

Abstract
The cyclooxygenase (COX) activity of prostaglandin H synthase-2 (PGHS-2) is implicated in colorectal cancer and is targeted by nonsteroidal anti-inflammatory drugs (NSAIDs) and dietary n – 3 fatty acids. We used purified, recombinant proteins to evaluate the functional impacts of the R228H, E488G, V511A and G587R PGHS-2 polymorphisms on COX activity, fatty acid selectivity and NSAID actions. Compared to wild-type PGHS-2, COX activity with arachidonate was ~20% lower in 488G and ~20% higher in 511A. All variants showed time-dependent inhibition by the COX-2-specific inhibitor (coxib) nimesulide, but 488G and 511A had 30–60% higher residual COX activity; 511A also showed up to 70% higher residual activity with other time-dependent inhibitors. In addition, 488G and 511A differed significantly from wild type in Vmax values with the two fatty acids: 488G showed ~20% less and 511A showed ~20% more discrimination against eicosapentaenoic acid. The Vmax value for eicosapentaenoate was not affected in 228H or 587R, nor were the Km values or the COX activation efficiency (with arachidonate) significantly altered in any variant. Thus, the E488G and V511A PGHS-2 polymorphisms may predict who will most likely benefit from interventions with some NSAIDs or n – 3 fatty acids.
Keywords: prostaglandin H synthase-2 polymorphisms, cyclooxygenase, nonsteroidal anti-inflammatory drugs, arachidonic acid, eicosapentaenoic acid
Introduction
The cyclooxygenase (COX) activity of prostaglandin H synthase isoforms 1 and 2 (PGHS-1 and PGHS-2) catalyzes a key step in the biosynthesis of prostaglandins from n – 6 and n – 3 polyunsaturated fatty acids such as arachidonic acid (AA) and eicosapentaenoic acid (EPA).1 The prostaglandins, which include prostaglandin E, prostacyclin and thromboxane, serve as potent signaling molecules in many pathophysiological processes.2 The two PGHS isoforms are functionally specialized, with PGHS-1 generally considered a constitutive, housekeeping enzyme, whereas PGHS-2 is strongly induced by cytokines and mitogens in a variety of cells and has been linked to the initiation and resolution phases of inflammation and to cell proliferation.3,4 PGHS-2 levels are elevated in many tumors and COX-2 activity has been implicated in the development of several types of cancer, including colorectal cancer.5,6
COX catalysis is strongly regulated by the availability of unesterified fatty acid substrate, the structure of the substrate (for example, n – 6 vs. n – 3) and the level of peroxide, which is required to generate the protein-based radical at Tyr385 that begins the catalytic cycle.1 In addition, COX catalysis is the primary target of NSAIDs (nonsteroidal anti-inflammatory drugs); these agents include aspirin, ibuprofen, naproxen and the COX-2 inhibitors (coxibs), which are selective for inhibition of COX-2.3 NSAID use is linked to significantly decreased risk of colorectal cancer in humans.5
A limited number of nonsynonymous polymorphisms have been identified in PTGS2, the gene that encodes human PGHS-2 (hPGHS-2); these polymorphisms include R228H, E488G, V511A and G587R.7,8 The prevalence of these polymorphisms is low, with a minor allele frequency ranging from 0.8 to 5% in the populations studied, suggesting that there is strong selective pressure on PTGS2. Epidemiological studies have examined the relation of the V511A polymorphism to the risks of colorectal adenoma or cancer,8–10 breast cancer11 and cardiovascular disease.12 Because these studies were generally small (N~ 400–1000) and the variant allele is rare (~4%), none of the studies found a statistically significant association with risk. However, two of the colorectal cancer studies reported inverse associations with the A allele and some suggestion of an NSAID interaction.8,10 Similarly, gene–NSAID interactions and pharmacogenetic relationships have been reported in noncoding PTGS2 single-nucleotide polymorphisms (SNPs).13 as well as in other genes related to prostaglandin synthesis.14,15
There have been few published studies on the nonsynonymous polymorphisms listed above,7–11,16,17 and the V511A polymorphism is the only nonsynonymous PTGS2 variant that has been studied for functional effects so far. Fritsche et al.7 observed no major differences between wild-type hPGHS-2 and the V511A variant for metabolism of AA or linoleic acid, nor any differences between the alleles for inhibition by indomethacin or celecoxib. Lin et al.8 reported that the major and minor alleles had similar Vmax and Km values, but noted it was premature to conclude that the V511A variant had no functional effect without a more thorough enzymological analysis. Several important aspects of COX-2 catalysis were not examined in the earlier studies. These aspects include: discrimination between n – 6 and n – 3 substrates, which has considerable potential for diet-driven modulation of prostanoid signaling;18 peroxide-dependent feedback activation, which contributes to regulation of COX activity;19 and time-dependent COX inhibition effects, which characterize the more potent pharmacological agents.20 The present studies were undertaken to address these important aspects of COX functionality in V511A and three other variants of hPGHS-2 using the purified recombinant proteins.
Materials and methods
High-purity AA and EPA were obtained in sealed ampules from Nu Chek Prep (Elysian, MN, USA) and handled under conditions found to minimize peroxide formation.21 Briefly, stocks of the fatty acids were dissolved in toluene, butylated hydroxytoluene (0.001 mol/mol) was added and the solutions stored at −20 °C. Working suspensions (3–30 mm) in 0.1 m Tris (pH 8.5) were prepared fresh each day and kept on ice. Tween 20 was from Anatrace (Maumee, OH, USA). Coxibs were from Cayman Chemical (Ann Arbor, MI, USA) or Tocris Bioscience (Ellisville, MO, USA). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA, USA). Oligonucleotides were from Integrated DNA Technologies (Coralville, IA, USA). Reagents for DNA manipulation were from Promega (Madison, WI, USA). Plasmid transfer vector pAcSG2 and BaculoGold linearized baculovirus DNA were from Pharmingen (San Diego, CA, USA). QuikChange single and multisite-directed mutagenesis kits and Escherichia coli strain XL-10 were from Stratagene (La Jolla, CA, USA). Sf9 insect cells (from Spodoptera frugiperda), E. coli strain DH5α, Grace’s supplemented medium and fetal bovine serum were from Invitrogen (Carlsbad, CA, USA). Ni-NTA agarose was purchased from Qiagen (Valencia, CA, USA). The integrity of each plasmid described below was verified by restriction enzyme digestion and by DNA sequencing at the Microbiology and Molecular Genetics Core Facility, UT Health Science Center at Houston. All other reagents were obtained from Sigma (St Louis, MO, USA).
Construction of plasmid for recombinant, 6 × His-tagged hPGHS-2
The 6 × His tag sequence was introduced after the signal peptide cleavage site near the N terminus of the hPGHS-2 wild type (major allele at all targeted positions), as previously reported.22 Nucleotides coding for the six histidine residues were inserted into the hPGHS-2/pAcSG2 vector using PCR with PfuUltra HF DNA polymerase (Stratagene) and the following 5′-primers: sense, 5′-pCAC CATCACCCTTGCTGTTCCCACCCATGTCAAAACC-3′; anti-sense, 5′-pATGGTGATGATTTGCTGTATGGCTGAGCGCCA GGACC-3′.
The PCR mixture was digested with DpnI; the resulting 7.4 kb product was separated by agarose gel electrophoresis and ligated with T4 DNA ligase. The resulting plasmid was transformed into E. coli strain DH5a, and ampicillin-resistant colonies were analyzed by restriction enzyme digest and DNA sequencing.
Construction of plasmid for hPGHS-2 with R228H, E488G, V511A or G587R minor alleles
Plasmids coding for hPGHS-2 with the desired point mutations were constructed with the QuikChange mutagenesis kit using the pAcSG2 plasmid containing the 6 × His-tagged, wild-type hPGHS-2 cDNA as template. Primer pairs were (base changes underlined) as follows: R228H–f, 5′-C TGGCTAGACAGCATAAACTGCGCCTTTTC-3′; R228H–r, 5′-G AAAAGGCGCAGTTTATGCTGTCTAGCCAG-3′; E488G–f, 5′-C ATCGATGCTGTGGGGCTGTATCCTGCCC-3′; E488G–r, 5′-GG GCAGGATACAGCCCCACAGCATCGATG-3′; V511A–f, 5′-GA AACCATGGTAGAAGCTGGAGCACCATTCTC-3′; V511A–r, 5′-G AGAATGGTGCTCCAGCTTCTACCATGGTTTC-3′; G587R–f, 5′-AGTTCTTCCCGCTCCCGACTAGATGATATC-3′; G587R–r, 5′-GATATCATCTAGTCGGGAGCGGGAAGAACT-3′.
Baculovirus generation and expression of recombinant proteins
Generation, amplification and titer determination of recombinant baculovirus containing the desired hPGHS-2 cDNA, and expression of the recombinant proteins used previously published techniques;23 procedures for expression of hPGHS-2 have been described.24
Purification of recombinant proteins
Purification of hPGHS-2 lacking a His tag has been described.24 The purification procedure for His-tagged hPGHS-2 wild-type and mutant proteins was modified from a published protocol.25 The Sf9 cells expressing the recombinant protein were suspended in 25 mm NaPi, 20 mm imidazole, 1.0 mm phenol (pH 7.4) and sonicated; the homogenate was centrifuged at 150 000 g at 4°C for 1 h. The resulting pellet was resuspended with a Dounce homogenizer in 100 ml of 25 mm NaPi, 100 mm NaCl, 20 mm imidazole, 0.1 mm phenol (pH 7.4), 1% v/v Tween 20 and stirred for 1 h at 4°C. Unextracted membrane residue was removed by centrifugation. The rest of the purification was as described,25 except that buffers were supplemented with 0.1 mm phenol. Holoenzyme was reconstituted by addition of heme.26
Protein characterization
Total protein concentrations were determined by a modified Lowry method.27 hPGHS-2 mutants were analyzed by polyacrylamide gel electrophoresis under denaturing conditions,28 visualization with Coomassie staining of the gel and densitometry using ImageJ29 to determine the fraction of total protein present as recombinant hPGHS-2.
COX activity
Oxygen uptake was measured polarographically in standard assays at 30°C in 100 mm KPi (pH 7.2) with 1.0 mm phenol, 1.0 µm heme and 0.025% Tween 20, using a ‘Standard’ electrode membrane (YSI, Yellow Springs, OH, USA).30 One unit of COX activity has an optimal velocity of 1 nmol O2 per min. The specific activity of each purified protein preparation was calculated by dividing the observed COX activity by the concentration of recombinant hPGHS-2 present. COX Km and Vmax values were determined by measuring the activity with 2–100 µm fatty acid at 25°C with a YSI ‘High Sensitivity’ electrode membrane, in the presence of 100 µm HOOH; these assay conditions ensure full activation and minimize dampening effects from the electrode membrane.31 The velocities were normalized to those for the same batch of enzyme with 100 µm AA without added peroxide and fitted to the Michaelis–Menten equation using KaleidaGraph (Synergy Software, Reading, PA, USA) to estimate the Vmax and Km values.
COX self-inactivation
COX self-inactivation rates for reactions in 100 µm AA were calculated by dividing the optimal velocity (nmol O2 per min) by the reaction extent at complete self-inactivation (nmol O2).32
COX activation efficiency
Feedback activation by peroxide was assessed by measuring the COX activity of a fixed amount of enzyme (45–55 U) in the presence of varying amounts of GPx-1.23 Data for the decline in the fraction of expressed COX activity with increasing peroxide scavenger were fitted to a straight line to obtain the slope; the slope obtained for hPGHS-2 wild type in a given experiment was divided by the slope obtained for hPGHS-2 with each minor allele to calculate the relative activation efficiency for that variant.
COX inhibition
COX inhibition kinetics were assessed at 30°C with a ‘Standard’ electrode membrane in the standard reaction mixture. Two experimental designs were used. The first focused on the forward reactions and involved timed preincubation of enzyme with inhibitor before the COX reaction was started by addition of AA. In this design, the enzyme (at ~10−8 m) was preincubated with the desired level of inhibitor in the reaction cuvette for various times before injection of 100 µ m AA to determine the amount of enzyme that had not progressed to tight inhibitor–protein complexes. Inhibitors were dissolved in ethanol; the final ethanol concentration was below 1% and did not affect activity. AA was suspended at 30 mm in 0.1 m Tris-HCl (pH 8.5). The second experimental design focused on the kinetics of conversion of tight inhibitor–protein complexes to looser complexes and dissociation of inhibitor. For this, the enzyme (~ 10−5 m) was first incubated with a small excess of inhibitor (50 µ m) to form an equilibrium mixture of tight and looser inhibitor complexes. Aliquots of this mixture were then diluted 1500-fold into the reaction cuvette and re-equilibrated for various lengths of time before the amount of enzyme in looser inhibitor complexes or without inhibitor bound was determined by addition of 100 µm AA.
Peroxidase activity
The stirred assay mixture contained 2.5 ml of 0.10 m Tris-HCl (pH 8.0) with 0.50 mm guaiacol, 1.0 µm heme and 0.40 mm HOOH at room temperature.33 Reaction was started by injection of enzyme and the initial velocity calculated from the rate of increase in A436 due to oxidized guaiacol (ε436 = 6.39 (mm oxidized guaiacol)−1cm−1). One unit of peroxidase (POX) activity has a velocity of 1 nmol guaiacol oxidized per min.
Statistical analysis
The results of replicate experiments are presented as mean ± s.d. of the mean. In most cases an unpaired, two-tailed t-test was used to assess statistical differences between individual variants and wild-type PGHS-2 (GraphPad Software, La Jolla, CA, USA). A P-value below 0.05 was considered statistically significant.
Results
Expression and purification
Several expression runs, totaling 3–41 of baculovirus-infected Sf9 cell culture, were pooled to prepare homogenates for purification of each recombinant protein. Wild-type hPGHS-2 and the four polymorphic proteins expressed well in the baculovirus system, as assessed by the yield of COX activity in the insect cell homogenate (Table 1). All of the proteins were readily solubilized by Tween 20 and purified to 75–90% electrophoretic homogeneity, with 38–64% recovery of COX activity (Table 1). The COX-specific activities of the purified enzymes, calculated based on the amount of recombinant PGHS protein determined by assay of total protein and electrophoretic analysis, are shown in Table 1. The specific activities of the 228H, 488G and 587R variants were 19–30% lower than the wild type, whereas the value for the 511A variant was 55% higher than the wild type. With the exception of the 511A variant, these specific activities fall in the range of 15–30 kU mg−1 observed for purified PGHS-2 in our laboratory31,34,35 To focus on possible effects of the polymorphic alleles that are specific for the COX activity, we normalized the COX activity for each of the purified proteins to the corresponding POX activity determined on the same day (Table 1). The COX/POX ratio was 2.7 for wild-type PGHS-2, and the values for the 228H and 587R variants were not significantly different. In contrast, the COX/POX ratio for the 511A variant was 20% higher, and the ratio for 488G ~20% lower, than the wild-type value; the difference was statistically significant for both of these variants. Thus, the COX-specific activity and the COX/POX ratio were both elevated for 511A and both decreased for 488G (Table 1). It is therefore likely that these two variant alleles have direct or indirect effects on COX catalytic efficiency.
Table 1.
Expression, recovery and enzymatic activities of recombinant hPGHS-2 proteins
| hPGHS-2 constructa |
COX in homogenate (kU/3 l culture) |
Yield of COX after purification (%) |
Electrophoretic homogeneity (%) |
COX-specific activityb (kU mg−1 PGHS-2) |
COX/POXb (U/U) |
|---|---|---|---|---|---|
| Wild typec | 440 | 64 | 87 | 30 ± 1 | 2.7 ± 0.1 |
| R228H | 480 | 38 | 87 | 22 ±0 | 2.5 ± 0.1 (P = 0.07) |
| E488G | 300 | 46 | 75 | 21 ± 1 | 2.1 ± 0.1 (P = 0.002) |
| V511A | 600 | 42 | 86 | 46 ± 1 | 3.2 ± 0.2 (P = 0.018) |
| G587R | 550 | 52 | 90 | 24 ± d2 | 2.5 ± 0.1 (P = 0.07) |
Variant alleles are underlined.
Mean ± s.d. for triplicate determinations in standard assays.
Major allele at all targeted residues.
COX Vmax and Km parameters
The COX kinetics of wild-type hPGHS-2 and the four variants were evaluated with prototypical substrates from the n – 6 (AA) and n – 3 (EPA) families, with precautions taken to ensure reliable COX activity measurements reflecting the dependence on substrate concentration.31 COX activity was measured at 25 °C with a ‘High-sensitivity’ oxygen electrode membrane to minimize dampening effects that can significantly attenuate velocity values. HOOH (100 µm) was included in each reaction to ensure full COX activation, avoiding complications due to partial COX activation; this is particularly important for reactions with EPA where artifactually low COX velocities occur in the absence of exogenous peroxide.31 Batch-to-batch variations in expression and purification of recombinant PGHS-2 protein can lead to differences in COX-specific activity and confound comparison of COX activity in wild-type and mutant proteins. To control for such variations in specific activity, we normalized the COX velocities for a particular protein reacted with a range of AA or EPA levels in the presence of HOOH to the average velocity of the same protein on the same day in replicate reactions with 100 µm AA without exogenous peroxide. This level of AA produces full COX activation without added peroxide.
Saturable responses of COX-2 reaction rate to AA and EPA concentration were observed for wild-type PGHS-2 and each of the variants, so values of Vmax and Km were estimated by fitting individual data sets to the Michaelis–Menten equation (examples are shown in Supplementary Figures S1–S5). The average Vmax values with AA for wild-type and polymorphic hPGHS-2 proteins were all near unity (Figure 1a), as expected because the rate with saturating AA was used for normalization. The average Km value for wild-type hPGHS-2 was 2.1 µm AA (Figure 1b), quite comparable to the value of 1.7 ± 0.4 µm reported under the same reaction conditions for hPGHS-2 without a His tag.31 None of the mutations produced a significant difference in the average Km for AA or for EPA (Figure 1b). The average Vmax value for wild-type hPGHS-2 with EPA was 0.78, or almost 80% of the Vmax with AA (Figure 1a). This shows that the fully activated enzyme oxygenates EPA more slowly than AA. The average Vmax values with EPA for the 228H and 587R variants were indistinguishable from the wild-type value (Figure 1a). However, the average Vmax value with EPA for 488G was 0.94, significantly higher than wild type and approaching the Vmax value obtained with AA, and 511A had an average Vmax value of 0.65 with EPA, significantly less than the wild-type protein (Figure 1a). Thus, the 488G variant led to less, and the 511A variant led to more, COX catalytic discrimination against EPA relative to AA.
Figure 1.

COX kinetic parameters for PGHS-2 wild type (WT) and variants with AA (open circles) and EPA (filled circles). Values for Vmax relative to the rate with 100 µm AA and no HOOH (a) and Km (b) represent the average ± s.d. for triplicate determinations. Asterisks indicate values significantly different from wild type (P < 0.05).
COX activation efficiency
Titrations with a peroxide scavenger, glutathione peroxidase, were used to quantitatively compare the COX activation efficiencies of the four variant PGHS-2 proteins to the wild-type enzyme. The results, shown in Table 2, indicate that the COX activation efficiency of the 511A and the 587R variants was the same as the wild type. Conversely, both 228H and 488G had slightly higher activation efficiencies, though the differences were not statistically significant.
Table 2.
COX activation efficiencies of purified hPGHS-2 proteins with AA as substrate
| hPGHS-2 constructa | Activation efficiencyb (relative to wild type) |
|---|---|
| Wild typec | 1.0 |
| R228H | 1.2 ± 0.2 (n = 3) P = 0.31 |
| E488G | 1.2 ± 0.2 (n = 4) P = 0.19 |
| V511A | 1.0 ± 0.1 (n = 4) P = 0.65 |
| G587R | 1.0 ± 0.1 (n = 2) P = 0.50 |
Variant alleles are underlined.
Values represent the average ± s.d. for the indicated number of determinations; P-values were determined by one-sample t-test.
Major allele at all targeted residues.
COX self-inactivation
The rate constant for self-inactivation was 2.5 min−1 for wild-type hPGHS-2 and the values for the variants ranged from 2.5 to 2.8 min−1, indicating that none of the variants caused a major change in the COX self-inactivation process.
COX inhibition kinetics
We analyzed the interactions of wild-type hPGHS-2 and the variant proteins with nimesulide, a COX-2-specific inhibitor that follows competitive, time-dependent kinetics and has proven useful as a probe of active site structure.36 The action of such inhibitors can be described by a two-step mechanism such as that shown in Scheme 1. The initial binding of inhibitor (I) to the COX site of enzyme (E) to form a lower affinity complex (EI) is governed by a forward rate constant (k1) and a reverse rate constant (k−1). This first interaction with inhibitor typically equilibrates very rapidly, and a typical Ki value (k−1/k1) is ~ 10−5 m. In the case of time-dependent inhibitors, the EI complex undergoes a relatively slow transition to a higher affinity complex (EI′), governed by rate constant k2. Unless a covalent bond is formed in EI′, the complex can revert back to EI by the k−2 process.
Scheme 1.
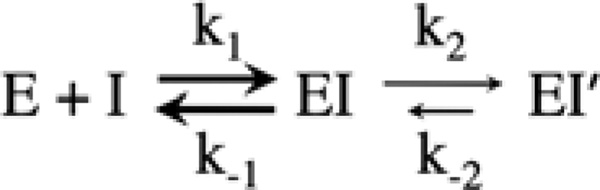
Mechanistic scheme for interaction of PGHS (E) with COX inhibitor (I).
Two types of experimental approach were used to analyze the interactions of wild-type hPGHS-2 and the variant proteins with coxibs. The first involves preincubation of enzyme with inhibitor in the absence of substrate for various lengths of time before injection of saturating level of AA to quantitate the surviving COX activity. With shorter preincubation periods, this approach focuses on the rate of progression to the EI′ complex, which is only slowly reversible and thus does not contribute to COX catalysis when AA is added. When the preincubation is long enough, this approach provides an indication of the distribution of enzyme between E/EI and EI′ at equilibrium. It is important to note that the full inhibitory potency of time-dependent agents, such as nimesulide against COX-2, cannot be reliably assessed from conventional IC50 measurements because the IC50 value is determined both by the affinity of the rapid, reversible initial binding (E + I ⇔ EI; Scheme 1) and by the rates of the slower EI ⇒ EI′ and EI ⇐ EI′ steps. On the other hand, the residual activity at long preincubation times is a useful measure of the maximal inhibition possible with a given agent, or conversely, what fraction of COX activity escapes inhibition even after equilibration with the agent (roughly mimicking the extended exposures in vivo). Thus, the ratio of the residual activities for variant and wild type gives a practical, quantitative index of any changes in COX-2 leakiness toward a given inhibitor.
Nimesulide was chosen for the screening because it has been established as a competitive, time-dependent inhibitor that is selective for PGHS-2,36 and it is readily available. Range-finding assays with wild-type hPGHS-2 at a variety of nimesulide concentrations indicated that equilibration was reached in a reasonable length of time with 2 µm inhibitor, so this level was used for the comparison with the variants. The results of this comparison are shown in Figure 2. The COX activity of each of the proteins was stable for at least 3 min of preincubation in control reactions without inhibitor. With nimesulide present, wild-type hPGHS-2 and the four variants all progressively lost COX activity with increasing preincubation time until the surviving activity plateaued as equilibrium was reached. The plateau fraction of surviving COX activity in the wild type was 0.17, and essentially the same values were found with the 228H and 587R variants (Figure 2 and Table 3). The surviving COX activity plateaued at a markedly higher level (0.27) for the 511A variant. This is some 60% higher than with wild-type hPGHS-2 and indicates that the structural change at residue 511 in the variant shifts the equilibrium significantly away from the tight EI′ complex toward the looser EI complex, weakening the COX inhibition. The plateau level of COX activity in the E488G protein equilibrated with 2 µm nimesulide was 0.22, also significantly above the wild-type value (Figure 2 and Table 3). Thus, formation of the EI′ complex was decreased in the 488G variant, though to about half the degree seen in the 511A variant. In all proteins examined, the initial decay in COX activity followed first-order kinetics (as indicated by the early linear drops in the diagnostic semi-log plot in Figure 2), as is expected from the mechanism shown in Scheme 1, but the decline was too rapid at this level of inhibitor for a detailed comparison of k2 values.
Figure 2.

Kinetics of COX inhibition in hPGHS-2 wild type (WT) and variants during preincubation with 2µm nimesulide. Aliquots of each protein (10 −8 m) were preincubated with (filled symbols) or without (open symbols) inhibitor for the indicated lengths of time before the surviving COX activity (i.e., enzyme not in the EI′ form) was assayed by injection of 100 µm AA. The values represent averages ± s.d. for triplicate measurements.
Table 3.
Residual COX activities of nimesulide complexes of hPGHS-2 and the four polymorphic variants
| hPGHS-2 constructa | Residual COX activity (fraction of control)b |
|---|---|
| Wild typec (n = 9) | 0.17 ± 0.01 |
| R228H (n = 9) | 0.18 ± 0.00 |
| E488G (n = 9) | 0.22 ± 0.01d |
| V511A (n = 6) | 0.27 ± 0.01d |
| G587R (n = 9) | 0.18 ± 0.01 |
Variant alleles are underlined.
After equilibration with 2 µm nimesulide, the average fraction (± s.d.) of COX activity surviving in the plateau region was calculated for each protein from the data in Figure 3.
Major allele at all targeted residues.
Significantly different from wild type (P < 0.05).
The same approach was used to assess the surviving COX activities of the wild-type and 511A hPGHS-2 proteins after equilibration with 2 µm levels of seven other coxibs. CAY-10404, tenidap, NS-398, valdecoxib and diclofenac exhibited time-dependent inhibition of COX-2 in semi-log plots similar to that in Figure 2, whereas COX-2 inhibition by FR122047 and SC-560 was not time dependent (data not shown). The residual COX-2 activities after equilibration with the inhibitors are presented in Table 4. With FR122047 and SC-560 the 511A variant was inhibited the same or slightly more than wild-type enzyme. Conversely, the residual activity for the V511A variant was 10–70% greater than wild type for all but one of the time-dependent agents tested. The exception to this pattern was diclofenac, which completely inhibited both enzymes.
Table 4.
Residual COX activities after equilibration of hPGHS-2 and the V511A variant with selected COX inhibitors
| Inhibitor | Wild-typea residual COX activity (fraction of control)b |
V511A residual COX activity (fraction of control)b |
V511A/wild type |
|---|---|---|---|
| FR122047 | 0.96 ± 0.03 (n = 5) | 0.97 ± 0.04 (n = 5) | 1.0 |
| CAY-10404c | 0.78 ± 0.03 (n = 6) | 0.88 ± 0.01d (n = 8) | 1.1 |
| Tenidapc | 0.45 ± 0.01 (n = 5) | 0.60 ± 0.01d (n = 5) | 1.3 |
| SC-560 | 0.40 ± 0.01 (n =12) | 0.37 ± 0.01d (n = 10) | 0.9 |
| Nimesulidec | 0.17 ± 0.01 (n = 9) | 0.27 ± 0.01d (n = 6) | 1.6 |
| NS-398c | 0.035 ± 0.003 (n = 6) | 0.058 ± 0.002d (n = 6) | 1.7 |
| Valdecoxibc | 0.035 ± 0.003 (n = 5) | 0.042 ± 0.001e (n = 5) | 1.2 |
| Diclofenacc | 0 (n = 2) | 0 (n = 2) | — |
Major allele at all targeted residues.
The values shown represent the averages from reactions with 2 µm inhibitor.
Time-dependent COX inhibition with PGHS-2.
P < 0.0001.
P < 0.002.
The second experimental approach to comparing interactions of the hPGHS-2 wild type and the 488G and 511A variants with inhibitor involved pre-equilibration of a concentrated solution of enzyme with a small excess of nimesulide to convert the enzyme to a mixture of EI and EI′ forms. This mixture was then diluted 1500-fold into buffer without substrate and preincubated for varying lengths of time to allow redistribution of the EI′ complex into EI and E forms. Finally, a saturating level of AA was added to assay the increase in EI and E. The results are presented in Figure 3, normalized to the corresponding reference enzyme/inhibitor mixture that was assayed immediately on dilution. After a lag of ~ 25 s, the wild-type COX activity increased in a first-order manner, with a rate constant of 4.6 × 10−3 s−1 and a maximal increase of 4.0-fold. The COX activity of the 488G variant increased at 3.5 × 10−3 s−1, ~ 25% slower than wild type, to eventually reach a 3.5-fold increase in activity. The increase in COX activity on dilution was ~40% faster in the V511A variant (6.4 × 10−3 s−1) than in wild type, reaching a 3.1-fold increase. The clear differences in the rate of recovery of COX activity on dilution of the enzyme–inhibitor mixtures indicate that redistribution from the EI′ complex is faster in the 511A variant, and slower in the 488G variant, compared to wild-type PGHS-2. It is worth noting that the magnitude of the COX increase observed on dilution of the enzyme–nimesulide mixtures decreased in the order, wild type > 488G > 511A, as might be expected from the opposite rank order of residual activity observed in the experiments in Figure 2. Overall, the results of the second approach (Figure 3) corroborate the results from the first approach (Figure 2 and Tables 3 and 4), and establish that the 488G and 511A variants alter the interaction of hPGHS-2 with one or more COX-2 selective inhibitors.
Figure 3.

Kinetics of re-equilibration of tightly bound nimesulide complexes of hPGHS-2 wild type (WT) and the E488G and V511A variants upon 1500-fold dilution. The diluted samples were subsequently incubated for the indicated lengths of time before the COX activity was assayed by injection of 100 µm AA. Activities were normalized to control values obtained by dilution into buffer already containing 100 µm AA. Averages ± s.d. for two separate experiments are presented. Each set of data points was fitted to a single exponential equation with a 25 s lag.
Discussion
The results of this study show that two (488G and 511A) of the four hPGHS-2 variants tested have significant functional effects on the COX activity with arachidonate, on the enzyme’s substrate selectivity and on the interactions with COX-2 selective inhibitors. These functional effects are in the 20–70% range, consistent with the selective pressures expected for an enzyme that figures in several major pathophysiological processes. The in vivo impacts of polymorphisms are more readily evaluated when they result in large changes in enzyme kinetics. Nevertheless, there are a number of cases where physiological or clinical effects are linked to variants whose kinetic parameters after purification are at least 30% of the wild-type Vmax and less than three times the wild-type Km. Examples include glucose-6-phosphate dehydrogenase L382P in Drosophila melanogaster,37,38 chicken amylase S,39,40 human ubiquitin C-terminal hydrolase L1 I93M,41 several variants of human propionyl-CoA carboxylase,42 human serine hydroxymethyl transferase S394N,43 human P450 oxidoreductase A284P and V605F,44 and human coproporphyrinogen oxidase A814C.45 Given these precedents and the importance of COX-2 to prostanoid signaling pathways, it would seem worthwhile to evaluate more closely the impact of the V511A and E488G variants on the effectiveness of therapeutic intervention with coxibs or elevated n – 3 fatty acid intake.
The V511A polymorphism has been previously examined for functional changes and no major differences with wild type in COX kinetic parameters, product profile or inhibition by NSAID were reported.7,8 Despite this outcome, it was recognized that more enzymological analysis was needed to fully characterize possible functional impacts of the 511A allele.8 The present studies incorporate several significant methodological improvements for examination of V511A and three other PGHS-2 polymorphisms. These include the use of purified recombinant proteins to avoid host cell lipids that might modify the interactions with hydrophobic substrates and inhibitors, quantitation of the COX/POX ratio as a sensitive index of COX-selective effects, examination of substrate discrimination between the two major prostanoid precursors (AA and EPA) and use of inhibitor assays focused on the time-dependent inhibition that is the major mode of action of selective coxibs.46 These new approaches revealed some functional effects of the 511A variant that could not be detected in the earlier studies.
As depicted in Figure 4a, the V511A polymorphism is in a segment that forms part of the COX active site.47 Although the side chain of residue 511 is not positioned to interact with bound fatty acid or inhibitor in crystallographic structures of PGHS-2,47 a Val ⇒ Ala substitution at this position could plausibly change the protein packing and the position of bound substrate with respect to the catalytic radical at Tyr371 (Tyr385 in conventional ovine PGHS-1 numbering), indirectly affecting the COX catalytic rate. AA and EPA have differences in binding interactions in PGHS-148 and the situation is likely to be similar in PGHS-2; such differences might well underlie the differential impact of V511A on the COX Vmax values with EPA and AA as substrate (Figure 1a). Most coxibs compete with fatty acid binding at the COX site,49 so the structural changes just described can be invoked to explain the observed changes in inhibitor interactions in the 511A variant. An important structural determinant for COX-2 time-dependent inhibition has been identified just two residues away at Val509 (Figure 4a).36,50 This may have some connection to the observation that the V511A/wild-type differences tended to be larger with time-dependent coxibs than with SC-560 and FR122047, which showed only simple reversible inhibition of COX-2 (Table 4).
Figure 4.

Crystallographic structure of murine PGHS-2 (6cox.pdb), with views focused on Val511 (a) and Glu488 (b). Residue numbers are for human PGHS-2, with ovine PGHS-1 equivalents in parentheses. Other highlights include heme, bound inhibitor (SC-558), the tyrosyl radical sites at Tyr371(385) and Tyr490(504), the coxib specificity determinant at Val509(523), and the Arg442(456) and Tyr481(495) residues that interact with Glu488.
Crystallographic data for PGHS-247 show that Glu488 (Glu502 in ovine PGHS-1 numbering) is positioned at some distance from the COX active site (Figure 4b). Thus, the perturbation of substrate selectivity and inhibitor interactions observed in the 488G variant is not readily explained by local structural changes. However, the side chain of Glu488 appears to form a salt bridge with Arg442 and to be hydrogen bonded to the side chain of Tyr481. These interactions would be lost in the 488G variant, with the potential for long-range structural changes affecting active site events, such as the EI ⇔ EI′ transition. In addition, Glu488 is just two residues away from Tyr490 (Tyr504 in ovine PGHS-1 numbering; Figure 4b), the site of the other tyrosyl radical in PGHS-2.34 It will thus be of interest to examine tyrosyl radical dynamics in the 488G variant.
Aspirin and coxibs are potent chemopreventive agents for colorectal adenoma recurrence;51–55 aspirin has also been shown to prevent colorectal cancer with about a 10-year delay.56 As PGHS-2 is a major target of NSAIDs and coxibs as well as an important mediator of inflammation, polymorphisms in PTGS2 have been extensively examined for their association with colorectal adenoma or cancer risk.8,9,13,57–71 Although several studies have shown associations with noncoding PTGS2 polymorphisms, few studies have evaluated potential SNP–NSAIDs interactions.57,59,60,63,67,70,71 The V511A polymorphism of PTGS2 has significant prevalence only in the African-American population (thus far), and the E488G polymorphism appears to be very rare.7,8 The polymorphisms studied here exist with allele frequencies between 0.8 and 5% in the African-American and Asian populations. In prior studies, the populations carrying a variant allele were up to 5%, suggesting that a significant proportion of African Americans can experience different NSAID pharmacokinetics. Differing effects among men and women were not observed in one study,61 but should be evaluated further. Nevertheless, these polymorphisms could have important pharmacogenetic implications, given the role of NSAIDs in colorectal adenoma prevention. Coxibs (PGHS-2-specific inhibitors) are potent chemopreventive agents for colorectal adenoma recurrence, but their use is associated with increased risk of myocardial infarction and other cardiovascular events.72 Evaluations of these polymorphisms in relation to coxibs may provide information on who is most likely to benefit from coxib use and who may be at risk of adverse events.6 Given that the incidence of colorectal cancer is elevated in African Americans compared to Caucasians73 and that Japan has one of the highest colorectal cancer incidences in the world,74 further study is required to determine whether nonsynonymous polymorphisms in PTGS2 can influence the chemo-preventive efficacy of NSAIDs in populations that are likely to carry these polymorphisms. This information may be very useful for the tailoring of therapy in these populations so as to minimize the serious adverse events associated with NSAID use.6
The 511A variant has a lower relative Vmax for EPA than does the wild type (Figure 1a), indicating that this variant discriminates more strongly against the n – 3 substrate relative to the n – 6 substrate. Increased dietary intake of n – 3 fatty acids such as EPA has been found to bring health benefits.75 One proposed mechanism is the lower COX activity for EPA compared to AA seen with PGHS-1 and to a lesser extent with PGHS-2.1 Accordingly individuals carrying the 511A variant of PTGS2 might be expected to benefit more from dietary intervention designed to increase the n – 3/n – 6 ratio in tissue lipids than would individuals with the wild-type allele. A recent study12 reported no association between the V511A polymorphism and risk of chronic heart disease or ischemic stroke, but dietary fatty acid intake was not examined as an independent variable.
In summary, our biochemical studies show that two genetic variants in PGHS-2 can affect enzyme activity, substrate selectivity and inhibition with coxibs. Further studies are needed to evaluate whether these functional differences affect the efficacy of dietary and pharmacological interventions, particularly in African Americans, the population in which these two variants have been observed.
Supplementary Material
Acknowledgments
We thank Ms Liren Xiao for assistance with the statistical analysis. This work was financially supported by National Institutes of Health, USA (Grant CA 114467).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on The Pharmacogenomics Journal website (http://www.nature.com/tpj)
References
- 1.Smith WL. Nutritionally essential fatty acids and biologically indispensable cyclooxygenases. Trends Biochem Sci. 2008;33:27–37. doi: 10.1016/j.tibs.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 2.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 3.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mann JR, DuBois RN. Cyclooxygenase-2 and gastrointestinal cancer. Cancer J. 2004;10:145–152. doi: 10.1097/00130404-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–140. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- 7.Fritsche E, Baek SJ, King LM, Zeldin DC, Eling TE, Bell DA. Functional characterization of cyclooxygenase-2 polymorphisms. J Pharmacol Exp Ther. 2001;299:468–476. [PubMed] [Google Scholar]
- 8.Lin HJ, Lakkides KM, Keku TO, Reddy ST, Louie AD, Kau IH, et al. Prostaglandin H synthase 2 variant (Val511Ala) in African Americans may reduce the risk for colorectal neoplasia. Cancer Epidemiol Biomarkers Prev. 2002;11:1305–1315. [PubMed] [Google Scholar]
- 9.Goodman JE, Bowman ED, Chanock SJ, Alberg AJ, Harris CC. Arachidonate lipoxygenase (ALOX) and cyclooxygenase (COX) polymorphisms and colon cancer risk. Carcinogenesis. 2004;25:2467–2472. doi: 10.1093/carcin/bgh260. [DOI] [PubMed] [Google Scholar]
- 10.Sansbury LB, Bergen AW, Wanke KL, Yu B, Caporaso NE, Chatterjee N, et al. Inflammatory cytokine gene polymorphisms, nonsteroidal anti-inflammatory drug use, and risk of adenoma polyp recurrence in the polyp prevention trial. Cancer Epidemiol Biomarkers Prev. 2006;15:494–501. doi: 10.1158/1055-9965.EPI-05-0763. [DOI] [PubMed] [Google Scholar]
- 11.Moorman PG, Sesay J, Nwosu V, Kane JG, de Cotret AR, Worley K, et al. Cyclooxygenase 2 polymorphism (Val511Ala), nonsteroidal anti-inflammatory drug use and breast cancer in African American women. Cancer Epidemiol Biomarkers Prev. 2005;14:3013–3014. doi: 10.1158/1055-9965.EPI-05-0291. [DOI] [PubMed] [Google Scholar]
- 12.Lee CR, North KE, Bray MS, Couper DJ, Heiss G, Zeldin DC. Cyclooxygenase polymorphisms and risk of cardiovascular events: the Atherosclerosis Risk in Communities (ARIC) study. Clin Pharmacol Ther. 2008;83:52–60. doi: 10.1038/sj.clpt.6100221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulrich CM, Whitton J, Yu JH, Sibert J, Sparks R, Potter JD, et al. PTGS2 (COX-2) −765G > C promoter variant reduces risk of colorectal adenoma among nonusers of nonsteroidal anti-inflammatory drugs. Cancer Epidemiol Biomarkers Prev. 2005;14:616–619. doi: 10.1158/1055-9965.EPI-04-0510. [DOI] [PubMed] [Google Scholar]
- 14.Ulrich CM, Bigler J, Sparks R, Whitton J, Sibert JG, Goode EL, et al. Polymorphisms in PTGS1 ( = COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2004;13:889–893. [PubMed] [Google Scholar]
- 15.Poole EM, Bigler J, Whitton J, Sibert JG, Potter JD, Ulrich CM. Prostacyclin synthase and arachidonate 5-lipoxygenase polymorphisms and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2006;15:502–508. doi: 10.1158/1055-9965.EPI-05-0804. [DOI] [PubMed] [Google Scholar]
- 16.Choi JH, Park HS, Oh HB, Lee JH, Suh YJ, Park CS, et al. Leukotriene-related gene polymorphisms in ASA-intolerant asthma: an association with a haplotype of 5-lipoxygenase. Hum Genet. 2004;114:337–344. doi: 10.1007/s00439-004-1082-1. [DOI] [PubMed] [Google Scholar]
- 17.Hamajima N, Takezaki T, Matsuo K, Saito T, Inoue M, Hirai T, et al. Genotype frequencies of cyclooxygenease 2 (COX2) rare polymorphisms for Japanese with and without colorectal cancer. Asian Pac J Cancer Prev. 2001;2:57–62. [PubMed] [Google Scholar]
- 18.Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, et al. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J Biol Chem. 2007;282:22254–22266. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- 19.Kulmacz RJ. Regulation of cyclooxygenase catalysis by hydroperoxides. Biochem Biophys Res Commun. 2005;338:25–33. doi: 10.1016/j.bbrc.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 20.Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Pawelek TR, Kulmacz RJ. Hydroperoxide dependence and cooperative cyclooxygenase kinetics in prostaglandin H synthase-1 and −2. J Biol Chem. 1999;274:20301–20306. doi: 10.1074/jbc.274.29.20301. [DOI] [PubMed] [Google Scholar]
- 22.Smith T, Leipprandt J, DeWitt D. Purification and characterization of the human recombinant histidine-tagged prostaglandin endoperoxide H synthases-1 and −2. Arch Biochem Biophys. 2000;375:195–200. doi: 10.1006/abbi.1999.1659. [DOI] [PubMed] [Google Scholar]
- 23.Bambai B, Rogge CE, Stec B, Kulmacz RJ. Role of Asn-382 and Thr-383 in activation and inactivation of human prostaglandin H synthase cyclooxygenase catalysis. J Biol Chem. 2004;279:4084–4092. doi: 10.1074/jbc.M304762200. [DOI] [PubMed] [Google Scholar]
- 24.Kulmacz RJ, Wang LH. Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and −2. J Biol Chem. 1995;270:24019–24023. doi: 10.1074/jbc.270.41.24019. [DOI] [PubMed] [Google Scholar]
- 25.MirAfzali Z, Leipprandt JR, McCracken JL, DeWitt DL. Fast, efficient reconstitution of the cyclooxygenases into proteoliposomes. Arch Biochem Biophys. 2005;443:60–65. doi: 10.1016/j.abb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Tsai A-L, Wu G, Palmer G, Bambai B, Koehn JA, Marshall PJ, et al. Rapid kinetics of tyrosyl radical formation and heme redox state changes in prostaglandin H synthase-1 and −2. J Biol Chem. 1999;274:21695–21700. doi: 10.1074/jbc.274.31.21695. [DOI] [PubMed] [Google Scholar]
- 27.Peterson GL. Determination of total protein. Methods Enzymol. 1983;91:95–119. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- 28.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 30.Kulmacz RJ, Lands W. Cyclo-oxygenase: measurement, purification and properties. In: Benedetto C, McDonald-Gibson RG, Nigam S, Slater TF, editors. Prostaglandins and Related Substances: A Practical Approach. Washington, DC: IRL Press; 1987. pp. 209–227. [Google Scholar]
- 31.Liu W, Cao D, Oh SF, Serhan CN, Kulmacz RJ. Divergent cyclooxygenase responses to fatty acid structure and peroxide level in fish and mammalian prostaglandin H synthases. FASEB J. 2006;20:1097–1108. doi: 10.1096/fj.05-5273com. [DOI] [PubMed] [Google Scholar]
- 32.Smith WL, Lands WE. Oxygenation of unsaturated fatty acids by soybean lipoxygenase. J Biol Chem. 1972;247:1038–1047. [PubMed] [Google Scholar]
- 33.Kulmacz RJ. Concerted loss of cyclooxygenase and peroxidase activities from prostaglandin H synthase upon proteolytic attack. Prostaglandins. 1989;38:277–288. doi: 10.1016/0090-6980(89)90133-0. [DOI] [PubMed] [Google Scholar]
- 34.Rogge CE, Liu W, Wu G, Wang LH, Kulmacz RJ, Tsai AL. Identification of Tyr504 as an alternative tyrosyl radical site in human prostaglandin H synthase-2. Biochemistry. 2004;43:1560–1568. doi: 10.1021/bi035717o. [DOI] [PubMed] [Google Scholar]
- 35.Rogge CE, Ho B, Liu W, Kulmacz RJ, Tsai AL. Role of Tyr348 in Tyr385 radical dynamics and cyclooxygenase inhibitor interactions in prostaglandin H synthase-2. Biochemistry. 2006;45:523–532. doi: 10.1021/bi051235w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo Q, Wang LH, Ruan KH, Kulmacz RJ. Role of Val509 in time-dependent inhibition of human prostaglandin H synthase-2 cyclooxygenase activity by isoform-selective agents. J Biol Chem. 1996;271:19134–19139. doi: 10.1074/jbc.271.32.19134. [DOI] [PubMed] [Google Scholar]
- 37.Eanes WF, Katona L, Longtine M. Comparison of in vitro and in vivo activities associated with the G6PD allozyme polymorphism in Drosophila melanogaster. Genetics. 1990;125:845–853. doi: 10.1093/genetics/125.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labate J, Eanes WF. Direct measurement of in vivo flux differences between electrophoretic variants of G6PD from Drosophila melanogaster. Genetics. 1992;132:783–787. doi: 10.1093/genetics/132.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gapusan RA, Yardley DG, Hughes BL. The amylase gene-enzyme system of chickens II Biochemical characterization of allozymes. Biochem Genet. 1990;28:553–560. doi: 10.1007/BF00553947. [DOI] [PubMed] [Google Scholar]
- 40.Hughes BL, Suniga RG, Yardley DG. Influence of amylase genotypes on growth rate and feed conversion of chickens. Poult Sci. 1994;73:953–957. doi: 10.3382/ps.0730953. [DOI] [PubMed] [Google Scholar]
- 41.Nishikawa K, Li H, Kawamura R, Osaka H, Wang YL, Hara Y, et al. Alterations of structure and hydrolase activity of parkinsonism-associated human ubiquitin carboxyl-terminal hydrolase L1 variants. Biochem Biophys Res Commun. 2003;304:176–183. doi: 10.1016/s0006-291x(03)00555-2. [DOI] [PubMed] [Google Scholar]
- 42.Jiang H, Rao KS, Yee VC, Kraus JP. Characterization of four variant forms of human propionyl-CoA carboxylase expressed in Escherichia coli. J Biol Chem. 2005;280:27719–27727. doi: 10.1074/jbc.M413281200. [DOI] [PubMed] [Google Scholar]
- 43.Fu TF, Hunt S, Schirch V, Safo MK, Chen BH. Properties of human and rabbit cytosolic serine hydroxymethyltransferase are changed by single nucleotide polymorphic mutations. Arch Biochem Biophys. 2005;442:92–101. doi: 10.1016/j.abb.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 44.Pandey AV. Biochemical analysis of mutations in P450 oxidoreductase. Biochem Soc Trans. 2006;34(Part 6):1186–1191. doi: 10.1042/BST0341186. [DOI] [PubMed] [Google Scholar]
- 45.Li T, Woods JS. Cloning, expression, and biochemical properties of CPOX4, a genetic variant of coproporphyrinogen oxidase that affects susceptibility to mercury toxicity in humans. Toxicol Sci. 2009;109:228–236. doi: 10.1093/toxsci/kfp066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Copeland RA, Williams JM, Giannaras J, Nurnberg S, Covington M, Pinto D, et al. Mechanism of selective inhibition of the inducible isoform of prostaglandin G/H synthase. Proc Natl Acad Sci USA. 1994;91:11202–11206. doi: 10.1073/pnas.91.23.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 48.Malkowski MG, Thuresson ED, Lakkides KM, Rieke CJ, Micielli R, Smith WL, et al. Structure of eicosapentaenoic and linoleic acids in the cyclooxygenase site of prostaglandin endoperoxide H synthase-1. J Biol Chem. 2001;276:37547–37555. doi: 10.1074/jbc.M105982200. [DOI] [PubMed] [Google Scholar]
- 49.Marnett LJ, Rowlinson SW, Goodwin DC, Kalgutkar AS, Lanzo CA. Arachidonic acid oxygenation by COX-1 and COX-2 Mechanisms of catalysis and inhibition. J Biol Chem. 1999;274:22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- 50.Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and −2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271:15810–15814. doi: 10.1074/jbc.271.26.15810. [DOI] [PubMed] [Google Scholar]
- 51.Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 52.Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 53.Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 54.Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131:1674–1682. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 55.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 56.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–1613. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 57.Siezen CL, Bueno-de-Mesquita HB, Peeters PH, Kram NR, van Doeselaar M, van Kranen HJ. Polymorphisms in the genes involved in the arachidonic acid-pathway, fish consumption and the risk of colorectal cancer. Int J Cancer. 2006;119:297–303. doi: 10.1002/ijc.21858. [DOI] [PubMed] [Google Scholar]
- 58.Ali IU, Luke BT, Dean M, Greenwald P. Allellic variants in regulatory regions of cyclooxygenase-2: association with advanced colorectal adenoma. Br J Cancer. 2005;93:953–959. doi: 10.1038/sj.bjc.6602806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox DG, Pontes C, Guino E, Navarro M, Osorio A, Canzian F, et al. Polymorphisms in prostaglandin synthase 2/cyclooxygenase 2 (PTGS2/COX2) and risk of colorectal cancer. Br J Cancer. 2004;91:339–343. doi: 10.1038/sj.bjc.6601906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira C, Pimentel-Nunes P, Brandao C, Moreira-Dias L, Medeiros R, Dinis-Ribeiro M. COX-2 polymorphisms and colorectal cancer risk: a strategy for chemoprevention. Eur J Gastroenterol Hepatol. 2010;22:607–613. doi: 10.1097/MEG.0b013e3283352cbb. [DOI] [PubMed] [Google Scholar]
- 61.Sansbury LB, Millikan RC, Schroeder JC, North KE, Moorman PG, Keku TO, et al. COX-2 polymorphism, use of nonsteroidal anti-inflammatory drugs, and risk of colon cancer in African Americans (United States) Cancer Causes Control. 2006;17:257–266. doi: 10.1007/s10552-005-0417-0. [DOI] [PubMed] [Google Scholar]
- 62.Gunter MJ, Canzian F, Landi S, Chanock SJ, Sinha R, Rothman N. Inflammation-related gene polymorphisms and colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1126–1131. doi: 10.1158/1055-9965.EPI-06-0042. [DOI] [PubMed] [Google Scholar]
- 63.Tan W, Wu J, Zhang X, Guo Y, Liu J, Sun T, et al. Associations of functional polymorphisms in cyclooxygenase-2 and platelet 12-lipoxygenase with risk of occurrence and advanced disease status of colorectal cancer. Carcinogenesis. 2007;28:1197–1201. doi: 10.1093/carcin/bgl242. [DOI] [PubMed] [Google Scholar]
- 64.Hubner RA, Muir KR, Liu JF, Logan RF, Grainge MJ, Houlston RS. Polymorphisms in PTGS1, PTGS2 and IL-10 do not influence colorectal adenoma recurrence in the context of a randomized aspirin intervention trial. Int J Cancer. 2007;121:2001–2004. doi: 10.1002/ijc.22942. [DOI] [PubMed] [Google Scholar]
- 65.Ueda N, Maehara Y, Tajima O, Tabata S, Wakabayashi K, Kono S. Genetic polymorphisms of cyclooxygenase-2 and colorectal adenoma risk: the Self Defense Forces Health Study. Cancer Sci. 2008;99:576–581. doi: 10.1111/j.1349-7006.2007.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xing LL, Wang ZN, Jiang L, Zhang Y, Xu YY, Li J, et al. Cyclooxygenase 2 polymorphism and colorectal cancer: −765G> C variant modifies risk associated with smoking and body mass index. World J Gastroenterol. 2008;14:1785–1789. doi: 10.3748/wjg.14.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ashktorab H, Tsang S, Luke B, Sun Z, Adam-Campbell L, Kwagyan J, et al. Protective effect of Cox-2 allelic variants on risk of colorectal adenoma development in African Americans. Anticancer Res. 2008;28:3119–3123. [PMC free article] [PubMed] [Google Scholar]
- 68.Gong Z, Bostick RM, Xie D, Hurley TG, Deng Z, Dixon DA, et al. Genetic polymorphisms in the cyclooxygenase-1 and cyclooxygenase-2 genes and risk of colorectal adenoma. Int J Colorectal Dis. 2009;24:647–654. doi: 10.1007/s00384-009-0656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson CL, Plummer SJ, Merkulova A, Cheng I, Tucker TC, Casey G, et al. No association between cyclooxygenase-2 and uridine diphosphate glucuronosyltransferase 1A6 genetic polymorphisms and colon cancer risk. World J Gastroenterol. 2009;15:2240–2244. doi: 10.3748/wjg.15.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barry EL, Sansbury LB, Grau MV, Ali IU, Tsang S, Munroe DJ, et al. Cyclooxygenase-2 polymorphisms, aspirin treatment, and risk for colorectal adenoma recurrence—data from a randomized clinical trial. Cancer Epidemiol Biomarkers Prev. 2009;18:2726–2733. doi: 10.1158/1055-9965.EPI-09-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoff JH, te Morsche RH, Roelofs HM, van der Logt EM, Nagengast FM, Peters WH. COX-2 polymorphisms −765G–> C and −1195A–> G and colorectal cancer risk. World J Gastroenterol. 2009;15:4561–4565. doi: 10.3748/wjg.15.4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bertagnolli MM. Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncol. 2007;8:439–443. doi: 10.1016/S1470-2045(07)70139-0. [DOI] [PubMed] [Google Scholar]
- 73.Horner MJ, Ries LAG, Krapcho M, Neyman N, Aminou R, Howlader N, et al., editors. SEER Cancer Statistics Review, 1975–2006. Bethesda, MD: National Cancer Institute; http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site, 2009. [Google Scholar]
- 74.Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59:366–378. doi: 10.3322/caac.20038. [DOI] [PubMed] [Google Scholar]
- 75.Ruxton CH, Reed SC, Simpson MJ, Millington KJ. The health benefits of omega-3 polyunsaturated fatty acids: a review of the evidence. J Hum Nutr Diet. 2004;17:449–459. doi: 10.1111/j.1365-277X.2004.00552.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.