

Abstract
Myelofibrosis (MF) is a manifestation of several disorders of hematopoiesis collectively referred to as myeloproliferative neoplasms (MPN). Allogeneic hematopoietic stem cell transplantation (ASCT) is the only therapy with proven curative potential. However, most patients with MF are in the 6th or 7th decade of life, and only some of these patients have been considered suitable transplant candidates. The development of reduced intensity conditioning regimens with limited toxicity has allowed clinicians to offer ASCT to a growing number of older patients. The availability of Janus Kinase (JAK)1/2 inhibitors allows clinicians to provide symptom relief and improved quality of life of MF patients. These drugs may also impact the decision regarding in particular the timing of ASCT. Future studies need to address the role of JAK1/2 inhibitors in patients who are transplant candidates and determine their role before and, possibly, after transplantation. The identification of indications for the use of JAK1/2 inhibitors in the context of transplantation may lead to new therapeutic strategies for patients with MF.
Keywords: Primary Myelofibrosis, Transplantation, Homologous, Janus Kinase 1, INCB018424
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN) characterized by expansion of clonal hematopoietic cells and the release of cytokines that trigger the development of marrow fibrosis, neo-angiogenesis, and osteosclerosis. PMF manifests with blood cytopenias, leukoerythroblastosis, extramedullary hematopoiesis, and progressive splenomegaly that may be accompanied by hepatomegaly. PMF is a rare disorder with an estimated annual incidence of approximately 1 per 100,000 and prevalence of 4–6 per 100,000 persons.1 The disease primarily affects older individuals (median age at presentation 67 years). The course of the disease varies considerably, ranging from indolent, with survival of more than a decade, to aggressive, with disabling constitutional symptoms, impaired quality of life, cachexia, and death within a year or two.2 Myelofibrosis (MF) can also arise from polycythemia vera (post-PV MF/PPV-MF) and essential thrombocythemia (post-ET MF/PET-MF). While the phenotype may be similar to that of PMF, PPV-MF and PET-MF represent distinct clinical entities.3
Conventional therapies such as erythropoietin, androgens, immunomodulatory drugs, interferon-alpha, cytoreductive agents, and non-pharmacological options such as blood transfusion, spleen irradiation, and splenectomy, have not significantly prolonged patient survival. Allogeneic stem cell transplantation (ASCT) is the only currently available therapy with curative potential for MF.4 However, because MF mainly affects older individuals, most MF patients have traditionally not been considered for ASCT. With the more recent adoption of reduced-intensity conditioning (RIC) regimens, ASCT has become applicable to a larger proportion of patients with MF.5–9 However, in older individuals, comorbidities (related or unrelated to MF) are common and may create challenges even with RIC, further affecting patient selection for ASCT, transplant timing, and conditioning strategy.10,11 Data on how the use of JAK1/2 inhibitors will impact transplant outcomes are only beginning to emerge.12–14
Risk-Scoring and Patient Selection
Therapeutic decisions surrounding ASCT for MF require a risk-adapted approach. The Lille score, based on hemoglobin level and white blood cell count, has been used to guide risk-adapted therapy, suggesting that ASCT should be considered with intermediate or higher risk disease (1 or 2 risk factors).15 More recently, new scoring systems have been developed.
The International Prognostic Scoring System (IPSS) estimates the expected survival from the time of MF diagnosis based on five risk factors16:
Age >65 years
Hemoglobin <100 g/l
Leucocyte count >25 × 109/l
Circulating myeloblasts ≥1%
Presence of constitutional symptoms
In the IPSS, patients are classified as low risk (score = 0, median survival 135 months), intermediate-1 risk (score = 1, median survival 95 months), intermediate-2 risk (score = 2, median survival 48 months), or high risk (score ≥3, median survival 27 months).16 A dynamic IPSS (DIPSS) score, proposed subsequently, uses the same five risk factors but allows for prognostic prediction at any time during the disease course. Under the DIPSS, hemoglobin concentration <100 g/L received a score of 2 points; the overall classification is as follows17:
Low risk (score = 0)
Intermediate-1 risk (score = 1 or 2)
Intermediate-2 risk (score = 3 or 4)
High risk (score = 5 or 6)
The DIPSS has been further refined as DIPSS Plus, which adds three additional risk factors — transfusion dependence, unfavorable karyotype, and platelet count <100 × 109/L — each assigned a 1-point score.18 All of these scoring systems were based on studies in patients with PMF only and were developed prior to the wider availability of JAK inhibitors.
Further refinement of risk stratification systems is expected by integrating somatic mutations in the models. One recent study has shown ASXL1, SRSF2, IDH1/2, and EZH2 mutations to be independently associated with poor survival.19 Using the mutation information of these 4 genes, a follow-up study by the same investigators showed that the hazard ratios for survival were 2.78 and 1.52, respectively, for patients who had ≥2 mutations or 1 mutation compared to patients without mutations.20 Recently, additional mutations involving the calreticulin (CALR) gene have been described in patients with PMF and ET.21,22 CALR mutations were mutually exclusive from mutations in JAK2. The data suggest that patients with PMF who harbor a CALR mutation have superior survival compared to those with a JAK2.21 It is not known at present how the presence of ASLX1, SRSF2, IDH1/2, and EZH2 mutations in CALR-positive patients may impact prognosis.
Factors Impacting Transplant Prognosis
Additional factors that may impact outcomes after ASCT include the presence of comorbidities, stem cell donor type, and conditioning regimen.2,11,23–25 Because patient comorbidities weigh heavily in transplant decisions, additional (not disease-specific) scoring systems have been developed, in particular the Hematopoietic Cell Transplantation Comorbidity Index (HCT-CI). This index assigns weighted scores to particular medical conditions that affect non-relapse mortality and survival. The highest scores are assigned to heart valve disease, severely impaired pulmonary function, moderate-to-severe hepatic disease, and a history of a solid tumor malignancy.23 While a formal validation in MF patients is pending, two recent analyses of transplant results in patients with MF showed an inverse correlation of HCT-CI scores and transplant success.26,27
The prognostic value of the Lille scoring system has been studied extensively in HCT recipients.4,11 While patients with low-risk disease have better outcomes compared with intermediate- and high-risk patients, these patients are generally not considered candidates for transplantation as their survival with supportive therapy alone is usually good. Relapse incidence appears to be higher in patients with high Lille scores.11 Studies on the use of new scoring systems in predicting outcomes after HCT have not shown consistent results. Two studies reported that post-HCT success was dependent on pre-HCT DIPSS scores;26,27 a large proportion of those patients received high intensity (“myeloablative”) conditioning. Two large studies from the European Group for Blood and Marrow Transplantation (EBMT) and the Center for International Blood and Marrow Transplant Research (CIBMTR), focusing mainly on reduced intensity conditioning, reported that DIPSS, although predictive, did not sufficiently differentiate between intermediate-1 and intermediate-2 risk populations.9,28
It has been controversial whether splenectomy prior to transplantation is associated with better outcomes, although several studies have shown that hematopoietic recovery is faster in splenectomized patients. A recent study from the CIBMTR failed to show any impact of splenectomy or splenic radiation on GVHD or survival in patients with myeloid malignancies, including myelofibrosis.29
The Challenge of Advanced Age and ASCT
One important factor in the decision-making process about transplantation is the advanced age of many patients with MF.2 One retrospective study analyzed the results of ASCT in 30 patients aged 60 to 78 years with PMF, PPV-MF, or PET-MF, some of whom had high HCT-CI scores.7 Donors were HLA-identical siblings or unrelated, and conditioning regimens ranged from very low (fludarabine plus 2 Gy total body irradiation) to high intensity (high dose busulfan plus cyclophosphamide). With a median follow-up of 22 months, 3-year overall survival (OS) and progression-free survival (PFS) were projected to be 45% and 40%, respectively. These results suggested that selected older patients with advanced MF can be treated successfully with ASCT.7
ASCT and Donor Sources
Only 25% to 30% of patients have an HLA identical sibling, and increasing numbers of transplants are carried out from unrelated donors (URD). Transplants from HLA-mismatched related (haploidentical) donors or with umbilical cord blood (UCB) are also being explored in MF.30,31 Several studies have reported results with URDs to be similar to those with HLA-identical siblings (outcomes with HLA-mismatched donors were inferior).4,32 However, data from the CIBMTR, showed a 1-year non-relapse mortality (NRM) of 27% for transplants from related donors, and 43% for URD ASCT.33 These data were confirmed by a recent update from the CIBMTR, showing adjusted probabilities of 5-year survival for matched sibling donors, well matched URDs, and partially matched URDs after reduced intensity conditioning of 56%, 48%, and 34%, respectively.9
The Italian registry also showed lower transplant-related mortality with transplants from related donors.34 The retrospective study of the Société Française de Greffe de Moelle et de Thérapie Cellulaire showed a lower probability of engraftment and survival with non-sibling donors compared to HLA-matched siblings.35 Differences in results between these studies could be attributable to small sample sizes, their retrospective nature, patient and disease heterogeneity, and the different regimens utilized for transplantation. In a prospective trial of RIC-ASCT in 66 patients, 78% of patients transplanted from related donors, compared to 44% of patients transplanted from URDs, survived at a median follow-up of 24 months.24 Data with HLA haploidentical transplants in MF are too limited for a detailed analysis.
Disease Risk and Patient Selection
Recommendations for ASCT in MPNs are to offer transplantation to eligible patients with life expectancy of <5 years. Patients in IPSS or DIPSS intermediate-2 and high risk categories should be considered for ASCT, as should be those with DIPSS-weighted RBC-transfusion dependence or unfavorable cytogenetics.2,24 An algorithm for ASCT is shown in Figure 1. However, there are patients, particularly of younger age, who do not fall into higher risk categories (e.g., who may have intermediate-1 risk disease) but deserve to be considered for transplantation.
Figure 1. Algorithm for hematopoietic cell transplantation in patients with MF based on DIPSS score.
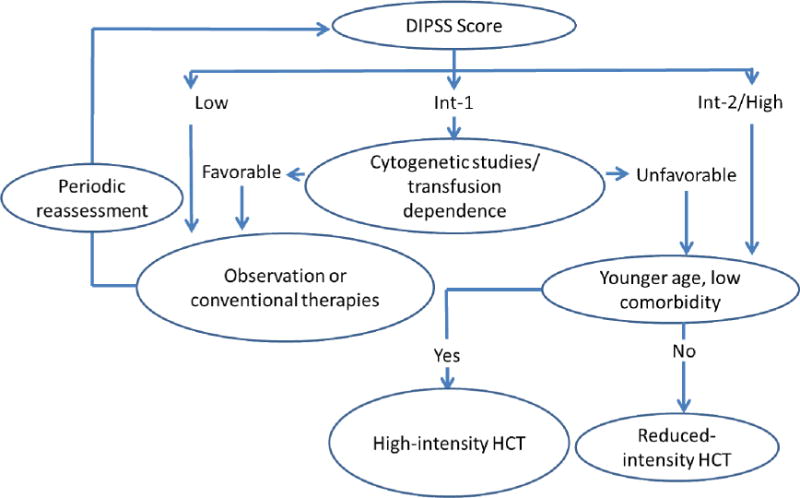
Platelet count, another parameter of the DIPSS Plus classification (in addition to karyotype and red cell transfusion dependence), also affects prognosis, with and without HCT. The selection of the intensity of the HCT conditioning regimen remains a matter of controversy. As discussed in the text, the ways in which the availability and use of JAK inhibitors will modify the decision about HCT have yet to be determined.
Post-transplant problems with ASCT for MF
Regimen-related toxicities
Patients with MF are at risk for liver injury, probably related to liver involvement by their disease (i.e., intrasinusoidal hematopoiesis, fibrosis, portal hypertension).36 One study showed significantly higher risks of post-transplant moderate/severe hyperbilirubinemia and veno-occlusive disease (sinusoidal obstruction syndrome) than seen, for example, in patients with myelodysplastic syndrome.36 Significant hyperbilirubinemia is associated with inferior survival. The incidence of hepatotoxicity has declined steeply in recent years, presumably related to the prophylactic use of ursodiol and novel conditioning regimens.37 Patients with MF may also be at risk of increased pulmonary complications due to extramedullary hematopoiesis in the lungs.
Graft Failure
The incidence of graft failure in patients with MF has been reported at 2% to 25%.11,24 In two prospective studies, the graft failure rate was 2% in patients conditioned with a combination of fludarabine, busulfan, and antithymocyte globulin (ATG),11 and, in a study from the Myeloproliferative Diseases Research Consortium (MPD-RC), 30% (primary or secondary graft failure) after URD ASCT and conditioning with fludarabine, melphalan, and ATG.24 In a CIBMTR analysis, day 100 TRM was 18% with HLA-matched related, 35% with unrelated, and 19% with HLA non-identical related donors.33
The actual risk factors for graft failure are poorly understood and need further study.5 It is currently not clear whether strategies such as the use of T-cell depleting therapies with ATG will reduce the risk of graft failure. As indicated earlier, a low risk of graft failure was seen in the German study using Fresenius ATG as part of the conditioning protocol. However, the risk of graft failure was high with unrelated donor transplantation in the MPD-RC study, which used the thymoglobulin preparation in the conditioning protocol. Most transplant physicians use mobilized peripheral blood stem cells as the preferred graft source for MF, although no formal comparison on the impact of various graft sources on outcome in MF has been presented. It is notable that a recent prospective trial in patients with MF, using conditioning with cyclophosphamide followed by busulfan, observed no graft failure.38
GVHD
GVHD remains the most frequent complication of ASCT.5 Data from the CIBMTR show grades II to IV acute GVHD in 43% of patients transplanted from HLA-matched related donors, 40% from URDs, and 24% from HLA non-identical related donors.30 The incidence of GVHD shows some correlation with the conditioning intensity.39 In one study, the rate of acute GVHD was significantly lower with RIC than with high intensity conditioning (18% vs. 78%, respectively).39 Inflammatory cytokines, which are constitutively dysregulated in MF, and are additionally released from injured tissue following transplant conditioning may contribute to the development of GVHD.5,40
Reduced Intensity vs. High Intensity (Myeloablative) Regimens
Early studies of ASCT for MF used myeloablative conditioning involving total body irradiation or high dose busulfan.41 The introduction of “targeted” busulfan (adjusting doses to predetermined plasma levels) reduced toxicity and improved survival.4 However, these regimens have generally not been used in older patients, for whom RIC has become the standard approach.34 RIC regimens have mostly been fludarabine-based and shown to be more immunosuppressive than myelosuppresive.42,43
An analysis of a CIBMTR cohort of 60 patients prepared with RIC regimens showed TRM of 15%. Relapse-free survival was 39%.30 However, there is currently no consensus on the use of RIC. In an analysis by the Italian transplant group, conditioning intensity did not have an important influence on outcomes, possibly related to the heterogeneity of drugs used within the trials. However, RIC was associated with a higher rate of graft failure compared to myeloablative regimens.34,44
While RIC regimens have played an important role in increasing the availability of ASCT and have been associated with reduced TRM, further studies are required to assess their relationship to improved overall survival.5,30 One such randomized trial, BMT CTN 0901, which is comparing high intensity and RIC, is currently ongoing in the United States in patients with acute myeloid leukemia or myelodysplastic syndrome.45
JAK1/2 Inhibitors in Myelofibrosis: Update on Clinical Trials
Ruxolitinib
Aberrant Janus kinase (JAK) activation is seen in the majority of patients with MF, irrespective of JAK2 (V617F) mutation. JAK inhibitors are compounds developed over the past decade for the treatment of MPNs and other conditions.2 Ruxolitinib is the first JAK inhibitor approved by the U.S. Food and Drug Administration (FDA) for patients with intermediate- or high-risk MF (primary MF, PPV-MF, or PET-MF).46–51 It is approved in Europe for MF patients with symptomatic splenomegaly, regardless of IPSS risk classification. Ruxolitinib, a JAK1/JAK2 inhibitor, showed early clinical benefits in patients with intermediate-2 and high risk MF, including reductions in spleen size and improvements in debilitating constitutional symptoms in a phase I/II (INCB18424-251) and in the phase III COMFORT-I and COMFORT-II trials.46,50,51 Analyses of both the COMFORT-I (ruxolitinib vs. placebo) and COMFORT-II (ruxolitinib vs. best available care) trials showed a survival benefit for patients treated with ruxolitinib.50,51
In the original INCB18424-251 study of 107 patients with intermediate-2 or high risk MF, 54% of patients still received ruxolitinib after a follow-up of 32 months, and survival was 69%. Reduction of splenomegaly and improvement of constitutional symptoms were sustained. Ruxolitinib was well tolerated, with cumulative discontinuation rates of 24%, 36%, and 46%, at 1, 2, and 3 years, respectively. Survival was significantly superior among patients treated with ruxolitinib than among 310 matched controls, mainly attributable to a highly significant difference in the high-risk group (P=0.006). Patients with ≥50% spleen size reduction survived longer than patients with <25% reduction. These data suggest that long-term ruxolitinib therapy may alter the natural course of the disease, though longer follow-up is needed to conclusively determine this effect.46
Fedratinib
Fedratinib (SAR302503; or TG101348) is a selective JAK2 inhibitor. In a phase I trial in intermediate- and high-risk patients with PMF or PPV MF/PET MF, fedratinib substantially relieved constitutional symptoms. After 6 to 12 cycles, a significant proportion of patients achieved spleen size reductions of39% and 47%, respectively, and most patients with leukocytosis or thrombocytosis normalized blood cell counts. Side effects included anemia, nausea and diarrhea. A decrease in JAK2 V617F allele burden observed at 6 months in mutation-carrying patients persisted after 12 months.52 Results of a phase III study with this compound were also encouraging; however, further development of fedratinib has been stopped due to several cases of a syndrome resembling Wernicke encephalopathy.53
SB1518/Pacritinib
In a phase I/II trial of pacritinib, another oral JAK2 inhibitor, in 21 patients, spleen size reduction of ≥50% occurred in 5 (24%), and of ≥35% in 9 (41%).54 A second study in 34 patients showed decreases in spleen size of ≥50% in 15 patients (44%), with 6 patients (18%) achieving complete clinical normalization. Constitutional symptoms were also reduced significantly. Myelosuppression was minimal, and the only relevant side effects were gastrointestinal (diarrhea).54 Pacritinib is being evaluated in two phase III trials, PERSIST-I and -II.55
Momelotinib
Momelotinib (CYT387) is yet another potent inhibitor of JAK 1/2. Results from a phase I/II multicenter study showed reduction of splenomegaly and constitutional symptoms, as well as reduced red cell transfusion requirements.56 In a longer-term phase I/II trial in 60 patients, anemia and spleen response rates were 59% and 48%, respectively, and 70% of transfusion dependent patients achieved transfusion independence of ≥12 weeks. In addition, 50% of patients had a decrease in spleen size of ≥50%, while 17% had a complete normalization.
Substantial improvements were also seen in constitutional symptoms.57 However, the true benefit was not clear in the absence of a control arm. Momelotinib will be compared to ruxolitinib in an upcoming phase III randomized study.
Several other JAK inhibitors (LY2784544, BMS911543, NS-018, and INCB-39110) are in various stages of clinical development.58
Disease Persistence with JAK2 Inhibitor Therapy
While JAK2 inhibitors improve MPN-associated splenomegaly and constitutional symptoms, they do not eliminate the MPN clone, and, in most patients, neither substantial reduction of JAK2 V617F allele burden nor marrow fibrosis has been observed. JAK inhibitors are not specific for the JAK2 V617F mutation; they likely control MF-related signs and symptoms via inhibition of the JAK-STAT pathway. After a period of response, symptoms may recur. This probably involves the reactivation of JAK-STAT signaling via heterodimerization between activated JAK1 and JAK2 or TYK2. This phenomenon is reversible, as demonstrated in cell lines, animal models, and patients treated with JAK2 inhibitors. Therapies that result in JAK2 degradation retain efficacy in persistent cells and may provide additional benefits in patients previously treated with JAK2 inhibitors.59
Defining the Role of JAK Inhibitors in ASCT
What Have We Learned from ASCT for Chronic Myeloid Leukemia (CML) in the Era of Tyrosine Kinase Inhibitors?
The arrival of the tyrosine kinase inhibitor (TKI) imatinib caused a dramatic change in the therapy of chronic phase CML. Prior to imatinib, the only curative therapy for CML was ASCT. Since the introduction of imatinib (and other TKIs), TKI therapy has replaced ASCT as front-line therapy for CML.60 The original 5-year randomized study comparing imatinib to standard combination therapy with cytarabine and interferon α showed complete cytogenetic responses in 69% of imatinib-treated patients by 12 months, and 87% by 60 months; only 7% of patients progressed to accelerated/blast phase.61 The second generation TKIs, nilotinib and dasatinib, show even greater short-term potency.62–64 While ASCT remains an important option for the management of CML, the number of ASCTs performed for CML has decreased significantly with the arrival of TKI therapy.60,65 National Cancer Center Network (NCCN) and the European Leukemia Net (ELN) CML guidelines promote ASCT in chronic phase disease only when patients are intolerant or resistant to all available TKIs, and for progression to accelerated/blast phase.66,67
Major lessons from CML that may be relevant to MF are as follows:
When new, effective therapy is available, treatment patterns may shift radically.
As this happens, the prevalence of disease will dramatically increase (since Prevalence = Incidence × Duration of disease).
Many studies showed that treating CML patients with imatinib before transplantation did not affect transplant outcomes.68 Similar studies will need to be performed in MF patients to assure that the policy of using JAK inhibitors as a bridge (or even, a wall) to transplant does not “boomerang” and ultimately interfere with favorable patient outcomes.
It is important to note, however, that, although CML and MF are both categorized as MPNs, the biology of MF is considerably more complex than CML. While JAK 1/2 inhibitors have salutary effects by decreasing the symptom burden in patients with MF, they are not comparable to BCR-ABL inhibitors. Reduction in JAK2 allele burden (a surrogate marker for the malignant clone) is modest with JAK 1/2 inhibitors, and the rates of discontinuation of JAK inhibitor therapy are much higher than observed with BCR-ABL inhibitors. An extended follow-up of the COMFORT-1 trial has shown that rates of discontinuation of ruxolitinib were 21%, 35%, and 51% at 1, 2, and 3 years, respectively.69 Moreover, JAK inhibitor therapy is not known to reduce the risk of leukemic transformation.
The Impact of the JAK Mutation on ASCT
The impact of the JAK mutation JAK2 (V617F) on ASCT outcomes is still uncertain, although it is present in 50% of patients with PMF, almost all patients with PPV-MF, and 40%–50% of patients with PET-MF. This mutation can serve, however, as a marker for residual or recurrent disease after ASCT. One study evaluated 139 of 162 patients with known JAK2 V617F mutation status after ASCT following RIC to assess the impact of JAK2 V617F allele burden and clearance on transplant outcome. After a median follow-up of 19 months, patients with the JAK2 V617F mutation showed superior survival (70% vs. 44% for those with wild-type JAK2). JAK2 V617F negativity was associated with significantly reduced incidence of relapse.70 However, this was not confirmed in a second study, which showed a lower probability of long-term survival in patients with the mutation.26 Thus, further investigations are needed to assess the impact of JAK2 V617F status and allele burden on transplant outcomes. In addition, studies will be required to understand how transplantation can modify the negative prognostic impact of ASLX1, SRSF2, IDH1/2, and EZH2 mutations.
The Potential Risks and Benefits of JAK1/2 Inhibitors in ASCT
The benefits of JAK2 inhibitors recognized so far include lowering of symptom burden by reducing splenomegaly and constitutional symptoms. JAK1/2 inhibitors may serve as an alternative to splenectomy in patients with significant splenomegaly as surgical splenectomy is associated with a high risk of perioperative complications (27.7%) and mortality (6–7%), and is not routinely recommended before ASCT.5 While a survival benefit has been observed in some trials with the use of JAK1/2 inhibitors, these survival benefit data must be confirmed in long-term follow-up studies and across the class of these drugs. JAK1/2 inhibitors reduce pro-inflammatory and pro-angiogenic cytokines, but have limited effects on marrow fibrosis, cytogenetic abnormalities, and leukemic transformation. MF-associated symptoms may also quickly return after discontinuation of therapy.5 Preliminary data suggest that the sequential use of JAK inhibitors before ASCT and ATG in the conditioning regimen may reduce the risk of GVHD, and, possibly, the incidence of rejection; however, patients will need to be monitored for infections and the GVL effect. The effect of JAK inhibition on bone marrow fibrosis is a focus of current investigations.71,72
As discussed for CML, it is likely that the commercial availability of JAK1/2 inhibitors will affect referral patterns for ASCT; it is currently not know whether this will affect outcomes after ASCT. Patients with clinically relevant responses to JAK1/2 inhibitors may delay a decision about transplantation and may have more advanced disease once referred for ASCT. One potential advantage of JAK1/2 inhibitors, in addition to decreasing constitutional symptoms, is the reduction of splenomegaly, which may hasten hematologic recovery following ASCT. Further, the down-regulation of inflammatory cytokines might favorably impact GVHD and, possibly, graft failure. This would be attractive as MF-associated symptoms are thought to be a risk factor for outcome after ASCT.30 However, only prospective trials can definitively address this question.
Clearly, data on the use of JAK1/2 inhibitors in the setting of transplantation are just beginning to emerge. A positive impact of the use of ruxolitinib prior to transplantation was recently suggested by two small retrospective studies from Germany.12,73 Of particular note, a significant improvement in performance status was observed with the use of ruxolitinib in patients who were not initially considered suitable candidates for transplantation.12 A second study from Germany reported the outcome of this strategy in 22 patients, and reported encouraging early results.73 However, preliminary results of a prospective multicenter study from France showed serious adverse events, such as cardiogenic shock and tumor lysis syndrome, with this approach.14 Due to multiple confounding factors, the reasons for poor results in the French study are not clear. All of the reported data so far are based on small numbers of patients. Therefore, although theoretically appealing, it will be important to investigate this approach as part of prospective studies and at experienced transplant centers.
There is little indication for the use of JAK1/2 inhibitors post-transplant, although they could be considered in patients who relapse (for overall symptom reduction) and, possibly, in patients with treatment-refractory GVHD.
While a case can be made for the use of JAK1/2 inhibitors as “bridging” therapy pre-transplant, other potential scenarios require further study:
Administration of JAK1/2 inhibitors throughout treatment, including post-ASCT
JAK1/2 inhibitors given post-transplant for control of persistent splenomegaly or constitutional symptoms
JAK1/2 inhibitors for GVHD modulation
The timing of transplantation in patients receiving JAK1/2 inhibitors is a crucial area of further investigation. Some of the dilemmas related to timing of transplantation in MF are highlighted in case studies in tables 1 and 2.
Table 1.
| Case 1 |
| A 68-year-old female was initially diagnosed with polycythemia vera in 2001, and initially treated with intermittent phlebotomy, low dose aspirin, and hydroxyurea. Her need for phlebotomy decreased in 2008 and was no longer required after 2009. In January 2012, she presented with abdominal fullness, decreased appetite, marked fatigue and a weight loss of 10 kg. |
| Her ECOG performance score was 2; the spleen was palpable by 20 cm below the left costal margin. The Hgb was 96g/L, WBC 26 × 109/L, and platelets 300 × 109/L. The differential count showed left shift, nucleated red cells and 2% myeloblasts. Her LDH was 985U/L (IULN 240). |
| A bone marrow biopsy showed grade 3/3 fibrosis, <5% myeloblasts, and normal cytogenetics. Cells expressed the JAK2V617F mutation. The patient has two healthy siblings, ages 64 and 66 years. |
Question 1: How would you treat this patient?
|
| Discussion: The patient’s physician started her on the JAK1/2 inhibitor ruxolitinib, and after 6 weeks, she had significant improvement in symptoms. Her blood counts were stable, and she was transfusion independent. HLA typing, meanwhile, shows one of her siblings to be HLA-identical. |
Question 2: What would you recommend at this point?
|
| Discussion: This patient had a gratifying response to JAK1/2 inhibition. As a 68-year-old patient, transplantation would, presumably, involve an RIC regimen. While some data suggest a higher probability of relapse with RIC than observed with high intensity conditioning, other reports have shown low relapse rates. While there is a 50–60% probability of long-term survival and remission, there is also approximately a 50% chance of developing GVHD requiring therapy, possibly long-term. Thus, with the present state of knowledge, it is difficult to provide an absolutely definitive recommendation. Some patients might prefer conservative management with continuation of ruxolitinib until there are signs of disease acceleration/progression. Other patients (and physicians) might prefer to proceed with HCT, acknowledging the risk of GVHD, but valuing the potential for cure higher than the possibility of relapse and risk of GVHD. |
Table 2.
| Case 2 |
| A 46-year-old female presents with symptoms of fatigue. Her spleen is palpable at the costal margin. Her Hgb is 112 g/L, WBC 27 × 109/L, and platelets 221 × 109/L. A smear shows 2% myeloblasts. |
| Bone marrow examination shows hypercellularity with megakaryocyte clustering. There is no dysplasia, myeloblasts are <5%, and a reticulin stain shows grade 2/3 fibrosis. Cytogenetics show del (5q) and t(1;21). Mutation analysis reveals the JAK2V617F mutation. |
| She does not complain of constitutional symptoms or symptoms related to splenomegaly. She has no siblings; an unrelated donor search shows 3 potential donors, HLA-matched by high-resolution typing. |
| Question: How would you treat this patient? |
|
| Discussion: While the presence of del(5q) suggests that lenalidomide may be a good option, there are no controlled prospective data in support of such an approach. She has neither constitutional symptoms nor significant splenomegaly, making a benefit from JAK1/2 inhibitor treatment unlikely. Her WBC is elevated and myeloblasts are circulating. |
| In view of the patient’s young age and the availability of HLA-matched donors, transplantation should be considered. Recent data indicate that the success rate with unrelated donors who are HLA matched by high resolution typing are comparable to those with HLA-identical siblings. |
The following questions must be considered:
When should ASCT be performed in patients responding to JAK1/2 inhibition?
In patients failing to respond to a given JAK1/2 inhibitors, should one go directly to ASCT or should other, perhaps investigational, inhibitors be given?
Should ASCT be carried out as soon as a donor is identified, regardless of JAK1/2 inhibitor response?
Conclusions
ASCT has a definitive role in the treatment of patients with MF and is the only modality with proven curative potential. However, many patients benefit symptomatically from treatment with JAK1/2 inhibitors, and there is evidence that JAK1/2 inhibitors may prolong survival (of non-transplanted patients). The impact of treatment with JAK1/2 inhibitors on ASCT, in particular timing of ASCT, is not clear and remains an essential area of research. Additional important questions (e.g., sequential use of multiple inhibitors, combinations of JAK inhibitors and other agents, selection of one JAK inhibitor vs. another) will need to be answered as novel JAK1/2 inhibitors become available. Whether there is a role for JAK1/2 inhibitors after ASCT remains to be determined. Finally, as JAK2 inhibitors have also been useful in patients without JAK2 mutations, it will be of interest to determine how the recent identification of CALR mutations in JAK1 negative patients with MF will impact treatment decisions.
Acknowledgments
The authors would like to thank Elizabeth Paczolt, MD, FACNM, for editorial assistance to the authors during preparation of this manuscript
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Conflict of Interest Disclosures
Vikas Gupta, MD, FRCP, FRC Path — Grant support through his institution from Incyte and Novartis; Consulting fees from Incyte and Novartis; Lecture fees from Novartis.
Jason Gotlib, MD, MS — Research funding from Incyte, Novartis, Sanofi-Aventis, and Gilead; Consulting fees from Incyte, Gilead, and Sanofi-Aventis; Steering committee for Novartis.
Jerald P. Radich, MD — Consulting fees from Novartis, Bristol-Myers Squibb, Pfizer, Ariad, Incyte, and Amgen; Research funding from Novartis; Honoraria from Novartis, Bristol-Myers Squibb, Pfizer, Ariad, Incyte, and Amgen.
Nicolaus M. Kröger, MD — Research funding from Novartis, Sanofi, Pierre Fabre, Celgene, and Fresenius Biotech; Lecture fees from Novartis, Sanofi, Pierre Fabre, and Johnson & Johnson.
Damiano Rondelli, MD — Honoraria from Celgene, Sanofi-Aventis, Bristol-Myers-Squibb, and Incyte.
Srdan Verstovsek, MD, PhD — Honoraria from Incyte Corporation, AstraZeneca, S*BIO, Lilly Oncology, Roche, Geron, Exelixis, NS Pharma, Bristol-Myers Squibb, Celgene, Infinity Pharmaceuticals, YM Biosciences, Gilead, and Seattle Genetics.
H. Joachim Deeg, MD — Research funding from Incyte; Advisory Board for medac.
References
- 1.Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2013 Jul 29; doi: 10.3109/10428194.2013.813500. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 2.McLornan DP, Mead AJ, Jackson G, Harrison CN. Allogenic stem cell transplantation for MF in 2012. Brit J Haematol. 2012;157:413–425. doi: 10.1111/j.1365-2141.2012.09107.x. [DOI] [PubMed] [Google Scholar]
- 3.Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for MF Research and Treatment. Leukemia. 2008;22:437–438. doi: 10.1038/sj.leu.2404914. [DOI] [PubMed] [Google Scholar]
- 4.Deeg HJ, Gooley TA, Flowers ME, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood. 2003;102:3912–3918. doi: 10.1182/blood-2003-06-1856. [DOI] [PubMed] [Google Scholar]
- 5.Gupta V, Hari P, Hoffman R. Allogeneic hematopoietic cell transplantation for MF in the era of JAK inhibitors. Blood. 2012;120:1367–1379. doi: 10.1182/blood-2012-05-399048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiele J, Kvasnicka HM, Dietrich H, et al. Dynamics of bone marrow changes in patients with chronic idiopathic myelofibrosis following allogeneic stem cell transplantation. Histol Histopat hol. 2005;20(3):879–889. doi: 10.14670/HH-20.879. [DOI] [PubMed] [Google Scholar]
- 7.Samuelson S, Sandmaier BM, Heslop HE, et al. Allogeneic haematopoietic cell transplantation for MF in 30 patients 60–78 years of age. Br J Haematol. 2011;153:76–82. doi: 10.1111/j.1365-2141.2011.08582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alchaby H, Badbaran A, Zabelina T, et al. Impact of JAK2V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood. 2010;116:3572–3581. doi: 10.1182/blood-2009-12-260588. [DOI] [PubMed] [Google Scholar]
- 9.Gupta V, Malone AK, Hari PN, et al. Reduced-intensity hematopoietic cell transplantation for patients with primary myelofibrosis: a cohort analysis from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2014;20(1):89–97. doi: 10.1016/j.bbmt.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rondelli D, Barosi G, Bacigalupo A, et al. Allogeneic hematopoietic stem-cell transplantation with reduced-intensity conditioning in intermediate- or high-risk patients with myelofibrosis with myeloid metaplasia. Blood. 2005;105(10):4115–4119. doi: 10.1182/blood-2004-11-4299. [DOI] [PubMed] [Google Scholar]
- 11.Kroger N, Holler E, Kobbe G, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2009;114:5264–5270. doi: 10.1182/blood-2009-07-234880. [DOI] [PubMed] [Google Scholar]
- 12.Jaekel N, Behre G, Behning A, et al. Allogeneic hematopoietic cell transplantation for myelofibrosis in patients pretreated with the JAK1 and JAK2 inhibitor ruxolitinib. Bone Marrow Transplant. 2013 Dec 2; doi: 10.1038/bmt.2013.173. [DOI] [PubMed] [Google Scholar]
- 13.Kröger N, Alchalby H, Ditschkowski M, et al. Ruxolitinib As Pretreatment Before Allogeneic Stem Cell Transplantation For Myelofibrosis ASH. 2013 doi: 10.1038/leu.2014.86. Abstract 392. [DOI] [PubMed] [Google Scholar]
- 14.Robin M, Francois S, Huynh A, et al. Ruxolitinib Before Allogeneic Hematopoietic Stem Cell Transplantation (HSCT) In Patients With myelofibrosis: a Preliminary Descriptive Report Of The JAK ALLO Study, a Phase II Trial Sponsored By Goelams-FIM In Collaboration With The Sfgmtc. ASH. 2013 Abstract 306. [Google Scholar]
- 15.Dupriez B, Morel, Demory JL, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88:1013–1018. [PubMed] [Google Scholar]
- 16.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary MF based on a study of the International Working Group for MF Research and Treatment. Blood. 2009;113:2895–2901. doi: 10.1182/blood-2008-07-170449. [DOI] [PubMed] [Google Scholar]
- 17.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment) Blood. 2010;115:1703–1708. doi: 10.1182/blood-2009-09-245837. [DOI] [PubMed] [Google Scholar]
- 18.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29:392–397. doi: 10.1200/JCO.2010.32.2446. [DOI] [PubMed] [Google Scholar]
- 19.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. doi: 10.1038/leu.2013.119. [DOI] [PubMed] [Google Scholar]
- 20.Guglielmelli P, Lasho TL, Biamonte F, et al. Effect of the number of prognostically relevant mutated genes on survival and leukemia progression in primary myelofibrosis. ASH. 2013 Abstract 104. [Google Scholar]
- 21.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 22.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorror ML, Maris MB, Storb B, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106:2912–2919. doi: 10.1182/blood-2005-05-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rondelli D, Goldberg JD, Marchioli R, et al. Results of Phase II Clinical Trial MPD-RC 101: Allogeneic Hematopoietic Stem Cell Transplantation Conditioned with Fludarabine/Melphalan in Patients with Myelofibrosis. Biol Blood Marrow Transplant. 2012;18(2):S216. [Google Scholar]
- 25.Bacigalupo A, Soraru M, Dominietto A, et al. Allogeneic hemopoietic SCT for patients with primary myelofibrosis: a predictive transplant score based on transfusion requirement, spleen size and donor type. Bone Marrow Transplant. 2010;45:458–463. doi: 10.1038/bmt.2009.188. [DOI] [PubMed] [Google Scholar]
- 26.Scott BL, Gooley TA, Sorror ML, et al. The Dynamic International Prognostic Scoring System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood. 2012;119(11):2657–64. doi: 10.1182/blood-2011-08-372904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ditschkowski M, Elmaagacli AH, Trenschel R, et al. Dynamic International Prognostic Scoring System scores, pre-transplant therapy and chronic graft-versus-host disease determine outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis. Haematologica. 2012;97(10):1574–81. doi: 10.3324/haematol.2011.061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alchalby H, Yunus DR, Zabelina T, et al. Br J Haematol. 2012;157(1):75–85. doi: 10.1111/j.1365-2141.2011.09009.x. [DOI] [PubMed] [Google Scholar]
- 29.Akpek G, Pasquini MC, Logan B, et al. Effects of spleen status on early outcomes after hematopoietic cell transplantation. Bone Marrow Transplant. 2013;48(6):825–31. doi: 10.1038/bmt.2012.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballen K. How to manage the transplant question in myelofibrosis. Blood Cancer J. 2012;2(3):e59. doi: 10.1038/bcj.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robin M, Giannotti F, Deconinck E, et al. Outcomes After Unrelated Cord Blood Transplantation For Adults With Primary Or Secondary Myelofibrosis: A Retrospective Study On Behalf Of Eurocord and Chronic Malignancy Working Party-EBMT. ASH. 2013 Abstract 306. [Google Scholar]
- 32.Kerbauy DM, Gooley TA, Sale GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant. 2007;13(3):355–65. doi: 10.1016/j.bbmt.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Ballen KK, Shrestha S, Sobocinski KA, et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2010;16:358–367. doi: 10.1016/j.bbmt.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patriarca F, Bacigalupo A, Sperotto A, et al. Allogeneic hematopoietic stem cell transplantation in myelofibrosis: the 20-year experience of the Gruppo Italiano Trapianto di Midollo Osseo (GITMO) Haematologica. 2008;93:1514–1522. doi: 10.3324/haematol.12828. [DOI] [PubMed] [Google Scholar]
- 35.Robin M, Tabrizi R, Mohty M, et al. Allogeneic haematopoietic stem cell transplantation for myelofibrosis: a report of the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC) Br J Haematol. 2011;152(3):331–339. doi: 10.1111/j.1365-2141.2010.08417.x. [DOI] [PubMed] [Google Scholar]
- 36.Wong KM, Atenafu EG, Kim D, et al. Incidence and risk factors for early hepatotoxicity and its impact on survival in patients with myelofibrosis undergoing allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18:1589–1599. doi: 10.1016/j.bbmt.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Ruutu T, Eriksson B, Remes K, et al. Nordic Bone Marrow Transplantation Group Ursodeoxycholic acid for the prevention of hepatic complications in allogeneic stem cell transplantation. Blood. 2002;100(6):1977–83. doi: 10.1182/blood-2001-12-0159. [DOI] [PubMed] [Google Scholar]
- 38.Rezvani AR, McCune JS, Storer BE, et al. Cyclophosphamide followed by intravenous targeted busulfan for allogeneic hematopoietic cell transplantation: pharmacokinetics and clinical outcomes. Biol Blood Marrow Transplant. 2013;19(7):1033–1039. doi: 10.1016/j.bbmt.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clift RA, Buckner CD, Appelbaum FR, et al. Long-term follow-up of a randomized trial of two irradiation regimens for patients receiving allogeneic marrow transplants during first remission of acute myeloid leukemia. Blood. 1998;92(4):1455–6. [PubMed] [Google Scholar]
- 40.Antin JH, Ferrara JL. Cytokine dysregulation and acute graft versus-host disease. Blood. 1992;80(12):2964–8. [PubMed] [Google Scholar]
- 41.Guardiola P, Anderson JE, Bandini G, et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Societe Francaise de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood. 1999;93:2831–2838. [PubMed] [Google Scholar]
- 42.Slavin S, Nagler A, Naparstek E, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood. 1998;91:756–763. [PubMed] [Google Scholar]
- 43.Baron F, Sandmaier BM. Chimerism and outcomes after allogeneic hematopoietic cell transplantation following nonmyeloablative conditioning. Leukemia. 2006;20(10):1690–700. doi: 10.1038/sj.leu.2404335. [DOI] [PubMed] [Google Scholar]
- 44.Patriarca F, Bacigalupo A, Sperotto A, et al. Outcome of allogeneic stem cell transplantation following reduced-intensity conditioning in patients with idiopathic myelofibrosis: the G.I.T.M.O. experience. Mediterr J Hematol Infect Dis. 2010;2(2):e2010010. doi: 10.4084/MJHID.2010.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.ClinicalTrials.gov. Reduced Intensity Conditioning Versus Myeloablative Conditioning for Acute Myeloid Leukemia or Myelodysplastic Syndrome (BMT CTN 0901) Available at http://clinicaltrials.gov/ct2/show/NCT01339910. Accessed July 29, 2013.
- 46.Verstovsek S, Kantarjian HM, Estrov Z, et al. Long-term outcomes of 107 patients with myelofibrosis receiving JAK1/JAK2 inhibitor ruxolitinib: survival advantage in comparison to matched historical controls. Blood. 2012;120:1202–1209. doi: 10.1182/blood-2012-02-414631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jakafi® prescribing information. Incyte Corporation; Wilmington, DE: Jun, 2013. http://www.incyte.com/sites/default/files/Jakafi_PI.pdf. Accessed July 2013. [Google Scholar]
- 48.Verstovsek S, Kantarjian H, Mesa R, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Eng J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Eng J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 50.Verstovsek S, Mesa RA, Gotlib JR, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Eng J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety and survival with ruxolitinib treatment in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013 Sep 13; doi: 10.3324/haematol.2013.092155. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pardanani A, Tefferi A, Jamieson C, et al. Long-Term Follow Up Of a Randomized Phase II Study Of The JAK2-Selective Inhibitor Fedratinib (SAR302503) In Patients With Myelofibrosis (MF) ASH. 2013 doi: 10.1038/bcj.2015.63. Abstract 4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komrokji R, Wadleigh M, Seymour J, et al. Results of a phase 2 study of pacritinib (SB1518) a novel oral JAK2 inhibitor, in patients with primary, post-polycythemia vera, and post-essential thrombycythemia myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2011 Abstract 282. [Google Scholar]
- 55.Clinicaltrials.gov. Oral Pacritinib Versus Best Available Therapy to Treat Myelofibrosis. Available at http://clinicaltrials.gov/show/NCT01773187. Accessed August 13, 2013.
- 56.Pardanani A, Gotlib J, Gupta V, et al. An expanded multicenter study of CYT387, a JAK-1/2 inhibitor for the treatment of myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2011 Abstract 3849. [Google Scholar]
- 57.Pardanani A, Gotlib J, Gupta V, et al. Phase I/II study of CYT387, a JAK1/JAK2 inhibitor for the treatment of myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2012 Abstract 3849. [Google Scholar]
- 58.Sonbol MB, Firwana B, Zarzour A, et al. Comprehensive review of JAK inhibitors in myeloproliferative neoplasms. Ther Adv Hematol. 2013;4(1):15–35. doi: 10.1177/2040620712461047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koppikar P, Bhagwat N, Kilipivaara O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489:155–159. doi: 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saussele S, Lauseker M, Grathwohl A, et al. German Study Group Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood. 2010;115:1880–1885. doi: 10.1182/blood-2009-08-237115. [DOI] [PubMed] [Google Scholar]
- 61.Druker BJ, Guilhot F, O’Brien SG, et al. IRIS Investigators Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Eng J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 62.Bacher U, KlyuchnIkov E, Zabelina T, et al. The changing scene of allogeneic stem cell transplantation for chronic myeloid leukemia—a report from the German Registry covering the period from 1998 to 2004. Ann Hematol. 2009;88:1237–1247. doi: 10.1007/s00277-009-0737-3. [DOI] [PubMed] [Google Scholar]
- 63.Saglio G, Kim DW, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 64.Kantarjian HM, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12:841–851. doi: 10.1016/S1470-2045(11)70201-7. [DOI] [PubMed] [Google Scholar]
- 65.Radich JP, Kopecky KJ, et al. A randomized trial of dasatinib 100 mg versus imatinib 400 mg in newly diagnosed chronic-phase chronic myeloid leukemia. Blood. 2012;120(19):3898–905. doi: 10.1182/blood-2012-02-410688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.National Comprehensive Cancer Network. Chronic myelologenous leukemia. Available at www.nccn.org/professionals/physician_gls/pdf/cml.pdf. Accessed on August 12, 2013.
- 67.Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. doi: 10.1182/blood-2013-05-501569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oehler VG, Gooley T, Snyder DS, et al. The effects of imatinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood. 2007;109(4):1782–1789. doi: 10.1182/blood-2006-06-031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verstovsek S, Mesa R, Gotlib J, et al. Long-Term outcomes of ruxolitinib therapy In patients with myelofibrosis: 3-Year update from COMFORT-I. ASH. 2013 Abstract 396. [Google Scholar]
- 70.Mesa RA, Nagomey DS, Schwager S, et al. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Blood. 2006;107:361–370. doi: 10.1002/cncr.22021. [DOI] [PubMed] [Google Scholar]
- 71.Kvasnicka HM, Thiele J, Bueso-Ramos CE, et al. Exploratory analysis of the effect of ruxolitinib on bone marrow morphology in patients with myelofibrosis. ASCO. 2013 Abstract 7030. [Google Scholar]
- 72.Kvasnicka HM, Thiele J, Bueso-Ramos CE, et al. Long-term intervention effects on bone marrow morphology in myelofibrosis: patients treated with ruxolitinib and best availabile therapy. 2013 Congress of the European Hematology Association. Abstract S591. [Google Scholar]
- 73.Stübig T, Alchalby H, Ditschkowski M, et al. JAK Inhibition with ruxolitinib as pretreatment for allogeneic stem cell transplantation in primary or post ET/PV myelofibrosis. Leukemia. 2014 Feb 26; doi: 10.1038/leu.2014.86. [DOI] [PubMed] [Google Scholar]