materials and Methods
Rat SAH model
Using a surgical approach, two injections of autologous unheparinized arterial blood (500 μL) were made 24 hours apart into the cisterna magna of anesthetized Sprague-Dawley (male, 10–12 week old) rats. After each injection, animals were placed head down at a 45° angle for 20 minutes prior to recovery from anesthesia, as previously described [12, 19]. Animals were euthanized 4 days after the first injection, and cortical brain slices (160-μm thick coronal sections, middle cerebral artery territory) were prepared using a vibratome. All experiments were conducted in accordance with The Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, revised 1996) and followed protocols approved by the Institutional Animal Use and Care Committee of the University of Vermont.
Brain slice studies
Brain slices were loaded for 1 hour at 29°C with the Ca2+ indicator dye, Fluo-4-AM (10 μM) and 0.05 % pluronic acid in artificial cerebrospinal fluid (aCSF) containing (in mM): 122 NaCl, 3 KCl, 18 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 5 glucose aerated with 5 % CO2 and 95 % O2. For simultaneous measurements of astrocyte endfoot Ca2+ and parenchymal arteriolar diameter, brain slices were superfused at 37°C with aCSF (aerated with 5 % CO2/95 % O2, pH ~7.35) containing 100 nM of the thromboxane analog, U46619. Similar experimental findings were obtained when brain slices were superfused with aCSF aerated with 5% CO2, 20 % O2 and 75 % N2 (Koide and Wellman, unpublished observations). Astrocyte endfoot Ca2+ was measured using a BioRad Radiance multiphoton imaging system coupled to a Chameleon Ti:Sapphire laser (Coherent) and an Olympus BX51WI upright microscope [10, 12]. Fluo-4 was excited at 820 nm and fluorescence emission was collected using a 575/150-nm bandpass filter. Calcium concentrations in astrocyte endfeet were determined using the maximal fluorescence (Fmax) method [10, 15]. Arteriole diameter images were simultaneously acquired using infrared-differential interference contrast (IR-DIC) microscopy. Arteriolar diameter was determined by averaging measurements obtained from three points along the length of the vessel on the same image and are expressed as percent change from the diameter recorded from the first image of the recording. For induction of neuronal activation, electrical field stimulation (EFS; 50 Hz; 0.3-ms alternating square pulse; 3-second duration) was applied using a pair of platinum wires.
Statistical Analysis
Data are expressed as mean ± SEM with n representing the number of recordings per group. Student’s paired t-test was used to determine statistical significance at the level of P < 0.05.
Results
Inhibition of TRPV4 channels does not alter the amplitude of spontaneous Ca2+ oscillations in astrocyte endfeet after SAH
SAH causes a marked increase in the amplitude of spontaneous Ca2+ oscillations leading to inversion of neurovascular coupling [12, 14]. However, the underlying mechanism responsible for the enhancement of this Ca2+ signaling modality after SAH is currently unclear. A recent study by Dunn et al. [6] found that the synthetic activator of TRPV4 channels, GSK 1016790A, increased the amplitude of spontaneous Ca2+ oscillations in endfeet imaged in brain slices from control mice. To examine whether SAH causes an increase in endfoot TRPV4 channel activity, studies were done using the TRPV4 antagonist, HC-067047. In the absence of this compound, the amplitude of spontaneous endfoot Ca2+ events was 474.5 ± 20.0 nM (n = 13 endfeet from 4 animals) (figure 1a, b) in brain slices from SAH model animals. These measurements are in agreement with our previous work, with peak amplitudes approximately 100 nM higher than spontaneous Ca2+ events recorded in endfeet from control animals. Incubation of brain slices from SAH animals with HC-067047 (10 μM for 25 min.) did not alter basal levels of Ca2+ (− HC-067047: 195.0 ± 7.1 nM; + HC-067047: 195.3 ± 8.5 nM; n = 13 endfeet from 4 brain slices) measured during the interval between spontaneous events (figure 1b). HC-067047 also did not change the frequency of these spontaneous Ca2+ events (figure 1c). Further, as illustrated in figure panels 1a and 1b, HC-067047 did not alter the amplitude of spontaneous Ca2+ events (471.7 ± 23.7 nM, n = 13 endfeet from 4 animals) in brain slices from SAH model animals. This data demonstrates that TRPV4 channel activity does not contribute to the enhanced amplitude of spontaneous Ca2+ events that occur in astrocyte endfeet after SAH.
Figure 1. TRPV4 inhibition does not alter the amplitude or frequency of spontaneous Ca2+ events in astrocyte endfeet after SAH.
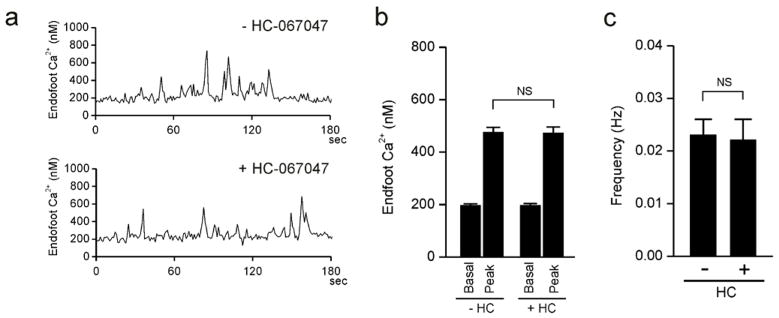
a) Spontaneous Ca2+ oscillations recorded from 1.2 × 1.2-μm regions of interest placed on distinct astrocyte endfeet in a brain slice from a SAH animal in the absence (upper trace) and presence (lower trace) of the TRPV4 antagonist HC-067047 (10 μM). (b) Summary data of endfoot Ca2+ levels in the absence and presence of HC-067047 (10 μM) obtained from brain slices of SAH model rats. The term “Peak” represents the average maximum Ca2+ concentration measured during individual spontaneous Ca2+ oscillations. c) Summary data of the frequency of spontaneous Ca2+ oscillations ± HC-067047 (10 μM) obtained from 4-minute recordings using brain slices from SAH model rats. For panels b and c, recordings were made from 13 endfeet in 4 brain slices from 4 animals. NS, not statistically significant (P > 0.05), using paired students t-test.
TRPV4 channels do not mediate SAH-induced inversion of neurovascular coupling
To examine the impact of TRPV4 channels on SAH-induced inversion of neurovascular coupling, electrical field simulation (EFS) was used to induce neuronal action potentials in brain slices from SAH animals. Consistent with our prior studies [12, 13], EFS resulted in an increase in endfoot Ca2+ from a resting level of 169.6 ± 8.6 nM to a peak of 348.9 ± 33.0 nM (n = 5) (figure 2a, c). Associated with this EFS-induced increase in Ca2+, parenchymal arterioles encased by these endfeet constricted by 21.2 ± 4.3 % (figure 2a, b). This vasoconstriction in response to neuronal activation after SAH represents an inversion of the physiological vasodilation observed in brain slices from healthy control animals [8, 12, 25]. Incubation of brain slices with the TRPV4 channel antagonist, HC-067047 (10 μM), did not alter EFS-evoked increases in endfoot Ca2+ (peak Ca2+ after EFS: 330 ± 30.5 nM) or the magnitude of ensuing vasoconstriction (18.6 ± 3.1 % decrease in diameter) (figure 2a–c). These findings indicate that TRPV4 channels do not alter EFS-induced increases in endfoot Ca2+ or SAH-induced inversion of neurovascular coupling.
Figure 2. TRPV4 channel inhibition does not influence SAH-induced inversion of neurovascular coupling.
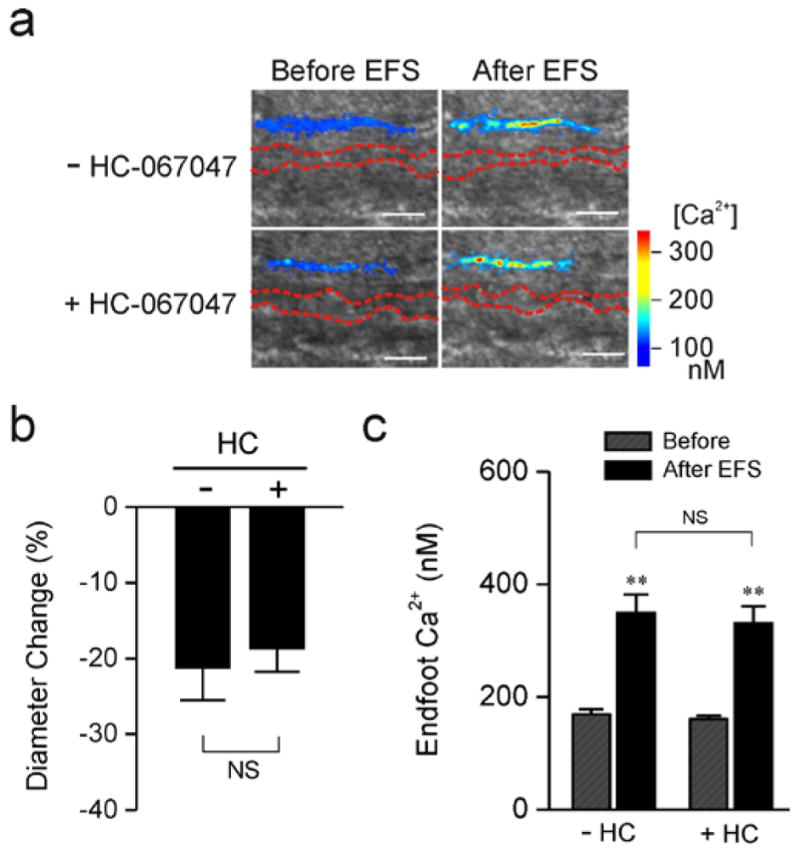
Parenchymal arteriolar diameter and astrocyte endfoot Ca2+ concentration were simultaneously measured using two-photon imaging and infrared-differential interference contrast (IR-DIC) microscopy. a) IR-DIC images in the absence (upper panels) and in the presence (lower panels) of the TRPV4 antagonist HC-067047 (10 μM) obtained from brain slices of SAH model rats. Red dashes outline the intraluminal diameter of parenchymal arterioles using IR-DIC microscopy. Overlapping pseudocolor-mapped endfoot Ca2+ levels were obtained using the fluorescent Ca2+ indicator Fluo-4 and two-photon imaging. Scale bars, 10 μm. b, c) Summary of EFS-evoked changes in arteriolar diameter (b) and astrocytic endfoot Ca2+ (c) obtained from SAH (n = 5 brain slices from 3 animals) model rats in the presence and absence of HC-067047. NS, not statistically significant (P > 0.05), −HC-067047 vs + HC-067047, using paired students t-test. ** P < 0.001, Before vs After EFS, using ANOVA followed by Tukey test.
Discussion
Neurovascular coupling is an important physiological process enabling increased local blood flow to metabolically active regions of the brain. Recent evidence indicates that subarachnoid blood causes a fundamental change in NVC that could lead to pathological decreases in blood flow, rather than the “normal” physiological increases in blood flow associated with localized, task-dependent increases in neuronal activity. Altered Ca2+ signaling in astrocyte endfeet, in the form of high-amplitude spontaneous Ca2+ oscillations, are responsible for this SAH-induced inversion of neurovascular coupling. In this present study, we hypothesized that enhanced Ca2+ entry via TRPV4 channels contributes to the increased amplitude of spontaneous Ca2+ events observed in astrocyte endfeet after SAH. However, this does not appear to be the case, as pharmacological inhibition of TRPV4 channels did not alter endfoot Ca2+ signaling or the inversion of neurovascular coupling in brain slices from SAH model animals.
Astrocytes exhibit a diverse array of Ca2+ signaling events, including nerve-evoked propagating Ca2+ transients [8, 13, 25], spontaneous intracellular and intercellular propagating Ca2+ waves [7, 22] and non-propagating spontaneous Ca2+ oscillations that can occur in either the cell body or in cell processes such as endfeet wrapping around intracerebral blood vessels [13, 17, 20]. Our recent evidence demonstrates that a marked elevation in the amplitude of spontaneous Ca2+ oscillations in endfeet rather than changes in nerve-evoked astrocyte Ca2+ signaling underlie inversion of neurovascular coupling after SAH [12]. In brain slices from SAH animals, high amplitude Ca2+ oscillations in endfeet lead to increased K+ efflux into the perivascular space via increased activity of large-conductance Ca2+-activated K+ (BK) channels. This increase in basal extracellular K+ when summed with nerve-evoked astrocyte K+ efflux elevates extracellular K+ in the microenvironment surrounding parenchymal arterioles above the constriction threshold leading to a polarity change in the neurovascular response from vasodilation to vasoconstriction.
In astrocytes from healthy animals, spontaneous Ca2+ oscillations reflect the release of Ca2+ stored in endoplasmic reticulum through activation of IP3-sensitive Ca2+ release channels (i.e. IP3 receptors) and occur independently of neuronal activity or metabotropic glutamate receptor activation [17, 20]. The activity of IP3 receptors is bimodally regulated by cytoplasmic Ca2+, with moderate increases in Ca2+ leading to an increase in IP3 receptor activation [9]. Thus, while requiring release of Ca2+ from intracellular stores, the amplitude of these spontaneous Ca2+ events in endfeet can be modulated by Ca2+ entering the cell through the plasma membrane. For example, Ca2+ permeable TRPV4 channels are present on astrocyte processes [3, 4] and activation of these channels caused an increase in the amplitude of spontaneous Ca2+ events in endfeet of brain slices prepared from healthy mice [6]. In addition, TRPV4 expression and function is upregulated in hippocampal astrocytes after cerebral ischemia [4]. However, our present data indicates that Ca2+ entry via TRPV4 channels does not contribute to the increase in amplitude of endfoot Ca2+ events occurring in SAH model animals. The present study does not exclude the possibility that Ca2+ entry through other TRP family members is upregulated in astrocytes after SAH. Although evidence suggests that normal native astrocytes do no express voltage-dependent Ca2+ channels, it is also possible that expression of these channels is upregulated in astrocytes after SAH. Alternatively, an increase in IP3 levels or other Ca2+-independent mechanisms leading to enhanced of IP3 receptor activity may underlie the increase in the amplitude of spontaneous Ca2+ events in endfeet after SAH. Altered astrocyte Ca2+ signaling has been reported to occur in brain pathologies other than SAH such as Alzheimer’s disease, ischemia/hypoxia and epilepsy [2, 4, 5, 23]. Further, reactive astrogliosis and microglia activation have also been associated with multiple forms of brain injuries including SAH [16, 21]. It has been postulated that Ca2+ oscillations in activated astrocytes may result in gliotransmitter release starting a cascade of events leading to excitotoxicity and brain damage [1]. Presently, the relationship between induction of reactive astrogliosis and the enhancement of astrocyte Ca2+ signaling is unclear and requires further investigation.
Conclusions
The inversion of neurovascular coupling from vasodilation to vasoconstriction represents a pathological response after SAH and is a likely contributor to poor outcome observed in SAH patients. Increased amplitude of spontaneously occurring Ca2+ oscillations underlying this SAH-induced inversion of neurovascular coupling are not affected by antagonism of TRV4 ion channels. Thus, future studies are required to elucidate the underlying mechanisms of the pathological changes in astrocyte Ca2+ signaling after SAH and pinpoint specific blood components involved in this response. A greater understanding of this pathway is likely to reveal new therapeutic targets that could benefit patients suffering from SAH.
Acknowledgments
This work was supported by the Totman Trust for Medical Research, the Peter Martin Brain Aneurysm Endowment and the NIH (P01 HL095488, R01 HL078983 and R01 HL078983-05S1). The authors would also like to acknowledge the use and assistance of the University of Vermont Neuroscience COBRE molecular biology core facility and input from Drs. Mark T. Nelson and Kathryn M. Dunn.
Footnotes
Conflict of interest statement: None.
References
- 1.Agulhon C, Sun MY, Murphy T, Myers T, Lauderdale K, Fiacco TA. Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front Pharmacol. 2012;3:139. doi: 10.3389/fphar.2012.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balazsi G, Cornell-Bell AH, Moss F. Increased phase synchronization of spontaneous calcium oscillations in epileptic human versus normal rat astrocyte cultures. Chaos. 2003;13:515–518. doi: 10.1063/1.1567652. [DOI] [PubMed] [Google Scholar]
- 3.Benfenati V, Amiry-Moghaddam M, Caprini M, Mylonakou MN, Rapisarda C, Ottersen OP, Ferroni S. Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience. 2007;148:876–892. doi: 10.1016/j.neuroscience.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 4.Butenko O, Dzamba D, Benesova J, Honsa P, Benfenati V, Rusnakova V, Ferroni S, Anderova M. The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS One. 2012;7:e39959. doi: 10.1371/journal.pone.0039959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding S, Wang T, Cui W, Haydon PG. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia. 2009;57:767–776. doi: 10.1002/glia.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. doi: 10.1002/glia.20396. [DOI] [PubMed] [Google Scholar]
- 8.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 9.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A. 2010;107:3811–3816. doi: 10.1073/pnas.0914722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 12.Koide M, Bonev AD, Nelson MT, Wellman GC. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc Natl Acad Sci U S A. 2012;109:E1387–E1395. doi: 10.1073/pnas.1121359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koide M, Bonev AD, Nelson MT, Wellman GC. Subarachnoid blood converts neurally evoked vasodilation to vasoconstriction in rat brain cortex. Acta Neurochir Suppl. 2013;115:167–71. doi: 10.1007/978-3-7091-1192-5_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koide M, Sukhotinsky I, Ayata C, Wellman GC. Subarachnoid hemorrhage, spreading depolarizations and impaired neurovascular coupling. Stroke Res Treat. 2013;2013:819340. doi: 10.1155/2013/819340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, Wellman GC. Subarachnoid Hemorrhage Induces Gliosis and Increased Expression of the Pro-inflammatory Cytokine High Mobility Group Box 1 Protein. Transl Stroke Res. 2011;2:72–79. doi: 10.1007/s12975-010-0052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nett WJ, Oloff SH, McCarthy KD. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J Neurophysiol. 2002;87:528–537. doi: 10.1152/jn.00268.2001. [DOI] [PubMed] [Google Scholar]
- 18.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;286:C195–C205. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 19.Nystoriak MA, O’Connor KP, Sonkusare SK, Brayden JE, Nelson MT, Wellman GC. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol. 2011;300:H803–H812. doi: 10.1152/ajpheart.00760.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parri HR, Crunelli V. The role of Ca2+ in the generation of spontaneous astrocytic Ca2+ oscillations. Neuroscience. 2003;120:979–992. doi: 10.1016/s0306-4522(03)00379-8. [DOI] [PubMed] [Google Scholar]
- 21.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 22.Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 24.Vergouwen MD, Vermeulen M, van GJ, Rinkel GJ, Wijdicks EF, Muizelaar JP, Mendelow AD, Juvela S, Yonas H, Terbrugge KG, Macdonald RL, Diringer MN, Broderick JP, Dreier JP, Roos YB. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41:2391–2395. doi: 10.1161/STROKEAHA.110.589275. [DOI] [PubMed] [Google Scholar]
- 25.Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]