

Abstract
Pancreatic ductal adenocarcinoma (PDA) is a lethal disease with etiological association with cigarette smoking. Nicotine, an important component of cigarettes, exists at high concentrations in the bloodstream of smokers. Osteopontin (OPN) is a secreted phosphoprotein that confers on cancer cells a migratory phenotype and activates signaling pathways that induce cell survival, proliferation, invasion, and metastasis. Here, we investigated the potential molecular basis of nicotine’s role in PDA through studying its effect on OPN. Nicotine significantly (p<0.02) increased OPN mRNA and protein secretion in PDA cells through activation of the OPN gene promoter. The OPN mRNA induction was inhibited by the nicotinic acetylcholine receptor antagonist, mechamylamine. Further, the tyrosine kinase inhibitor genistein inhibited the nicotine-mediated induction of OPN, suggesting that mitogen activated protein kinase signaling mechanism is involved.
Nicotine activated the phosphorylation of ERK1/2, but not p38 or c-Jun NH2-terminal MAP kinases. Inhibition of ERK1/2 activation reduced the nicotine-induced OPN synthesis. Rats exposed to cigarette smoke showed a dose-dependent increase in pancreatic OPN that paralleled the rise of pancreatic and plasma nicotine levels. Analysis of cancer tissue from invasive PDA patients, the majority of whom were smokers, showed the presence of significant amounts of OPN in the malignant ducts and the surrounding pancreatic acini. Our data suggest that nicotine may contribute to PDA pathogenesis through upregulation of OPN. They provide the first insight into a nicotine-initiated signal transduction pathway that regulates OPN as a possible tumorigenic mechanism in PDA.
Keywords: pancreatic cancer, nicotine, cigarette smoke, osteopontin
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is the fourth leading cause of adult cancer mortality in the United States (1). Because of its aggressive nature and the inability to detect it early, the disease is often far advanced in most patients by the time diagnosis is established. The overall 5-year survival rate is less than 5%, and more than 85% of the tumors are diagnosed after the tumor has infiltrated into adjacent organs or when distant metastases are present. There is, therefore, an urgent need for an improved understanding of the basic molecular mechanisms that contribute to the aggressive nature of PDA and for the design of therapies that are more effective than the current regimens.
Osteopontin (OPN), a secreted noncollagenous, sialic acid-rich, phosphoprotein, is a member of the SIBLING gene family (Small Integrin-Binding LIgand, N-linked Glycoproteins) (2). It functions as both an extracellular matrix component and a cytokine found in all body fluids that signals through its binding cell adhesion molecules, integrins and CD44 variants (3, 4). OPN was initially discovered as an inducible marker of transformation of epithelial cells and later was shown to be frequently overexpressed in many human cancers (5). In cultured cells, overexpression of OPN renders cells more tumorigenic and/or metastatic, while its down-regulation by an antisense approach reduces cell growth in soft agar and in mice both for primary tumors and for experimental metastatic foci (6). Although high levels of OPN have been reported in PDA (7–10), the mechanistic regulation of OPN by PDA risk factors has not been studied.
Tobacco smoking is the most important environmental risk factor for developing pancreatic cancer (11,12), with the risk increasing with higher levels of tobacco use and more years of exposure. A survey on the association between cigarette smoking and pancreatic cancer showed that cigarette smokers had a significantly higher risk (70%) of developing pancreatic cancer in comparison with non-smokers (13–16). When compared with non-smokers, subjects who smoked filtered cigarettes had a 50% elevated risk. The proportion of pancreatic cancer attributable to cigarette smoking was 29% in blacks and 26% in whites (17). Nicotine, a major component of tobacco and cigarette smoke, is an addictive agent and has been characterized as a drug of abuse by the U.S. Surgeon General (18). The association of nicotine exposure through cigarette smoking with the increased incidence of pancreatic cancer (13–16) has been reported. Most of the data linking cigarette smoke/nicotine to pancreatic diseases were gathered in humans (10–20). Studies conducted with animals have shown that nicotine or its metabolites could induce pathological and functional changes in the pancreas (21–24). However, very few studies have suggested mechanisms as to how these functional or morphological changes could contribute to the progression of pancreatic cancer. Moreover, none of these studies have identified reliable nicotine-induced factors that could mediate the carcinogenic effects of nicotine, which could be explored as novel molecular targets for the treatment of PDA.
In this study, we tested the hypothesis that nicotine may contribute to pancreatic carcinogenesis through induction of OPN in PDA cells. We analyzed its effect on OPN synthesis and protein production in three human pancreatic cancer cell lines: AsPC-1, HS766T, and PK9, and evaluated the signaling mechanisms involved. We also examined the effect of cigarette smoke on pancreatic structure and OPN expression in rats. Finally, we analyzed the expression of OPN in normal human pancreata and in pancreata containing premalignant and malignant lesions, and correlated OPN expression with patients’ smoking history.
MATERIALS AND METHODS
Cell Culture
The human PDA cell line AsPC-1 was purchased from the American Type Culture Collection (Manassas, VA). HS766T and PK9 cells were generously donated by Dr. Scott Kern, Johns Hopkins University School of Medicine, Baltimore, MD. Cells were cultured at 1×104 to near confluence in 96-well plate and maintained in DMEM supplemented with 10% fetal bovine serum in a humid atmosphere of 5% CO2/95% air. Cells were treated with nicotine (3×10−9 – 3×10−7 mol/L), which is in the physiological range of blood levels of nicotine in smokers. Cells were incubated for 24 and 72 h, and were evaluated for the expression of OPN mRNA by real time PCR. To examine OPN protein secretion in the media, cells were treated with nicotine for 48 and 72 h, after which the media were harvested and analyzed. We then examined the detailed signaling involved in the nicotine-mediated increase in OPN in AsPC-1 cells. To evaluate whether nicotine binding to its nicotinic acetylcholine receptor (nAChR) results in induction of OPN mRNA, AsPC-1 cells were preincubated with a receptor antagonist, mechamylamine, for 1 hour, before stimulation by nicotine. The concentrations of nicotine and antagonist used in these experiments were selected based on dose-response studies.
RNA Extraction and Real Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from PDA cells or pancreata using Trizol reagent (Life Technologies, Gaitherburg, MD). RNAs were quantified and input amounts were optimized for each amplicon. Primers and probes were designed with the help of Primer Express Software (Applied Biosystems; Foster City, CA). The specificity of OPN primers was validated using semi quantitative PCR. GAPDH was used as the house keeping gene. cDNA was prepared, diluted, and subjected to real-time PCR using the TaqMan technology (7500 Sequence Detector; Applied Biosystems). Probes were labeled with a reporter and a quencher. Each sample was analyzed in at least two independent assays with duplicate samples and the corresponding no-reverse transcriptase (RT) mRNA sample was included as a negative control. The relative mRNA levels were presented as unit values of 2^[CT (GAPDH) - CT (OPN)], where CT is the threshold cycle value defined as the fractional cycle number at which the target fluorescent signal passes a fixed threshold above baseline.
Protein Isolation and Western Blot Analysis
Cell lysates were analyzed as described elsewhere (25). Anti- total and -phospho-ERK1/2 MAPK, anti-total and-phospho-p38 MAPK, and anti-total and – phospho SAPK/JNK, and anti–β-actin antibodies were purchased from Cell Signaling Technology and Sigma, respectively.
ELISA
Human OPN concentration in the cell culture media were measured using human-specific ELISA kit (Assay Design, Ann Arbor, MI). Spectrophotometric evaluation of OPN levels were made by Synergy HT multi-detection microplate reader (BioTeck, Winooski, VT).
MTT assay
To examine the effect of nicotine, mechamylamine, genistein, or the MAPKK inhibitor PD980509 on in vitro cell growth, AsPC-1 cells were plated in quintuplicate in 96-well plates and incubated in full growth media at 37°C and 5% CO2. Cells were treated with or without nicotine (3×10−9 – 3×10−7 mol/L), mechamylamine (1–100 µM), genistein (15–60 µM), PD98050 (0.1–10 µM) for 24 and 72 hours. Cell viability was examined using the MTT (3-(4, 5-methylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide) conversion Assay. MTT (Sigma) was added (50 µg/well) for 4 hours. Formazan products were solubilized with acidic isopropanol, and the optical density was measured at 570 nm. Optical density is directly correlated with cell quantity. Experiments were made in triplicate and repeated 3 times.
Transient Transfection and OPN Promoter Studies
The OPN promoter, (−1984luc) (GenBank™ accession number AF017274) in a luciferase expression vector pGL3 basic (Promega), was kindly provided by Dr. S Mori, Chiba University, Japan (26, 27). Cells were seeded into 24-well culture plates (105). At ~80% confluence they were co transfected by TransFast reagent (Promega) and 0.5 µg of pGL3 vectors containing the luciferase-labeled OPN promoter and 0.1 µg of GFP as transfection control. Two hours later, serum-containing medium was overlaid and the cells were incubated for additional 24 hr. The cells then were incubated with serum free medium for 18 h followed by addition of nicotine (3×10−9 – 3×10−7 mol/L). To determine the effect of the different inhibitors on OPN transcription, mechamylamine (1–100 µM), genestein (15–60 µM), or PD98050 (0.1–10 µM) were added to AsPC cells. Luciferase activities were assayed with the Dual-Luciferase Reporter Assay System (Promega) in a Veritas Microplate Luminometer (Turner Designs, Sunnyvale, Calif., USA). Transfection efficiency was normalized using the total protein concentration of the cell lysates. The results for nicotine-treated cells were expressed as a fold-induction of the luciferase activity of the same construct in control condition, taking control (no nicotine added) value as 1.
Rats
Thirty-six female Sprague Dawley rats were exposed to two doses of environmental tobacco smoke (100 mg or 160 mg/m3 total suspended particulate matter per m3 for 70 min twice daily for 12 weeks). All animal studies were performed in accordance with guidelines set forth by the Animal Resource Committee of Creighton University. The smoke exposure system was composed of smoke generator, mixing and conditioning chamber, rodent exposure chamber, exhaust chamber, and air sampler. A group of sham-treated animals received the same treatment without being exposed to cigarette smoke. Blood nicotine levels were measured by gas chromatography before sacrifice (28). Pancreata were isolated, cleaned from surrounding fat and lymph nodes and were either snap frozen in liquid nitrogen, or covered with OCT compound (Sakura Finetek, Torrance, California).
Nicotine Levels
An aliquot (135 µL) of the cell lysate was used and the pH was adjusted by adding 13.5 µL of 1 N NaOH. The sample was extracted four times with 0.5 mL of ethyl acetate. The organic extracts were dried over Na2SO4 and filtered, mixed with 250 µL of 2-propanol and concentrated to approximately 200 µL. Samples were analyzed using a GC/MSD system consisting of a Hewlett Packard Model 6890 gas chromatograph (Hewlett-Packard, Palo Alto, CA) interfaced with a HP Model 5973 Mass Selective Detector. The GC/MSD ion source pressure was approximately 3 × 105 torr and the emission current and ionization energy were 39.6 µA and 70 eV. Injections of 1 µL were made. Nicotine was analyzed on a DB-5MS fused silica capillary column (30 m × 0.32 mm, 0.25-µm film thickness, J & W Scientific, Folsom, CA). The injection port was maintained at 250°C. For detection and quantitation, a selected ion monitoring technique was used as monitoring and the ions of m/z 162 for nicotine.
Histology and Immunofluorescence Staining
Five µm cryostat cut frozen sections were stained with H&E after acetone fixation. To localize OPN in the pancreas, frozen sections from the sham and different experimental groups were stained with immunofluorescence using a monoclonal antibody against OPN (2A1, Santa Cruz Biotechnology, Santa Cruz, CA) (1:100). Negative controls were incubated with blocking solution without primary antibody. Sections were incubated with Texas Red conjugated goat anti-mouse IgG, diluted 1:200 in PBS (Vector Laboratories, Inc., Burlingame, California), as the secondary antibody, and viewed by fluorescence microscopy. For quantitative assessment of OPN staining, images were captured using a color SPOT camera (Diagnostic instruments, Inc, Sterling Heights, MI) and analyzed using ImagePro plus software. Digital images were captured and color segmentation was performed to highlight the stained area. The software then calculated this as a percentage of the total defined area, after exclusion of the islets areas. All histological assessment and image analysis was performed on coded, randomized sections by a blinded observer.
Human Tissue Acquisition and Analysis
Human PDA (n=25) and premalignant specimens (n=11) and normal pancreatic tissue were obtained from patients who underwent surgical resection at Thomas Jefferson University Hospital between 2005 and 2007. All patients signed an appropriate consent for tissue acquisition and study. The study was approved by the Institutional Review Board of Thomas Jefferson University. Patients’ smoking history was examined and correlated with OPN expression levels.
Tissue samples were stored in RNA Later for RNA analysis or fixed in neutral formaline for histological processing. Sections at 5 µm were stained with H&E. To localize OPN, sections from the different tissues were analyzed by immunohistochemistry using a monoclonal antibody against OPN (2A1, Santa Cruz) (1:100). A vectastain universal elite ABC kit and 3,3'-diaminobenzidine tetrahydrochloride chromogenic substrate (Vector Laboratories Inc.) was used according to the manufacturer protocol to visualize the tissue reaction. Antibody specificity was validated with nonimmune isotype serum. Negative control sections, where the primary or secondary antibodies were omitted were also prepared.
Statistical analysis
All experiments were performed 4 to 6 times. Data were analyzed for statistical significance by ANOVA with post-hoc student t test analysis. These analyses were performed with the assistance of a computer program (JMP 5 Software SAS Campus Drive; Cary, NC). Differences were considered significant at P≤0.05.
RESULTS
Nicotine induces OPN mRNA accumulation and secretion in cultured PDA cells
To investigate whether nicotine can directly increase OPN mRNA accumulation in PDA cells, AsPC-1, HS766T and PK9 cells were treated with or without nicotine (3×10−9 – 3×10−7 mol/L) for 24 and 72 hr. Significant induction of OPN mRNA expression was seen with a maximum increase at 24 hr at 3×10−8 mol/L of nicotine in ASPC-1 cells. Nicotine at 3×10−7 mol/L induced a time-dependent increase of OPN mRNA (Fig 1A). In HS766T cells, the increase in OPN mRNA levels could be detected after 24 hr of nicotine stimulation, and returned to baseline levels at 72 hr after nicotine treatment (Fig 1B). In PK9 cells, nicotine induced a significant increase of OPN mRNA at 24 and 72 hr, with more significant increase at 24 hr (Fig 1C). To examine whether the increase in OPN mRNA levels in response to nicotine was associated with OPN production, OPN protein levels in the media were determined by ELISA. Cells were treated with nicotine at its most effective concentration (3×10−8 mol/L). Extracellular OPN protein concentration increased markedly from 0.9 ng/ml to 7.79 ng/ml and 10.17 ng/ml after 48 hours and 72 hours of nicotine stimulation in AsPC-1 cells, respectively (Fig 1D). In HS766T cells, OPN basal levels of 12.66 ng/ml were significantly increased by nicotine to 19.56 ng/ml and 29.90 ng/ml after 48 and 72 hours of nicotine stimulation, respectively (Fig 1E). In PK9 cells, OPN levels were increased from 160.18 ng/ml to 194.90 ng/ml and 258.85 ng/ml after 48 and 72 hours of nicotine stimulation, respectively (Fig 1F). These data indicate that while basal OPN levels may vary between PDA cell lines, OPN induction by nicotine is a general phenomenon seen in all tested PDA cells lines.
Figure 1.






Nicotine induces OPN accumulation in cultured PDA cells. AsPC-1 (A), Hs766T (B), and PK9 (C) cells were treated with nicotine (3×10−9 – 3×10−7 mol/L) for 24 and 72 hr. Significant induction of OPN mRNA expression is seen with the maximum induction after 24 hr at 3×10−8 mol/L of nicotine.
OPN protein in culture media was measured using human-specific ELISA kit. Significant induction of OPN protein secretion is seen in AsPC-1 cells (D), HS766T (E), and PK9 (F) with a maximum at 72h. Each experiment was repeated three times for reproducibility. Values are expressed as mean ± SEM of three experiments. * p < 0.05 # p<0.005 vs. control levels, using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test.
Nicotine induces OPN promoter activity in PDA cells
AsPC-1 cells were transfected with rat OPN promoter/luciferase gene construct. After 24 h of transfection, the cells were incubated with nicotine (3×10−9 – 3×10−7 mol/L) for 3 hr, after which the luciferase activity in the cell lysates was measured. A significant increase in OPN promoter activation was seen after incubation with nicotine (Fig 2A). These data show that the OPN promoter responds directly to nicotine.
Fig 2.


A. Nicotine induces OPN promoter activity in AsPC-1 cells. After 24 h of transfection, the cells were incubated with nicotine (3×10−9 – 3×10−7 mol/L) for 3 hours. After incubation, the luciferase activity in the cell lysates was measured. Nicotine increased OPN promoter activity with maximum induction at 3×10−9 mol/L. Relative luciferase activity was calculated after deduction of the activity levels with pGL3 vector alone. Results represent mean ± SEM of triplicate determinations. All experiments were repeated at least three times to confirm the reproducibility of the observations. B. nAChR antagonist blocks nicotine-induced OPN mRNA expression in AsPC-1 cells. Cells were preincubated for 1 h with nAChR antagonist mechamylamine before addition of nicotine (3×10−8 mol/L) for 3 h. Mechamylamine induced a dose-dependent reduction in OPN mRNA levels. Values are expressed as mean ± SEM of three experiments. *p < 0.05 vs. nicotine alone treated cells using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test.
Since nicotine has been shown to increase cellular proliferation in pancreatic acinar cells (29,30), we tested whether it has similar effect in PDA cells. MTT assay was performed on AsPC-1 cells treated with or without nicotine (3×10−9 – 3×10−7 mol/L) for 24 and 72 hr. Addition of nicotine did not affect cellular proliferation when compared to control values (data not shown). These data suggest that the nicotine-mediated increase in OPN is independent of cellular proliferation.
Nicotine-induced OPN mRNA expression in AsPC-1 cells is blocked by nAChR antagonist
To determine the receptor that mediates the nicotine-induced OPN gene expression in PDA cells, the nAChR antagonist, mechamylamine (1–100 µM), was added for 1 hour prior to addition of nicotine to AsPC-1 cells. Mechamylamine dose dependently prevented the increase in OPN by nicotine (Fig 2B). These data demonstrate that PDA cells express nAChR and suggest that nicotine-OPN gene expression is mediated through nicotine receptor.
To examine whether mechamylamine itself can directly affect OPN transcription, AsPC-1 cells were incubated with mechamylamine (1–100 µM) after their transfection with luciferase-labeled OPN construct. Mechamylamine had no effect on OPN transcription (data not shown). Mechamylamine also did not influence cell proliferation when MTT assay was performed on AsPC-1 cells treated with or without nicotine (3×10−8 mol/L) for 24 and 72 hr (data not shown).
Nicotine-induced OPN gene expression requires tyrosine kinase activity. Nicotine signals through the nicotinic receptors and induces protein tyrosine phosphorylation in many cell types (31, 32). We investigated whether a similar signaling pathway mediates nicotine-induced OPN gene expression in AsPC-1 cells. To determine whether an increase in tyrosine phosphorylation induces OPN mRNA accumulation, cells were exposed to the protein tyrosine phosphatase inhibitor, sodium orthovanadate (10–50 µM) for 3 h. Sodium orthovanadate induced an increase in OPN mRNA accumulation in AsPC-1 at 25 µM with a maximal effect at 50 µM (Fig 3A). This suggests that increased protein tyrosine phosphorylation can induce OPN mRNA accumulation. To determine the involvement of specific protein kinases in nicotine-induced OPN expression, ASPC-1 cells were pretreated with or without the specific tyrosine kinase inhibitor genistein (60 µM) for 30 minutes. Nicotine-induced OPN mRNA accumulation was significantly inhibited by genistein (Fig 3B). Genistein alone also reduced the basal OPN mRNA levels (Fig 3B). To examine whether genistein has a direct effect on OPN transcription, AsPC-1 cells were incubated with genistein (15–60 µM) after their transfection with luciferase-labeled OPN construct for 3 h. Genistein dose-dependently reduced OPN promoter activity (Fig 3C), suggesting that tyrosine phosphorylation plays an important role in the signaling events that lead to activation of the OPN promoter.
Figure 3.

Nicotine-induced OPN gene expression requires both tyrosine kinase and ERK1/2 MAP kinase activity. A. The protein tyrosine phosphatase inhibitor sodium orthovanadate induces OPN mRNA accumulation in AsPC-1 cells. Cells were treated with sodium orthovanadate (10–50 uM) for 3 h. OPN mRNA levels were determined by real time PCR. *p < 0.05 # p< 0.02 vs. control untreated cells using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test. B. The tyrosine kinase inhibitor genistein blocks nicotine-induced OPN mRNA accumulation in AsPC-1 cells. Cells were pretreated with genistein for 1 h (60 µM) then exposed to nicotine (3×10−8mol/L) for 3 h. Three independent experiments showed similar results. *p < 0.05 vs. nicotine treated cells using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test. C. Genistein reduces OPN promoter activity in AsPC-1 cells. After 24 h of transfection, the cells were incubated with genistein (15–60 µM) for 3 hours. After incubation, the luciferase activity in the cell lysates was measured. Genistein reduced OPN promoter activity with maximum reduction at 60 µM. Relative luciferase activity was calculated after deduction of the activity levels with pGL3 vector alone. Results represent mean ± SEM of triplicate determinations. All experiments were repeated at least three times to confirm the reproducibility of the observations. D. Genistein reduces AsPC-1 cell proliferation. Cells were incubated with genistein (15–60 µM) for 24 and 72 h. Genistein dose dependently (30–60 µM) reduced cellular proliferation at the two time points. *p < 0.05 vs. control, using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test.
Since genistein has been shown to reduce cellular proliferation (33), we performed MTT assay in AsPC-1 cells after their treatment with or without with genistein (15–60 µM) for 24 and 72 h. Genistein dose-dependently reduced cellular proliferation at 24 and 72 h (Fig 3D), suggesting that there is a correlation between the reduction in OPN transcription and inhibition of cell proliferation.
Nicotine-induced OPN gene expression requires ERK1/2 MAP kinase activity
Nicotine signaling through nicotine receptor activates members of the MAPK family in different tissues (34,35). We tested whether nicotine-induced OPN mRNA expression involves activation of MAP kinases in AsPC-1 cells. After washing, the cells were lysed and 40 µg protein aliquots were subjected to Western blot analysis. We probed the blots with antibodies specific for phosphorylated ERK1/2(Thr183/Tyr185), phosphorylated p38 (Thr180/Tyr182), and phosphorylated JNK/SAPK (Thr183/Tyr185).
Nicotine increased ERK1/2 phosphorylation within 5 min of treatment (Fig 4A), but not JNKs or p38 (data not shown). Preincubation of the cells with the MAPKK inhibitor PD098059 abolished the nicotine-induced OPN gene expression (Fig 4B). This suggests that specific activation of ERK1/2 kinase may play an important role in nicotine-induced OPN expression in AsPC-1 cells.
Fig 4.




Time dependent activation of ERK1/2 MAP kinase signaling pathway by nicotine in AsPC-1 cells. A. Representative Western blot probed with phospho antibody against the activated from of ERK1/2 showing increased phosphorylation of ERK1/2 after incubation of nicotine (3×10−8 mol/L) 5–60 min. Blots were stripped and developed with anti- total ERK1/2 as control for equal protein loading. B. Effect of MAPKK inhibitor, PD098059, on nicotine induced increase in OPN mRNA. Cells were pretreated with the inhibitor (0.1–10 µM) for 10 minutes before incubation with nicotine (3×10−8mol/L) for 3 h. Data represent three independent experiments. *p < 0.05 # p<0.002 vs. nicotine treated cells using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test. C. PD098059 reduces OPN promoter activity in AsPC-1 cells. After 24 h of transfection, the cells were incubated with PD098059 (0.1–10 µM) for 3 hours. After incubation, the luciferase activity in the cell lysates was measured. PD098059 reduced OPN promoter activity with maximum reduction at 10 µM. Relative luciferase activity was calculated after deduction of the activity levels with pGL3 vector alone. Results represent mean ± SEM of triplicate determinations. All experiments were repeated at least three times to confirm the reproducibility of the observations. D. PD098059 reduces AsPC-1 cell proliferation. Cells were incubated with PD098059 (0.1–10 µM) for 24 and 72 h. PD098059 (10 µM) reduced cellular proliferation at the two time points at. *p < 0.05 vs. control, using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test.
To examine whether PD098059 has a direct effect on OPN transcription, AsPC-1 cells were incubated with PD098059 (0.1–10 µM) for 3 h after their transfection with luciferase-labeled OPN construct. PD098059 at the highest dose of (10 µM) reduced OPN promoter activity (Fig 4C), suggesting that ERK1/2 MAP kinase may play a role in the signaling events that lead to activation of the OPN promoter.
Since PD098059 has been shown to reduce cellular proliferation (36), we performed MTT assay in AsPC-1 cells treated with or without with PD098059 (0.1–10 µM) for 24 and 72 h. PD098059 the highest dose of (10 µM) reduced cellular proliferation at 24 and 72 h (Fig 4C), suggesting that there maybe a correlative relation between the reduction of OPN transcription and inhibition of cell proliferation.
Nicotine is detectable in the blood and pancreatic tissue of rats
To evaluate the effect of cigarette smoke on blood nicotine levels in smoke-exposed rats, blood nicotine levels were measured in samples collected 16–20 h after the last smoke exposure. The average nicotine concentration in the serum of animals from the control group was less than 1 ng/ml, but was 7.36 ± 0.72 ng/ml in animals exposed to low dose smoke, which was not significantly different than the high-dose-exposed group due to the short half life of nicotine. To detect and quantify nicotine in the pancreas, a selected ion monitoring technique was used, and ions of m/z162 were monitored for nicotine (28). Based on these parameters, nicotine levels in the pancreatic lysates obtained from the control and low dose smoke groups were below detectable levels. However, a nicotine concentration of 36 pg/µl in pancreas was detected in the animals exposed to the high dose of smoke. These data indicate that nicotine is detectable in the pancreata of the high dose smoke-exposed animals.
High dose cigarette smoke causes structural changes in the rat pancreas and increased OPN expression
H&E staining of frozen sections of pancreata from the different groups revealed the presence of areas of necrosis and connective tissue deposition in the rats exposed to the high dose of cigarette smoke. The changes were noticed in the interlobular regions in the areas surrounding the ducts (Fig 5A). In the normal pancreas, OPN could be detected in the islets (Fig 5B), while very faint OPN immunofluorescence could be seen in the inter- and intra-lobular connective tissue (CT) (Fig 5C). Immunofluorescence staining of rat pancreata from the different groups revealed increased expression of OPN protein, especially in the inter- and interlobular connective tissue (Fig 5C). Figure 5D shows quantitation of pancreatic OPN in the control and smoke-exposed groups. Significantly higher immunofluorescence reactivity was seen in the pancreata from rats exposed to the high dose of cigarette smoke. Analysis of quantitative PCR data of OPN mRNA, corrected with GAPDH as an internal control, showed a significant increase in OPN mRNA in the high dose group (Fig 5F).
Figure 5.




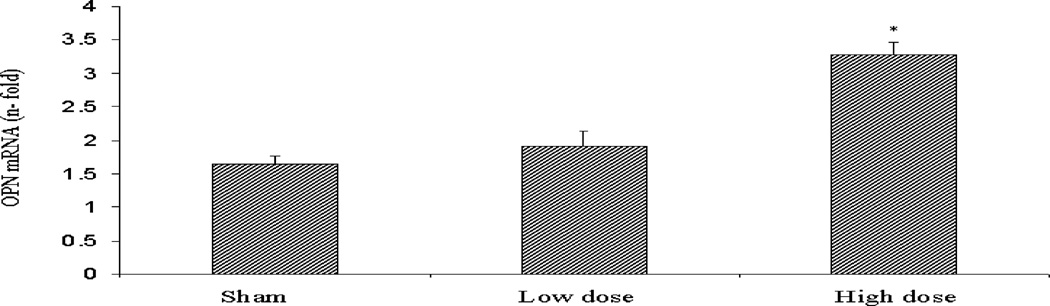
High dose cigarette smoke causes structural changes in the rat pancreas. A. Staining of frozen sections of pancreata from different group revealed the presence of areas of necrosis and connective tissue deposition in the rats treated with high dose of cigarette smoke. The changes were noticed in the interlobular regions in the areas surrounding the ducts (arrows). B. Representative immunofluorescence staining using OPN antibody shows expression of OPN in the islets of the normal rat pancreas. Negative control samples where the primary antibody was omitted did not show non-specific reaction. X 200 original magnification. C. Representative immunofluorescence staining of rat pancreata from the different groups revealed increased expression of OPN protein in the CT in the rats exposed to high dose of cigarette smoke, especially in the inter- and interlobular connective tissue. X 200 original magnification. D. Pancreatic staining of OPN. Staining was analyzed by image analysis and expressed as percentage area stained as described in Materials and Methods. There was minimal staining in the sham and low dose groups. High dose group showed markedly significant increase in staining. E. Real time PCR analysis of OPN mRNA expression in the different rat groups shows significant increases in OPN mRNA in the pancreata of rats exposed to high dose of smoke. * p<0.05 vs. sham levels using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test.
Expression of OPN in human PDA
We next examined the endogenous levels and localization of OPN in premalignant and malignant lesions. By real time PCR, the average OPN levels in PDA lesions were significantly higher than that in the non malignant or premalignant lesions (Fig 6A). Interestingly, only 27% of the patients with premalignant lesions were smokers (3 of 11), whereas of the examined malignant lesions, 64% belonged to patients who were smokers (16 of 25) (Fig 6A). Using immunohistochemical staining, we found that in the normal pancreatic ducts, OPN was focally present and mostly on the apical surface of the ductal epithelium (Fig 6B). In Pancreatic Intraepithelial Neoplasia PanIN-3 lesions, intense expression of OPN could be seen in the transforming cells (Fig 6C). In PDA (Fig 6D), OPN ductal epithelial staining was intensified and localized to the membrane and cytoplasm of the tumor cells. Interestingly, OPN was also intensified in the apparently normal-looking acini, especially in the areas surrounding the malignant lesions (Fig 6E).
Figure 6.





Expression of OPN in human tissue. A. Real time PCR analysis of OPN mRNA in non-malignant pancreatic tissues, premalignant lesions, and invasive PDA. Significantly higher OPN mRNA levels are seen in invasive PDA. Analysis of patient history of the samples used for OPN analysis shows that invasive PDA patients were mostly (64%) smokers, while a the majority (73%) of patients with premalignant lesions were non smokers* p<0.05 vs. non-malignant levels using one-way repeated ANOVA with subsequent all pairwise comparison procedure by student t-test.
B. Representative immunohistochemical staining for OPN in non-malignant pancreatic tissue. Paraffin embedded pancreatic sections were stained with OPN antibody. In the non malignant ducts OPN is focally present and mostly on the apical surface of the ductal epithelium (× 400 original magnification). C. In PanIN lesions OPN staining was seen in the transforming ducts (× 100 original magnification). D. In invasive PDA, intense OPN is localized to the membrane and cytoplasm of the tumor cells (×200 original magnification). E. Compared to non-malignant tissue, intense OPN staining is seen in the acini neighboring the malignant lesions (×100 original magnification). Negative control (Neg C) sections where the primary antibody was not added did not show non-specific reaction
DISCUSSION
In this study, we evaluated the potential molecular basis of the role of nicotine as a major risk factor in PDA. We show for the first time an interesting relationship between nicotine and OPN, a phosphoprotein that confers on cancer cells a migratory phenotype and activates signaling pathways that contribute to cell survival, invasion, and metastasis (5).
Our data show that nicotine induced OPN accumulation rapidly and with significant magnitude in three PDA cell lines (Fig 1A–1F). Dose- and time-response studies demonstrated a significant induction of OPN mRNA and protein levels at the physiological range of blood levels of nicotine in smokers (3×10−9 – 3×10−7 mol/L). The maximal effective concentration at (3×10−8 mol/L) is similar to other nicotine actions that have been reported (37). The process of nicotine-mediated upregulation of OPN transcription and secretion was independent of cell proliferation since the same doses that induced OPN failed to increase cell proliferation. Although previous studies have shown that nicotine induces cellular proliferation (29,30,38), these studies used pancreatic acinar cells and chronic exposure protocols, which were not used in our current studies.
We also demonstrate that nicotine-induced OPN gene expression occurs through a nicotinic acetylcholine receptor (nAChR)-mediated mechanism (Fig 2B) and via enhancement of its promoter activity, as demonstrated by our promoter studies (Fig 2A). Addition of the nAChR antagonist, mechamylamine, prevented the nicotine mediated-OPN production (Fig 2B), without affecting cell proliferation or OPN transcription. nAChR is a cholinergic receptor that belongs to the superfamily of receptors that form ligand-gated ion channels in the plasma membrane. Since increased expression of nAChR has been shown to result in elevation of the functional response to nicotine (39), we assumed that nicotine itself could have a regulatory effect on nAChR in PDA cells, which could consequently contribute to nicotine-OPN induction. Indeed, addition of nicotine to AsPC-1 cells resulted in a concomitant increase in nAChR (α-7) mRNA expression (unpublished observation), confirming observations from previous studies in rat aortic smooth muscle cells (39). Thus, it is possible that nicotine induces OPN transcripts in PDA cells directly through acting on its promoter, and indirectly through nAChR upregulation.
Our data further showed that the OPN promoter was significantly stimulated as early as after 3 h after exposure to nicotine (Fig 2A). The implications of this acute response to nicotine could be critical in conditions where pancreatic ductal cells are exposed continuously to high levels of nicotine. Further studies are required to analyze the nicotine specific cis-elements on the OPN promoter, and the transcription factor(s) involved in the nicotine-mediated OPN upregulation. Studies in this regard are currently ongoing in our lab.
MAP kinases encoded by the extracellular signal regulated kinase (ERK) genes are a family of serine/threonine protein kinases activated as early responses to a variety of stimuli involved in cell growth, transformation, and differentiation (40). Two isoforms of ERK referred to as p44 (ERK1), and p42 (ERK2), are activated by phosphorylation of threonine and tyrosine residues by MAP kinase kinase (41,42). Nicotine activates MAP kinases in aortic smooth muscle cells (34) and in lung cancer cells (42). Using a selective inhibitor for MAPK activation, PD908905, we demonstrated that nicotine-induced OPN mRNA occurs through a MAPK-sensitive mechanism (Fig 4B). Nicotine had no effect within this time period on the phosphorylation of either p38 or SAPK/JNK. Further studies are now required to fully delineate the specific signaling pathway by which nicotine ultimately modulates OPN synthesis in PDA cells.
Our data also show that tyrosine phosphorylation and ERK1/2 may play an important role in the activation of OPN transcription by nicotine, as well as the signaling events that lead to activation of OPN promoter. Although we demonstrated that genestein and PD098059 reduce cell proliferation, it is not clear whether a casual or a correlative relationship exists between the decrease in OPN transcription and the reduction in cell proliferation. Studies in this regard are currently ongoing in our laboratory.
No data have been previously reported concerning the regulation of OPN expression in the pancreas during the exposure to environmental tobacco smoke. We show, consistent with our previously published data, that OPN is present in the normal rat islets (Fig 5B) with very faint expression in the interlobular connective tissue (43). In this study, we demonstrate for the first time that exposure of rats to cigarette smoke for 12 weeks resulted in significant increase in OPN mRNA and protein expression levels, which paralleled the rise of pancreatic and plasma nicotine levels. OPN expression was particularly intensified in the interlobular connective tissue (Fig 5C). We also detected structural changes in the pancreas (Fig 5A) in the form of connective tissue deposition and necrosis, concurring with previously reported data (31). Our results suggest that short periods of exposure to tobacco could affect pancreatic structure and OPN expression. Whether this early increase in OPN expression contributes to the initiation and progression of the carcinogenic process is yet to be determined.
Numerous studies have correlated high levels of OPN expression with tumor progression and metastasis in many cancers, including pancreatic cancer (7). OPN promotes cell survival and facilitates metastatic cell behavior through activation of the PI-3 Kinase/AKT-NF-κB pathways (44), urokinase plasminogen activator (45), and matrix melloproteinase-2 (46). OPN also induces the expression of vascular endothelial growth factor (47) and promotes integrin-mediated endothelial cell migration (48). In tumor microenvironment macrophages, OPN downregulates of the activity of inducible nitric oxide synthase leading to protection of tumor cells from the macrophage nitric oxide-mediated cytotoxicity (49). In our studies, we demonstrate that OPN mRNA levels are significantly higher in invasive PDA lesions, the majority of which were resected from smokers (Fig 6A). This is the first report to link OPN expression in invasive PDA to cigarette smoking. Still, the presence of high OPN levels in invasive lesions in patients who were non-smokers suggests that other factors are involved in the overexpression of OPN in invasive PDA as compared to premalignant lesions among non-smokers. We also show that OPN protein is intensely expressed in the premalignant lesion, the pancreatic intraepithelial neoplasms (PanINs) (Fig 6C). The presence of high levels of OPN in these premalignant lesions supports a possible role in cancer progression. We also demonstrate that OPN was intensely expressed in the acini adjacent to the PDA lesions (Fig 6D&E). Although these acini looked normal by H&E staining, the long-term autocrine/paracrine impact of these high levels of OPN on the normal acini phenotype is unknown.
Our study demonstrates that nicotine elicits a pro metastatic response in PDA cells by stimulation of OPN production through a nAChR-ERK1/2-dependent mechanism. Exposure of animals to environmental cigarette smoke and the patients’ history of cigarette smoking correlated with increased expression levels of OPN. The existence of OPN as a downstream effector of nicotine that is capable of mediating its carcinogenic effects in PDA cells is novel and could provide a unique potential target to control pancreatic cancer aggressiveness, especially in the cigarette smoking population.
Acknowledgements
This work was supported by NIH grant 1R21 CA133753-01
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Thun MJ. Cancer Statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Bellahcène A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008;8:212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Denhardt DT, Noda M, O'Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–1061. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan SA, Cook AC, Kappil M, Günthert U, Chambers AF, Tuck AB, Denhardt DT. Enhanced cell surface CD44 variant (v6, v9) expression by osteopontin in breast cancer epithelial cells facilitates tumor cell migration: novel post-transcriptional, post-translational regulation. Clin Exp Metastasis. 2005;22(8):663–673. doi: 10.1007/s10585-006-9007-0. [DOI] [PubMed] [Google Scholar]
- 5.Jain S, Chakraborty G, Bulbule A, Kaur R, Kundu GC. Osteopontin: an emerging therapeutic target for anticancer therapy. Expert Opin Ther Targets. 2007;1:81–90. doi: 10.1517/14728222.11.1.81. [DOI] [PubMed] [Google Scholar]
- 6.Gardner HA, Berse B, Senger DR. Specific reduction in osteopontin synthesis by antisense RNA inhibits the tumorigenicity of transformed Rat1 fibroblasts. Oncogene. 1994;9:2321–2326. [PubMed] [Google Scholar]
- 7.Koopmann J, Rosenzweig CN, Zhang Z, Canto MI, Brown DA, Hunter M, Yeo C, Chan DW, Breit SN, Goggins M. Serum markers in patients with resectable pancreatic adenocarcinoma: macrophage inhibitory cytokine 1 versus CA19-9. Clin Cancer Res. 2006;12:442–446. doi: 10.1158/1078-0432.CCR-05-0564. [DOI] [PubMed] [Google Scholar]
- 8.Sedivy R, Peters K, Klöppel G. Osteopontin expression in ductal adenocarcinomas and undifferentiated carcinomas of the pancreas. Virchows Arch. 2005;446:41–45. doi: 10.1007/s00428-004-1142-x. [DOI] [PubMed] [Google Scholar]
- 9.Van Heek NT, Maitra A, Koopmann J, Fedarko N, Jain A, Rahman A, Iacobuzio-Donahue CA, Adsay V, Ashfaq R, Yeo CJ, Cameron JL, Offerhaus JA, et al. Gene expression profiling identifies markers of ampullary adenocarcinoma. Cancer Biol Ther. 2004;3:651–656. doi: 10.4161/cbt.3.7.919. [DOI] [PubMed] [Google Scholar]
- 10.Koopmann J, Fedarko NS, Jain A, Maitra A, Iacobuzio-Donahue C, Rahman A, Hruban RH, Yeo CJ, Goggins M. Evaluation of osteopontin as biomarker for pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2004;13:487–491. [PubMed] [Google Scholar]
- 11.Lowenfels AB, Maisonneuve P. Risk factors for pancreatic cancer. J Cell Biochem. 2005;95:649–656. doi: 10.1002/jcb.20461. [DOI] [PubMed] [Google Scholar]
- 12.Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:197–209. doi: 10.1016/j.bpg.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Mack TM, Yu MC, Hanisch R, Henderson BE. Pancreas cancer and smoking, beverage consumption and past medical history. J Natl Cancer Inst. 1986;76:49–60. [PubMed] [Google Scholar]
- 14.Farrow DC, Davis S. Risk of pancreatic cancer in relation to medical history and use of tobacco, alcohol and coffee. J Natl Cancer Inst. 1986;76:49–60. doi: 10.1002/ijc.2910450504. [DOI] [PubMed] [Google Scholar]
- 15.Silverman DT, Dunn JA, Hoover RN, Schiffman M, Lillemoe KD, Schoenberg JB, Brown LM, Greenberg RS, Hayes RB, Swanson GM, Wacholder S, Schwartz AG, et al. Cigarette smoking and pancreas cancer: a case control study based on direct interviews. J Natl Cancer Inst. 1986;86:1510–1516. doi: 10.1093/jnci/86.20.1510. [DOI] [PubMed] [Google Scholar]
- 16.Miller BA, Silverman DT, Kaplan R. Pancreas. In: Miller BA, Ries LA, Hanky BF, et al., editors. Cancer Statistics Review, 1973–1990. Bethesda, MD: NIH Publication, NCI; 1993. pp. 2789–2793. [Google Scholar]
- 17.Nicotine addiction as a disease. In: Miller NS, editor; Cocores JA, editor. The Clinical Management of Nicotine Dependence. New York: Springer-Verlag; 1991. pp. 66–78. [Google Scholar]
- 18.Surgeon General. The Health Consequences of Using Smokeless Tobacco. Bethesda, MD: USPHS; 1986. NIH Publication #86-2874. [Google Scholar]
- 19.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L. Pancreatitis and risk of pancreatic cancer. The International Pancreatic Study Group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 20.Mack TM, Yu MC, Hanisch R, Henderson BE. Pancreas cancer and smoking, beverage consumption and past medical history. J Natl Cancer Inst. 1986;76:49–60. [PubMed] [Google Scholar]
- 21.Gold EB, Cameron JL. Chronic pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;323:1485–1486. doi: 10.1056/NEJM199305203282010. [DOI] [PubMed] [Google Scholar]
- 22.Chowdhury P, Rayford PL, Chang LW. Inductions of pancreatic acinar cell pathology via inhalation of nicotine. Proc Soc Exp Biol Med. 1992;20:159–164. doi: 10.3181/00379727-201-43494b. [DOI] [PubMed] [Google Scholar]
- 23.Chowdhury P, Hosotani R, Chang L, Rayford PL. Metabolic and pathologic effects of nicotine on the gastrointestinal tract and pancreas of rats. Pancreas. 1990;5:222–229. doi: 10.1097/00006676-199003000-00016. [DOI] [PubMed] [Google Scholar]
- 24.Dubick MA, Palmer R, Lau PP, Morril PR, Geokas MC. Altered exocrine pancreatic function in rats treated with nicotine. Toxicol Appl Pharmacol. 1988;96:132–139. doi: 10.1016/0041-008x(88)90255-4. [DOI] [PubMed] [Google Scholar]
- 25.Chowdhury P, Doi R, Tangoku A, Rayford PL. Structure and functional changes of rat exocrine pancreas exposed to nicotine. Int J Pancreatol. 1995;18:257–264. doi: 10.1007/BF02784950. [DOI] [PubMed] [Google Scholar]
- 26.Chipitsyna G, Gong Q, Gray CF, Arafat HA. Induction of monocyte chemoattractant protein-1 expression by angiotensin II in the pancreatic islets and beta-cells. Endocrinology. 2007;148:2198–2208. doi: 10.1210/en.2006-1358. [DOI] [PubMed] [Google Scholar]
- 27.Takemoto M, Yokote K, Nishimura M, Shigematsu T, Hasegawa T, Kon S, Uede T, Matsumoto T, Saito Y, Mori S. Enhanced expression of osteopontin in human diabetic artery and analysis of its functional role in accelerated atherogenesis. Arteriosclerosis Thrombosis and Vascular Biology. 2000;3:624–628. doi: 10.1161/01.atv.20.3.624. [DOI] [PubMed] [Google Scholar]
- 28.Asaumi S, Takemoto M, Yokote K, Ridall AL, Butler WT, Fujimoto M, Kobayashi K, Kawamura H, Take A, Saito Y, Mori S. Identification and characterization of high glucose and glucosamine responsive element in the rat osteopontin promoter. Journal of Diabetes Complications. 2003;17:34–38. doi: 10.1016/s1056-8727(02)00189-7. [DOI] [PubMed] [Google Scholar]
- 29.Bose C, Zhang H, Udupa KB, Chowdhury P. Activation of p-ERK1/2 by nicotine in pancreatic tumor cell line AR42J: effects on proliferation and secretion. Am J Physiol Gastrointest Liver Physiol. 2005;289:G926–G934. doi: 10.1152/ajpgi.00138.2005. [DOI] [PubMed] [Google Scholar]
- 30.Chowdhury P, Bose C, Udupa KB. Nicotine-induced proliferation of isolated rat pancreatic acinar cells: effect on cell signalling and function. Cell Prolif. 2007;40:125–141. doi: 10.1111/j.1365-2184.2007.00418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittel UA, Pandey KK, Andrianifahanana M, Johansson SL, Cullen DM, Akhter MP, Brand RE, Prokopczyk B, Batra SK. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am J Gastroenterol. 2006;101:148–159. doi: 10.1111/j.1572-0241.2006.00405.x. [DOI] [PubMed] [Google Scholar]
- 32.Moffett J, Kratz E, Stachowiak MK. Increased tyrosine phosphorylation and novel cis-acting element mediate activation of the fibroblast growth factor-2 (FGF-2) gene by nicotinic acetylcholine receptor. New mechanism for trans-synaptic regulation of cellular development and plasticity. Brain Res Mol Brain Res. 1998;55:293–305. doi: 10.1016/s0169-328x(98)00010-2. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar FH, Adsule S, Padhye S, Kulkarni S, Li Y. The role of genistein and synthetic derivatives of isoflavone in cancer prevention and therapy. Mini Rev Med Chem. 2006;6:401–407. doi: 10.2174/138955706776361439. [DOI] [PubMed] [Google Scholar]
- 34.Dasgupta P, Chellappan SP. Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle. 2006;5(20):2324–2328. doi: 10.4161/cc.5.20.3366. Epub 2006 Oct 16. [DOI] [PubMed] [Google Scholar]
- 35.Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006;20:2093–2101. doi: 10.1096/fj.06-6191com. [DOI] [PubMed] [Google Scholar]
- 36.Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000;22:818–826. doi: 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 37.Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- 38.Chu M, Guo J, Chen CY. Long-term exposure to nicotine, via ras pathway, induces cyclin D1 to stimulate G1 cell cycle transition. J Biol Chem. 2005;280:6369–6379. doi: 10.1074/jbc.M408947200. [DOI] [PubMed] [Google Scholar]
- 39.Wada T, Naito M, Kenmochi H, Tsuneki H, Sasaoka T. Chronic nicotine exposure enhances insulin-induced mitogenic signaling via up-regulation of alpha7 nicotinic receptors in isolated rat aortic smooth muscle cells. Endocrinology. 2007;148(2):790–799. doi: 10.1210/en.2006-0907. [DOI] [PubMed] [Google Scholar]
- 40.Messersmith WA, Hidalgo M, Carducci M, Eckhardt SG. Novel targets in solid tumors: MEK inhibitors. Clin Adv Hematol Oncol. 2006;4:831–836. [PubMed] [Google Scholar]
- 41.Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 42.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 43.Katakam AK, Chipitsyna G, Gong Q, Gabbeta J, Dafoe DC, Arafat HA. Streptozotocin (STZ) mediates acute upregulation of serum and pancreatic osteopontin (OPN): a novel islet-protective effect of OPN through inhibition of STZ-induced nitric oxide production. J Endocrinol. 2005;18:237–247. doi: 10.1677/joe.1.06411. [DOI] [PubMed] [Google Scholar]
- 44.Lin YH, Yang-Yen HF. The osteopontin-CD44 survival signal involves activation of the phosphatidylinositol 3. kinase/Akt signaling pathway. J Biol Chem. 2001;276:46024–46030. doi: 10.1074/jbc.M105132200. [DOI] [PubMed] [Google Scholar]
- 45.Tuck AB, Hota C, Chambers AF. Osteopontin (OPN)-induced increase in human mammary epithelial cell invasiveness is urokinase (uPA)-dependent. Breast Cancer Res and Treat. 2001;70:197–204. doi: 10.1023/a:1013095329825. [DOI] [PubMed] [Google Scholar]
- 46.Mi Z, Guo H, Wai PY, Gao C, Kuo PC. Integrin linked kinase regulates osteopontin-dependent MMP-2 and uPA expression to convey metastatic function in murine mammary epithelial cancer cells. Carcinogenesis. 2006;27:1134–1145. doi: 10.1093/carcin/bgi352. [DOI] [PubMed] [Google Scholar]
- 47.Chakraborty G, Jain S, Kundu GC. Osteopontin promotes vascular endothelial growth factor-dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Res. 2008;68:152–161. doi: 10.1158/0008-5472.CAN-07-2126. [DOI] [PubMed] [Google Scholar]
- 48.Tuck AB, Hota C, Wilson SM, Chambers AF. Osteopontin-induced migration of human mammary epithelial cells involves activation of EGF receptor and multiple signal transduction pathways. Oncogene. 2003;22:1198–1205. doi: 10.1038/sj.onc.1206209. [DOI] [PubMed] [Google Scholar]
- 49.Wai PY, Guo L, Gao C, Mi Z, Guo H, Kuo PC. Osteopontin inhibits macrophage nitric oxide synthesis to enhance tumor proliferation. Surgery. 2006;40:132–140. doi: 10.1016/j.surg.2006.02.005. [DOI] [PubMed] [Google Scholar]