

Summary
Voltage-gated potassium (KV) channels regulate cerebral artery tone and have been implicated in subarachnoid hemorrhage (SAH)-induced pathologies. Here, we examined whether matrix metalloprotease (MMP) activation contributes to SAH-induced KV current suppression and cerebral artery constriction via activation of epidermal growth factor receptors (EGFRs). Using patch clamp electrophysiology, we observed that KV currents were selectively decreased in cerebral artery myocytes isolated from SAH model rabbits. Consistent with involvement of enhanced MMP and EGFR activity in SAH-induced KV current suppression, we found that: 1) OxyHb and/or the exogenous EGFR ligand, HB-EGF, failed to induce further KV current suppression after SAH and 2) gelatin zymography detected significantly higher MMP-2 activity after SAH. The removal of reactive oxygen species (ROS) by combined treatment with superoxide dismutase (SOD) and catalase partially inhibited OxyHb-induced KV current suppression. However, these agents had little effect on OxyHb-induced MMP-2 activation. Interestingly, in the presence of a broad spectrum MMP inhibitor (GM6001), OxyHb failed to cause KV current suppression. These data suggest OxyHb suppresses KV currents through both ROS-dependent and ROS-independent pathways involving MMP activation. The ROS-independent pathway involves activation of MMP-2, whereas the ROS-dependent pathway involves activation of a second unidentified MMP or ADAM (a disintegrin and metalloprotease domain).
Key words and/or reference phrases: K+ channels, heparin-binding EGF-like growth factor (HB-EGF), parenchymal arteriole, patch clamp, vascular smooth muscle, vasospasm
Introduction
Subarachnoid hemorrhage (SAH) following cerebral aneurysm rupture is associated with substantial morbidity and mortality and existing therapeutic options have limited efficacy. A major contributor to poor outcome is delayed cerebral ischemia (DCI) manifesting 4-10 days after aneurysm rupture. Despite decades of study, mechanisms contributing to SAH-induced DCI remain controversial. Factors contributing to the development of DCI after SAH may include early brain injury, cortical spreading depression, disruption of the blood-brain barrier, activation of inflammatory pathways, and enhanced constriction of brain surface arteries/arterioles and intracerebral arterioles [5, 8, 16, 17, 18].
The membrane potential of cerebral artery myocytes is a key regulator of vascular diameter, with membrane potential depolarization leading to an increase in the open-state probability of voltage-dependent Ca2+ channels, enhanced Ca2+ entry and vasoconstriction [15]. Studies using intracellular microelectrodes to measure smooth muscle membrane potential in intact cerebral arteries have found enhanced membrane potential depolarization concomitant with enhanced constriction in tissue from SAH model animals [6, 16, 22]. Voltage-gated potassium (KV) channels play an important role in the regulation of smooth muscle membrane potential and arterial diameter with decrease KV channel activity leading to membrane potential depolarization [1, 3, 4]. Evidence indicates that KV current suppression contributes to enhanced membrane potential depolarization and constriction of cerebral arteries isolated from SAH model animals [7, 11, 20, 22]. Further, we have previously demonstrated that acute application of the blood component oxyhemoglobin (OxyHb) leads to matrix metalloprotease (MMP) activation, shedding of heparin binding EGF-like growth factor (HB-EGF), epidermal growth factor receptor (EGFR) activation and KV channel suppression via internalization [11]. However, the mechanism underlying OxyHb-induced MMP activation and HB-EGF shedding is unclear. The objective of this study was to examine the contribution of reactive oxygen species (ROS) on enhanced MMP activity and KV current suppression in cerebral artery myocytes following SAH.
Materials and Methods
Rabbit double-hemorrhage SAH model
As previously described, two injections of unheparinized autologous arterial blood (3 mL) were delivered via the cistern magna onto the brain surface of anesthetized rabbits at an interval of 48 hours [7, 8, 10]. Five days after the initial surgery, rabbits were euthanized and posterior cerebral and cerebellar arteries (100-200 μm diameter) were isolated from the brain surface for in vitro studies.
Artery diameter measurements
Freshly isolated arteries were cannulated and pressurized to 60 mmHg, superfused with artificial cerebrospinal fluid (aCSF) and diameter measurements obtained using video edge detection equipment [8, 16]. Constriction (tone) is expressed as percent decrease from maximum diameter obtained using Ca2+-free aCSF with 100 μM diltiazem and 1 μM forskolin.
Patch clamp electrophysiology
Whole cell K+ currents were measured using the conventional whole cell configuration of the patch-clamp technique [7, 10, 11]. Outward currents were elicited by 800-msec depolarizing voltage steps from a holding potential of -70 mV to +50 mV [7, 11]. The bath solution contained (in mM): 134 NaCl, 6 KCl, 1 MgCl2, 0.1 CaCl2, 10 Glucose, 10 HEPES (pH 7.4). Patch pipettes (3-5 MΩ) were filled with an internal solution that contained (in mM): 87 Potassium aspartate, 20 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES (pH 7.2). Inwardly rectifying K+ (KIR) channel currents were measured as 100 μM barium-sensitive currents using voltage ramps from -100 mV to +40 mV [23]. For KIR recordings the bath solution contained (in mM): 140 KCl, 1 MgCl2, 0.1 CaCl2, 10 glucose and 10 Hepes (pH 7.4) and patch pipette contained (in mM): 87 Potassium aspartate, 20 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 10 EGTA, 25 KOH (pH 7.2). For all recordings, cell capacitance was not different between groups (control: 10.9 ± 0.4 pF, n=37; SAH: 11.3 ± 0.3 pF, n=30). Current density was calculated by dividing K+ current by cell capacitance.
Zymography
MMP activity was measured using gelatin zymography [11]. Cerebral arteries were homogenized in gel loading buffer (100 mM Tris, 2% SDS and 20 % glycerol). Lysate (15 μg protein) was applied to a 10 % polyacrylamide gel copolymerized with the MMP2/9 substrate, gelatin (1 mg/mL). Following electrophoresis, the gel was rinsed overnight then incubated at 37 °C for 20 hours to allow gelatinolytic activity. The gel was stained with Coomassie Brilliant Blue and MMP activity was detected as unstained bands against the background of blue stained gelatin [11].
PCR
Expression of mRNA was examined by semi-quantitative RT-PCR [9]. Total RNA was extracted from intact cerebral arteries, and converted into cDNA. Amplification of cDNA was performed using Taq DNA polymerase (GenScript) and primers for MMP-2 (Forward: 5′-CCG TGT GAA GTA TGG CAA TG-3′, Reverse: 5′-CGT AGA GCT CTT GAA TGC CC-3′). Band intensity in the linear range of amplification was normalized to band intensity for 18S ribosomal RNA.
Statistical Analysis
Data are expressed as mean ± SEM with n representing the number of cells or samples per group. Student's paired or unpaired t-test were used to determine statistical significance at the level of P < 0.05 (*) or P < 0.01 (**).
Results
Selective suppression of KV channel function after SAH involves EGF receptor activation
Whole cell voltage-dependent and inwardly rectifying K+ currents were examined in cerebral artery myocytes freshly isolated from control and SAH model rabbits using the conventional whole-cell configuration of the patch clamp technique. Whole cell voltage-dependent K+ currents were measured during 10 mV voltage steps from a holding potential of -70 mV and represent the combined activity of large-conductance Ca2+-activated (BK) and delayed rectifier (KV) K+ channels. As illustrated in figure 1a, current amplitude of whole cell composite K+ currents was reduced in cerebral artery myocytes from SAH model animals. To separate BK and KV currents, selective blockers of BK channels (paxilline, 1 μM) and KV channels (4-aminopyridine, 4-AP, 10 mM) were applied to myocytes isolated from control and SAH model animals. The current densities of paxilline-sensitive BK currents were similar in myocytes isolated from both groups; controls: 6.5 ± 0.7 pA/pF at +50 mV (n = 6) and SAH: 5.6 ± 0.6 pA/pF at +50 mV (n = 4). In marked contrast, 4-AP-sensitive KV currents were significantly decreased in myocytes from SAH animals (12.1 ± 1.4 pA/pF at +50 mV, n = 4) compared to myocytes from control animals (16.3 ± 0.6 pA/pF at +50 mV, n = 6) (figure 1b). Inwardly rectifying K+ (KIR) channel currents determined as inward currents sensitive to 100 μM Ba2+ using voltage ramps from -100 mV to +40 mV were not significantly different between groups. For example, Ba2+- sensitive current densities at -100 mV were -5.4 ± 0.9 pA/pF and -5.2 ± 0.6 pA/pF in myocytes from control (n = 24) and SAH (n = 23) animals, respectively. This data demonstrates that KV currents are selectively suppressed in cerebral artery myocytes from SAH model rabbits. Consistent with SAH-induced KV current suppression, constrictions to 4-AP (10 mM) were significantly reduced in arteries isolated from SAH (17.4 ± 3.7% decrease in diameter, n = 4) model animals compared to control (38.9 ± 6.2 % decrease in diameter, n = 4) animals.
Figure 1. OxyHb and HB-EGF suppressed KV currents of cerebral artery myocytes from control, but not SAH model animals.
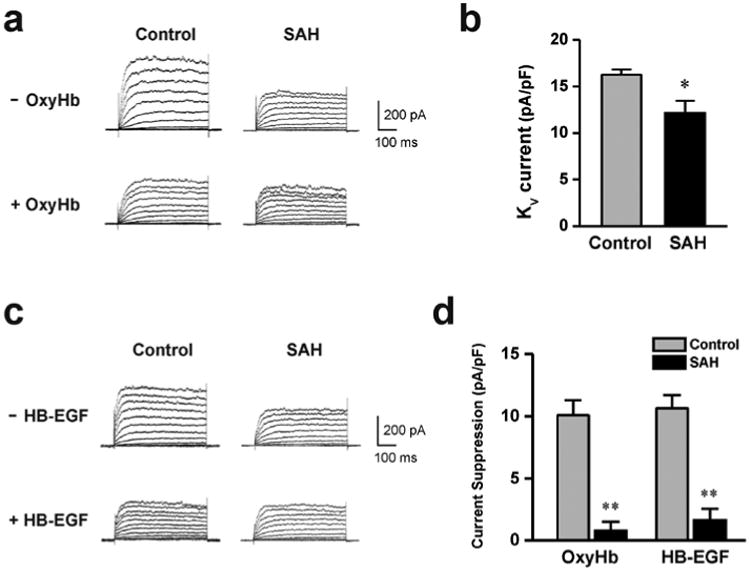
a) Example of whole-cell K+ currents before and after 10 minute application of OxyHb (10 μM) to cerebral artery myocytes isolated from a control and SAH model animal. b) Summary of 4-AP-sensitive KV currents obtained from control (n = 6) and SAH model (n= 4) animals. c) Examples of whole cell K+ currents before and after 10 minute application of HB-EGF (30 ng/ml) to cerebral artery myocytes isolated from control and SAH model animals. d) Summary data demonstrating that OxyHb and HB-EGF significantly suppressed KV currents in cerebral artery myocytes from control, but not SAH model animals. OxyHb treatment; control: n = 7, SAH: n = 4, HB-EGF treatment; control: n = 6, SAH: n = 5. ** P < 0.01 vs control, unpaired students t-test.
Our previous work has demonstrated that acute application of the blood component OxyHb decreased KV currents in cerebral artery myocytes through a mechanism involving HB-EGF shedding and EGF receptor activation [11]. As with this previous work, OxyHb decreased KV currents in myocytes isolated from control animals. However, OxyHb failed to reduce KV currents obtained from SAH animals (figure 1a, d); indicating that acute application of OxyHb and four day exposure of subarachnoid blood in vivo may work through a common mechanism to decrease KV channel activity. Further, exogenous application of HB-EGF mimicked the actions of acute application of OxyHb, causing a reduction in KV currents in myocytes obtained from control, but not SAH model animals (figure 1c, d). This data suggests SAH-induced KV suppression involves HB-EGF shedding and EGF receptor activation in a manner similar to that caused by OxyHb.
MMP-2 activity is enhanced in cerebral arteries from SAH model animals
The matrix metalloprotease subtype, MMP-2, has been implicated in HB-EGF shedding and mesenteric artery constriction [12]. In addition, our previous work has demonstrated that OxyHb increases MMP-2 activity in cerebral artery homogenates from control animals [11]. To examine if increased MMP-2 activity may be involved in SAH-induced HB-EGF shedding and KV current suppression, zymography using the MMP-2 substrate gelatin was done using cerebral artery homogenates from control and SAH model animals. Gelatin zymography from cerebral artery homogenates produced a single 65 kDa band similar to commercially purified MMP-2 (figure 2a). Interestingly, MMP-2 band intensity was significantly greater in homogenates from SAH model animals (figure 2b, c). Although MMP-2 activity was increased after SAH, mRNA levels of MMP-2 were similar in cerebral arteries from control and SAH model animals (figure 2d). This data demonstrates that MMP-2 activity, but not expression, is increased in cerebral arteries after SAH in a manner similar to that observed with acute application of OxyHb.
Figure 2. MMP-2 activity is enhanced in cerebral arteries from SAH model animals.
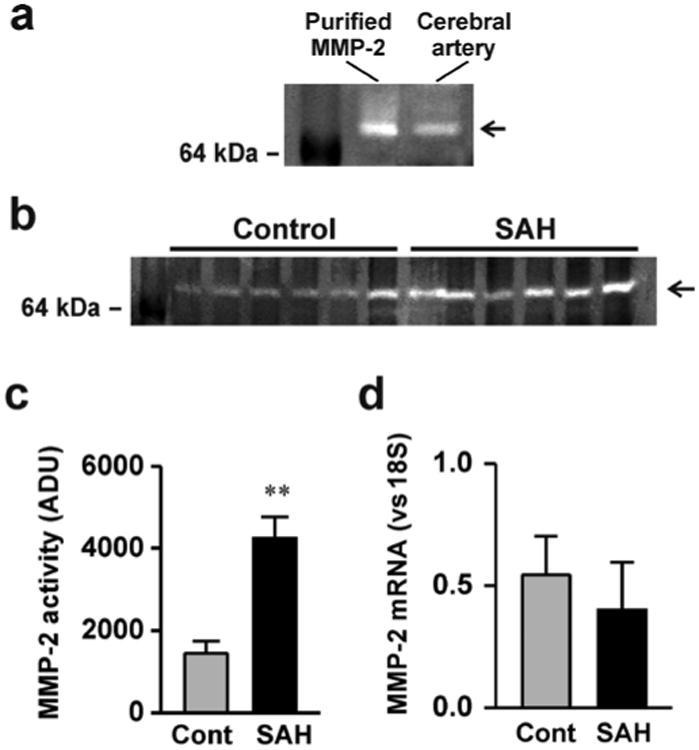
a) An example of gelatin zymography demonstrating activity of commercially purified MMP-2 and cerebral artery homogenate from a control animal. B) Gelatin zymography demonstrating enhanced MMP-2 activity in homogenates obtained from SAH model animals. c) Summary data showing significantly greater MMP-2 activity in cerebral artery homogenates from SAH model animals compared to cerebral artery homogenates from un-operated control animals. (n = 8 for each) d) Summary data demonstrating that MMP-2 mRNA levels are similar in cerebral artery homogenates obtained from control and SAH model animals (n = 7 for each). ** P < 0.01 vs control, unpaired students t-test.
Oxyhemoglobin activates MMPs and suppresses KV currents via ROS-dependent and ROS-independent pathways
To examine if reactive oxygen species (ROS) such as super oxide anions (O2-) contribute to increased MMP activity caused by OxyHb, studies were done in cerebral artery myocytes from control animals using a combination of superoxide dismutase and catalase. Superoxide dismutase catalyzes the conversion of super oxide anions (O2-) into oxygen (O2) and hydrogen peroxide (H2O2), while catalase converts H2O2 to H2O and O2. The combination of superoxide dismutase (150 U/mL) and catalase (500 U/mL) decreased OxyHb-induced KV suppression by approximately 40% (figure 3a, b). In comparison, the broad-spectrum MMP inhibitor, GM6001 (10 μM), caused a substantially greater decrease in OxyHb-induced KV suppression of nearly 80%. These findings suggest ROS partially mediates OxyHb-induced KV suppression; however, ROS-independent MMP activation also contributes to suppression of KV currents by OxyHb. Further, as illustrated in figure 3c, OxyHb-induced MMP-2 activity was not altered by superoxide dismutase and catalase. These findings indicate that both ROS-dependent and ROS-independent MMP activation contribute to OxyHb-induced KV current suppression and that OxyHb increases MMP-2 activity independently of ROS generation.
Figure 3. ROS-dependent and ROS-independent MMP activation and KV current suppression caused by exogenous OxyHb.
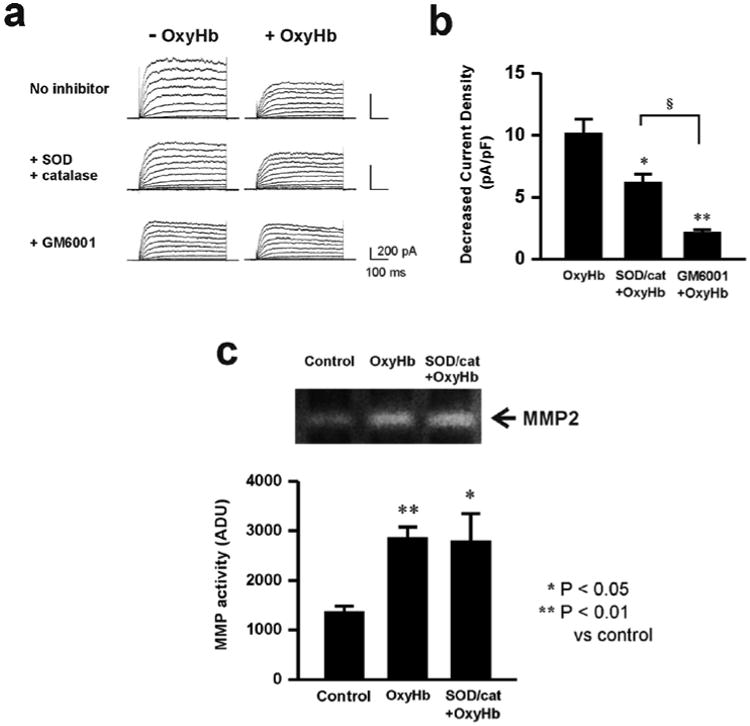
a) Representative KV current recordings demonstrating the ability of the free radical scavengers superoxide dismutase (SOD) and catalase or the MMP inhibitor, GM6001 to reduce OxyHb-induced KV current suppression. b) Summary data demonstrating that GM6001 caused a greater inhibition of OxyHb-induced KV current suppression than SOD/catalase. * P < 0.05; ** P < 0.01 SOD/catalase and GM6001 on OxyHb-induced KV suppression (n = 7); § P < 0.05 GM6001 (n = 5) vs. SOD/catalase treatmtent on OxyHb-induced KV suppression (n = 5). ANOVA followed by Tukey test. c) Representative gel and summary of zymography data demonstrating that SOD/catalase treatment did not prevent OxyHb-induced MMP-2 activation. * P < 0.05; ** P < 0.01 versus control. (Control: n = 8, OxyHb: n = 8, SOD/cat + OxyHb: n = 4) ANOVA followed by Tukey test.
Discussion
The present study indicates that in vivo administration of subarachnoid blood or acute ex vivo application of OxyHb act via multiple pathways that converge to induce HB-EGF shedding and KV current suppression in cerebral artery myocytes. We provide evidence that one of these pathways involves activation of MMP-2 via a mechanism independent of ROS generation and that a second pathway involves ROS-dependent activation of a matrix metalloprotease (MMP) or a disintegrin metalloprotease (ADAM) distinct from MMP-2. The combination of these ROS-dependent and ROS-independent mechanisms and the resultant shedding of HB-EGF and EGF receptor activation account for the selective suppression of KV channels in cerebral artery myocytes from SAH model animals.
Our observation that OxyHb does not cause additional suppression of KV currents in myocytes isolated from SAH model animals indicates that OxyHb is the blood component largely responsible for reduced KV currents leading to enhanced cerebral artery constriction after SAH. Consistent with this concept, both OxyHb and subarachnoid blood suppress KV currents through a pathway involving MMP activation, HB-EGR shedding and EGFR activation. Our present findings also indicate that OxyHb activates at least two distinct MMPs or ADAMs responsible for HB-EGF shedding— MMP-2 and an as of yet unidentified additional MMP/ADAM. It also appears that ROS are involved in the activation of the unidentified MMP/ADAM, but not MMP-2. The oxidation of OxyHb to methemoglobin releases O2- and secondarily leads to the production of hydroxyl radicals [14, 21]. Other studies [2, 13, 19] have demonstrated that these reactive oxygen species can increase activity and expression of MMPs, including MMP-2. However in the present study, super oxide dismutase and catalase, scavengers of O2- and H2O2, did not prevent OxyHb-induced stimulation of MMP-2 activity (figure 3). This finding indicates that OxyHb can also act independently of ROS to enhance MMP-2 activity. Future studies are needed to determine the mechanism of ROS-independent MMP-2 activation and to determine the identity of additional MMPs/ADAMs involved in OxyHb-induced KV current suppression.
Conclusions
Enhanced cerebral artery constriction represents one component of the multi-factorial and inter-related series of pathological events leading to delayed cerebral ischemia in patients after aneurysmal SAH. Our data indicate that OxyHb contributes to SAH-induced cerebral artery constriction via activation of multiple MMPs/ADAM, leading to HB-EGF shedding and KV current suppression in cerebral artery myocytes. Evidence is also provided that ROS-dependent and ROS-independent pathways are involved in OxyHb-induced MMP activation. These findings suggest that SAH-induced MMP/ADAM activation may play an important role in the development of DCI after SAH and represent a new target for therapies to alleviate the detrimental consequences of cerebral aneurysm rupture.
Acknowledgments
This work was supported by the Totman Trust for Medical Research, the Peter Martin Brain Aneurysm Endowment, the National Institutes of Health (NIH) (P01 HL095488, R01 HL078983 and R01 HL078983-05S1) and the American Heart Association (0725837T). The authors would also like to acknowledge the use and assistance of the University of Vermont Neuroscience COBRE molecular biology core facility.
Footnotes
Conflict of interest statement: None.
References
- 1.Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, Rusch NJ. Voltage-gated K+ channels in rat small cerebral arteries: molecular identity of the functional channels. J Physiol. 2003;551:751–763. doi: 10.1113/jphysiol.2003.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali MA, Kandasamy AD, Fan X, Schulz R. Hydrogen peroxide-induced necrotic cell death in cardiomyocytes is independent of matrix metalloproteinase-2. Toxicol In Vitro. 2013;27:1686–1692. doi: 10.1016/j.tiv.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Amberg GC, Santana LF. KV2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol. 2006;291:C348–C356. doi: 10.1152/ajpcell.00086.2006. [DOI] [PubMed] [Google Scholar]
- 4.Chen TT, Luykenaar KD, Walsh EJ, Walsh MP, Cole WC. Key role of KV1 channels in vasoregulation. Circ Res. 2006;99:53–60. doi: 10.1161/01.RES.0000229654.45090.57. [DOI] [PubMed] [Google Scholar]
- 5.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- 6.Harder DR, Dernbach P, Waters A. Possible cellular mechanism for cerebral vasospasm after experimental subarachnoid hemorrhage in the dog. J Clin Invest. 1987;80:875–880. doi: 10.1172/JCI113146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishiguro M, Morielli AD, Zvarova K, Tranmer BI, Penar PL, Wellman GC. Oxyhemoglobin-induced suppression of voltage-dependent K+ channels in cerebral arteries by enhanced tyrosine kinase activity. Circ Res. 2006;99:1252–1260. doi: 10.1161/01.RES.0000250821.32324.e1. [DOI] [PubMed] [Google Scholar]
- 8.Ishiguro M, Puryear CB, Bisson E, Saundry CM, Nathan DJ, Russell SR, Tranmer BI, Wellman GC. Enhanced myogenic tone in cerebral arteries from a rabbit model of subarachnoid hemorrhage. Am J Physiol Heart Circ Physiol. 2002;283:H2217–H2225. doi: 10.1152/ajpheart.00629.2002. [DOI] [PubMed] [Google Scholar]
- 9.Koide M, Bonev AD, Nelson MT, Wellman GC. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc Natl Acad Sci U S A. 2012;109:E1387–E1395. doi: 10.1073/pnas.1121359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koide M, Nystoriak MA, Krishnamoorthy G, O'Connor KP, Bonev AD, Nelson MT, Wellman GC. Reduced Ca2+ spark activity after subarachnoid hemorrhage disables BK channel control of cerebral artery tone. J Cereb Blood Flow Metab. 2011;31:3–16. doi: 10.1038/jcbfm.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koide M, Penar PL, Tranmer BI, Wellman GC. Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H1750–H1759. doi: 10.1152/ajpheart.00443.2007. [DOI] [PubMed] [Google Scholar]
- 12.Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–3593. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Lemus LA, Zhao G, Galinanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol. 2011;300:H2005–H2015. doi: 10.1152/ajpheart.01066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Misra HP, Fridovich I. The generation of superoxide radical during the autoxidation of hemoglobin. J Biol Chem. 1972;247:6960–6962. [PubMed] [Google Scholar]
- 15.Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 16.Nystoriak MA, O'Connor KP, Sonkusare SK, Brayden JE, Nelson MT, Wellman GC. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol. 2011;300:H803–H812. doi: 10.1152/ajpheart.00760.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, Kasuya H, Wellman G, Keller E, Zauner A, Dorsch N, Clark J, Ono S, Kiris T, Leroux P, Zhang JH. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009;31:151–158. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- 19.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–C60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 20.Sobey CG, Faraci FM. Subarachnoid haemorrhage: what happens to the cerebral arteries? Clin Exp Pharmacol Physiol. 1998;25:867–876. doi: 10.1111/j.1440-1681.1998.tb02337.x. [DOI] [PubMed] [Google Scholar]
- 21.Steele JA, Stockbridge N, Maljkovic G, Weir B. Free radicals mediate actions of oxyhemoglobin on cerebrovascular smooth muscle cells. Circ Res. 1991;68:416–423. doi: 10.1161/01.res.68.2.416. [DOI] [PubMed] [Google Scholar]
- 22.Wellman GC. Ion channels and calcium signaling in cerebral arteries following subarachnoid hemorrhage. Neurol Res. 2006;28:690–702. doi: 10.1179/016164106X151972. [DOI] [PubMed] [Google Scholar]
- 23.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]