

Abstract
BACKGROUND
Optimal prevention of late cytomegalovirus (CMV) disease remains poorly defined.
OBJECTIVE
To compare valganciclovir prophylaxis with PCR-guided preemptive therapy.
DESIGN
Randomized, double-blind trial. Patients were randomized to valganciclovir or placebo. ClinicalTrials.gov registration number: NCT00016068
SETTING
Multicenter trial involving 8 centers.
PATIENTS
184 HCT recipients at high risk for late CMV disease (valganciclovir N=95, placebo N=89).
INTERVENTIONS
6 months of valganciclovir (900 mg/day) or placebo. Patients with PCR positivity at ≥ 1000 copies/mL (1000 IUs/mL) were treated with ganciclovir or valganciclovir (5 mg/kg or 900 mg twice daily, respectively). Hematopoietic growth factors were prescribed for neutropenia.
MEASUREMENTS
Composite primary endpoint was death or CMV disease or other invasive infections by day 270 after HCT. Secondary endpoints were CMV disease, CMV DNAemia, death, other infections, resource utilization, ganciclovir resistance, quality of life, immune reconstitution, and safety.
RESULTS
The primary composite outcome occurred in 20% of valganciclovir vs. 21% of placebo-preemptive therapy) recipients (treatment difference −0.01, 95% confidence interval −0.13, 0.10, P=0.86). There was no difference in the primary endpoint and its components at day 640 after HCT. CMV DNAemia ≥ 1000 copies/mL was reduced in the valganciclovir group (11% versus 36%, P < 0.001). Neutropenia was not significantly different at the absolute neutrophil count level of <0.5 × 109 per L (P=0.57), however, more subjects received hematopoietic growth factors in the valganciclovir group (25% versus 12%, P=0.026). No statistically significant differences were observed in other secondary outcomes.
LIMITATIONS
Some high-risk patients were not included.
CONCLUSIONS
Valganciclovir prophylaxis did not improve the CMV disease- free and invasive infection-free survival composite endpoint when compared to PCR-guided preemptive therapy. Both strategies performed similarly with regard to most clinical outcomes.
PRIMARY FUNDING SOURCE
Roche Laboratories
Introduction
Ganciclovir effectively prevents CMV disease during the first 3 months after hematopoietic cell transplantation (HCT) when given prophylactically at engraftment or pre-emptively for pp65 antigenemia or detection of CMV DNA by PCR, and improves survival in selected high-risk patients (1, 2). However, the majority of CMV disease now occurs after discontinuation of ganciclovir (3-7). Most cases of late CMV disease, occur between day 100 and day 270 after transplantation (3). In the absence of preventive strategies, both late CMV infection and disease are independent predictors for mortality after HCT (3). Although preemptive therapy based on virologic surveillance is the most commonly used strategy to prevent CMV disease during the first 3 months after HCT (8), maintaining surveillance is often difficult late after HCT because patients often return to remote locations and regular blood draws may be difficult to perform. The rationale for studying a prophylactic approach is supported by the observation that even asymptomatic CMV infection was associated with increased mortality, suggesting a role of indirect effects of CMV in the late period (3). However, the benefits of (val)ganciclovir prophylaxis are theoretically counterbalanced by its most common toxicity (neutropenia), which is also independently associated with mortality early after HCT (9, 10).
Methods
Design
This was an investigator-initiated, multicenter, double-blind, placebo-controlled and randomized clinical trial. The Fred Hutchinson Cancer Research Center (FHCRC) held the IND and served as the coordinating center. Patients were randomized to receive valganciclovir (900 mg once per day) or matching placebo (Appendix Figure 1) between 1999 and 2008. Study subjects, study personnel, and all clinical personnel were blinded. Study drug was discontinued when CMV viral load was greater than 1000 copies per mL (1000 IU per mL) or a greater than 5 times the baseline value, and preemptive therapy was started with intravenous ganciclovir (5 mg/kg twice daily) or valganciclovir (900 mg twice daily); foscarnet (90 mg/kg twice daily) was used instead if indicated due to neutropenia. All doses were adjusted to the creatinine clearance as per manufacturer recommendations. Weekly study samples were mailed to Seattle and tested at the University of Washington clinical laboratories. CMV and chemistry testing results were made available in real-time to study sites to allow initiation of open-label preemptive treatment for CMV and dose adjustment, drug discontinuation or start of hematopoietic growth factors as pre-specified in the protocol (Appendix). Study drug was held and growth factors were started if the absolute neutrophil count dropped below 1.0 × 109 per L.
CMV DNA at > 1000 copies/ml or consecutive positive results with increasing levels was used to discontinue study drug and to start preemptive treatment with open label intravenous ganciclovir treatment. The protocol was amended half way through the study to make valganciclovir open label treatment available to participants for breakthrough preemptive therapy (instead of requiring intravenous treatment) and to formally include G-CSF treatment of neutropenia at a level of 1.0 × 109 per L.
Setting and Participants
Allogeneic HCT recipients ≥ 16 years of age who were CMV seropositive pre-transplant or had a seropositive donor were eligible. Study subjects also had to meet one of the following criteria: (i) seropositive recipients had to have either CMV infection with appropriate treatment course before randomization, a history of GvHD after transplantation requiring treatment with systemic corticosteroids at doses of > 0.5 mg/kg at any time before enrollment, chronic clinically-extensive GvHD requiring treatment with corticosteroids, or receipt of ganciclovir, valganciclovir, foscarnet or cidofovir prophylaxis between engraftment and randomization; (ii) seronegative recipients with seropositive donors had to have a CMV infection with appropriate treatment course before randomization. A complete listing of inclusion and exclusion criteria is shown in Appendix Listing 1.
Randomization and Interventions
Randomization occurred once subjects were identified as eligible for the study at a median of 97 and 98 days post HCT for the valganciclovir and placebo arms, respectively (Table 1). We employed an adaptive randomization scheme implemented using a statistical program written by an FHCRC statistician and run by staff of the FHCRC protocol office. Randomization was stratified by study site, prior neutropenia (presence/absence of ANC<1000/mm3 after initial engraftment) and presence/absence of refractory GVHD (requiring secondary therapy) at study enrollment (11, 12).
Table 1.
Demographics of the study cohort by arm (subjects who were randomized and received at least one dose of study drug)
| Characteristics | Valganciclovir (n=95) | Placebo (n=89) |
|---|---|---|
| N (%) | N (%) | |
| Median (range) Age, years | 50 (17-70) | 49 (16-69) |
| Gender | ||
| Male | 57 (60.0%) | 47 (52.8%) |
| Female | 38 (40.0%) | 42 (47.2%) |
| Type of Transplant | ||
| Peripheral blood stem cell | 84 (88.4%) | 70 (78.7%) |
| Unrelated cord blood | 1 (1.1%) | 1 (1.1%) |
| Marrow | 10 (10.5%) | 18 (20.2%) |
| Donor Relationship | ||
| Related | 46 (48.4%) | 45 (50.6%) |
| Unrelated | 49 (51.6%) | 44 (49.4%) |
| Disease Status at Randomization | ||
| Remission | 94 (98.9%) | 86 (96.6%) |
| Cytogenetic Relapse | 1 (1.1%) | 2 (2.2%) |
| Molecular Relapse | 0 (0.0%) | 1 (1.1%) |
| Intensity of Conditioning Regimen | ||
| Myeloablative | 69 (72.6%) | 62 (69.7%) |
| Non-Myeloablative/Reduced Intensity | 26 (27.4%) | 27 (30.3%) |
| Recipient CMV Status | ||
| Positive | 86 (90.5%) | 82 (92.1%) |
| Negative | 9 (9.5%) | 7 (7.9%) |
| Donor CMV Status | ||
| Positive | 56 (58.9%) | 46 (51.7%) |
| Negative | 39 (41.1%) | 43 (48.3%) |
| CMV serostatus | ||
| D−/R+ | 2 (41.0%) | 43 (48.3%) |
| D+/R− | 9 (9.5%) | 7 (9%) |
| D+/R+ | 47 (49.5%) | 39 (43.8%) |
| Patient HSV Status | ||
| Positive | 77 (81.1%) | 70 (78.7%) |
| Negative | 10 (10.5%) | 13 (14.6%) |
| Unknown | 8 (8.4%) | 6 (6.7%) |
| Patient VZV Status | ||
| Positive | 85 (89.5%) | 80 (89.9%) |
| Negative | 0 (0.0%) | 1 (1.1%) |
| Unknown | 10 (10.5%) | 8 (9.0%) |
| High Risk Underlying Disease at Transplant | ||
| No | 46 (48.4%) | 41 (46.1%) |
| Yes | 49 (51.6%) | 48 (53.9%) |
| CD34 selection | ||
| No | 86 (90.5%) | 78 (87.6%) |
| Yes | 9 (9.5%) | 11 (12.4%) |
| Refractory GVHD pre-randomization | ||
| No | 91 (95.8%) | 84 (94.4%) |
| Yes | 4 (4.2%) | 5 (5.6%) |
| Neutropenia pre-randomization | ||
| No | 69 (72.6%) | 63 (70.8%) |
| Yes | 26 (27.4%) | 26 (29.2%) |
| Sites | ||
| FHCRC | 76 (83.2%) | 75 (84.3%) |
| Others | 19 (16.8%) | 14 (15.7%) |
| Median (range) Time to randomization after transplant, days | 97 (82-121) | 98 (83-121) |
| Median (range) Length of time on study drug, days | 150 (5-187) | 120 (2-187) |
| Median (range) Length of follow-up, days | 537 (36-651) | 540 (34-679) |
| Randomization before 1/1/2004 | 44 (46) | 42 (47) |
| Randomization after 1/1/2004 | 51 (54) | 47 (53) |
Outcomes and Follow-up
The primary endpoint was a composite outcome consisting of CMV disease or invasive bacterial or fungal infections or death (whichever occurred first), thereby assessing the net-effect of the strategy including consequences of neutropenia (9). Since most cases of late CMV diseaseoccur between 4 and 9 months after HCT (3-5) the primary study period for the intervention was until day 270 after HCT. Follow-up was extended until day 640 post HCT for CMV disease, mortality and CMV-specific immune reconstitution. Secondary endpoints were CMV disease [defined as per international guidelines (13)], death, invasive bacterial/fungal infections [defined as published (1, 14), Appendix 2], use of invasive tests (i.e. endoscopy procedures including bronchoscopies), number of days alive without hospitalization during the active study period, treatment-emergent ganciclovir resistance, quality of life (QOL) as determined by the EORTC QLQ 30 questionnaire (15), herpesvirus-specific T cell function, and adverse events (clinical and laboratory).
Laboratory Methods
CMV viral load was determined at the University of Washington using a validated laboratory-developed quantitative PCR method (16). CMV specific T cell function was determined by a multicolor flow cytometry assay designed to detect polyfunctional CD4 and CD8 T cells (17, 18). Ganciclovir resistance was examined by testing subjects with persistent or increasing viral load (>1000 copies/mL) while on open label treatment for mutations in the UL97 gene, using a rapid PCR- and sequencing-based assay as previously described (19). Safety laboratory testing (serum creatinine, alanine aminotransferase, blood differential, platelet count) was done using established methods and limits for normal/abnormal at the University of Washington clinical laboratories.
Statistical Analysis
The frequency of the primary composite endpoint was 53% in our previous cohort (3). Our study was designed to test the superiority of the prophylactic strategy. In order to demonstrate a 45% reduction of the primary endpoint (which was deemed clinically meaningful), 184 randomized patients (92 per treatment group) were needed to provide approximately 87% power (allowing for one interim analysis at the 0.005 alpha level and a final analysis at the 0.048 level). All statistical tests are two-sided.
All patients who were randomized and received at least one dose of study medication were included in the analysis. Descriptive statistics were used to summarize demographic and baseline characteristics of study subjects. Comparison between study arms of time to the primary endpoint from HCT to day 270 after HCT was evaluated using Cox regression models adjusted for the stratification factors of pre-randomization neutropenia presence/absence of ANC<0.1 × 109 per L after initial engraftment and study site (FHCRC vs. Other). There were not sufficient numbers of patients with prior refractory GVHD to include this stratification factor. Secondary time-to-event outcomes were evaluated similarly; some to day 640 as noted in Table 2. Cumulative incidence curves were also used to evaluate time-to-event outcomes, with death as a competing risk for all outcomes except mortality (20). All time to event analyses were censored at the time of last contact in the absence of a competing risk or event of interest. Absolute differences and associated 95% confidence intervals (CI) were evaluated using standard methods for risk differences for proportions (21) but utilizing standard errors of cumulative incidence to construct confidence intervals for cumulative incidence. Differences in medians were with 95% CIs were evaluated using Hodges-Lehmann estimation (22). Other secondary outcomes were compared between study arms with chi-square, Fisher's exact or Wilcoxon rank-sum tests, as appropriate. Subset analyses of efficacy and neutropenia endpoints were performed post-hoc and displayed in forest plots. These were carried out within stratum defined by unique categories of each of the following variables: gender, CMV seropositive recipient/donor status, HCT conditioning type, donor relation, underlying disease risk, cell source, pre-randomization neutropenia, absence of pre-randomization refractory GVHD, randomized pre or post 1/1/2004. Missing data in our study was minimal, only present to any extent for QOL data at 6 months and 9 months where it was 78% and 79% complete, respectively. Multiple imputation methodology was used to generate 10 replicate values for each missing QOL measurement at each time point using the method of partial mean matching (23) using study arm and baseline covariates, including baseline QOL for the latter time points, for the prediction model Reported analytic results were obtained by combining analysis results across imputed data sets with standard formulas (24) Linear regression models were used to compare QOL between study arms at each time point.
Table 2.
Primary and Secondary Outcomes (time-to-event analysis of endpoints by arm).
| Outcome* | Cumulative Incidence Estimate | Difference† (V-P) | 95% CI‡ | HR V vs. P | 95% CI | p-value§ | |||
|---|---|---|---|---|---|---|---|---|---|
| Valganciclovir | Placebo | ||||||||
| N† | Cum Inc† | N† | Cum Inc† | ||||||
| Primary endpoint (day 270) | |||||||||
| Primary endpoint (composite) | 18 | 0.20 | 18 | 0.21 | −0.01 | −0.13, 0.10 | 0.9 | 0.5, 1.8 | 0.86 |
| Secondary endpoints | |||||||||
| CMV Disease day 270 | 2 | 0.02 | 2 | 0.02 | −0.001 | −0.04, 0.04 | 0.9 | 0.1, 6.2 | 0.88 |
| Mortality day 270 | 6 | 0.07 | 6 | 0.07 | −0.005 | −0.08, 0.07 | 0.9 | 0.3, 2.9 | 0.89 |
| Invasive bacterial and fungal infections day 270 | 17 | 0.18 | 15 | 0.17 | 0.01 | −0.10, 0.12 | 1.0 | 0.5, 2.1 | 0.94 |
| CMV Infection day 270 | 10 | 0.11 | 31 | 0.36 | −0.25 | −0.37, −0.13 | 0.3 | 0.1, 0.5 | <0.001 |
| Primary endpoint (composite) at day 640 | 32 | 0.35 | 34 | 0.40 | −0.04 | −0.19, 0.10 | 0.8 | 0.5, 1.3 | 0.45 |
| CMV Disease day 640 | 5 | 0.06 | 5 | 0.06 | −0.004 | −0.07, 0.07 | 0.9 | 0.3, 3.3 | 0.93 |
| Mortality day 640 | 17 | 0.19 | 16 | 0.19 | −0.001 | −0.12, 0.12 | 1.0 | 0.5, 1.9 | 0.96 |
| Invasive bacterial and fungal infection day 640 | 21 | 0.23 | 24 | 0.28 | −0.05 | −0.18, 0.08 | 0.8 | 0.4, 1.4 | 0.40 |
| HSV-VZV infection day 640∥ | 8 | 0.09 | 3 | 0.04 | 0.05 | −0.02, 0.12 | 2.3 | 0.6, 8.7 | 0.22 |
| Neutropenia <1000 per mm3 while on study drug | 34 | 0.43 | 19 | 0.29 | 0.14 | −0.02, 0.30 | 1.9 | 1.1, 3.4 | 0.025 |
| Neutropenia <500 per mm3 while on study drug | 5 | 0.07 | 6 | 0.10 | −0.03 | −0.13, 0.07 | 0.7 | 0.2, 2.3 | 0.57 |
| Neutropenia <200 per mm3 while on study drug | 2 | 0.02 | 3 | 0.04 | −0.02 | −0.08, 0.04 | 0.6 | 0.1, 3.8 | 0.62 |
| Relapse at day 270 | 10 | 0.11 | 10 | 0.11 | −0.007 | −0.10, −0.08 | 1.0 | 0.4, 2.3 | 0.92 |
| Immunosuppression discontinuation at day 270 | 45 | 0.47 | 43 | 0.49 | −0.01 | −0.16, 0.13 | 0.9 | 0.6, 1.4 | 0.76 |
Relapse was considered a competing risk for the primary endpoint and mortality. Death was considered a competing risk for CMV infection, CMV disease, HSV-VZV infection, relapse, immunosuppression discontinuation and neutropenia endpoints while on study drug.
N = Number of events, Cum Inc = Cumulative incidence estimates, V = Valgancyclovir, P = Placebo, (V-P) = Difference in cumulative incidence estimates.
95% confidence interval for difference in cumulative incidence estimates.
P-value from cox proportional hazard model adjusted for pre-randomization neutropenia (Yes vs. No) and sites (FHCRC vs. Others).
All except 2 HSV/VZV infections occurred after day 270, therefore the incidence is estimated at day 640 instead of day 270 as stated by the protocol.
An interim analysis was conducted and reviewed by the independent Data Safety Monitoring Board after 50% of patients (N=92) had completed 90 days of study. Toxicity and the primary endpoints and its components were reviewed. The study was permitted to continue. All statistical analyses were carried out using SAS, version 9.3 (SAS Institute, Cary, North Carolina).
Role of the Funding Source
Roche Laboratories provided the study drug and placebo and provided funding for the conduct of the clinical trial, but had no role in the design, conduct, analysis, or decision to submit this manuscript for publication. All analyses (statistical, laboratory) were done at FHCRC, and investigators had full access to the data and data analysis. Partial support was obtained by National Institutes of Health grant CA 18029 for resistance and T cell immunity testing.
Results
Study Population
The Consort diagram is shown in Figure 1. Table 1 shows balanced clinical characteristics between the study arms.
Figure 1.

Study enrollment and randomization (Consort Diagram).
Primary Endpoint
There were no statistically significant differences between the groups for the composite endpoint (death or CMV disease or other serious invasive infections by day 270 after HCT) or for its individual components (Figure 2, Tables 1, 2). Analyses of the primary endpoint at day 270 and at day 640 were performed within subsets of the study population (see Appendix Figure 2 for details), which also did not suggest differential treatment effects within specific subgroups.
Figure 2.

Cumulative incidence of the primary endpoint (CMV disease, invasive bacterial fungal infections or death) (panel A) and overall survival (panel B).
Secondary Efficacy Endpoints
CMV endpoints
The cumulative incidence of initiation of preemptive therapy, any CMV DNA detection (CMV DNA > 1000 copies/ml of plasma or rising levels of CMV DNA of > 5 times the baseline level), and any PCR positivity and CMV DNA > 10,000 copies/mL are shown in Table 2, Table 3, and Figure 3. As expected per study design there was significantly more CMV infection in the placebo arm. The hazards of CMV disease did not differ between the study groups throughout the entire study period (Table 2); there was also no apparent difference in the clinical manifestations of CMV (Appendix Table 1). The 6 week and 6 month mortality following CMV disease was not different between patients who developed CMV disease in the valganciclovir or placebo-preemptive therapy arm (Appendix Table 1).
Table 3.
Other Secondary Outcomes between randomization and day 270 after HCT.
| Outcome | Valganciclovir N=95 | Placebo N=89 | P-value* | Difference† (V-P) | 95% CI |
|---|---|---|---|---|---|
| Biopsy and BAL procedures (%) | 30.5% | 36.0% | 0.43 | −0.05 | −0.19, 0.08 |
| Bone marrow biopsy procedures (%) | 12.6% | 16.9% | 0.42 | −0.04 | −0.14, 0.06 |
| Days alive and not hospitalized, median (range) | 161 (27-190) | 166 (6-185) | 0.32 | −5 | −3.00, 7.00 |
| Days in the intensive care unit, median (range) | 0 (0-22) | 0 (0-9) | 0.88 | 0 | −3.00, 13.0 |
| Received RBC Transfusion, n (%) of patients | 20 (21.1%) | 22 (24.7%) | 0.69 | −0.04 | −0.16, 0.08 |
| RBC Transfusion Units, median (range) | 2.5 (1-9) | 1.5 (1-21) | 0.58 | 1 | −1.00, 1.00 |
| Received Platelets Transfusion, n (%) of patients | 9 (9.5%) | 10 (11.2%) | 0.55 | −0.02 | −0.11, 0.07 |
| Platelets Transfusion Units, median (range) | 4 (1-21) | 7 (1-17) | 0.67 | −3 | −8.00, 5.00 |
| GCSF use, n (%) of patients€ | 24 (25%) | 11 (12%) | 0.026 | 0.13 | 0.02, 0.24 |
| GCSF use, median doses (range) | 1 (1-18) | 1 (1-7) | 0.98 | 0 | −1.00, 1.00 |
| Receipt of any transfusion, n (%) | 23 (24.2%) | 22 (24.7%) | 0.94 | −0.005 | −0.13, 0.12 |
| Total Transfusion Units, median (range) | 2 (1-26) | 2 (1-30) | 0.88 | 0 | −1.00, 2.00 |
| Serum Creatinine > 2.0 mg/dL (%) | 19.0% | 19.1% | 0.97 | −0.002 | −0.12, 0.11 |
| Serum Creatinine > 2.5 mg/dL (%) | 6.3% | 9.0% | 0.54 | −0.03 | −0.10, 0.05 |
P-values from chi-square test, Fisher's exact or Wilcoxon rank-sum test as appropriate.
(V-P) = Difference in cumulative incidence estimates.
‡ During the first part of the study G-CSF was given at the site attending physician's discretion (valganciclovir 7/44 [16%] vs. placebo 2/42 [5%]); after 1/1/2004 the protocol included routine administration for ANC levels of < 1000/mm3 (valganciclovir 17/51 [33%] vs. placebo 9/47 [19%]). All G-CSF doses were given during the double-blind phase of the trial.
Figure 3.


Cumulative incidence of CMV DNA positivity (A: > 1000/mL or 5× increase over baseline [study drug discontinuation], P=0.004; B: any PCR positivity, P=0.005; C: >10,000 copies/mL, P=0.025).
HSV and VZV
Hazards of HSV and VZV infections between randomization and day 640 were not statistically significantly different between arms (Table 2). There were no cases of HSV infection in the valganciclovir arm and one case in the placebo arm before day 270; by day 640, 7 cases of HSV occurred in valganciclovir arm (7.4%) versus 2 in the placebo arm (2.2%). There was one case of VZV infection in each arm (valganciclovir: day 383; placebo: day 224).
Invasive bacterial and fungal infections
There was no difference in the incidence of invasive bacterial and fungal infections between randomization and day 270 and 640 after HCT (Table 2).
Survival
Mortality from all causes was compared between valganciclovir and placebo arms (day 270 and 640) and was not statistically significant different between the arms (Figure 2).
Safety Endpoints
Adverse Events (AEs) and Serious adverse events (SAEs)
There was no difference in the number of patients with AEs and SAEs (Appendix Table 3). A higher proportion of patients with drug-related grade 2 AEs were reported in the valganciclovir arm, which was driven by neutropenia (40% in the placebo vs 55% in the valganciclovir group, P=0.043), but there were no differences between groups at grade 3 or higher levels (data not shown). There was no statistically significant difference between the study arms for the proportion of patients with any of the AE categories by organ system, with the exception of grade 2 events before day 270 in the Blood and Marrow category where the difference approached statistical significance (placebo: 46%, valganciclovir: 53%, P=0.052). No differences in the proportion of patients with GI or renal AEs were demonstrated between arms.
Hematologic parameters
Neutropenia was analyzed at different levels (Table 2). The analysis was done during the double-blind phase of the study drug and for the entire active treatment period (randomization until day 270 after transplantation). There was more neutropenia at the 1.0 × 109/L level in valganciclovir recipients during the double-blind phase; this trend did not persist at more severe levels of neutropenia (< 0.5 × 109/L). More subjects received G-CSF in the valganciclovir group (Table 3). In a post-hoc analysis to determine whether neutropenia was more common in specific risk groups, no statistically significant predisposition to neutropenia at moderate and severe levels of neutropenia was found with the exception of donor status (Appendix Figure 3). Use of preemptive G-CSF (introduced for subjects randomized after 1/1/2004) appeared to eliminate the difference in severe neutropenia (<0.5 × 109/L) between the two arms. There was no difference in thrombocytopenia or in the requirement for blood product support between the groups (Table 3).
Resource Utilization
We compared various parameters of resource utilization between groups. There was no statistically significant difference for any of these parameters with the exception of G-CSF use (Table 3).
Resistance
Subjects who met the criteria for resistance testing had both the first and last plasma sample analyzed (N=24; 6 randomized to valganciclovir, 18 to placebo). No UL97 mutations were found that confer marker-transfer confirmed phenotypic resistance (23).
Immune Reconstitution
Virus-specific T cell immunity was assessed at baseline and longitudinally. No differences were found between study arms for lymphoproliferative responses to CMV, HSV and VZV (Figure 4). and for IFN-γ CD4+ intracellular cytokine responses (data not shown); a sensitivity analysis using >0.1% as the threshold for a positive response also did not show differences between the study groups.
Figure 4.

Virus-specific T cell reconstitution in Placebo vs Valganciclovir (VGCV) arms. Box-whisker plots show the first, second (median) and third quartiles, whiskers show the min and max. Upper panel: CD4+ T helper function by lymphoproliferation to CMV, HSV, and VZV in all patients. Stimulation index (SI) = (mean cpm antigen-stimulated cells/mean cpm unstimulated cells) shown on log-scale. Lower panel: Polyfunctional CD8+ CMV-specific cytotoxic T cell function in a subset of 20 patients with complete follow-up to day 640 as measured by production of cytokines interferon-gamma (IFNg, denoted by G on plots), tumor necrosis factor-alpha (TNFa) and interleukin 2 (IL-2) or degranulation marker CD107a. G columns indicate monofunctional CD8+ cells that produced IFNg alone; +1, +2, +3 columns indicate polyfunctional CD8+ cells that produced IFNg + 1, 2, or 3 additional markers.
Longitudinal testing of polyfunctional intracellular cytokines was performed in a subset of patients who had complete follow-up. There were no differences in overall functionality (data not shown) and polyfunctionality among subjects who had positive IFN-γ (Figure 4) and IL-2 responses (data not shown).
Quality of Life
Quality of Life (QOL) was assessed at baseline, day 180 and day 270 after transplantation using the validated EORTC QLQ-30 questionnaire (version 3). Overall, there were no differences in QOL parameters between the arms. There was an imbalance in the category ‘Emotional functioning’ at baseline and this difference persisted at the final study evaluation (Appendix Table 2).
Discussion
Our study failed to show superiority of valganciclovir prophylaxis for prevention of late CMV disease- and invasive infection-free survival after HCT when compared to PCR-guided CMV preemptive therapy. Both strategies appeared to perform similarly well in preventing CMV disease during the active study period and for 1 year thereafter, and appeared to perform significantly better than historical cohorts that used no prevention strategies (4, 5, 7, 26, 27). Valganciclovir prophylaxis was associated with similar rates of hematologic and other toxicities compared to PCR-guided preemptive therapy. No differences were observed between the groups with regard to other herpesvirus infection, the tempo of T cell immune reconstitution, or the rate of UL97 mutations.
We chose the primary endpoint of CMV disease- and invasive infection-free survival in order to capture both the putative benefits and risks of valganciclovir prophylaxis. There was no statistically significant difference in the composite endpoint between the groups (Tables 2, 3).
One possible reason for the failure of valganciclovir prophylaxis to improve outcomes compared to preemptive therapy is the excellent performance of the preemptive strategy. Prevention of CMV disease with PCR-guided preemptive therapy was near complete, with a CMV disease rate of 2% at the end of the active study period at day 270. A similar effectiveness of anti-CMV preemptive therapy was reported in a recent multicenter clinical trial during the first 3 months after HCT (28). Preemptive therapy appears to work well even when administered to patients that live far away from comprehensive cancer centers, as in this clinical study where laboratory testing was done by means of overnight shipment and clinical feedback was provided via the phone. However, this strategy is resource intensive: in our study, virologic testing was closely monitored by dedicated study personnel, and study subjects were contacted when a sample did not arrive. Our monitoring procedures were primarily designed to ensure regular safety monitoring, as half of the subjects were on valganciclovir prophylaxis; however, it also proved to be an effective approach for CMV monitoring. Whether high adherence to weekly testing can be replicated in usual clinical practice is unknown but limited evidence exists (29).
One concern with prolonged valganciclovir prophylaxis in HCT recipients is hematotoxicity (2, 30-31). We found relatively high rates of neutropenia in both groups. Although there was more neutropenia at a level of less than 1.0 × 109/L in prophylaxis recipients, there were essentially similar rates of severe neutropenia (< 0.5 × 109/L) in both groups. We attribute these similar rates to the routine preemptive administration of G-CSF in the second part half the study. While this protocol change resulted in increased use of G-CSF in the valganciclovir group, we achieved the desired effect of comparable rates of invasive bacterial and fungal infections between the groups (Table 3).
A delay of recovery of T cell immunity has been observed with effective long-term viral suppression (34, 35), and a delay in T cell recovery was associated with prolonged risk periods for herpesvirus complications (3, 4, 36). However, the association between antiviral prophylaxis and delayed immune reconstitution is inconsistent (7, 38). Our study examined T cell immunity to CMV, HSV and VZV longitudinally and found no difference in the immune reconstitution rates throughout the study period. Thus, extended valganciclovir prophylaxis did not affect virus-specific T cell recovery during or after drug administration.
The study also examined several other clinically relevant endpoints (Table 3). Both strategies showed similar outcomes using all of these measures. We also performed post-hoc subset analyses to determine if efficacy and toxicity outcomes were equally distributed between arms within participant subgroups. We found no differences between study groups in terms of efficacy or toxicity within subgroups (Appendix Tables 2 and 3).
This is the first study that attempted and successfully completed real-time management of CMV preemptive treatment and toxicity management across 34 US states. However,due to complexity of the design and study requirements it took eight years to complete the trial.
One limitation of the study is that, similar recent phase III maribavir trial (28), highest risk patients for CMV complications (i.e. those with persistent viral replication during the enrollment window) were excluded from the trial. Another limitation is that no formal cost-effectiveness analysis was performed.
In conclusion, valganciclovir prophylaxis was not superior in reducing the composite endpoint of CMV disease, invasive bacterial and fungal disease and death when compared to PCR-guided preemptive therapy. Both CMV prevention strategies used in this study proved to be extremely effective, and our trial extends the evidence base for management of patients at high risk for late CMV disease.
Acknowledgements
We thank all study participants and study site personnel for their contributions.
Study sites
Participating Centers, Investigators, Research Coordinators and Research Personnel:
Fred Hutchinson Cancer Research Center, Seattle, WA: Michael Boeckh, M.D., Garrett Nichols, M.D.
University of Texas M.D. Anderson Cancer Center, Houston, TX: Roy F. Chemaly, M.D., M.P.H.
Memorial Sloan Kettering Cancer Center, New York, NY: Genovefa A. Papanicolaou, M.D.
University of Florida, FL: John Wingard, M.D.
Mayo Clinic, Rochester, MN: Mark Litzow, M.D.
University of Michigan, Ann Arbor, MI: Voravit Ratanatharathorn M.D.
City of Hope National Medical Center, Duarte, CA: John Zaia, M.D.
Duke University, NC: Nelson Chao, M.D.
Virologic Testing
MeeiLi Huang, Tracy Santos, Linda Cook (University of Washington)
T cell immunity Testing
Terry Stevens-Ayers (Fred Hutchinson Cancer Research Center).
DSMB
David Snydman, MD (chair), Richard Whitley, MD, Elisabeth Reed, MD, Jacqueline Benedetti, PhD (statistician).
Potential conflicts of interest:
M.B. received research funding from Roche Pharmaceuticals, Viropharma Inc., Vical Inc., Merck, Astellas, and Chimerix Inc.; he served as a consultant for Genentech/Roche, Viropharma Inc., Vical Inc., Chimerix Inc., Merck, Microbiotix, Theraclone, Astellas, and Glaxo-Smith-Kline.
W. Garrett Nichols is currently an employee of GlaxoSmithKline.
Roy F. Chemaly received research funding from Chimerix Inc., AiCuris Inc., Viropharma Inc., Roche Pharmaceuticals, and Merck; he served as a consultant for Astellas, and Viropharma Inc.
Genovefa A. Papanicolaou received research funding from Viropharma Inc., Vical Inc., Merck, Astellas, Glaxo-Smith-Kline and Chimerix Inc.; she served as a consultant for Viropharma Inc, Chimerix Inc., Merck and Astellas.
John R. Wingard served as a consultant to Astellas and received speaker fees from Pfizer.
Hu Xie: No relevant conflicts.
Mary E.D. Flowers: No relevant conflicts.
Terry Stevens-Ayers: No relevant conflicts.
Karen Syrjala: No relevant conflicts.
Keith Jerome: No relevant conflicts.
Wendy Leisenring: No relevant conflicts
Appendix Listing 1: Inclusion and exclusion criteria
Patient Inclusion Criteria (all criteria must be met).
Patients ≥ 16 years of age
Patients undergoing allogeneic peripheral blood stem cell, cord blood, or marrow transplantation (related and unrelated, T cell –depleted and non-T cell depleted, CD34 selected and non-selected, myeloablative and non-myeloablative)
Positive pre-transplant CMV serology of recipients and/or donor
- Either one of the following criteria:
- Seropositive recipients: either
-
(i)CMV infection with appropriate treatment course before randomization. This may occur as early as day 80 or as late as day 120. CMV infection is defined as a pp65 antigenemia at any level, CMV DNA in plasma, PBL or whole blood at any level detected by PCR or hybrid capture, CMV pp67 mRNA, CMV viremia by blood culture, surveillance BAL (culture or cytology); or CMV disease greater than 6 weeks prior to enrollment. or
-
(ii)History of GvHD after transplantation (defined as acute GvHD that required treatment with systemic corticosteroids of doses of > 0.5 mg/kg at any time before enrollment or chronic clinical-extensive GvHD requiring treatment with corticosteroids [see 5.A.8.]), or
-
(iii)Receipt of ganciclovir, valganciclovir, foscarnet or cidofovir prophylaxis from engraftment until randomization or development of toxicity.
-
(i)
- Seronegative recipients with seropositive donors:
-
(i)CMV infection with appropriate treatment course before randomization. This may occur as early as day 80 or as late as day 120. CMV infection is defined as a pp65 antigenemia at any level, CMV DNA in plasma, PBL or whole blood at any level detected by PCR or hybrid capture, CMV pp67 mRNA, CMV viremia by blood culture, surveillance BAL (culture or cytology); or CMV disease greater than 6 weeks prior to enrollment.
-
(i)
Serum creatinine < 2.5 mg/ml
Written informed consent
Proficiency in English language (to allow phone contacts as required by protocol)
Note: Patient may have received ganciclovir, foscarnet, cidofovir, high-dose acyclovir, or valacyclovir prior to study entry (either as prophylaxis or as preemptive therapy).
Patient Exclusion Criteria
Documented hypersensitivity to ganciclovir or valganciclovir
Neutropenia (ANC < 1000/mm3) within one week of study enrollment
Severe renal insufficiency (serum creatinine > 2.5 mg/ml)
Uncontrolled diarrhea or severe gastrointestinal disease preventing oral medication
CMV disease within 6 weeks prior to randomization (for definition of disease see Appendix A)
Rising or uncontrolled CMV load (via pp65 antigenemia or PCR for CMV DNA) at time of evaluation; pp65 antigenemia levels of ≤ 1/slide or ≤ 100 copies of CMV DNA per mL of plasma or per 106 peripheral blood leukocytes are permissible
Prophylactic use of high-dose acyclovir (doses of > 800 mg twice daily), valacyclovir (doses of > 500 mg twice daily), or famciclovir (doses > 500 mg/day); limited treatment courses at higher doses for VZV infections are permissible.
Ongoing prophylactic use of foscarnet, cidofovir or ganciclovir (IV or oral); limited treatment courses of low-dose cidofovir (≤ 0.5 mg/kg per week) are permissible
Leukemic relapse; cytogenetic and molecular relapse are permissible
Pregnancy
Nursing mothers
Refusal to use birth control
Imminent demise (expected survival < 2 weeks)
HIV infection (baseline HIV test prior to transplant acceptable)
Home residence outside of North America (precludes study sample shipments as required by protocol)
Appendix Listing 2: Definitions of Invasive fungal and bacterial infections
Aspergillus spp. or other mold infection.
Proven: Clinical signs and symptoms plus a tissue biopsy revealing growth of an organism or positive histopathology.
Probable: Clinical signs and symptoms with bronchoalveolar lavage (BAL) yielding positive growth or positive histopathology.
Possible: at least 3 clinical signs or symptoms and growth of an organism from non-sterile fluid (i.e. sputum).
Dissemination: proven dissemination only if confirmed by biopsy (or autopsy) and probable if clinical (skin lesions) or suggestive radiographic findings were apparent.
Candida spp. or other yeast (e.g. T. glabrata).
Proven fungemia: Any single positive blood culture that is culture positive for Candida spp. or T. glabrata. [Note: Removal of indwelling catheters is strongly encouraged.]
Tissue documented: Clinical signs and symptoms compatible with invasive yeast infection and a positive culture from a normally sterile site with histologic evidence of tissue invasion (definite) or positive culture from sterile site without histologic evidence of invasion (probable).
Bacteremia. Any single positive blood culture that is culture positive for bacterial pathogens consistent with a serious bloodstream infection. Results will be analyzed in groups (gram negative, gram positive with and without coagulase-negative staphylococci and non-JK corynebacteria). Urinary tract infections will not be captured.
Invasive Bacterial Tissue Infection. Clinical signs and symptoms compatible with disease (sinusitis, pneumonia, intra-abdominal abscess) and radiographic evidence of disease and pure or predominant culture or pathogen detection from a sterile site biopsy. Pathogen detection in respiratory secretion or sinus aspirates or CSF specimens will be considered if they are predominant and compatible with the clinical picture. Typhlitis (neutropenic enterocolitis) is defined as clinical signs and symptoms compatible with disease and typical radiographic evidence of disease with or without culture confirmation.
Infection Recurrence. Recurrent infections, in contradistinction to new infectious episodes, are defined as follows:
Mold infections diagnosed < 6 weeks following previous mold infection with same mold species
Yeast infections diagnosed < 6 weeks following previous yeast infection with same yeast species
Bacterial infections diagnosed < 3 weeks following previous bacterial infection with same bacterial species
- Viral infections diagnosed < 3 weeks following previous viral infection with same viral species.
Appendix Figure 1.
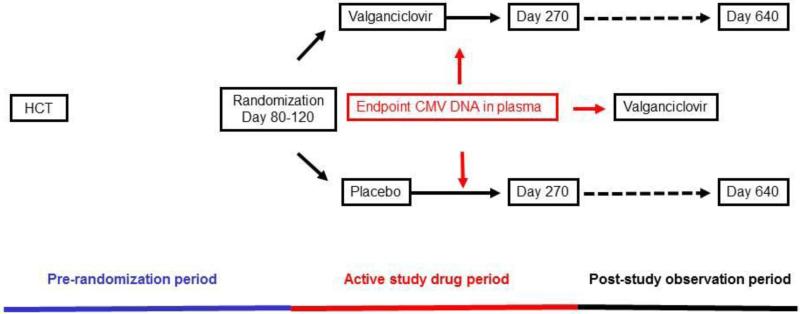 Study design.
Study design.Appendix Figure 2.
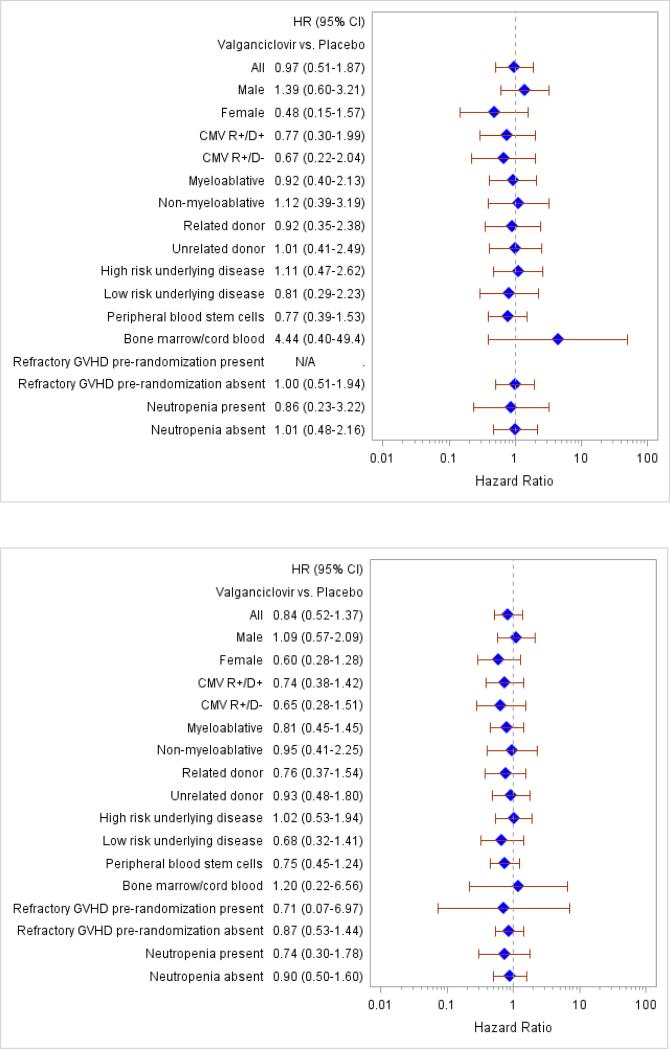 Analyses comparing the primary endpoint between study arms within subsets at day 270 (upper panel) and day 640 (lower panel). Reference=placebo. Each row of figure represents a subgroup.
Analyses comparing the primary endpoint between study arms within subsets at day 270 (upper panel) and day 640 (lower panel). Reference=placebo. Each row of figure represents a subgroup.Appendix Figure 3.
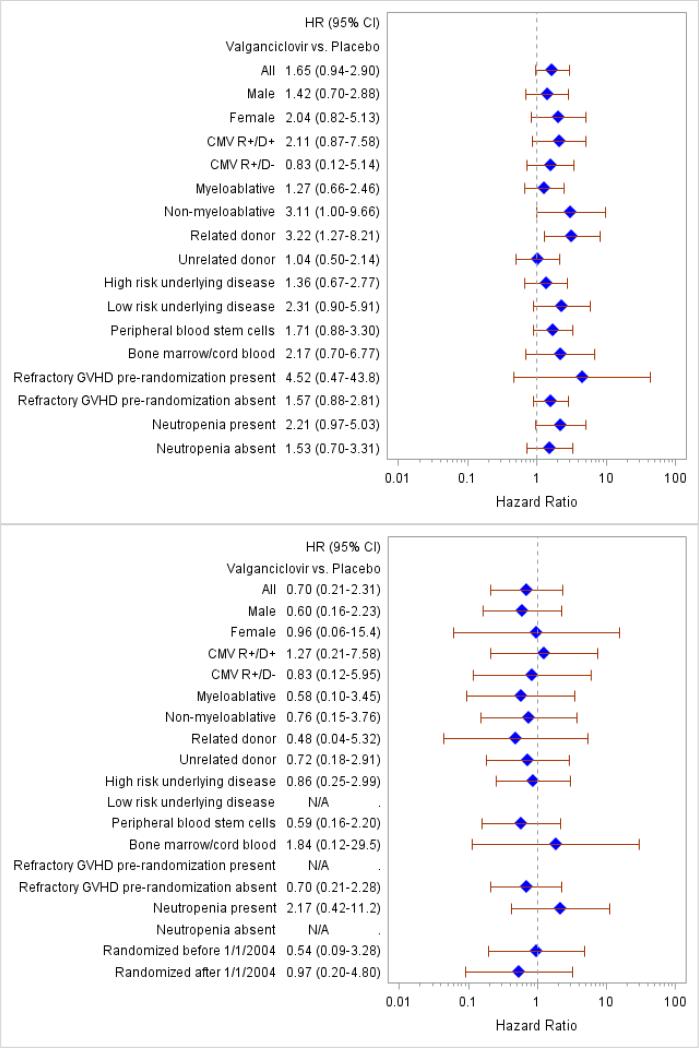 Subset analyses comparing study arms for the neutropenia endpoints: absolute neutrophil counts < 1000/mm3 (upper panel) and < 500/mm3 (lower panel). Reference=placebo. Each row of figure represents a subgroup.
Subset analyses comparing study arms for the neutropenia endpoints: absolute neutrophil counts < 1000/mm3 (upper panel) and < 500/mm3 (lower panel). Reference=placebo. Each row of figure represents a subgroup.Appendix Table 1.
Characteristics of CMV disease.Subject # Randomization Before Day 270 Refractory CMV at baseline Received cGVHD therapy Days from randomization to CMV disease On Study Drug at time of CMV Days from last study drug to disease Reason study drug stopped Low Grade (<1000) PCR Result Prior to disease (closest to onset date) Days from PCR to disease Missed PCR tests prior to disease onset? Died < 3months from onset 1 Plac X 395 334 Study drug completed 150 322 no yes 2 Plac 183 137 Study drug completed 190 42 week 21 no 3 Plac X 22 17 CMV disease, presenting with diarrhea 95 8 no yes 4 VGCV X X 49 5 AE - thrombocytopenia 460 22 no yes 5 VGCV 290 107 AE no CMV weeks 16 & 21 no 6 VGCV X 35 X 0 CMV disease 590 7 no no 7 VGCV 516 358 no CMV week 23 no 8 VGCV 169 154 Study drug completed 100 15 no no 9 Plac X 38 5 Lack of efficacy (PCR > 1000 copies/ml) 100 36 no no Appendix Table 2.
Quality of Life analysis: comparison of functional categories by study arm at baseline, mid-study [day 180] and end-of-treatment analyses [day 270].Valganciclovir N = 95 Mean (Std) Placebo N = 89 Mean (Std) P-value Difference** (V-P) 95% CI* Time period 1 (Baseline) N = 95* N = 89* Physical Functioning 77.1 (1.8) 76.4 (2.2) 0.81 0.7 −4.9, 6.3 Role Functioning 63 (3.3) 62.2 (3.4) 0.86 0.8 −8.5, 10.2 Emotional Functioning 77.5 (2) 82.9 (1.6) 0.037 −5.4 −10.5, −0.3 Cognitive Functioning 77.6 (2.4) 80.3 (2.1) 0.41 −2.7 −9.1, 3.7 Social Functioning 59 (2.7) 57.3 (2.9) 0.66 1.8 −6.1, 9.7 Global health status / QoL 60.5 (2.1) 62.1 (2.2) 0.59 −1.7 −7.8, 4.5 Fatigue 44.8 (2.8) 41.6 (2.6) 0.40 3.2 −4.2, 10.6 Nausea / Vomiting 12.1 (1.8) 10.8 (1.9) 0.61 1.4 −3.9, 6.6 Pain 21.3 (2.5) 22.7 (3.0) 0.72 −1.4 −9.2, 6.3 Dyspnea 18.3 (2.4) 17.9 (2.4) 0.91 0.4 −6.3, 7.1 Insomnia 31.2 (3) 32.3 (2.9) 0.78 −1.2 −9.4, 7 Appetite loss 25.9 (3) 20.1 (2.8) 0.162 5.8 −2.3, 13.9 Constipation 3.5 (1.2) 4.2 (1.4) 0.70 −0.7 −4.2, 2.9 Diarrhea 14.2 (2.4) 15.1 (2.6) 0.79 −0.9 −7.8, 6 Financial Problems 37.5 (3.2) 41.7 (3.5) 0.38 −4.1 −13.4, 5.2 Time period 2 (Day 180) N = 84* N = 80* Physical Functioning 83 (2.1) 78.8 (2.4) 0.20 4.2 −2.2, 10.6 Role Functioning 69.2 (3.3) 66 (3.6) 0.49 3.3 −6.1, 12.7 Emotional Functioning 81.4 (2.2) 81.4 (2.1) 0.98 −0.1 −6.1, 5.9 Cognitive Functioning 80.7 (2.5) 79.9 (2.5) 0.82 0.8 −6.2, 7.8 Social Functioning 69.7 (3.5) 67.9 (3.2) 0.70 1.8 −7.2, 10.8 Global health status / QoL 66.4 (2.2) 64.9 (2.6) 0.69 1.4 −5.6, 8.5 Fatigue 36.7 (2.7) 38.9 (3.0) 0.56 −2.2 −9.7, 5.3 Nausea / Vomiting 9.1 (1.8) 12.8 (2.4) 0.23 −3.7 −9.7, 2.3 Pain 20.5 (3.6) 22.5 (3.4) 0.70 −2.0 −12, 8.1 Dyspnoea 16.2 (2.9) 16.3 (2.8) 0.97 −0.1 −8.2, 7.9 Insomnia 24.6 (3.4) 27 (3.2) 0.62 −2.3 −11.6, 7 Appetite loss 20.4 (3.6) 21.8 (3.8) 0.80 −1.4 −11.7, 9 Constipation 5.9 (2.0) 7.9 (2.3) 0.51 −2.0 −7.8, 3.9 Diarrhea 12 (2.7) 10.3 (2.5) 0.66 1.7 −5.8, 9.2 Financial Problems 29.4 (4.4) 38.6 (4.5) 0.140 −9.3 −21.6, 3.1 Time period 3 (Day 270) N = 75* N = 77* Physical Functioning 81.2 (2.5) 82.4 (2.5) 0.73 1.2 −5.5, 7.9 Role Functioning 70.1 (3.6) 67.3 (3.6) 0.59 −2.8 −13.1, 7.5 Emotional Functioning 85.7 (1.9) 79.7 (2.2) 0.051 −5.9 −11.9, 0 Cognitive Functioning 80.7 (2.4) 82.1 (2.4) 0.69 1.3 −5.2, 7.9 Social Functioning 73.9 (3.7) 74 (3.3) 0.99 0.0 −10.4, 10.5 Global health status / QoL 67.2 (2.5) 65.4 (2.2) 0.60 −1.8 −8.5, 4.9 Fatigue 33.7 (3.1) 40.6 (2.9) 0.111 6.9 −1.6, 15.3 Nausea / Vomiting 8.4 (2.0) 7.9 (1.9) 0.87 −0.5 −5.9, 5 Pain 24.1 (3.8) 23.3 (3.4) 0.88 −0.8 −11.1, 9.5 Dyspnoea 16.4 (3.1) 19.8 (3.3) 0.46 3.4 −5.6, 12.3 Insomnia 21 (3.2) 28.6 (3.7) 0.134 7.6 −2.4, 17.6 Appetite loss 12.9 (3.1) 17.8 (3.9) 0.34 4.9 −5.3, 15 Constipation 4.8 (1.7) 7.3 (2.7) 0.44 2.5 −3.9, 8.8 Diarrhea 13.3 (3.0) 10.5 (2.5) 0.49 −2.8 −10.7, 5.1 Financial Problems 31.7 (4.7) 30 (4.3) 0.79 −1.7 −14.5, 11.1 *Includes imputed data for 9 subjects at Time 1, 35 at Time 2 and 29 at Time 3, among those alive and eligible for assessment at each time: 184, 166 and 155 at Times 1, 2 and 3, respectively. Two subjects at Time 2 and 3 at Time 3 were not imputed due to missing Time 1 QoL assessment for the imputation model.Appendix Table 3.
Deaths, Adverse Events, Serious Adverse Events and Discontinuations due to Adverse Events (within each category the proportion of subjects with 0, 1-5 and 5+ events is shown).Patients, n (%)
Variable Valganciclovir (n=95) Placebo (n=89) Death by day 270 6 (6) 6 (7) Any adverse event 83 (87) 70 (79) Any adverse event Grade 2 before day 270 0 17 (18) 24 (27) 1-4 63 (66) 51 (57) 5+ 15 (16) 14 (16) Adverse event Grade 2 after day 270 0 90 (95) 84 (94) 1-4 5 (5) 5 (6) 5+ 0 (0) 0 (0) Adverse event Grade 3 before day 270 0 47 (49) 42 (47) 1-4 41 (43) 42 (47) 5+ 7 (7) 5 (6) Adverse event Grade 3 after day 270 0 94 (99) 87 (98) 1-4 1 (1) 2 (2) 5+ 0 (0) 0 (0) Adverse event Grade 4 before day 270 0 81 (85) 78 (88) 1-4 14 (15) 10 (11) 5+ 0 (0) 1 (1) Adverse event Grade 4 after day 270 0 95 (100) 89 (100) 1-4 0 (0) 0 (0) 5+ 0 (0) 0 (0) Any serious adverse event 0 53 ( 56) 48 (54) 1-4 29 (31) 34 (38) 5+ 13 (14) 9 (10) Any discontinuation due to adverse event* 29 (31) 15 (17) Neutropenia, thrombocytopenia 13 (14) 8 (9) Hematological or underlying disease relapse 8 (8) 7 (8) Nausea, vomiting, diarrhea 3 (3) 0 Other† 4 (4) 2 (2) *P = 0.03†including edema, elevated liver function tests, headache, and polymicrobial bacteremia in the valganciclovir group and enterococcal sepsis and fatigue in the placebo group.



Footnotes
Author contributions:
Conception and design: M. Boeckh, W.G. Nichols, W. Leisenring.
Analysis and interpretation of the data: M. Boeckh, W.G. Nichols, H. Xie, W. Leisenring.
Drafting of the article: M. Boeckh. W. Leisenring.
Quality of life testing design and analysis: K Syrjala.
Virologic and immunologic testing: Keith Jerome, Terry Stevens-Ayers
Critical revision of the article for important intellectual content: all authors.
Final approval of the article: all authors.
Provision of study materials or patients: M. Boeckh, W.G. Nichols, M. Flowers, R. Chemaly, G. Papanicolaou, J. Wingard.
Statistical expertise: H. Xie, W. Leisenring.
Obtaining funding: M. Boeckh.
Collection and assembly of data: M. Boeckh, W.G. Nichols, H. Xie.
References
- 1.Boeckh M, Gooley TA, Myerson D, Cunningham T, Schoch G, Bowden RA. Cytomegalovirus pp65 antigenemia-guided early treatment with ganciclovir versus ganciclovir at engraftment after allogeneic marrow transplantation: a randomized double-blind study. Blood. 1996;88(10):4063–71. [PubMed] [Google Scholar]
- 2.Einsele H, Ehninger G, Hebart H, Wittkowski KM, Schuler U, Jahn G, et al. Polymerase chain reaction monitoring reduces the incidence of cytomegalovirus disease and the duration and side effects of antiviral therapy after bone marrow transplantation. Blood. 1995;86(7):2815–20. [PubMed] [Google Scholar]
- 3.Boeckh M, Leisenring W, Riddell SR, Bowden RA, Huang M, Myerson D, et al. Late Cytomegalovirus Disease and Mortality in Allogeneic Hematopoietic Stem Cell Transplant Recipients: Importance of Viral Load and T Cell Immunity. Blood. 2003;101:407–14. doi: 10.1182/blood-2002-03-0993. [DOI] [PubMed] [Google Scholar]
- 4.Krause H, Hebart H, Jahn G, Muller CA, Einsele H. Screening for CMV-specific T cell proliferation to identify patients at risk of developing late onset CMV disease. Bone Marrow Transplantation. 1997;19:1111–6. doi: 10.1038/sj.bmt.1700801. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen Q, Champlin R, Giralt S, Rolston H, Raad I, Jacobson K, et al. Late cytomegalovirus pneumonia in adult allogeneic blood and marrow transplant recipients. Clinical Infectious Diseases. 1999;28(3):618–23. doi: 10.1086/515146. [DOI] [PubMed] [Google Scholar]
- 6.Green ML, Leisenring W, Stachel D, Pergam SA, Sandmaier BM, Wald A, et al. Efficacy of a Viral Load-Based, Risk-Adapted, Preemptive Treatment Strategy for Prevention of Cytomegalovirus Disease after Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant. 2012;18(11):1687–99. doi: 10.1016/j.bbmt.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaia JA, Gallez-Hawkins GM, Tegtmeier BR, ter Veer A, Li X, Niland JC, et al. Late Cytomegalovirus Disease in Marrow Transplantation is Predicted by Virus Load in Plasma. Journal of Infectious Diseases. 1997;176:782–5. doi: 10.1086/517301. [DOI] [PubMed] [Google Scholar]
- 8.Pollack M, Heugel J, Xie H, Leisenring W, Storek J, Young JA, et al. An international comparison of current strategies to prevent herpesvirus and fungal infections in hematopoietic cell transplant recipients. Biol Blood Marrow Transplant. 2011;17(5):664–73. doi: 10.1016/j.bbmt.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamae H, Storer B, Sandmaier BM, Maloney DG, Davis C, Corey L, et al. Cytopenias after day 28 in allogeneic hematopoietic cell transplantation: impact of recipient/donor factors, transplant conditions and myelotoxic drugs. Haematologica. 2011;96(12):1838–45. doi: 10.3324/haematol.2011.044966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzberger B, Bowden RA, Hackman R, Davis C, Boeckh M. Neutropenia in allogeneic marrow transplant recipients receiving ganciclovir for prevention of CMV disease: risk factors and outcome. Blood. 1997;90:2502–8. [PubMed] [Google Scholar]
- 11.Hannigan JF, Brown BW. Adaptive randomization biased coin-design: experience in a cooperative group clinical trial. Stanford University: 1982. Division of Biostatistics (Technical report no.74) [Google Scholar]
- 12.Efron B. Randomizing and balancing a complicated sequential experiment. In: Miller RG Jr., Efron B, Brown BW Jr., Moses LE, editors. Biostatistics Casebook. J. Wiley and Sons, Inc.; New York: 1980. pp. 19–30. [Google Scholar]
- 13.Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis. 2002;34(8):1094–7. doi: 10.1086/339329. [DOI] [PubMed] [Google Scholar]
- 14.De Pauw B, Walsh TJ, Donnelly JP, Stevens DA, Edwards JE, Calandra T, et al. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis. 2008;46(12):1813–21. doi: 10.1086/588660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopp M, Schweigkofler H, Holzner B, Nachbaur D, Niederwieser D, Fleischhacker WW, et al. EORTC QLQ-C30 and FACT-BMT for the measurement of quality of life in bone marrow transplant recipients: a comparison. Eur J Haematol. 2000;65(2):97–103. doi: 10.1034/j.1600-0609.2000.90143.x. [DOI] [PubMed] [Google Scholar]
- 16.Boeckh M, Huang M, Ferrenberg J, Stevens-Ayers T, Stensland L, Nichols WG, et al. Optimization of quantitative detection of cytomegalovirus DNA in plasma by real-time PCR. J Clin Microbiol. 2004;42(3):1142–8. doi: 10.1128/JCM.42.3.1142-1148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerrero A, Riddell SR, Storek J, Stevens-Ayers T, Storer B, Zaia JA, et al. Cytomegalovirus viral load and virus-specific immune reconstitution after peripheral blood stem cell versus bone marrow transplantation. Biol Blood Marrow Transplant. 2012;18(1):66–75. doi: 10.1016/j.bbmt.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maris M, Boeckh M, Storer B, Dawson M, White K, Keng M, et al. Immunologic recovery after hematopoietic cell transplantation with nonmyeloablative conditioning. Exp Hematol. 2003;31(10):941–52. doi: 10.1016/s0301-472x(03)00201-7. [DOI] [PubMed] [Google Scholar]
- 19.Castor J, Cook L, Corey L, Jerome KR. Rapid detection directly from patient serum samples of human cytomegalovirus UL97 mutations conferring ganciclovir resistance. J Clin Microbiol. 2007;45(8):2681–3. doi: 10.1128/JCM.00526-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prentice RL, Kalbfleisch JD. The analysis of failure times in the presence of competing risks. Biometrics. 1978;34:541–54. [PubMed] [Google Scholar]
- 21.Rothman KJ. Modern Epidemiology. 2nd ed. Boston: Little Brown. 1986:242–3. [Google Scholar]
- 22.Hodges JL, Lehman EL. Hodges-Lehmann estimators. In: Kotz S, Johnson NL, Read CB, editors. Encyclopedia of Statistical Sciences. John Wiley and Sons; New York: 1983. pp. 463–5. [Google Scholar]
- 23.Schenker N, Taylor JMG. Partially parametric techniques for multiple imputation. Computational Statistics & Data Analysis. 1996;22:425–46. [Google Scholar]
- 24.Rubin DB. Multiple Imputation for Nonresponse in Surveys. John Wiley & Sons; New York: 1987. [Google Scholar]
- 25.Hakki M, Chou S. The biology of cytomegalovirus drug resistance. Curr Opin Infect Dis. 2011;24(6):605–11. doi: 10.1097/QCO.0b013e32834cfb58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Junghanss C, Boeckh M, Carter RA, Sandmaier BM, Maris MB, Maloney DG, et al. Incidence and outcome of cytomegalovirus infections following nonmyeloablative compared with myeloablative allogeneic stem cell transplantation, a matched control study. Blood. 2002;99(6):1978–85. doi: 10.1182/blood.v99.6.1978. [DOI] [PubMed] [Google Scholar]
- 27.Almyroudis NG, Jakubowski A, Jaffe D, Sepkowitz K, Pamer E, O'Reilly RJ, et al. Predictors for persistent cytomegalovirus reactivation after T-cell-depleted allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis. 2007;9(4):286–94. doi: 10.1111/j.1399-3062.2007.00235.x. [DOI] [PubMed] [Google Scholar]
- 28.Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect Dis. 2011;11(4):284–92. doi: 10.1016/S1473-3099(11)70024-X. [DOI] [PubMed] [Google Scholar]
- 29.Peggs KS, Preiser W, Kottaridis PD, McKeag N, Brink NS, Tedder RS, et al. Extended routine polymerase chain reaction surveillance and pre-emptive antiviral therapy for cytomegalovirus after allogeneic transplantation. Br J Haematol. 2000;111(3):782–90. [PubMed] [Google Scholar]
- 30.Goodrich JM, Bowden RA, Fisher L, Keller C, Schoch G, Meyers JD. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Annals of Internal Medicine. 1993;118(3):173–8. doi: 10.7326/0003-4819-118-3-199302010-00003. [DOI] [PubMed] [Google Scholar]
- 31.Winston DJ, Ho WG, Bartoni K, Du Mond C, Ebeling DF, Buhles WC, et al. Ganciclovir prophylaxis of cytomegalovirus infection and disease in allogeneic bone marrow transplant recipients. Results of a placebo-controlled, double-blind trial. Annals of Internal Medicine. 1993;118(3):179–84. doi: 10.7326/0003-4819-118-3-199302010-00004. [DOI] [PubMed] [Google Scholar]
- 32.Palmer SM, Limaye AP, Banks M, Gallup D, Chapman J, Lawrence EC, et al. Extended valganciclovir prophylaxis to prevent cytomegalovirus after lung transplantation: a randomized, controlled trial. Ann Intern Med. 2010;152(12):761–9. doi: 10.7326/0003-4819-152-12-201006150-00003. [DOI] [PubMed] [Google Scholar]
- 33.Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One. 2009;4(5):e5512. doi: 10.1371/journal.pone.0005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reusser P, Riddell SR, Meyers JD, Greenberg PD. Cytotoxic T-lymphocyte response to cytomegalovirus after human allogeneic bone marrow transplantation: pattern of recovery and correlation with cytomegalovirus infection and disease. Blood. 1991;78(5):1373–80. [PubMed] [Google Scholar]
- 35.Ljungman P, Wilczek H, Gahrton G, Gustavsson A, Lundgren G, Lonnqvist B, et al. Long-term acyclovir prophylaxis in bone marrow transplant recipients and lymphocyte proliferation responses to herpes virus antigens in vitro. Bone Marrow Transplantation. 1986;1(2):185–92. [PubMed] [Google Scholar]
- 36.Reusser P, Cathomas G, Attenhofer R, Tamm M, Thiel G. Cytomegalovirus (CMV)-specific T cell immunity after renal transplantation mediates protection from CMV disease by limiting the systemic virus load. J Infect Dis. 1999;180(2):247–53. doi: 10.1086/314879. [DOI] [PubMed] [Google Scholar]
- 37.Hakki M, Riddell SR, Storek J, Carter R, Stevens-Ayers T, Sudour P, et al. Immune Reconstitution to Cytomegalovirus after Allogeneic Hematopoietic Stem Cell Transplantation: Impact of Host Factors, Drug Therapy, and Subclinical Reactivation. Blood. 2003;102(8):3060–7. doi: 10.1182/blood-2002-11-3472. [DOI] [PubMed] [Google Scholar]
- 38.Boeckh M, Kim HW, Flowers ME, Meyers JD, Bowden RA. Long-term acyclovir for prevention of varicella zoster virus disease after allogeneic hematopoietic cell transplantation--a randomized double-blind placebo-controlled study. Blood. 2006;107(5):1800–5. doi: 10.1182/blood-2005-09-3624. [DOI] [PMC free article] [PubMed] [Google Scholar]