

Abstract
Systems for glucose monitoring based on resonance energy transfer (RET) and competitive binding using Concanavalin A (Con A) are problematic as a result of problems of toxicity, aggregation, and irreversible binding. This paper presents an improved RET assay wherein Con A was replaced by apo-glucose oxidase (apo-GOx). The basic principle for transduction is identical to that used in assays based on Con A–dextran: a reduction in RET from fluorescein isothiocyanate (FITC) to tetramethyl rhodamine isothiocyanate (TRITC) occurs when FITC–dextran (donor) is displaced from TRITC–apo-GOx (acceptor) as a result of the competition of glucose. Fluorescence measurements confirm that the apo-GOx/dextran complexes are highly sensitive to glucose, measured as an increase in the donor peak relative to acceptor due to stepwise addition of glucose. The solution-phase assay showed strong signals and excellent repeatability, with a sensitivity of 0.0163 (ratio units)/mM over the range of 0–90 mM glucose. If properly encapsulated, these sensors can potentially be used for in vivo sensing without the concern of toxicity associated with Con A.
Introduction
Glucose monitoring systems are of tremendous commercial interest because the incidence of diabetes mellitus is increasing uncontrollably worldwide.1 Frequent monitoring of glucose levels, followed by appropriate actions to maintain normoglycemia, is necessary to minimize the adverse affects of diabetes. A noninvasive glucose sensor, the so-called “holy grail” of medical devices, would help improve the lives of many people by reducing the pain associated with the testing, thus improving the quality of the treatment. The minimally invasive glucose sensors commercially available still require extraction of blood samples.2 Fully implantable systems with wireless communication are an ideal solution; however, while many such devices are under development, controlling the stability of the enzyme-electrochemical system used for the sensors remains a major challenge.
Optical sensors provide an attractive alternative because they enable analyte monitoring through spectroscopic characterization of complex and sensitive samples.3–17 Glucose sensors based on fluorescence spectroscopy are being investigated by many groups because of the advantages of high sensitivity and superior specificity provided by molecular recognition.3–14,16–18 One of the more thoroughly investigated approaches for monitoring glucose with fluorescence involves competitive binding and fluorescence resonance energy transfer (RET). In a series of careful studies, the association of dextran and Concanavalin A (Con A) has been demonstrated to be sensitive to glucose concentration. Schultz has reported several variations of sensors based on the same basic principle: with the addition of glucose, fluoroscein isothiocyanate (FITC)–dextran is displaced from tetramethyl rhodamine isothiocyanate (TRITC)–Con A, resulting in measurable changes in energy transfer between FITC and TRITC.4–7 Thus, glucose concentration can be estimated from RET efficiency, and the ratiometric nature of the RET analysis method allows compensation for variations in instrumental parameters, assay component concentrations, and measurement configuration.19 Glucose assays based on the Con A/dextran system were also demonstrated using fluorescence lifetime measurements to monitor the changes in RET.8,9 Glucose sensors using RET were also demonstrated using fluorescence steady-state and lifetime measurements on genetically engineered glucose-binding proteins.10–12
The RET-based sensors described above were demonstrated in solution as well as with chemistry immobilized within membranes of fiber probes. A more practical method, closer to a form useful for clinical application, was proposed by Coté et al.: a novel “smart tattoo” system, wherein the assay chemistry is immobilized within hydrogel microspheres which may be implanted in the skin and interrogated transdermally.13 The promise of this concept, even for the short-wavelength FITC–TRITC RET pair, has been demonstrated by modeling and experimental studies of tissue optics and transdermal measurements.14,15 Recently, experimental work on sensors was extended into the near-infrared (NIR) by labeling the lectin and ligand with an NIR donor–acceptor pair.16 While the Con A/dextran assay has been encapsulated and response to glucose has been observed, problems remain in the production of glucose-sensitive microspheres, including fabrication uniformity, leaching stability, and reversibility.
Previously, we reported on a nanoengineered polymeric microcapsule comprising multilayer films of TRITC–Con A and FITC–dextran using affinity binding and layer-by-layer (LbL) self-assembly technique.3 These glucose-sensitive thin films packaged in a microcapsule were sensitive to glucose, as observed by a change in the energy transfer (11%) with the addition of 0.05 M glucose.3 However, it is important to note that all of the above-mentioned Con A/dextran-based sensing systems carry concerns over toxicity and nonspecific binding.
To overcome the limitations of Con A, we have developed an alternative approach to the competitive-binding glucose assay. The system employs an inactive form of the enzyme apo-glucose oxidase (apo-GOx) as the glucose-binding protein, which is highly specific to β-D-glucose. The design of the sensor is illustrated in Figure 1. Briefly, apo-GOx is prepared from GOx by removing FAD cofactor.17 When apo-GOx is labeled with TRITC and exposed to FITC–dextran, strong fluorescence peaks due to significant energy transfer between FITC and TRITC are observed. Because of the high affinity of apo-GOx for glucose, addition of glucose results in the displacement of dextran from apo-GOx. This change in physical proximity is manifested as a decrease in the energy transfer efficiency (Figure 1), as evidenced by a stronger FITC peak relative to TRITC. In this study, the glucose sensitivity of the TRITC–apo-GOx/FITC–dextran was demonstrated by measuring changes in RET between FITC and TRITC resulting from titration of glucose. This system retains the advantages of the competitive binding approach, including selectivity to the analyte of interest and elimination of reaction byproducts, and there is no consumption of the analyte during the sensing process.
Figure 1.
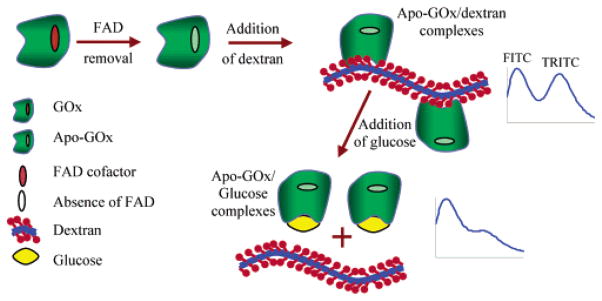
Schematic of a glucose assay based on competitive binding between dextran and glucose for binding sites on apo-GOx.
Apo-GOx was previously used as a biosensor based on its ability to reconstitute into a holoenzyme with the addition of semi-artificial cofactors.20,21 Apo-GOx and apo-glucose dehydrogenese were also demonstrated as direct glucose sensors in which the intrinsic fluorescence increases with the addition of glucose.17,22 Apo-GOx was also tagged with an environmentally sensitive fluorophore, which exhibited a decrease in intensity with the addition of glucose due to partial shielding of tryptophan residues.17 The disadvantages of these approaches include the short wavelengths required to excite intrinsic fluorescence and the inability to correct for inner filter, dilution, or other nonspecific effects due to nonratiometric operation.
This communication demonstrates, for the first time, a fluorescent RET assay which utilizes the glucose affinity of an inactive form of GOx. The system is proven to be stable and highly sensitive to glucose. This approach may potentially be used for in vivo glucose monitoring, if properly encapsulated. Furthermore, these sensors may be used in other applications, such as drug delivery, wherein the apo-GOx/dextran dissociation results in changing material properties that determine drug release in response to glucose.
Experimental Section
Materials
FITC–dextran (molecular weight 2 MDa, 0.003–0.020 mol FITC/mol of glucose), GOx (G-2133), β-D-glucose, sodium bicarbonate, dimethyl formamide, ammonium sulfate, sodium acetate buffer, and peroxidase were obtained from Sigma Chemical Co. (St Louis, MO). TRITC (Molecular Probes) was used to label apo-GOx. All reagents were used as received.
Instrumentation
A UV–vis absorbance spectrometer (Perkin-Elmer Lambda 45) was used to collect absorbance spectra and perform catalytic activity tests. The slit size (4 nm) and scanning speed (480 nm/min) were held constant throughout all the experiments. A scanning fluorescence spectrometer (QM1, Photon Technology International) was used to collect fluorescence emission spectra by exciting the sample at 488 nm. The slit size and integration time were 5 nm and 0.5 s, respectively.
Methods
Preparation of Apo-GOx
The basic procedure for apo-GOx preparation was followed as previously described.17,23 GOx (20 mg) was dissolved in 1 mL of sodium acetate buffer, and 10 mL of prepared ammonium sulfate (25% saturated, pH 1.4) solution was added. The sample was then incubated on an ice bath with stirring for 2 h. Excess (NH4)2SO4 salt was added to the solution to separate protein and FAD. The protein without FAD cofactor (apo-GOx) was precipitated by centrifuging (twice) at 4500 rpm and 4 °C. The supernatant was then removed, and sodium acetate buffer was added to redissolve and neutralize. Finally the apo-GOx was labeled with TRITC using a standard amine labeling procedure.24 Upon analysis, the conjugated TRITC–apo-GOx (TAG) was found to have 1.67 mol of apo-GOx/mol of TRITC.
Assessment of FAD Cofactor Removal
Absorbance measurements were performed on the prepared apo-GOx solution to quantify the amount of FAD, which has a characteristic peak at 300 nm. In addition, apo-GOx activity measurements were performed to observe the decrease in activity compared to native GOx. Apo-GOx activity was monitored through a standard colorimetric assay based on the oxidation of o-dianisidine through a peroxidase coupled system on an UV–vis spectrometer. During continuous stirring with a magnetic bar, apo-GOx was added to the assay to achieve a 0.17 μM concentration, and the absorbance at 500 nm was monitored as a function of time, resulting in a catalytic profile for the apo-GOx. This experiment was repeated for GOx and TAG.
Energy Transfer Experiments
All RET experiments were performed in PBS (0.01 M phosphate buffer, 2.7 mM potassium chloride, 0.137 M sodium chloride) maintained at pH 7.4. Observations of RET between FITC–dextran and TAG were performed with a fluorescence spectrometer using 488 nm excitation, with emission scans collected across the range of 500–625 nm. Initially, ~2.5 pmol of FITC–dextran was added to 1.1 mL of PBS, corresponding to 260 μmol of glucose moieties. Fluorescence spectra were then collected after each titration of TAG into the sample solution.
To determine the nature of the energy transfer (nonradiative, radiative, or both), fluorescence measurements were also performed after dilution with deionized (DI) water. Finally, to assess the relative affinity of apo-GOx for glucose and FITC–dextran, RET changes were observed during stepwise addition of aliquots of 100 mg/mL β-D-glucose solution into the sample containing FITC–dextran/TAG complexes.
Results and Discussion
Assessment of FAD Cofactor Removal
The activity measurements were performed at regular time intervals after FAD removal to ensure that apo-GOx did not regain activity with time, and similar measurements were performed with TAG and GOx to determine the relative catalytic rate of the molecules. The results of these activity measurements are shown in Figure 2. The catalytic activity of apo-GOx and TAG was observed to be three and four orders of magnitude lower than that of the native GOx, respectively. The activity of apo-GOx approximately doubled over 4 weeks, but remained two orders of magnitude lower than the native GOx. The TAG activity was observed to be less than the apo-GOx activity, likely a result of the labeling procedure employed to conjugate apo-GOx with TRITC.
Figure 2.
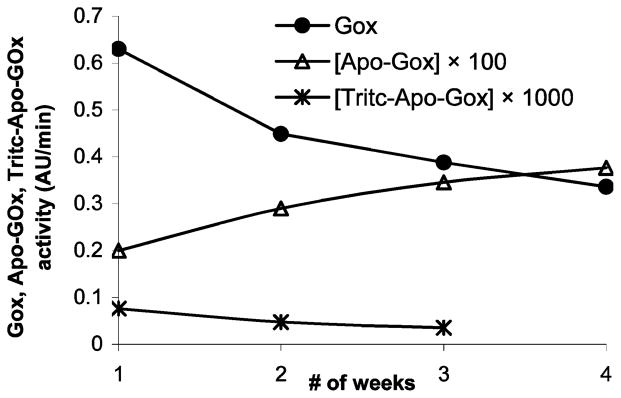
GOx, apo-GOx, and TAG activities with respect to time.
Energy Transfer Experiments
Energy transfer from FITC to TRITC was monitored by collecting fluorescence spectra after each titration of TAG into the FITC–dextran sample solution. Energy transfer was observed to increase with each titration of TAG as shown in Figure 3a. The spectra were normalized to the FITC peak at 515 nm to accentuate the changes in the TRITC fluorescence. From the data in Figure 4, it can be observed that the FITC/TRITC peak intensity ratio decreases with each addition of TAG. This does not directly indicate that energy transfer is nonradiative, because the same effect could be observed with radiative energy transfer. Fluorescence spectra for measurements made after diluting the sample are presented in Figure 3a, and the change in the FITC/TRITC peak intensity ratios for these spectra is given in Figure 4. The constant peak intensity ratio seen in Figure 4 after each addition of DI water confirms that the energy transfer between FITC–dextran and TAG is nonradiative.
Figure 3.

(a) Change in the FITC/TRITC peak intensity ratio with the titration of FITC–dextran and DI water. (b) Fluorescence spectra with the titration of FITC–dextran into TAG, DI water, and glucose into FAD/TAG complexes.
Figure 4.

Average change in the FITC/TRITC peak intensity ratio and standard deviation values with the titration of (a) TAG into the sample containing FITC–dextran and (b) DI water and (c) glucose into the sample containing FITC–dextran/TAG complexes; units for the x-axis scale are mentioned in the graph.
The glucose sensitivity of TAG/FITC–dextran complexes was demonstrated by monitoring RET changes with the addition of glucose. The energy transfer was observed to decrease regularly with the addition of glucose solution, as indicated by the increase in the FITC peak relative to that of TRITC (Figure 3b). It was found that equilibrium was reached within 1–2 min for glucose concentrations in the range of 5–60 mM, proving that displacement of dextran from apo-GOx occurs on a sufficiently short time scale.
The glucose sensitivity of the TAG/FITC–dextran samples was measured three separate times, and the average FITC/TRITC peak ratio is plotted versus glucose concentration in Figure 4. The data clearly show a decrease in the energy transfer with increasing glucose concentration up to approximately 140 mM glucose. A 30% decrease in energy transfer was observed for a concentration of 90 mM glucose. These measurements verify that the apo-GOx and dextran molecules dissociated in the presence of glucose.
The Förster distance (R0) for FITC and TRITC fluorophores has been previously measured to be 55 Å.25 Taking this value and the experimentally determined change in energy transfer efficiency, eq 1 was used to compute the change in the average distance (R) between FITC–dextran and TAG as ~11 Å with the addition of 90 mM glucose.
| (1) |
where E is the energy transfer efficiency and Fda and Fd represent the donor fluorescence intensities with and without the addition of acceptor, respectively.
Over the 0–90 mM range, the sensitivity curve shows an approximately linear increase in the FITC/TRITC fluorescence peak intensity ratio with glucose concentration. The slope of the sensitivity curve in the linear region was calculated to be 0.0163 (ratio units)/mM, which corresponds to ~5% of the total ratio change for each step of 100 mg/dL of glucose. Above the linear range, the signal appears to plateau, indicating the saturation of apo-GOx binding sites with glucose. Using measurements from multiple experiments, the dissociation constant was found to vary between 30 and 36 mM, which is comparable to the native GOx (33mM). From Figure 4 it can be seen that almost 88.35% of the FITC–dextran initially bound to apo-GOx was released, indicating that the glucose sensor is almost completely reversible. Dissociation of glucose from apo-GOx has also been observed in preliminary experiments, though this requires approximately twice as long to reach steady state as the forward reaction. This issue will be more thoroughly addressed in future studies.
As the glucose sensitivity of the apo-GOx/dextran complexes was successfully demonstrated, future work with these smart assemblies will be focused on developing nano-engineered microdevices. These sensing devices can potentially be fabricated using LbL26 self-assembly after cross-linking apo-GOx. The schematic of a proposed assembly process on thin film and spherical substrates is shown in Figure 5. On the basis of our previous findings, the ultrathin films layered on the substrates are expected to retain their binding capabilities;3 thus, apo-GOx/dextran thin films will dissociate in the presence of glucose.
Figure 5.
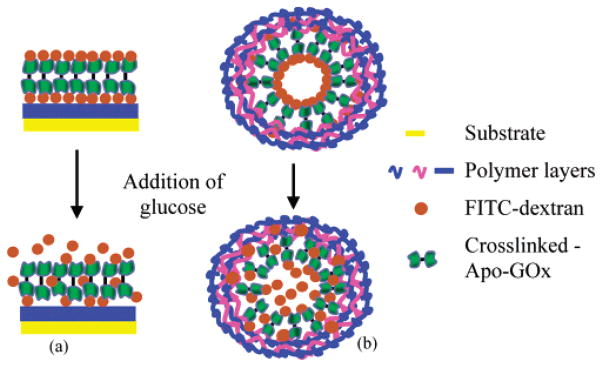
Schematic of LbL self-assembly of glucose sensitive-materials on (a) thin films and (b) spherical substrates.
Conclusions
This paper demonstrates a highly specific, sensitive, and reversible fluorescent biosensor for monitoring glucose. The sensor design is based on energy transfer between two fluorophores and on the competition between glucose and dextran for the binding sites on apo-GOx. This sensor showed a very sensitive response with the addition of glucose in the interested physiological range (0–70 mM). It was observed from fluorescence spectra that the apo-GOx/dextran complexes dissociated in the presence of glucose, manifested by an increase in the FITC peak relative to TRITC with the stepwise addition of glucose. The sensitivity of the sensor was estimated to be 0.0163 (change in ratio units)/mM with the addition of 90 mM glucose, which is three times higher compared to that of previously published data, and the repeatability of the sensor response was excellent. These sensors may eventually be deployed for in vivo glucose sensing with superior sensitivity and without the concern of toxicity.
Acknowledgments
The authors gratefully acknowledge the National Science Foundation for a Nanoscale Interdisciplinary Research Team Grant (0210298). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the view of the National Science Foundation.
References and Notes
- 1.DiCesare N, Lakowicz JR. Proc SPIE. 2002;4625:152. [Google Scholar]
- 2.Pickup J, McCartney L, Rolinski O, Birch D. Br Med J. 1999;319:1289. doi: 10.1136/bmj.319.7220.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chinnayelka S, McShane MJ. J Fluoresc. in press. [Google Scholar]
- 4.Meadows D, Schultz JS. Biotechnol Bioeng. 1991;27:1066. doi: 10.1002/bit.260371112. [DOI] [PubMed] [Google Scholar]
- 5.Meadows DL, Schultz JS. Anal Chim Acta. 1993;280:21. [Google Scholar]
- 6.Ballerstadt R, Schultz JS. Anal Chim Acta. 1997;345:203. [Google Scholar]
- 7.Ballerstadt R, Schultz JS. Anal Chem. 2000;72:4185. doi: 10.1021/ac000215r. [DOI] [PubMed] [Google Scholar]
- 8.Tolosa L, Szmacinski H, Rao G, Lakowicz JR. Anal Biochem. 250:102. doi: 10.1006/abio.1997.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tolosa L, Malak H, Rao G, Lakowicz JR. Sens Actuators, B. 1997;45:93. doi: 10.1016/S0925-4005(97)00275-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye K, Schultz JS. Anal Chem. 2003;75:3451. doi: 10.1021/ac034022q. [DOI] [PubMed] [Google Scholar]
- 11.Tolosa L, Gryczynski I, Eichorn LR, Dattelbaum JD, Castellano FN, Rao G, Lakowicz JR. Anal Biochem. 1999;267:114. doi: 10.1006/abio.1998.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge X, Tolosa L, Rao G. Anal Chem. 2004;76:1403. doi: 10.1021/ac035063p. [DOI] [PubMed] [Google Scholar]
- 13.Russell RJ, Pishko MV, Gefrides CC, McShane MJ, Cote GL. Anal Chem. 1999;71:3126. doi: 10.1021/ac990060r. [DOI] [PubMed] [Google Scholar]
- 14.McShane MJ, Rastegar S, Pishko MV, Cote GL. IEEE Trans Biomed Eng. 2000;47:624. doi: 10.1109/10.841334. [DOI] [PubMed] [Google Scholar]
- 15.McShane MJ, Russell RJ, Pishko MV, Cote GL. IEEE-EMBS Magazine. 2000;19:36. doi: 10.1109/51.887244. [DOI] [PubMed] [Google Scholar]
- 16.Ballerstadt R, Polak A, Beuhler A, Frye J. Biosens Bioelectron. 2004;19:905. doi: 10.1016/j.bios.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 17.D’Auria S, Herman P, Rossi M, Lakowicz JR. Biochem Biophys Res Commun. 1999;263:550. doi: 10.1006/bbrc.1999.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers KR. Mol Biotechnol. 2000;14:109. doi: 10.1385/MB:14:2:109. [DOI] [PubMed] [Google Scholar]
- 19.Birch DJS, Rolinski O. Res Chem Intermed. 2001;27:425. [Google Scholar]
- 20.Willner I, Heleg-Shabtai V, Blonder R, Katz E, Tao G, Bückmann AF, Heller A. J Am Chem Soc. 1996;118:10321. [Google Scholar]
- 21.Xiao Y, Patolsky F, Katz E, Hainfeld JF, Willner I. Science. 2003;299:1877. doi: 10.1126/science.1080664. [DOI] [PubMed] [Google Scholar]
- 22.D’Auria S, Cesare ND, Gryczynski Z, Gryczynski I, Rossi M, Lakowicz JR. Biochem Biophys Res Commun. 2000;274:727. doi: 10.1006/bbrc.2000.3172. [DOI] [PubMed] [Google Scholar]
- 23.Swoboda BEP. Biochim Biophys Acta. 1969;175:365. doi: 10.1016/0005-2795(69)90014-2. [DOI] [PubMed] [Google Scholar]
- 24.Svensson HP, Kadow JF, Vrudhula VM, Wallace PM, Senter PD. Bioconjugate Chem. 1992;3:176. doi: 10.1021/bc00014a013. [DOI] [PubMed] [Google Scholar]
- 25.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Press; New York: 1999. [Google Scholar]
- 26.Lvov YM, Ariga JK, Ichinose I, Kunitake T. J Chem Soc, Chem Commun. 1995:2313. [Google Scholar]