

Abstract
Background
Statins are potent inhibitors of cholesterol biosynthesis and are clinically beneficial in preventing cardiovascular diseases, however, the therapeutic utility of these drugs is limited by myotoxicity. Here, we explored the mechanism of statin-mediated activation of ERK5 in the human endothelium with the goal of identifying compounds that confer endothelial protection but are nontoxic to muscle.
Methods
An ERK5-one hybrid luciferase reporter transfected into COS-7 cells with pharmacological and molecular manipulations dissected the signaling pathway leading to statin activation of ERK5. qRT-PCR of HUVEC cells documented the transcriptional activation of endothelial-protective genes. Lastly, morphological and cellular ATP analysis, and induction of atrogin-1 in C2C12 myotubes were used to assess statin-induced myopathy.
Results
Statin activation of ERK5 is dependent on the cellular reduction of GGPPs. Furthermore, we found that the combination of FTI-277 (inhibitor of farnesyl transferase) and GGTI-298 (inhibitor of geranylgeranyl transferase I) mimicked the statin-mediated activation of ERK5. FTI-277 and GGTI-298 together recapitulated the beneficial effects of statins by transcriptionally upregulating anti-inflammatory mediators such as eNOS, THBD, and KLF2. Finally, C2C12 skeletal myotubes treated with both FTI-277 and GGTI-298 evoked less morphological and cellular changes recognized as biomarkers of statin-associated myopathy.
Conclusions
Statin-induced endothelial protection and myopathy are mediated by distinct metabolic intermediates and co-inhibition of farnesyl transferase and geranylgeranyl transferase I confer endothelial protection without myopathy.
General Significance
The combinatorial FTI-277 and GGTI-298 drug regimen provides a promising alternative avenue for endothelial protection without myopathy.
Keywords: Pitavastatin, ERK5, GGTI-298, FTI-277, myopathy, endothelial protection
1. Introduction
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, or statins, can substantially reduce cardiovascular morbidity and mortality [1-4]. Statins mechanistically reduce low-density lipoprotein (LDL) by inhibiting the conversion of HMG-CoA to mevalonate, the rate-limiting step in the biosynthesis of cholesterol [5]. Although originally prescribed to reduce LDL, it has been suggested that the therapeutic efficacy of statins in preventing cardiovascular disorders may be unrelated to the decrease in cholesterol levels [6, 7]. While a number of different signaling pathways have been shown to contribute to these pleiotropic effects of statins [8], one line of evidence suggests that activation of the mitogen-activated protein kinase (MAPK) ERK5 and its downstream targets may be responsible for the endothelial protective effects of these drugs [9].
Analogous to the related ERK1/2 MAPKs, ERK5 activation requires a dual phosphorylation of a TEY consensus motif by an upstream mitogen-activated protein kinase kinase (MAPKK), MEK5 [10]. MEK5 phosphorylates ERK5, which translocates to the nucleus and stimulates transcription through direct phosphorylation of transcription factors such as Myocyte Enhancing Factor (MEF) 2C and Kruppel-Like Factor (KLF) 2 [11]. Additionally, ERK5 can directly influence transcriptional activity (transactivation) through a poorly defined mechanism that requires a unique C-terminal domain [12]. Knockout studies have demonstrated an indispensable role of the ERK5 gene in the development of the cardiovascular system and the maintenance of vascular integrity in mature blood vessels [13, 14]. In mature animals the MEK5/ERK5 pathway mediates shear stress-dependent repression of inflammatory responses via interaction with PPARγ1 [15] and additionally regulates flow-mediated expression of KLF2, thus controlling anti-thrombotic and anti-inflammatory responses to laminar flow [16-19].
Statins have been shown to activate the phosphorylation of ERK5 [9]. This activation of ERK5 leads to the de novo synthesis of both KLF2 and KLF4 [9, 19]. Statin-mediated induction of KLF2 through ERK5 has been demonstrated to drive the expression of endothelial nitric oxide synthase (eNOS) and thrombomodulin (THBD), important enzymes influencing the properties of the endothelium [9, 19, 20].
A common adverse effect of statin therapy is the muscle myalgia associated with long-term use [21]. Multiple lines of evidence suggest that the depletion of isoprenoid precursors is implicated in the etiology of this adverse drug response [21, 22]. Furthermore, the myotoxic effects of statins on muscle fibers have recently been attributed to the prenylation of protein substrates targeted by geranylgeranyl transferase II [23].
In light of these latest results, we sought to determine the signaling responsible for statin-mediated myotoxicity in C2C12 myofibers—an established cellular model of muscle myopathy. First, we found that co-administration of inhibitors of farnesyl transferase I, (e.g., FTI-277) and geranylgeranyl transferase I (e.g., GGTI-298) activated ERK5 and its downstream anti-inflammatory targets, such as eNOS, THBD, and KLF2. Second, in C2C12 myofibers, these compounds were nontoxic in comparison to pitavastatin, as measured by the lack of cellular ATP depletion and atrogin-1 transcriptional activation. Together, these results demonstrated that co-administration of FTI-277 and GGTI-298 provided an alternative pharmacological strategy for activating ERK5 that may be effective as a therapeutic tool for the treatment of vascular diseases but devoid of myotoxicity.
2. Materials and Methods
2.1 Reagents
Statins were obtained from various sources: atorvastatin (Sigma-Aldrich, St. Louis, MO, Cat. No. PZ0001), cerivastatin (American Radiolabeled Chemicals, Inc., St. Louis, MO, Cat. No. ARCD 0403), fluvastatin (American Radiolabeled Chemicals, Inc., St. Louis, MO, Cat. No. ARCD 0405), lovastatin (American Radiolabeled Chemicals, Inc., St. Louis, MO, Cat. No. ARCD 0242), mevastatin (Sigma-Aldrich, St. Louis, MO, Cat. No. M2537), pitavastatin (kindly provided by Kowa Pharmaceutical, Inc., Japan), pravastatin (Sigma-Aldrich, St. Louis, MO, Cat. No. P4498), rosuvastatin (Selleck Chemicals, Houston, TX, Cat. No. S2169), simvastatin (Sigma-Aldrich, St. Louis, MO, Cat. No. S6196). Intermediates in the mevalonate pathway were purchased from Sigma-Aldrich: (R)-Mevalonic acid sodium salt (Cat. No. 41288), farnesyl pyrophosphate (FPP) (Cat. No. F6892), and geranylgeranyl pyrophosphate (GGPP) (Cat. No. G6025). Cholesteryl hemi-succinate (CHEMS) was purchased from MP Biomedical (Cat. No. ICN10137905). Inhibitors of farnesyl transferase (FTI-277) (Cat. No. F9803) and geranylgeranyl transferase I (GGTI-298) (Cat. No. G5169) were also purchased from Sigma-Aldrich). An inhibitor of geranylgeranyl transferase II (2-(3-pyridinyl)-1-hydroxyethylidene-1,1-phosphonocarboxylic acid or 3-PEHPC) was donated by the laboratory of Dr. Kirill Alexandrov from the University of Queensland, St. Lucia, Australia. The MEK5 inhibitor BIX 02189 (Cat. No. S1531) was obtained from Selleck Chemicals, Houston, TX.
2.2. ERK5 expression plasmids
Mus musculus ERK5 cDNA (NM_011841) cloned into a pBIND vector (Promega, Madison, WI, Cat. No. E2440) between the BamH I and Not I sites with a 22 amino acid linker separating the Gal4 DNA Binding Domain and the ERK5 start methionine (pBIND-ERK5) was provided by Dr. Junichi Abe (University of Rochester, New York). The ERK5 catalytically dead mutant (KM mutant) was created by replacing lysine 84 with methionine as reported previously [24] and the non-phosphorylatable mutant of ERK5 was created by mutating the TEY motif to AEF—both by site-directed mutagenesis accomplished by the two-step PCR method. The pBIND vector co-expresses Renilla luciferase that can be used for normalization of transfection efficiency. Constitutively active (CA) -MEK5 and dominant negative (DN) -MEK5 subcloned into the pCI/neo (Promega, Madison, WI, Cat No. E1841) eukaryotic expression vector were reported previously by Cameron et al. [25].
2.3. Cell culture
Clonally selected COS-7 cells stably expressing pG5luc Vector (Promega, Madison, WI, Cat. No. E2440) were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (v/v) fetal bovine serum and 100 U/ml penicillin/100 μg/mL streptomycin (Life Technologies Inc., Carlsbad, CA) (i.e. growth media) with the addition of 250 μg/mL hygromycin. Mouse C2C12 myoblast cells were obtained from ATCC (ATCC, Manassas, VA, Cat. No. CRL-1772) and maintained in a similar growth medium, except without hygromycin. Human umbilical vein endothelial cells (HUVEC) (Promocell, Heidelberg, Germany, Cat. No. C-12200) were grown in Endothelial Cell Growth Medium (Promocell, Heidelberg, Germany, Cat. No. C-22110) with the addition of 1% antibiotic/antimycotic (Life Technologies Inc., Carlsbad, CA, Cat. No. 15240-096). Experiments requiring transfection of plasmids were done using COS-7 cells while endogenous signaling was studied in the more physiologically relevant HUVEC or C2C12 cells. All cells were kept in a humidified incubator at 5% CO2 and 37°C.
2.4. Transient transfection
All endotoxin-free plasmids used for transfection were prepared with PureYield™ Plasmid Midiprep System (Promega, Madison, WI, Cat. No. A2492). COS-7 cells were seeded at 2.0 × 105 cells per well in a 6-well plate 24 h prior to transfection. All transfections of COS-7 cells were performed using Lipofectamine 2000 (Life Technologies Inc., Carlsbad, CA, Cat. No. 11668019) with 1 mg DNA following the manufacturer's recommendation. The ratio of Lipofectamine reagents to DNA concentration was fixed at 3.3 μL to 1 μg DNA. Twenty-four hours after transfection media were switched to DMEM containing 100 U/ml penicillin and 100 μg/mL streptomycin (i.e., serum-free media) with the indicated drug treatment regimen. Transfection of siRNA against ERK5 was performed similarly using Lipofectamine RNAi/Max Reagent (Life Technologies Inc., Carlsbad, CA, Cat. No. 13778030) using the manufacturer's recommendation. 48 hours post-transfection cells were stimulated with drug treatments and harvested 6 hours post-drug treatment for qPCR analyses.
2.5. Luciferase assays
COS-7 cells stably expressing the pGL4.31 Vector (luc2P containing the protein degradation PEST sequence resulting in a destabilized reporter protein with reduced half-life) were transfected with pBIND-ERK5 as described above. Twenty-four hours after transfection, cells were trypsinized, counted and seeded into 96-well plates (at ∼5,000 cells/well density) containing the indicated concentrations of drug or combination of drugs. Each drug treatment condition was performed in triplicate. Twenty-four hours after drug treatments, luciferase assays were carried out using the Dual-Glo® Luciferase Assay System (Promega, Madison, WI, Cat. No. E2920). Briefly, Dual-Glo® Luciferase Substrate (100 μL) was added to each well and incubated at RT for 10 min before luminescence measurements were taken using the BioTek Synergy2 Microplate Reader. Subsequently, Stop & GLO® Substrate/Buffer (100 μL) was added to each well and the luminescence was measured after 10 min incubation at RT. The firefly/Renilla luciferase ratio was calculated individually from each replicate well and averaged. The data are expressed as the fold induction relative to the equivalent DMSO only control. Concentrations of metabolites used for these experiments follow those used by others [23].
The saturation curve for pitavastatin was determined by first normalizing the fold luciferase induction against 0.1% DMSO as negative control and the highest concentration (100 μM) of pitavastatin as 100%. The EC50 value for pitavastatin was calculated by taking the half max of the sigmoidal dose response determined from GraphPad Prism (San Diego, CA). Saturation curves for FTI-277 and GGTI-298 were plotted similarly, except the dose response curves for each were determined in the presence of a fixed concentration (10 μM) of the opposite compound.
2.6. Western blot analyses
HUVEC subjected to a drug treatment regimen were lysed with RIPA (10 mM Tris-Cl pH 7.5, 50 mM NaCl, 1 mM sodium orthovanadate, 30 mM sodium pyrophosphate, 50 mM NaF, 1% NP40, 0.1% SDS, 1 mM PMSF, 1% Triton X-100, and 0.5% sodium deoxycholate) and sonicated for 5 s on 10% power (Fisher Scientific microtip sonicator, model 150I) to obtain total cell lysates. Cell lysates were cleared by centrifugation at a maximum speed on a bench top centrifuge (10 min at 4°C). Protein concentrations of the cleared supernatant were determined by micro BCA Assay (Thermo Fisher, Waltham, MA, Cat. No. 23223) and 15-20 μg of each sample were separated on a 10% SDS polyacrylamide gel at 150 V. Proteins were transferred to a nitrocellulose membrane at a constant amperage of 0.5 for 2 h at 4°C and subsequently blocked with 5% nonfat dry milk for 1 h at RT. The membrane was probed with 1:1,000 dilution of rabbit anti-ERK5 antibody (EMD Millipore, Billerica, MA, Cat. No. 07-039) in 1% nonfat dry milk/Tris Buffered Saline containing 0.2% Tween-20 (TBST) overnight at 4°C (approximately18 h). The following morning, the membrane was washed for 15 min × 3 in TBST followed by incubation with HRP-conjugated secondary antibody in 1% milk/TBST at RT for 1 h. After rinsing 3 times with TBST, each for 15 min, proteins were detected by the enhanced chemiluminescence method (Western Lightning chemiluminescence reagent plus, PerkinElmer, Waltham, MA, Cat. No. NEL105001EA) and imaged with the Bio-Rad ChemiDoc XRS+ (Bio-Rad, Hercules, CA). Quantitation of western results were performed using NIH Image J and the data are expressed as percent of p-ERK5 relative to total ERK5 proteins.
2.7. Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from either HUVEC or C2C12 myotubes using the IBI Scientific Total RNA Mini Kit (IBI Scientific, Peosta, IA, Cat. No. IB47321) according to the manufacturer's instructions. cDNA was reverse transcribed from 1-5 μg of total RNA using the Applied Biosystems cDNA Synthesis Kit (Life Technologies Inc., Carlsbad, CA, Cat. No. 4368814). Templates were amplified in a 25 μL reaction mixture containing 10 μL of Eva Green qPCR Mastermix (MIDSCI, Valley Park, MO, Cat. No. BEQPCR-LR), 0.2 μM of a mixture of forward and reverse primers, and 1 μL of cDNA reaction solution. Real-time polymerase chain reaction analyses were performed using the Stratagene MX3000P instrument with the following amplification conditions: 95°C for 10 min, 40 cycles of 15 s at 95°C and 60 s at 60°C. PCR specificity was confirmed by melting curve analysis after the amplification. Relative expression was quantified using the ΔΔCt method, in which GAPDH was used as the internal standard. Validated qPCR primers were obtained from Qiagen SABiosciences, (Valencia, CA): human GAPDH (PPH00150F), human KLF2 (PPH02566A), human NOS3 (PPH01298F), human THBD (PPH02576A), mouse GAPDH (PPM02946E) and mouse KLF4 (PPM25088B).
2.8. C2C12 myotubes differentiation conditions
C2C12 cultures were expanded in DMEM supplemented with 10% (v/v) fetal bovine serum (Invitrogen) and 100 U/ml penicillin/100 μg/mL streptomycin at 5% CO2 and 37°C. For C2C12 differentiation, 5 × 104 cells were seeded in 6-well plates and cultured in growth media until reaching 70-80% confluence. Cell media were then replaced with DMEM supplemented with 2% (v/v) horse serum (Life Technologies, Inc.) containing 100 U/ml penicillin and 100 μg/mL streptomycin (differentiation medium) [26]. Cells were fed with fresh differentiation medium every 24 h, and the myocyte fusion into myotubes was monitored daily using an inverted microscope. On day five 10 μM drugs in differentiation medium were administered for 36 hours and assayed.
2.9. Immunocytochemistry
36 hours after drug treatment cells were washed in PBS and fixed in 4% paraformaldehyde for 40 minutes and immuno-stained with anti-myosin heavy chain (MHC) antibody (1:50, Clone MF20, #MAB4470 R&D Systems, Minneapolis, MN) in PBST (PBS with 0.2% Triton-100) for 2 hrs followed by an Alexa-488-conjugated secondary antibody for 1 hr, both at room temperature. Nuclear staining was performed using 100 μg/mL Hoechst in PBST for 10 minutes, and a final wash in PBS.
2.10. Quantitation of myotubes diameters
Fluorescence microscopic images (20× objective lens) were captured at the indicated time-points and analyzed offline. For quantification of myotube diameter, 10 cross-sectional measurements per myotube were manually obtained using NIH Image J and approximately 10 myotubes per image were collected. For each drug condition, an average of 30 images totaling 300 myotubes were quantified by an experimentally blinded observer. * P value < 0.05, one-way ANOVA followed by Dunnett's multiple comparisons test, n=3. Representative images for each drug condition were taken and are shown in Figure 5A.
Fig. 5.
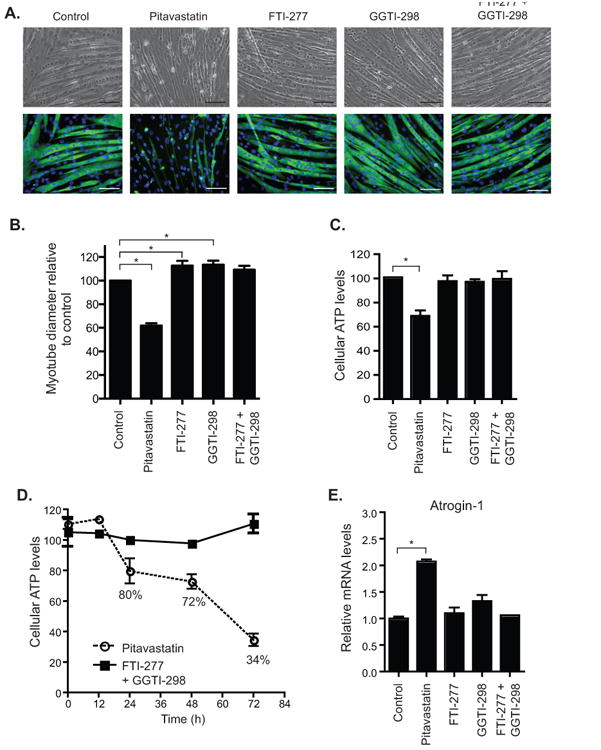
FTI-277 and GGTI-298 in combination did not produce a myopathic phenotype in C2C12 myotubes. A. Bright field and anti-MHC stained images of C2C12 myotubes after treatment with indicated compound or combination of compounds for 36 h: control (0.2% DMSO), pitavastatin (10 μM), FTI-277 (10 μM), GGTI-298 (10 μM). Scale bars represent 100 μm. B. Myotube diameters were determined from the measurements of 300 myotubes per condition and statistical analyses were performed by one-way analysis of variance followed by Dunnett's multiple comparison test. Myotube diameters are expressed as percent relative to control where the mean myotube diameter, 21.9 ± 0.6 micrometers, was set as 100%. * P< 0.05. C. Pitavastatin caused a marked decrease in cellular ATP levels while FTI-277 and GGTI-298 combinations were comparable to controls. Data are expressed as percent of control; bar graphs were obtained from the average of 3 separate experiments, each performed in triplicate. * P< 0.05, t-test. D. Time-dependent decrease in cellular ATP levels measured in the presence of FTI-277 and GGTI-298 combination as compared to pitavastatin. E. Pitavastatin produced a marked increase in the levels of atrogin-1 mRNA, a cellular biomarker of statin-associated myopathy while FTI-277 and GGTI-298 together failed to induce a significant increase in atrogin-1. qRT-PCR results were obtained from the average of 3 separate experiments, each performed in duplicate. All values are mean ± S.E.M. * P value < 0.05, t-test.
2.11. Cell-based assay for measuring ATP levels
C2C12 cells were seeded at 5000-10,000 cells/well in 96-well plates. Differentiation occurred over 4-6 days as described earlier. Myotubes were treated with 10 μM drugs (pitavastatin, FTI-277, GGTI-298 or FTI-277 in combination with GGTI-298, or 0.2% DMSO as a control). After incubation for the indicated times, Cell Titer-Glo reagent (100 μL) was added to each well without removing the cell-culture medium. Luminescence was measured after 10 min incubation using the BioTek Synergy2 Microplate Reader.
2.12. Statistics
Data were obtained from the luciferase/Renilla signal reporter assay for ERK5 activation and qRT-PCR quantitation of mRNA levels were assayed in triplicate from at least three biological experiments. Initial ANOVA demonstrated the triplicate assays to be a significant source of variation. Therefore, the grand mean was determined as the overall mean including all data from the triplicates and biological replicates, and the standard error of the means was calculated from the average of the three means. Statistical significance of the means was evaluated by the student t-test corrected for multiple comparisons (Bonferroni adjustment) where appropriate and P < 0.05 was considered significant.
3. Results
3.1. Statin activation of ERK5
Previously, it was reported that pitavastatin activates ERK5 and reduces endothelial dysfunction [27]. As an extension of this work, we screened a series of commercially available statins using a reporter assay system with a Gal4 DNA binding domain fused to Mus musculus ERK5 (Fig. 1A). At 1 μM, cerivastatin was the most effective of the tested compounds (with a 4.2 ± 0.7 fold induction), while fluvastatin, lovastatin, pitavastatin, and simvastatin showed a statistically significant but less dramatic ERK5 activation (Fig. 1B), which may be explained by their respective affinities at HMG-CoA reductase [5]. At 10 μM, cerivastatin, fluvastatin, lovastatin, mevastatin, pitavastatin, and simvastatin induced ERK5 activation by at least two-fold (Fig. 1B), while atorvastatin, pravastatin, and rosuvastatin did not. At 100 μM, only atorvastatin induction of the GAL-4 reporter was greater than the lower concentration of 10 μM. In fact, 100 μM cerivastatin, fluvastatin, and simvastatin were toxic to the cells (Supplementary Fig. 1) providing an explanation on why the reporter induction was reduced (denoted by # in Fig. 1B) at the higher concentration. Pravastatin and rosuvastatin with known low cell permeability failed to induce the activation of ERK5 even at 100 μM which may be due to its requirement of membrane transporters for its cellular uptake [28]. We found that the EC50 of pitavastatin activation of ERK5 was 1.3 ± 0.2 μM (Fig. 1C). We chose pitavastatin, a statin recently approved for clinical use in the United States, at a concentration of 10 μM as the drug and concentration to elucidate the signaling of statin-mediated ERK5 activation.
Fig. 1.
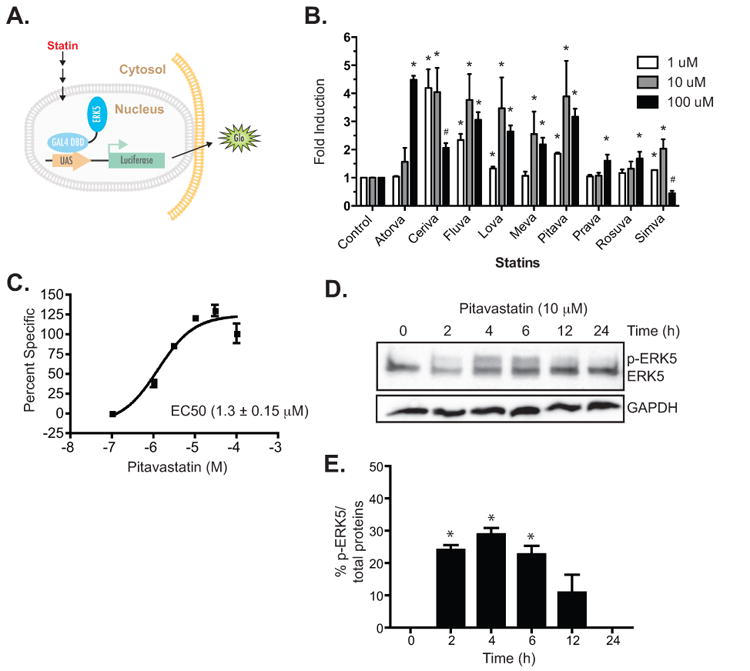
Statin-mediated activation of ERK5. A. A schematic representation of the reporter assay. The Gal4 DNA binding domain - ERK5 fusion was transfected into COS-7 cells and used to drive the expression of firefly luciferase as a measure of ERK5 activation. B. Activation of ERK5 by a series of statins at different concentrations (1 μM, 10 μM, and 100 μM). Data are first normalized to protein expression by dividing firefly luciferase signals over Renilla and the data are plotted as fold induction relative to control (0.1% DMSO) as 1. Data are representative of at least 3 experiments, each performed in triplicate. * P value < 0.05, t-test. # indicates statistically significant reduction in the reporter activity at 100 μM compared to 10 μM, P value < 0.05, t-test, n=3. C. A representative dose response curve of pitavastatin-mediated activation of ERK5. Values are plotted as percent specific and EC50 was calculated as the half max ± SEM. EC50 from three separate experiments for pitavastatin dose response is 1.3 ± 0.15 μM. D. Time course of pitavastatin-mediated phosphorylation of endogenous ERK5 in HUVEC as detected by the pan-ERK5 antibody. The mobility-shifted slower migrating band is considered the p-ERK5 while the lower faster migrating band is the non-phosphorylated ERK5. E. The level of ERK5 phosphorylation was quantitated by dividing the p-ERK5 band over total protein (p-ERK5 band + ERK5) and expressed as percentage of total ERK5 protein. The summary bar diagram shows the mean ± SEM of densitometric quantitation from 3 independent experiments. * P value < 0.05, t-test.
3.2. Role of MEK5 in statin activation of ERK5
We next determined the phosphorylation status of endogenous ERK5 in the presence of pitavastatin in HUVEC cells. We found that pitavastatin induced transient ERK5 phosphorylation over the course of 24 h and maximized at approximately 4 h (Fig. 1D and 1E) in agreement with the previously reported statin-induced phosphorylation of ERK5 [9]. The increase in phosphorylation of ERK5 is apparent by the presence of a higher molecular weight mobility-shifted band on western blots (Fig. 1D and 1E). According to the canonical signaling scheme MEK5 an upstream member of the MAPKK family, phosphorylates ERK5 on the TEY motif, leading to N-terminal kinase activation and autophosphorylation of its C-terminal domain [11]. Hence, ERK5 TEY phosphorylation is synonymous with both functional N-terminal kinase activity and transactivation of the C-terminal tail. It should be noted that the mobility shift of ERK5 due to multiple sites of phosphorylation often appear smeared with no single dominant mobility-shifted species. Pharmacological inhibition of MEK5 with BIX02189 (10 μM) reduced the phosphorylation of ERK5 induced by pitavastatin (Fig. 2A and 2B), however, residual statin-induced pERK5 not blocked by BIX02189 remained. Pitavastatin in the presence of BIX02189 (10 μM) reduced the pitavastatin-mediated transactivation of the ERK5 reporter (Fig. 2C). Cells transfected with the EYFP-ERK5 plasmid demonstrated pitavastatin-induced nuclear translocation blocked by BIX02189 (Fig. 2D). Next we used functionally inactive mutants of the ERK5 to confirm the pharmacological results observed with the small molecule MEK5 inhibitor. As shown in Fig. 2E, while pitavastatin was able to activate wild-type ERK5, it failed to activate the kinase-dead KM-mutant [29]. Expression of the DN-MEK5 also reduced the pitavastatin-induced ERK5 transactivation reporter readout. Together, these data demonstrate that the pitavastatin-mediated activation of ERK5 is dependent on ERK5 kinase activity with a likely role of the upstream MEK5.
Fig. 2.
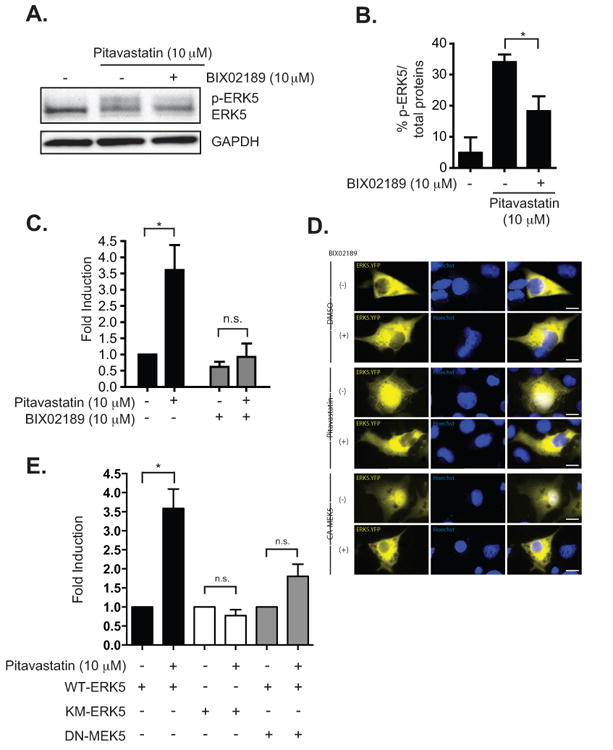
Statin-mediated activation of ERK5 in COS-7 cells requires MEK5. A. A representative western blot of mobility-shifted ERK5 after drug treatments. Pitavastatin (10 μM) -mediated phosphorylation of ERK5 is inhibited by the co-treatment with a MEK5 specific inhibitor, BIX02189 (10 μM). B. Relative levels of p-ERK5 compare to control after drug treatments. The level of ERK5 phosphorylation was quantitated by dividing the p-ERK5 band over total protein (p-ERK5 band + ERK5) and expressed as percentage of total ERK5 protein. The summary bar diagram shows the mean ± SEM of densitometric quantitation from 3 independent experiments. C. Activation of ERK5 by pitavastatin, as assessed by the wt ERK5-pBIND luciferase reporter assay, was inhibited by the co-treatment with BIX02189 (10 μM). Data are normalized by dividing firefly luciferase over Renilla activity and presented as fold induction relative to the control untreated condition. D. An ERK5.YFP plasmid encoding a fusion protein was transfected and the cells treated with DMSO (top two rows) or pitavastatin (10 μM) (middle two rows) without or with BIX02189 (10 μM). Fluorescent images were captured to localize the ERK5.YFP and overlaid with the nucleus stained with the Hoechst stain. The lower two rows is a parallel experiment in cells co-transfected with the CA-MEK5 plasmid to serve as a positive control for ERK5 activation. Calibration bar=15 μm. E. Pitavastatin failed to activate the kinase dead KM-ERK5 (i.e., pBIND reporter with the KM-ERK5 instead of wt ERK5). The co-expression of DN-MEK5 with wt ERK5-pBIND reporter also prevented the pitavastatin-mediated increase in the reporter activity. For panels B,C, and E, error bars were obtained from an average of 3 separate experiments performed in triplicate. * P value < 0.05, t-test.
3.3. ERK5 activation by statin is caused by depletion of geranylgeranyl pyrophosphate
We next explored the upstream mechanism of statin-mediated activation of ERK5. Statins inhibit the metabolic conversion of HMG-CoA to mevalonic acid, a precursor to the biosynthesis of cholesterol, squalene, Coenzyme Q10 (CoQ10), and isoprenoids such as FPP and GGPP (Wagner et al., 2011) (Fig. 3A). Since the inhibition of HMG-CoA reductase leads to a drastic reduction of cholesterol, CoQ10, and isoprenoids, the pitavastatin-mediated activation of ERK5 may be an indirect consequence of the dampening of signaling networks that require physiological levels of these metabolic intermediates. For example, a depletion of isoprenoids leads to a reduction in post-translational modifications and trafficking of small GTPases including the Ras, Rac/Rho and Rab families of GTPases (for a review, refer to Buhaescu and Izzedine [30], all of which have important signaling functions. To determine whether the reduction of mevalonic acid by pitavastatin is responsible for the activation of ERK5, COS-7 cells stably expressing pGL5-luc reporter vector and transfected with Gal4-ERK5 were treated with pitavastatin and various intermediates in the HMG-CoA pathway. As shown in Fig. 3B, both mevalonate and GGPP, at 100 μM and 10 μM respectively, were able to reverse the pitavastatin-mediated transactivation of ERK5. On the contrary, FPP, CHEMS (an amphipathic ester derivative of cholesterol with physicochemical properties that allow it to incorporate into cellular membranes [31]), and CoQ10, all at 10 μM, failed to reverse the pitavastatin-mediated activation of ERK5. The inability of FPP, by itself, to reverse the effects of pitavastatin on ERK5 activation may be due to its faster conversion to cholesterol and CoQ10 [32, 33]. However, the reversal of pitavastatin-mediated ERK5 activation by GGPP points to the prenylation of either the Rac/Rho or Rab family of GTPases as possible downstream signaling molecule(s) that affect the activity of ERK5.
Fig. 3.

Statin-mediated signaling leading to ERK5 activation in COS-7 cells. A. A schematic diagram depicting metabolic pathways downstream of HMG-CoA leading to the synthesis of cholesterol, CoQ10 and isoprenoids (FPP and GGPP) based on a scheme from [23]. Chemical inhibitors of steps of the pathway experimentally manipulated are denoted in red. Farnesyl-PP, farnesyl pyrophosphate; geranylgeranyl-PP, geranylgeranyl pyrophosphate; FT, farnesyl transferase; GGT-I, geranylgeranyl transferase I; GGT-II, geranylgeranyl transferase II; GGTI-298, GGT-I inhibitor; 3-PEHPC, GGT-II inhibitor. B. Statin-mediated activation of ERK5 was inhibited by mevalonate and partly by GGPP but not FPP, CoQ10, and CHEMS. C. The suppression of ERK5 activation by GGPP can be reversed by a co-treatment with an inhibitor of GGT-I (GGTI-298) but not by an inhibitor of GGT-II (3-PEHP). D. Activation of ERK5 can be recapitulated by inhibition of both farnesyl transferase with FTI-277 and geranylgeranyl transferase I with GGTI-298. Note that this is not statistically significant but a trend towards activation of the reporter by GGTI-298 alone. Data are representative of at least 3 experiments, each experiment performed in triplicate. For B, C, and D, * P value < 0.05, t-test; n.s., nonsignificant E. A representative dose response curve of GGTI-298-induced ERK5 activation in the presence of FTI-277. Shown is the EC50 of GGTI-298 from the representative dose response curve; the EC50 from three separate experiments, each experiment performed in triplicate, is 8.6 ± 0.15 μM. F. As in E but with a representative dose response curve of FTI-277 in the presence of GGTI-298. Shown is the EC50 of FTI-277 from the representative dose response curve; the EC50 from three separate experiments, each experiment performed in triplicate, is 16.0 ± 0.2 μM.
3.4. Inhibition of farnesyl transferase by FTI-277 and geranylgeranyl transferase I by GGTI-298 mimic the activation of ERK5 by pitavastatin
Geranylgeranylation by geranylgeranyl transferase I has been demonstrated for the Rac/Rho family of GTPases, while the Rab family of GTPases requires geranylgeranyl transferase II [34]. To determine whether one or both pathways are important for ERK5 activation, we treated the ERK5-activation reporter COS-7 cells with 10 μM pitavastatin and 10 μM GGPP, a condition which reversed ERK5 activation. Simultaneously, we added specific small-molecule inhibitors of either GGT-I or GGT-II; the inability to transfer geranylgeranyl groups to the relevant protein(s) should therefore abolish GGPP's reversal of statin-induced ERK5 activation. We found that inhibition of GGT-II by 3-PEHPC had no effect on the reporter readout, whereas inhibition of GGT-I by GGTI-298 abolished the effect of GGPP, reestablishing ERK5 activation similar to pitavastatin treatment alone (Fig. 3C). These results suggest that blockage of a Rac/Rho/Cdc42 prenylation by pitavastatin plays a role in the activation of ERK5. While we could isolate the GGT-I pathway as partly responsible for the statin-mediated activation of ERK5 in the presence of pitavastatin, application of Rho and Rac inhibitors by themselves in the absence of pitavastatin, however, did not seem to have any effect on ERK5 activation (data not shown) as was previously reported [35].
As expected, the inhibition of GGT- I with GGTI-298 was necessary but not sufficient for ERK5 activation (Fig. 3D) as GGTI-298 by itself did not lead to full ERK5 activation. In order to determine whether prenylation by farnesyl transferase and geranylgeranyl transferase together leads to the pitavastatin-mediated maximal activation of ERK5, we inhibited both farnesyl transferase and geranylgeranyl transferase with their specific pharmacological inhibitors. The combined inhibition of both FT and GGT-I with FTI-277 and GGTI-298, respectively, led to ERK5 activation as measured by the induction of firefly luciferase (Fig. 3D). Since the presence of one compound was insufficient to activate ERK5 (Fig. 3D), we determined the EC50 values for each of the compounds while holding the concentration of the other drug constant. As shown in Fig. 3E, the average EC50 values for GGTI-298 in the presence of a fixed concentration of FTI-277 (10 μM) was 8.6 ± 0.2 μM. Similarly, the average EC50 of FTI-277 in the presence of GGTI-298 (10 μM) was 16.0 ± 0.2 μM (Fig. 3F). Both of the EC50 values for FTI-277 and GGTI-298 are within the effective biologically-relevant concentrations determined in other systems (4 μM for GGTI-298 and 10 μM for FTI-277) [36]. The activation of endogenous ERK5 by FTI-277 and GGTI-298 was confirmed by the time-dependent increase in the mobility-shifted ERK5 in HUVEC as shown by Western analyses with the pan-ERK5 antibody (Fig. 4A and 4B).
Fig. 4.
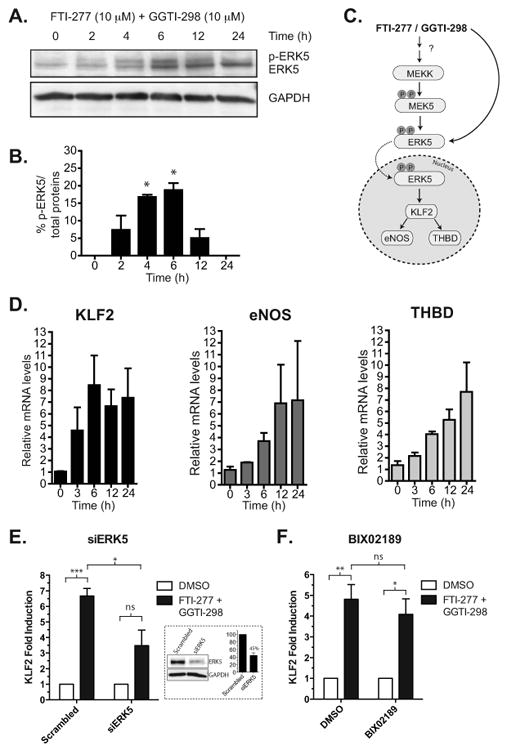
Activation of ERK5 and its downstream targets in the presence of both FTI-277 and GGTI-298 in HUVEC. A. FTI-277 and GGTI-298 (at 10 μM each) in combination increases the levels of p-ERK5 and maximizes at 4 hrs as detected by the pan-ERK5 antibody. B. The level of ERK5 phosphorylation was quantitated by dividing the p-ERK5 band over total protein (p-ERK5 band + ERK5) and expressed as the percentage of total ERK5 protein. The summary bar diagram shows the mean ± SEM of densitometric quantitation from 3 independent experiments. * P value < 0.05, t-test. C. Diagram of the ERK5 signaling pathway. FTI-277 and GGTI-298 could activate the MEKK upstream of MEK5 or directly activate MEK5. Alternatively drug combination could result in alternative direct ERK phosphorylation independent from canonical MAPK signaling (see Discussion). Once activated, ERK5 transcriptionally upregulates eNOS, KLF2, and THBD. D. FTI-277 and GGTI-298 together induced the transcriptional upregulation of known ERK5 targets, including KLF2, eNOS, and THBD, in a time dependent manner. E. siRNA knock down of ERK5 reduces FTI-2777 and GGTI-298 dual drug-induction of KLF2. Cells were transfected with 10 nM scramble or ERK5 siRNA and after 48 hrs treated with drugs or DMSO for an additional 6 hrs. KLF2 mRNA level was assessed by qRT-PCR. Mean ± SEM, ns, nonsignificant; *P<0.05; ***P<0.0005, t-test, n=3. The inset shows a Western blot confirming ERK5 protein knockdown (average of 55% knockdown by densitometry from n=3 experiments). F. Same as in E but with and without BIX02189 (10 μM). Mean ± SEM, ns, non-significant; *P<0.05; **P<0.001, t-test, n=3. All qRT-PCR results were obtained from the average of 3 separate experiments, each performed in duplicate.
3.5. Activation of ERK5 by FT-277 and GGTI-298 lead to increase in eNOS, THBD, and KLF2
Multiple lines of evidence suggested that the pleiotropic beneficial effects of statins are attributable to their upregulation of anti-inflammatory and anti-atherogenic mediators in the endothelium (for a review, refer to Jain and Ridker [37]). As outlined in Fig. 4C, ERK5 activation leads to the upregulation of members of the KLF family of transcription factors (KLF2 and KLF4), eNOS and THBD [9]. We showed here that similar to pitavastatin (Supplemental Fig. 2), FTI-277 and GGTI-298 in combination were able to increase the mRNA expression of KLF2, eNOS, and THBD in HUVEC over the course of 24 h (Fig. 4D) and siRNA knockdown of ERK5 reduced the induction of KLF2 (Fig 4E). However, BIX 02189 only minimally inhibited KLF2 induction by the dual drug treatment (Fig. 4F) hinting of a possible ERK5-dependent but MEK-5-independent signaling underlying the gene induction.
3.6. FTI-277 and GGTI-298 are not toxic in a C2C12 cell model of muscle myopathy
While statins provide beneficial effects to the endothelium, one of the major concerns with their use is their myotoxic effect. To determine whether the combination of FTI-277 and GGTI-298 exhibited similar muscle toxicity comparable to the statins, we employed the C2C12 murine myoblasts as an in vitro cellular model to measure the potential myotoxic effects of these compounds [38]. Differentiated C2C12 myotubes were treated with the drug combination for 36 h before measuring their fiber diameter size by a blinded observer. As shown in Fig. 5A and B pitavastatin treatment resulted in a marked decrease in the mean myotube diameter while FTI-277 and GGTI-298 in combination showed attenuated effects. Since phase contrast microscopy does not allow an unambiguous identification of myotubes from myocytes, we employed immuno-staining for MHC that selectively identifies myotubes. The results indicated less morphological evidence of myotoxicity upon treatment with the FTI-277 and GGTI-298 drug combination compared to pitavastatin.
Previously, Wagner et al. [39] demonstrated that statins drastically reduce the cellular level of ATP in C2C12 myotubes, and this correlated with the myopathic phenotypes observed in the zebrafish embryo. We employed the same assay to determine whether the combination of FTI-277 and GGTI-298 affects the cellular ATP level of C2C12 myotubes. As shown in Fig. 5C, despite the extended applications of FTI-277 and GGTI-298 up to 48 h, cellular ATP levels remained intact as compared to pitavastatin treatment alone. The FTI-277 and GGTI-298 co-administration did not affect the cellular ATP level over time in contrast to pitavastatin treatment (Fig. 5D). Lastly, we compared the effects of the pitavastatin and FTI-277/GGTI-298 drug regimen on the upregulation of a biomarker for myopathy. Previously, Hanai et al. [40] showed that the muscle toxicity associated with statins is partly due to an upregulation of an E3 ubiquitin ligase, atrogin-1, in C2C12 myotubes. Here, we showed that at 24 h post drug treatment, while pitavastatin caused a twofold induction of atrogin-1 mRNA levels, FTI-277 and GGTI-298 alone or together had no effect on the transcriptional regulation of atrogin-1 in C2C12 myotubes (Fig. 5E). Together with the ATP depletion assay and the myotube diameter measurements, we concluded that the drug combinations are nontoxic to skeletal muscle cells.
4. Discussion
The HMG-CoA inhibitors, statins, are highly effective in preventing the development of cardiovascular diseases independent of their cholesterol-lowering effects. However, 5-15% of patients undergoing statin therapy develop muscle myalgia. Although the beneficial effects of statins on the cardiovascular system are multifactorial, multiple lines of evidence suggest that its effects may be partly mediated via the activation of the ERK5 pathway in the endothelium [9, 41]. In order to examine the feasibility of capitalizing on the beneficial effects of statins and at the same time averting the myotoxicity associated with this class of drug, we first determined the signaling molecules responsible for statin-mediated ERK5 activation with the goal of identifying small molecule activators of the ERK5 pathway that have minimal toxicity on muscle cells. We showed that the primary mechanism of statin-mediated activation of ERK5 involves the reduction of substrates metabolized by farnesyl transferase and geranylgeranyl transferase I. Co-application of FTI-277 (to inhibit farnesyl transferase) and GGTI-298 (to inhibit geranylgeranyl transferase I) mimicked statin-mediated activation of ERK5 and functionally upregulated previously identified anti-inflammatory and anti-thrombogenic ERK5 targets including KLF2, eNOS, and THBD while exhibiting minimal toxicity on myotubes.
The mechanism of ERK5 activation and downstream gene induction by FTI-277/GGTI-298 treatment parallels that of statins since FTI-277/GGTI-298 increased pERK5 in a time-dependent manner and siRNA inhibition of ERK5 reduced the gene induction by either treatments. BIX02189, an alleged MEK5 specific inhibitor, abrogated ERK5 phosphorylation but did not affect the KLF2 gene induction. Although BIX02189 is regarded as a selective MEK5 inhibitor, this small molecule also directly inhibits the N-terminal kinase activity of ERK5 (IC50 1.5 and 59 nM, for MEK5 and ERK5, respectively [42]). Therefore pharmacological experiments alone cannot untangle the role of MEK5 vs. ERK5 in statin-induced downstream gene induction. However siRNA knockdown of ERK5 reduced KLF2 induction by both pitavastatin and the dual drug treatment confirming the role of ERK5 itself.
According to the canonical signaling scheme, MEK5 specifically activates ERK5 by phosphorylation of the ERK5 TEY motif. However, ERK5 can be phosphorylated in the C-terminal domain through non-canonical signaling involving the cyclin-dependent kinase independent of MEK5 [43, 44], and the signaling pathways upstream of MEK5 are poorly defined. Statin and dual-drug activation of ERK5 likely involves complex upstream crosstalk between isoprenylation of multiple GTPases. Our observation that GGTI-298 by itself trends towards ERK5 activation but requires a combination treatment with FT-277 for full statin-like effect (Fig. 3D) alludes to such a complex regulation. Mechanistic elucidation of the mediators of ERK5 activation by FTI-277 and GGTI-298 together is beyond the scope of the present work, and future studies will be necessary to explore the finer details of this drug-combination-induced signaling that ultimately leads to ERK5 activation and endothelial-protective phenotype. An in vitro kinase assay described in a paper published by Le et al [45] while the present manuscript was under review suggests that statin induction of ERK5 is through a direct effect of statin on ERK5 not involving MEK5 or other kinases.
The regulation of the ERK5 pathway and its downstream targets appear to be cell type dependent. For example, in C2C12 mouse myoblast, ERK5 activation of KLF4 was shown to be essential for the cell fusion into myotubes with little effect on the differentiation program [46]. In HUVEC, statin upregulation of both KLF2 and KLF4 were implicated as the mechanism of endothelial protection afforded by these drugs. Importantly, Sen-Barnerjee et al. [35] demonstrated that MEF2 binds to the promoter region of KLF2 (from -114 to -221) and regulates the expression of KLF2 in a statin-dependent manner. siRNA knockdown of KLF2 reduced the statin-mediated induction of eNOS and THBD [35], indicating that the transcription factor KLF2 regulates the levels of eNOS and THBD in endothelial cells. MEF2 factors (-A, -B, -C, -D) belong to a superfamily of transcription factors that regulate genes important in a number of cellular processes, notably muscle development [47] and stress response during cardiac hypertrophy [48]. MEF2C and MEF2D have been shown to directly interact and can be phosphorylated by ERK5 in vitro [49]. Thus, it is mechanistically possible that statin- and FTI-277/GGTI-298-mediated activation of ERK5, leading to the phosphorylation of MEF2, transcriptionally activates KLF2 and its downstream targets such as eNOS and THBD.
A major impediment to the widespread therapeutic use of statins for hypercholesterolemia is the undesirable statin-associated myopathy seen in some patients. The extent of the statin-associated myopathy ranges from patient-reported muscle weakness and cramps to life-threatening rhabdomyolysis which occurs in 0.1% to 0.5% of patients [50]. Vigorous exercise is a risk factor for these side effects [21]. The hypotheses on the mechanism of statin-associated myopathy have changed over time; while cholesterol and CoQ10 were previously identified as the main determinants of statin-associated myalgia, emerging evidence suggests that depletion of isoprenoids may be the main cause for the toxicity associated with HMG-CoA reductase inhibitors. In fact, multiple lines of evidence demonstrated that supplementation with GGPP in the presence of statin can rescue the cellular markers associated with statin toxicity. Specifically, Wagner et al. [23, 39] demonstrated that statin toxicity is associated with alteration in mitochondrial function that was the result of the inhibition of geranylgeranyl transferase II. In another study, an E3 ubiquitin ligase, atrogin-1, was shown to be upregulated under chronic statin treatment but abrogated by co-supplementation with GGPP [40, 51]. Furthermore, atrogin-1 knockdown C2C12 cells appeared to be unaffected by statin when myofiber diameters were compared [40].
5. Conclusion
The observation that FTI-277 and GGTI-298 in combination mimicked the beneficial effects of statins but were nontoxic to C2C12 myotubes may provide the foundation for novel strategies in the treatment of vascular diseases. Several farnesyl transferase and geranylgeranyl transferase I inhibitors, some highly selective and others inhibiting both, have been explored as cancer therapeutics with many showing acceptable toxicity profiles in human clinical trials [49]. Future directions of this work include characterization of FTI-277/GGTI-298 combination drug treatment in whole animal models of atherosclerosis and myotoxicity, and screening of small molecule libraries to identify other novel drugs capable of inducing ERK5 activation to provide endothelial protection while exhibiting little myotoxicity.
Supplementary Material
Highlights.
-Myopathy due to statins is a significant side-effect often limiting the use of this class of drugs.
-Statin-initiated anti-thrombogenic and the pathogenic myopathy pathways are distinct.
-Co-inhibition of FT and GGT I mimics the anti-thrombogenic actions of statins but without myopathy.
Acknowledgments
We thank Dr. Kirill Alexandrov from the University of Queensland, Australia, for the generous donation of 3-PEHPC and Kowa Pharmaceuticals (Japan) for providing pitavastatin. We also thank Dr. Junichi Abe (University of Rochester) for providing the mERK5-pBIND construct and helpful discussions. This work was supported by the Betty Bamforth Endowment Fund and NIH R01 HL108551 to JY, the PhRMA Foundation postdoctoral fellowship to UC, and the Clinical Translational Cardiovascular Training pre-doctoral fellowship Grant T32 HL07936 to TD.
Abbreviations
- CoQ10
Coenzyme Q10
- eNOS
endothelial Nitric Oxide Synthase
- ERK5
extracellular regulated kinase 5
- FT
farnesyl transferase
- GGPP
geranylgeranyl pyrophosphate
- GGT
geranylgeranyl transferase
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HUVEC
human umbilical vein endothelial cell
- KLF
Kruppel-Like Factor
- L DL
low-density lipoprotein
- MEF2
Myocyte Enhancing Factor 2
- MEK5
mitogen activated protein kinase kinase 5
- PPAR
peroxisome proliferator activated receptor
- THBD
thrombomodulin
Footnotes
Conflicts of Interest: There are no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pedersen TR, Kjekshus J, Berg K, Haghfelt T, Faergeman O, Faergeman G, Pyorala K, Miettinen T, Wilhelmsen L, Olsson AG, Wedel H. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Atheroscler Suppl. 2004;5:81–87. doi: 10.1016/j.atherosclerosissup.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 2.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 3.The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 4.Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM., Jr Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- 5.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 6.Wilson SH, Simari RD, Best PJ, Peterson TE, Lerman LO, Aviram M, Nath KA, Holmes DR, Jr, Lerman A. Simvastatin preserves coronary endothelial function in hypercholesterolemia in the absence of lipid lowering. Arterioscler Thromb Vasc Biol. 2001;21:122–128. doi: 10.1161/01.atv.21.1.122. [DOI] [PubMed] [Google Scholar]
- 7.Bonetti PO, Wilson SH, Rodriguez-Porcel M, Holmes DR, Jr, Lerman LO, Lerman A. Simvastatin preserves myocardial perfusion and coronary microvascular permeability in experimental hypercholesterolemia independent of lipid lowering. J Am Coll Cardiol. 2002;40:546–554. doi: 10.1016/s0735-1097(02)01985-x. [DOI] [PubMed] [Google Scholar]
- 8.Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, Najib-Fruchart J, Glineur C, Staels B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest. 2001;107:1423–1432. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohnesorge N, Viemann D, Schmidt N, Czymai T, Spiering D, Schmolke M, Ludwig S, Roth J, Goebeler M, Schmidt M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4) J Biol Chem. 2010;285:26199–26210. doi: 10.1074/jbc.M110.103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006;7:782–786. doi: 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi M, Lee JD. Role of the BMK1/ERK5 signaling pathway: lessons from knockout mice. J Mol Med (Berl) 2004;82:800–808. doi: 10.1007/s00109-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 18.Lin Z, Kumar A, Sen Banerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, Garcia-Cardena G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 19.Sen Banerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, Garcia-Cardena G, Jain MK. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morikawa S, Takabe W, Mataki C, Wada Y, Izumi A, Saito Y, Hamakubo T, Kodama T. Global analysis of RNA expression profile in human vascular cells treated with statins. J Atheroscler Thromb. 2004;11:62–72. doi: 10.5551/jat.11.62. [DOI] [PubMed] [Google Scholar]
- 21.Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- 22.Vaklavas C, Chatzizisis YS, Ziakas A, Zamboulis C, Giannoglou GD. Molecular basis of statin-associated myopathy. Atherosclerosis. 2009;202:18–28. doi: 10.1016/j.atherosclerosis.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Wagner BK, Gilbert TJ, Hanai J, Imamura S, Bodycombe NE, Bon RS, Waldmann H, Clemons PA, Sukhatme VP, Mootha VK. A small-molecule screening strategy to identify suppressors of statin myopathy. ACS Chem Biol. 2011;6:900–904. doi: 10.1021/cb200206w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondoh K, Terasawa K, Morimoto H, Nishida E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol Cell Biol. 2006;26:1679–1690. doi: 10.1128/MCB.26.5.1679-1690.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron SJ, Itoh S, Baines CP, Zhang C, Ohta S, Che W, Glassman M, Lee JD, Yan C, Yang J, Abe J. Activation of big MAP kinase 1 (BMK1/ERK5) inhibits cardiac injury after myocardial ischemia and reperfusion. FEBS Lett. 2004;566:255–260. doi: 10.1016/j.febslet.2004.03.120. [DOI] [PubMed] [Google Scholar]
- 26.Velica P, Bunce CM. A quick, simple and unbiased method to quantify C2C12 myogenic differentiation. Muscle Nerve. 2011;44:366–370. doi: 10.1002/mus.22056. [DOI] [PubMed] [Google Scholar]
- 27.Takei Y, Le NT, Lee H, Heo KS, Hurley C, Smrcka AV, Miller B, Ko KA, Morrell C, Fujiwara K, Akaike M, Abe J. Abstract 34: Statins directly activate ERK5 and reduce endothelial dysfunction and acute allograft rejection. Circ Res. 2012;111:A34. [Google Scholar]
- 28.Shirasaka Y, Suzuki K, Nakanishi T, Tamai I. Intestinal absorption of HMG-CoA reductase inhibitor pravastatin mediated by organic anion transporting polypeptide. Pharm Res. 2010;27:2141–2149. doi: 10.1007/s11095-010-0216-5. [DOI] [PubMed] [Google Scholar]
- 29.Xu BE, Stippec S, Lenertz L, Lee BH, Zhang W, Lee YK, Cobb MH. WNK1 activates ERK5 by an MEKK2/3-dependent mechanism. J Biol Chem. 2004;279:7826–7831. doi: 10.1074/jbc.M313465200. [DOI] [PubMed] [Google Scholar]
- 30.Buhaescu I, Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem. 2007;40:575–584. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 31.Massey JB. Effect of cholesteryl hemisuccinate on the interfacial properties of phosphatidylcholine bilayers. Biochim Biophys Acta. 1998;1415:193–204. doi: 10.1016/s0005-2736(98)00194-1. [DOI] [PubMed] [Google Scholar]
- 32.Furfine ES, Leban JJ, Landavazo A, Moomaw JF, Casey PJ. Protein farnesyltransferase: kinetics of farnesyl pyrophosphate binding and product release. Biochemistry. 1995;34:6857–6862. doi: 10.1021/bi00020a032. [DOI] [PubMed] [Google Scholar]
- 33.Lo Grasso PV, Soltis DA, Boettcher BR. Overexpression, purification, and kinetic characterization of a carboxyl-terminal-truncated yeast squalene synthetase. Arch Biochem Biophys. 1993;307:193–199. doi: 10.1006/abbi.1993.1578. [DOI] [PubMed] [Google Scholar]
- 34.Zverina EA, Lamphear CL, Wright EN, Fierke CA. Recent advances in protein prenyltransferases: substrate identification, regulation, and disease interventions. Curr Opin Chem Biol. 2012;16:544–552. doi: 10.1016/j.cbpa.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–726. doi: 10.1161/CIRCULATIONAHA.104.525774. [DOI] [PubMed] [Google Scholar]
- 36.Miquel K, Pradines A, Sun J, Qian Y, Hamilton AD, Sebti SM, Favre G. GGTI-298 induces G0-G1 block and apoptosis whereas FTI-277 causes G2-M enrichment in A549 cells. Cancer Res. 1997;57:1846–1850. [PubMed] [Google Scholar]
- 37.Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–987. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 38.Mullen PJ, Luscher B, Scharnagl H, Krahenbuhl S, Brecht K. Effect of simvastatin on cholesterol metabolism in C2C12 myotubes and HepG2 cells, and consequences for statin-induced myopathy. Biochem Pharmacol. 2010;79:1200–1209. doi: 10.1016/j.bcp.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 39.Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, Golub TR, Mootha VK. Large-scale chemical dissection of mitochondrial function. Nat Biotechnol. 2008;26:343–351. doi: 10.1038/nbt1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP, Lecker SH. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest. 2007;117:3940–3951. doi: 10.1172/JCI32741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu K, Tian S, Zhou H, Wu Y. Statins protect human endothelial cells from TNF-induced inflammation via ERK5 activation. Biochem Pharmacol. 2013;85:1753–1760. doi: 10.1016/j.bcp.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Takake RJ, O'Neal MM, Kennedy CA, Wayne AL, Jakes S, Wu D, Kugler SZ, Kashem MA, Kaplita P, Snow RJ. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophsy Res Comm. 2008;377:120–125. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- 43.Diaz-Rodriguez E, Pandiella A. Multiple phosphorylation of ERK5 in mitosis. J Cell Sci. 2010;123:3146–3156. doi: 10.1242/jcs.070516. [DOI] [PubMed] [Google Scholar]
- 44.Inesta-Vaquera RA, Campbell DG, Tournier C, Gomez N, Lizcano JM, Cuenda A. Alternative ERK5 regulation by phosphorylation durig the cell cycle. Cell Signalling. 2010;22:1829–1827. doi: 10.1016/j.cellsig.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 45.Le NE, Takei Y, Izawa-Ishizawa Y, Heo KS, Lee H, Smrcka AV, Miller BL, Ko KA, Ture S, Morrell C, Fujiwara K, Akaike M, Abe JA. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by stains and antimalarialagents. J Immunol. 2014;193:3803–3815. doi: 10.4049/jimmunol.1400571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sunadome K, Yamamoto T, Ebisuya M, Kondoh K, Sehara-Fujisawa A, Nishida E. ERK5 regulates muscle cell fusion through Klf transcription factors. Dev Cell. 2011;20:192–205. doi: 10.1016/j.devcel.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 47.Olson EN. MyoD family: a paradigm for development? Genes Dev. 1990;4:1454–1461. doi: 10.1101/gad.4.9.1454. [DOI] [PubMed] [Google Scholar]
- 48.Kolodziejczyk SM, Wang L, Balazsi K, DeRepentigny Y, Kothary R, Megeney LA. MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Curr Biol. 1999;9:1203–1206. doi: 10.1016/S0960-9822(00)80027-5. [DOI] [PubMed] [Google Scholar]
- 49.Yang CC, Ornatsky OI, McDermott JC, Cruz TF, Prody CA. Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids Res. 1998;26:4771–4777. doi: 10.1093/nar/26.20.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Graham DJ, Staffa JA, Shatin D, Andrade SE, Schech SD, La Grenade L, Gurwitz JH, Chan KA, Goodman MJ, Platt R. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585–2590. doi: 10.1001/jama.292.21.2585. [DOI] [PubMed] [Google Scholar]
- 51.Cao P, Hanai J, Tanksale P, Imamura S, Sukhatme VP, Lecker SH. Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect. FASEB J. 2009;23:2844–2854. doi: 10.1096/fj.08-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors in cancer therapy: important mechanistic and bench to bedside issues. Expert Opin Investig Drugs. 2000;9:2767–2782. doi: 10.1517/13543784.9.12.2767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.