

Abstract
Sepsis, defined as a clinical syndrome brought about by an amplified and dysregulated inflammatory response to infections, is one of the leading causes of death worldwide. Despite persistent attempts to develop treatment strategies to manage sepsis in the clinical setting, the basic elements of treatment have not changed since the 1960s. As such, the development of effective therapies for reducing inflammatory reactions and end-organ dysfunction in critically ill patients with sepsis remains a global priority. Advances in understanding of the immune response to sepsis provide the opportunity to develop more effective pharmaceuticals. This article details current information on the modulation of the lipopolysaccharide (LPS) receptor complex with synthetic Lipid A mimetics. As the initial and most critical event in sepsis pathophysiology, the LPS receptor provides an attractive target for antisepsis agents. One of the well-studied approaches to sepsis therapy involves the use of derivatives of Lipid A, the membrane-anchor portion of an LPS, which is largely responsible for its endotoxic activity. This article describes the structural and conformational requirements influencing the ability of Lipid A analogues to compete with LPS for binding to the LPS receptor complex and to inhibit the induction of the signal transduction pathway by impairing LPS-initiated receptor dimerization.
Keywords: Endotoxin, Immune response, Inflammatory reactions, Lipid A, Lipo-polysaccharide, LPS receptor complex, Mimetics, Sepsis, Toll-like receptor
I. Introduction
Interactions between the mammalian and microbial worlds are intimate, complex, and vital to the good health of both.1 Over the course of almost a billion years, the mutual accommodation between microorganisms and multicellular hosts has enabled us both to survive and adapt to a changing environment. The evolution of multicellular organisms would not have been possible without intracellular microbial parasites, that is, the basic processes of cellular respiration in eukaryotes are possible only because of the presence of a microbial parasite in the cell called the mitochondrion.2 However, this intricate, symbiotic relationship between humans and microbes has its darker side. From parasitic and acute infections to chronic illnesses such as peptic ulcer disease, cancer, and coronary heart disease, microorganisms have triggered a plethora of other human diseases.3 In response, the innate immunity in humans has evolved into a complex system that enables it to respond to a microbial threat and achieve a survival advantage. The latter is often accomplished by exploiting features unique to the threat, just as the microbes have used the same defensive and subversive strategies to circumvent the human immune system.1
Accurate recognition of pathogen-associated molecular patterns (PAMPs) by pattern-recognition receptors (PRRs) is the cornerstone of the innate immune response.4 A systemic activation of PRRs usually implies loss of control of the host’s immune-response mechanisms and a rapid and vast nonspecific response known as the systemic inflammatory response syndrome (SIRS) may result. SIRS is a clinical condition defined by at least two of the four following criteria: (1) temperature below 36 or above 38 °C, (2) heart rate above 90 beats/min, (3) respiratory rate above 20 breaths/min or pCO2 above 4.3 kPa, (4) white blood-cell count below 4 × 109/L or above 12 × 109/L, or more than 10% immature neutrophils.5 When SIRS occurs in response to an infection, it is defined as sepsis. The disruption of homeostatic balance as a consequence of sepsis leads to massive production of proinflammatory mediators and dysregulation of the anti-inflammatory mechanisms. Sepsis, therefore, can be described as a pro- and anti-inflammatory disequilibrium syndrome.6 The high mortality from sepsis is mostly due to an evolving multiorgan dysfunction resulting from local changes in blood flow, namely, sepsis-induced hypotension, diffuse intravascular coagulation, and cytokine-induced abnormalities to microcirculation.7 A study by Angus et al. linking hospital discharge records from seven large states in the USA identified 192,980 cases of severe sepsis out of the 6,621,559 patients admitted.8 The average cost per case was $22,100 and it adds to annual total costs of $16.7 billion in the USA alone. Despite many attempts to develop treatment strategies to manage sepsis in the clinical setting, the basic elements of treatment have not changed since the 1960s. As such, development of effective therapies for reducing inflammatory reactions and end-organ dysfunction in critically ill patients with sepsis remains a global priority.
Understanding the mechanisms underlying the recognition of invading pathogens through PRRs is of great interest when considering potential treatments. In the past four decades, our insight into how the immune system senses and identifies infective microorganisms to trigger the innate immune response, along with the link between innate and adaptive immunity, has improved dramatically. This understanding began with Hoffmann’s pioneering discovery of the role of the Toll receptor in the defense against infections of the fruit fly Drosophila melanogaster.9 Fruit flies lack an adaptive immune system, and its host defense against infections relies solely on the innate immune response. Using flies having mutations in genes of the Toll signaling pathway, Hoffmann and coworkers were able to demonstrate that Toll controlled the expression of antimicrobial peptide genes, and that deficient Toll signaling dramatically reduced survival after fungal infection. Subsequently, Toll was also found to be involved in the antibacterial defense of Drosophila.10
These results were extended to mammals after Beutler’s discovery of homologous receptors, named Toll-like receptors (TLRs), in humans and in mice.11 While searching for the receptor of the Gram-negative bacterial product involved in endotoxic shock (endotoxins, also known as lipopolysaccharides or LPSs), Beutler and coworkers observed that mice which displayed resistance to the LPS challenge carried constitutive mutations in a gene similar to the Drosophila Toll gene. This gene coded for a receptor now known as TLR4, and the latter was found subsequently to be a key player in the LPS receptor complex.
II. Immune Response to LPSs
In the 1890s, Pfeiffer and Centanni independently described a heat-stable pyrogenic toxin intrinsic to Vibrio cholerae and Salmonella typhi.12 Pfeiffer initially called it “endotoxin,” and in the 1930s, Boivin was able to extract and purify it using trichloroacetic acid.13 Endotoxin purified through Boivin’s method, however, was essentially a crude fraction containing many cell-wall proteins. This slowed the progress towards understanding the role of endotoxins and their biological impact remained inconclusive. In 1946, Westphal and Lüderitz were finally able to develop a method for obtaining pure active fractions of “endotoxin,” which was shown to be LPS.14
LPSs are composed of three genetically, structurally, and antigenically distinct regions namely: (1) a hydrophobic membrane anchor called Lipid A; (2) a short chain of sugar residues with multiple phosphoryl substituents referred to as the core oligosaccharide; and (3) a structurally diverse, serospecific polymer composed of oligosaccharide repeat-units called the O-antigen (Fig. 1). While the LPSs of mucosal pathogens, for example, of Haemophilus and Neisseria species, lack the typical long-chain repeating O-antigen, the basic tripartite LPS framework stands for all characterized Gram-negative bacteria.
Fig. 1.
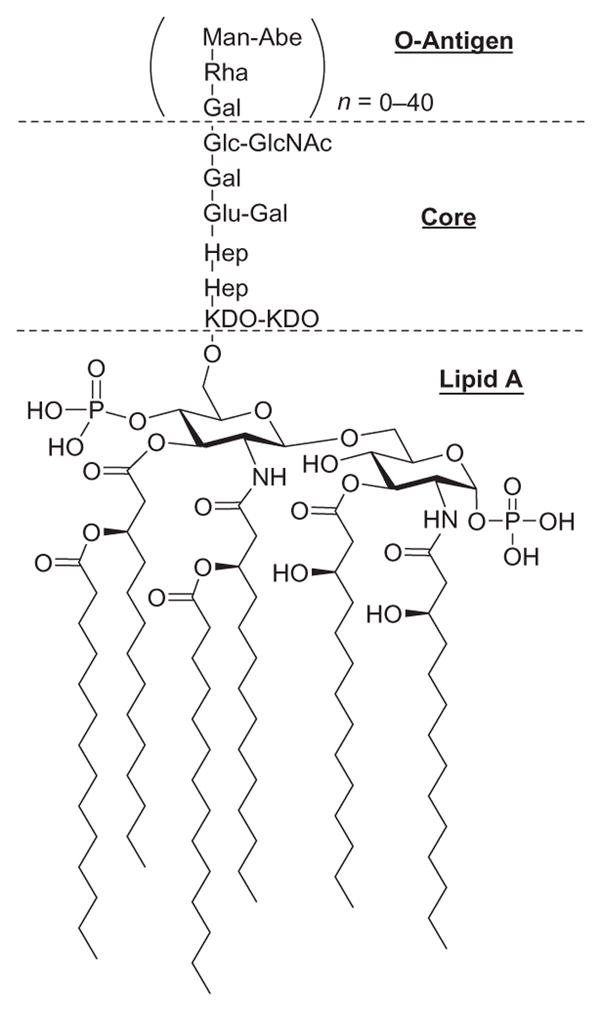
Structure of LPS and its typical antigenic regions: Lipid A, core oligosaccharide, and O-antigen.
Lipid A, the covalently linked lipid component of LPSs, is composed of six or more fatty acid residues linked to two phosphorylated glucosamine residues.15 Four of the fatty acids have an (R)-3-hydroxyl group and the other two are devoid of it. Each Gram-negative bacterial species has a unique Lipid A composition, and the structural features that differentiate each molecule are as follows. First, the acylation pattern on each glucosamine residue can have either a symmetric (3+3) or an asymmetric (4+2) arrangement. Second, three or four different fatty acids can be present in the molecule, with a chain length that can be anywhere between 10 and 16 carbon atoms. Finally, a 4-amino-deoxy-L-arabinose and/or phosphonoethanolamine can be linked to the C-1 axial or C-4′ phosphate groups on the glucosamine residues.
The core region is a short polysaccharide chain showing moderate interbacterial variability. The inner core consists of two or more 3-deoxy-D-manno-octulosonic acid (Kdo) residues linked to C-6′ of the two glucosamine residues (Lipid A) on one side and two or three L-glycero-D-manno-heptose residues on the other.16 In should be noted that both Kdo and L-glycero-D-manno-heptose are unique to bacterial species.17 Under natural conditions, the smallest LPS produced by Gram-negative bacteria is Re-LPS—it consists of Lipid A with one or two Kdo residues—but longer LPSs are more common.18 The Rd1- and Rd2-LPS serotypes contain a complete inner core and an inner core without two heptose residues, respectively.19 The outer core, on the other hand, consists of common sugars and is more variable than the inner core. It is normally two to three residues long and has one or more covalently bound polysaccharides as side chains.20 LPSs consisting of the Lipid A and the complete inner and outer core are denoted Ra-LPS, whereas the Rb- and Rc-LPS serotypes only contain a part of the outer core.
The O-antigenic outer-core portion is the most variable part and consists of repeating units of oligosaccharides. Attached to the terminal sugar of the inner core, this portion extends from the bacterial surface and is highly immunogenic.21 The chemical composition and structure of the O-antigen can be strain-specific (interstrain LPS heterogeneity), or it can vary within one bacterial strain (intra-strain LPS heterogeneity).22 The inter- and intra-strain heterogeneity is characterized by variations at different levels. The first variation can occur through nonstoichiometric modification of the O-antigens with sugar moieties, that is, with glucosyl and fucosyl residues. The second variation occurs via addition of noncarbohydrate substituents—such as acetyl or methyl groups—to the O-antigen. This may arise with regularity, although, in most cases, these modifications are also nonstoichiometric. The length of the O-antigen may vary from 0 to as many as 40 repeating units, but it generally consists of 20 to 40 repeating units.
LPSs participate in physiological functions of the membrane and are therefore essential for bacterial growth and viability.23 They contribute to low membrane permeability and enhance the resistance toward hydrophobic agents. LPSs are not toxic while they remain incorporated in the bacterial outer membrane. When released from the bacterial surface—either following cell division or death, as a consequence of antibacterial action of the immune system, or interaction with antibacterial agents—LPSs may form aggregates and interact with the cells of the immune system. Following this interaction, LPSs elicit multiple acute pathophysiological effects, such as fever, toxicity, Schwartzman reactivity, macrophage, and B-lymphocyte activation, among others.24 In 1954, it was proposed that the Lipid A portion alone is responsible for the endotoxic properties of LPSs, and that the polysaccharide portion is dispensable.25 After Shiba and coworkers completed the first targeted synthesis of the Lipid A of Escherichia coli,26 comparative experiments between the natural LPS and the synthetic Lipid A confirmed that it is Lipid A that constitutes the source of toxicity of LPSs.27
Under physiological conditions, the immune cells are continuously exposed to low levels of LPSs derived from gastrointestinal bacteria. These LPSs are taken up by macrophages and may be essential to maintain a basal level of attentiveness of the immune system. It was originally believed that LPSs activated the immune cells through a nonspecific mechanism that involved intercalation of Lipid A into the mammalian lipid bilayer.28 In the early 1980s, reports emerged suggesting that the biological actions of LPSs were facilitated by their binding to endogenous proteins. Indeed, the response of a host cell to LPSs is highly dependent on whether it encounters the latter in free or bound form.29
Over the past 20 years, one of the major aims in LPS research has been to elucidate the exact sequence of events from when the LPS binds to the cell to when it elicits a response from it. As mentioned earlier, distinct plasma membrane proteins mediate the initial interaction between LPSs (Lipid A) and phagocytes (monocytes, macrophages, polymorphonuclear leukocytes (PMNs)). Some of these interactions may be solely involved in the removal and eventual degradation of LPSs, whereas others may play a critical role in transmembrane signaling. The receptors involved in transmembrane signaling are shown in Fig. 2 and are discussed in the succeeding sections.
Fig. 2.
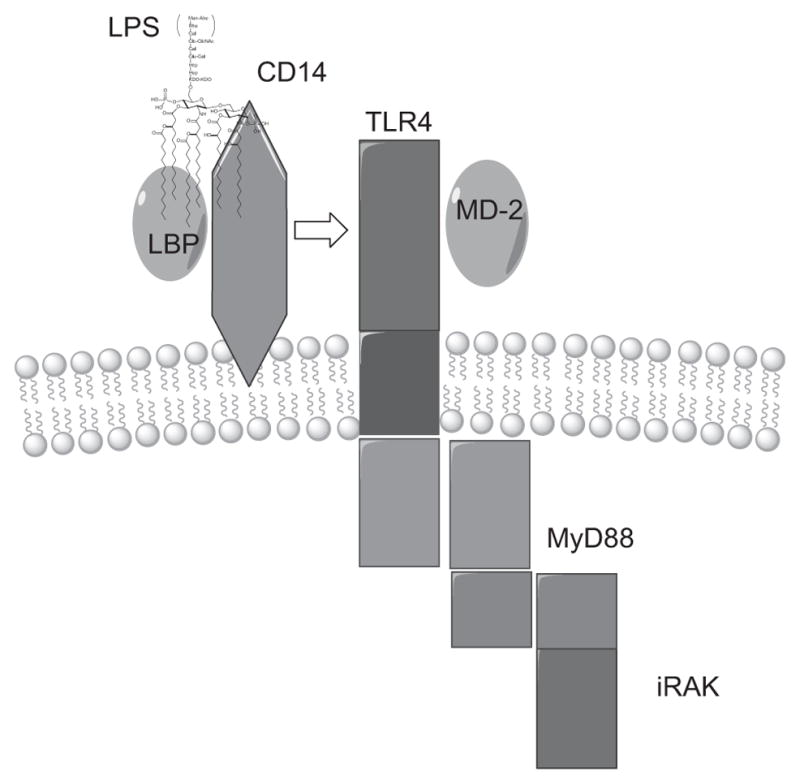
Recognition of LPS on the surface of phagocytes. LPS is opsonized by lipopolysaccharide-binding protein (LBP), and the LPS–LBP complex is recognized by cluster of differentiation 14 (CD14). CD14 is incapable of generating a transmembrane signal, and subsequently, the LPS–LBP–CD14 ternary complex activates toll-like receptor 4 (TLR4). TLR4, in turn, signals through the Myeloid differentiation primary response gene (88) (MyD88) and the interleukin-1 receptor-associated kinase (IRAK). Myeloid differentiation 2 (MD-2) is a secreted protein that binds to the extracellular domain of TLR4 and is an important component of its signaling pathway.30 (See the color plate.)
III. The LPS Receptor Complex
1. LPS-Binding Protein
While studying binding of LPSs to high-density lipoproteins (HDLs) in normal and acute-phase rabbit serum, Tobias and coworkers observed that LPS was mainly complexed to a protein in the acute-phase serum.31 Isolation of this protein from the rabbit serum led to the discovery of the LPS-binding protein (LBP). LBP was recovered as a glycoprotein having molecular weight of 58 and 60.5 kDa, wherein the difference in molecular mass reflects different degrees of glycosylation.31,32 It is synthesized primarily by hepatocytes and released into the bloodstream after glycosylation.33 Other sources of LBP include epithelial cells of the skin, the lung, the intestine, and human gingival tissues, as well as the small-muscle cells of the lung arteries, heart muscle cells, and renal cells.34
Human LBP consists of 452 amino acids and has the characteristic 25-amino acid signal sequence of secreted proteins.33 Its amino acid sequence revealed a sequence homology to bactericidal/permeability-increasing protein (BPI), cholesteryl ester transfer protein (CETP), and phospholipid transfer protein (PLTP) of 45%, 23%, and 25%, respectively. It has also suggested a similarity in the tertiary structure of these proteins. After the three-dimensional structure of BPI was determined by X-ray crystallography, it provided a useful framework for modeling the three-dimensional structure of the LT/LBP family.35 BPI appears as a boomerang-shaped molecule and consists of two symmetrical barrel domains connected by a proline-rich linker region. Each domain is composed of an antiparallel β-stranded layer twisted around a long α-helix and forms a hydrophobic pocket that can incorporate one phosphatidyl choline molecule. From these observations, Beamer et al. proposed a simulated three-dimensional model for LBP that is very similar to the structurally and functionally related BPI.36 Analogously with BPI, the LPS-binding domain of LBP is located at the N-terminal region. Indeed, the three-dimensional LBP model showed that the cationic cluster of the LPS-binding site is fully exposed at the N-terminal tip.36
To verify the veracity of the claim that the LPS-binding site of LBP is at the N-terminus, Lamping et al. performed mutagenesis experiments.37 LBP mutants with amino acid exchanges within the N-terminal region were expressed and tested in five different functional assays—binding to immobilized LPSs, facilitation of binding of LPS aggregates to monocytes, transfer of LPS monomers from aggregates to other LPS receptors, transfer of LPS monomers to HDL, and enhancement of LPS-induced cell activation. The double mutant glutamic acid 94/95 was completely lacking LPS binding, transfer, and cell-stimulatory activity, indicating that the integrity of amino acids 94 and 95 is required for LBP function.37 While mutations of the amino acids Arginine-94 or Lysine-95 into alanine diminished the LPS-binding activity of LBP dramatically, the ability to facilitate binding of LPS aggregates to membrane-bound cluster of differentiation 14 (mCD14) at the cell surface was retained. These findings emphasize the distinction between binding of LPS aggregates to cells and the binding of LPS monomers to CD14—the former is not associated with cell stimulation and the latter leads to cell stimulation.
Studies show that LBP has a dual role in interactions with LPSs. At low LPS concentrations, LBP enhances LPS signaling by extracting it from the bacterial membranes (LPS monomerization) and transferring the LPS monomers to CD14.38 At high concentrations, it inhibits the LPS signaling by shuttling the LPS to the serum lipoproteins and by forming aggregates with LPS.39 Indeed, the increased secretion of LBP as a result of LPS stimulation serves as an inhibitor of excessive response to LPS in the serum of septic patients.
The two domains of LBP have different functions. As with LBP and its homologue BPI, the amino-terminal domain has a high affinity for LPSs.40 The C-terminal domain, on the other hand, is required for the interaction with CD14 or the cell membrane.41 Adding LBP to a serum-free cell system enhances the LPS-mediated stimulation of CD14-positive cells by 100- to 1000-fold.33,42 In addition, LBP also transfers LPS to soluble CD14 (sCD14), resulting in the activation of mCD14-negative cells (endothelial and epithelial cells). Thus, the ability of LBP to transfer disaggregated LPS to both mCD14 and sCD14 supports the view that LBP has a central role in mediating LPS responses. It was proposed that a single LBP molecule is able to transport hundreds of LPS molecules to sCD14, and that LBP is not consumed by this reaction.43 Consequently, Yu and Wright demonstrated first-order kinetics for this enzymatic transfer and were further able to define catalytic constants for this reaction.38b,43 To explain the catalytic reaction mechanism for the transfer of LPS to sCD14, two models were proposed. The “binary complex” model proposes that the initial step in the transfer involves a bimolar reaction between LBP and an LPS micelle. Following dissociation from the micelle with one molecule of LPS bound, LBP then binds to sCD14.43 The “ternary complex” model, on the other hand, suggests a simultaneous interaction among LBP, LPS micelles, and sCD14.44
2. Cluster of Differentiation 14
While it was clear that the CD18 complex interacts with LPS by bridging bacteria to the surface of phagocytes,45 it was not clear whether this interaction actually triggers cellular responses. To elucidate the exact role that CD18 plays in the cellular activation by LPS, Wright et al. performed experiments on mononuclear cells from CD18-deficient patients.46 From these experiments, it became clear that CD18-deficient cells can bind LPS and that the binding event can result in cellular activation. Clearly, additional receptors must be present on the surface of macrophages and PMNs. Subsequently, Wright and coworkers identified this unknown receptor to be CD14—a differentiation antigen of monocytes.47 Based on their report, CD14 binds complexes of LPS and LBP, and the blockade of CD14 with anti-CD14 antibodies prevents further binding of LPS-coated erythrocytes to macrophages. With the absence of a binding event, macrophages are unable to produce an LPS-induced inflammatory response. Golenbock and coworkers corroborated these findings by demonstrating that LPS-induced responsiveness can be transferred to a heterologous nonresponder cell type by expression of a single leukocyte-specific gene product.48 Thus, transfection of human CD14 into Chinese hamster ovary (CHO) fibroblasts and treatment of CD14-bearing CHO cells with LPS led to a macrophage-like responsiveness in otherwise LPS-unresponsive cells. Similarly, Lee and coworkers showed that CD14-bearing 70Z/3 cells bind LPS, and when LPS is complexed with LBP, the binding activity is even higher.49 Consequently, Kirkland and coworkers determined the binding affinity of the LPS–LBP complex to CD14-transfected CHO cells and THP-1 cells and found Kd values of 2.7 × 10−8 to 4.8 × 10−8 M.50
CD14, a serum/cell surface glycoprotein and the first PRR to be described, is usually found in two forms: membrane bound (mCD14) and soluble (sCD14).47,51 Since sCD14 lacks the glycosyl phosphatidylinositol (GPI) anchor, mCD14 and sCD14 have molecular masses of 53 and 48 kDa, respectively.52 To determine the amino acid composition of CD14, Ferrero and Goyert cloned the CD14 gene and revealed a transcript encoding a 356-amino acid protein.53 It was also found to have high leucine content (15.5%) and four putative N-glycosylation sites.53,54 The site involved with LPS binding, as well as the sites involved in the interaction of human CD14 with supposed accessory receptors, has been identified in the N-terminal part of CD14.55 This was determined by generating and transfecting 23 mutants in the N-terminal 152 amino acids of human CD14.56 In each mutant, a block of 5 amino acids was substituted with Ala. Thus, Stelter and coworkers found that the region between amino acids 39 and 44 forms an essential part of the LPS-binding site of human CD14.56 Moreover, for human sCD14, two other regions were found to be essential for eliciting LPS-induced responses from endothelial and smooth muscle cells: aa 9–13 and aa 91–101.57
CD14 is expressed by various cells such as cells of the myeloid lineage (monocytes, macrophages, PMNs), B cells, parenchymal cells of the liver, gingival fibroblasts, and microglial cells.58 Each source expresses CD14 differentially: peritoneal and pleural macrophages exhibit a high level of constitutive CD14 expression, while (murine) Kupffer cells, alveolar macrophages, monocytes, and PMNs have a low level of constitutive CD14 expression.58a,59 In addition, LPS and tumor necrosis factor-α (TNFα) induce the release of sCD14 by mononuclear cells and PMNs in a dose-dependent manner, whereas interferon-γ (IFN-γ) and interleukin 4 (IL-4) inhibit the release of sCD14.60 In the steady state, human serum contains 2–6 μg/mL of sCD14.61 This level increases in response to the presence of LPS, and consequently, Landmann et al. suggested the use of sCD14 levels as a diagnostic marker in patients with severe infections.62 The level of sCD14 in human milk also explains why newborn infants are innately immune to bacteria with their hitherto sterile intestines. Thus, Labeta and coworkers found that the concentration of sCD14 is 10-fold higher in human milk than that in serum.63
Binding of LPS to a cell does not result in immediate response—a time lapse of 15–30 min is usually observed between LPS binding and LPS-induced cellular responses. Detmers and coworkers, along with Lichtman and coworkers, suggested that monomeric LPS is internalized in vesicles, and uptake may be required for signaling.64 Indeed, several studies have revealed that blocking the internalization or endosome fusion also blocks LPS-induced signaling.64,65 Although the precise mechanisms of this blocking event are not completely understood, it has been shown that monomeric LPS is transported into the cell to the Golgi complex and activates the cell from there on.66 To determine if mCD14 directs the movement of LPS to the Golgi apparatus, Vasselon and coworkers used an mCD14 chimera containing enhanced green fluorescent protein (mCD14-EGFP) to follow trafficking of mCD14 in stable transfectants.67 It was found that monomeric LPS is transferred out of mCD14 at the plasma membrane and traffics within the cell independently of mCD14 involvement. In contrast, particulate (bacterium) and aggregated (micelles) LPSs were internalized to the lysosomes via a CD14-dependent pathway called macropinocytosis—a process resembling that of phagocytosis.65b After internalization, LPS induces mononuclear phagocytes (MPs) to produce three groups of powerful mediators: the reactive oxygen intermediates (O2, H2O2, OH, and singlet oxygen), the proinflammatory cytokines, and a number of arachidonic acid metabolites, including prostaglandins and leukotrienes.
Since CD14 is a glycophosphatidylinositol-linked receptor that lacks a transmembrane domain, it was anticipated that it requires an accessory molecule for signal transduction.68 This hypothesis was confirmed using different anti-CD14 antibodies that either blocked LPS binding to CD14 or did not block LPS binding while preventing LPS-induced cell activation.44,69 This accessory receptor has been identified as a member of the TLR family.
3. Toll-Like Receptor 4
Nearly a decade after the importance of LBP and CD14 was initially delineated in seminal discoveries, the next main advance in understanding the mechanism of innate immunity emerged—the identification of the putative transmembrane protein that acted with CD14 to generate a transmembrane signal for LPS-induced cell activation. Two highly original and influential discoveries gave the impetus for this advance. First, it was found that TLRs play an important role in the innate immune response of Drosophila flies.9,70 Second, a TLR homologue was identified as the gene responsible for LPS responses in two natural mouse mutants.11,71 These results formed the basis for understanding how the innate immune system regulates responses to infection and how plasma membrane receptors control adaptive immune responses.30
It was well known that, despite the lack of an adaptive immune system, Drosophila flies are very resistant to microbial infections. The only rationale for this attribute, at the time, had been its demonstrated ability to synthesize potent antimicrobial peptides. Then, in the early 1980s, Anderson and coworkers conducted a mutagenesis screen for genes involved in dorsoventral patterning of the Drosophila embryo.72 Their studies revealed a mutant gene that had an unusual appearance. Consequently, the authors named this gene “Toll,” meaning weird. The Toll gene, which encodes a single-pass transmembrane receptor, became highly important after it was found that it activates the signaling pathways that induce the synthesis of drosomycin, an antifungal peptide in Drosophila flies.9,72
Twelve years after Anderson’s discovery, Williams et al. showed that 18-wheeler, another TLR gene found in Drosophila, could induce the release of attacin,70 one of the potent antibacterial peptides synthesized by Drosophila. As a result, it was established that the activation of a proteolytic cascade that produces peptidic ligands for the TLRs leads to the induction of these antimicrobial responses. It remains unanswered whether this mechanism is unique to Drosophila, or whether it is conserved in mammalian cells. What was remarkably inferred from these results, however, was that Drosophila TLRs were capable of discriminating between fungi and bacteria and, consequently, of inducing an appropriate and distinct antimicrobial response. Subsequently, Imler and coworkers showed that the activation of TLR-induced pathways in Drosophila initiates an intracellular kinase cascade that ultimately produces a translocation of transcription factors, Dif and Relish, from cytoplasm to nucleus.73 Dif and Relish are homologous to nuclear factor-kappa B (NF-κB), a transcription factor known to activate inflammatory mediators in humans, thereby linking Drosophila TLRs to the study of LPS biology.72
The apparent importance of TLRs, as well as the observation that the Toll gene shares a certain homology with the human IL-1 receptor, provided impetus for the field of Toll biology to move beyond flies.74 Thus, in the mid-1990s, Janeway and coworkers began a search for dToll-related proteins in mammalian gene sequences. As a result of their efforts, the first human homologue of Drosophila Toll, initially termed human Toll and subsequently termed TLR4, was identified.75 Human TLR is an 841-amino acid protein with a molecular mass of 92 kDa.75 After cloning and characterization, human Toll was found to be a type I transmembrane protein, the cytoplasmic domain of which bears a structural homology to the human IL-1 receptor. Janeway and coworkers also determined that similarly with Drosophila Toll, human Toll could induce activation of NF-κB and subsequently induce the expression of NF-κB-controlled genes for the inflammatory cytokines. Finally, their observation that TLR4 could induce members of the B7 family—molecules that are required for the activation of naive T cells by antigen-presenting cells—provided a potentially important link between pathogen detection and induction of the adaptive immune response.
Other compelling evidence on the importance of TLRs in LPS-induced responses came when TLRs addressed the issue of why some strains of mice were unresponsive to LPS. For years, LPS has been known to be a very active mediator of inflammation in most mammalian system.76 It was found, however, that LPS is relatively ineffective at inducing responses in the C3H/HeJ or C57BL/10ScCr strains of mice.77 Then in 1998, Beutler and coworkers showed via positional cloning techniques that mutations of a gene termed the “LPS gene” selectively reduced the ability of C3H/HeJ and C57BL/10ScCr mice to sense LPS. The codominant Lpsd allele of the C3H/HeJ strain was a result of a mis-sense mutation in the third exon of TLR4, a mutation that was predicted to result in a Pro712→His substitution.11,71a When this mutation was introduced into wild-type TLR4, the receptor was converted into a dominant-negative mutant that inhibited LPS-dependent responses in a transfected macrophage cell line.78 Similarly, Hoshino et al. demonstrated that C3H/HeJ mice have a single-point mutation of the amino acid that is conserved among the IL-1/Toll receptor family.71b Using genetically modified mice in which the TLR4 gene was deleted, the latter showed that TLR4 was essential for sensing LPS and mutations in this gene explained the lack of responsiveness in C3H/HeJ mice. Together, these seminal publications provided the first direct connection between TLRs and the physiological responses to LPS. Heine and coworkers provided further proof of this connection by showing that Chinese hamsters respond normally to LPS even though they carry a null allele for TLR2.79 Their results implied that expression of TLR2 is sufficient, but not essential, for mammalian responses to endotoxin. Finally, it was shown that a dominant-negative mutant of TLR2 did not cause LPS responsiveness in transfected macrophages.78
4. Myeloid Differentiation Antigen 2
Despite the fact that several groups have already shown evidence of LPS-induced signal transduction through TLRs, direct binding of LPS to the latter is yet to be demonstrated.80 Moreover, it was found that in vitro transfection of TLR4 cDNA did not confer LPS responsiveness on two LPS-unresponsive cell lines: human embryonic kidney-derived and a mouse IL-3-dependent pro-B cell-line Ba/F3.81 Then in 1999, Shimazu and coworkers reported and characterized a novel LBP called myeloid differentiation antigen-2 (MD-2).81b In a series of experiments, they showed that MD-2 physically associated with TLR4 on the cell surface and confers responsiveness to LPS. In a similar fashion, Da Silva Correia determined that LPS binds directly to each of the members of a tripartite LPS receptor complex.80b Using modified and radio-iodinated LPS, they showed that LPS is cross-linked specifically to TLR4 and MD-2 when coexpressed with CD14. Thus, maximal cellular activation by LPS must be a cascade of events that likely involves transferring of LPS by LBP to CD14 and then to TLR4-MD-2. Moreover, although CD14 and LBP enhance cellular activation, activation of TLR4 by LPS was found to absolutely require MD-2.82
MD-2 is a 20- to 25-kDa extracellular glycoprotein that belongs to the MD-2-related lipid-recognition family of lipid-binding receptors.83 Since MD-2 lacks a transmembrane domain that would anchor it to the cell membrane, several groups performed studies to verify the process by which MD-2 associates with TLR4—whether it is a soluble intracellular protein that binds to TLRs in the endoplasmic reticulum (ER) or it is first secreted into the medium and then binds to TLRs on the cell surface. To this end, Visintin et al. found that, in some cells, MD-2 is synthesized in large excess to TLR4 and it saturates all available TLR4 molecules in the ER. The excess MD-2 is then secreted into the medium.84 Although proper glycosylation and trafficking of TLR4 to the cell surface require intracellular association with MD-2,85 functional TLR4 can be presented on the cell surface without MD-2 in both transfected86 and epithelial cells of the human airway.87 For reporter cells that expressed TLR4, but not MD-2, secreted MD-2 (sMD2) was found to restore LPS responsiveness.84 Thus, even at concentrations as low as 50 pM, Visintin and colleagues showed that MD-2 significantly enhances LPS reactivity and suggested that TLR4 has a functional affinity constant for MD-2 in the range of 50–500 pM.84
Human MD-2 contains 160 amino acid residues, prominent regions of which are the 17-amino acid sequence at the N-terminus, 7 cysteine residues, and 2 N-glycosylation sites.88 To identify the regions of functional importance on human and mouse MD-2, common analytical methods—namely, analysis of peptide fragments,89 mutation analysis,90 and computational modeling91—have been utilized. Computer modeling suggests that MD-2 is capable of forming a barrel-like structure with a hydrophobic cavity sufficient to accommodate the fatty acid moieties of Lipid A.91,92 In addition, Visintin and coworkers reported that a positively charged region flanking the hypothetical hydrophobic cavity of MD-2 is required for stable binding to LPS.93 On the other hand, site-directed mutagenesis identified the regions of human MD-2 involved in TLR binding, and consequently, in conferring LPS responsiveness.90d Thus, Re and coworkers found that MD-2 binding to TLR4 took place via Cys95 and Cys105, probably through the formation of an intermolecular disulfide bond.90d Several studies predict that Cys95 is located on the surface of the hypothetical barrel, along with the other Cys residues, except for Cys133.84,94 This prediction is consistent with the idea that MD-2 is capable of forming covalently bound oligomers, but it does not preclude the existence of a monomeric form. Indeed, monomeric MD-2 has been reported to bind preferentially to a recombinant soluble TLR4 ectodomain.95 Hydrophilic and charged residues surrounding this area, such as R90, K91, D100, and Y102, also contributed to the formation of the TLR4–MD-2 complex.90d Re and Strominger found, however, that a different region of MD-2 was responsible for conferring LPS responsiveness.90d This region is not involved in TLR4 binding and is rich in basic and aromatic residues, several of which contribute to LPS responsiveness and might represent an LPS-binding site. Consequently, mutations in the lysine residues of this region are correlated with the loss of LPS binding and, as a result, the loss of activity.
Finally, it was found that binding of MD-2 by Lipid A was greatly enhanced by serum components that had long been known to enhance LPS responses, namely, sCD14 and LBP.86,94a,96 MD-2 is unstable at 37 °C, but the binding of LPS to MD-2 has been reported to dramatically stabilize its activity.97 Overall, the evidence supports a model in which LPS interacts with the MD-2/TLR4 surface heterodimer. The interaction of LPS with the receptor complex occurs with high affinity, and the Kd is estimated to be 3–10 nM.98 The binding of LPS to MD-2 is then responsible for the aggregation of TLR4 and the recruitment of intracellular signal transducers.
IV. Host-Derived Mediators and the Pathogenesis of Sepsis
Once TLR4 binds to its LPS ligand, two possible pathways of cellular activation can occur—either through the myeloid differentiation factor 88 (MyD88) or through the TLR-domain-containing adapter-inducing interferon-β (TRIF) pathway.99 In each pathway, signaling events lead to the sequential activation of specific tyrosine and threonine/serine kinases. This signaling cascade ultimately results in phosphorylation, ubiquitination, and degradation of inhibitory kappa-B (IκB) and other transcriptional activators. IκB degradation leads to translocation of NF-κB into the nucleus. Once NF-κB is translocated into the nucleus, it binds to specific DNA sequences located in the promoter regions and participates in the activation of a large variety of genes including cytokines, chemokines, stress-response proteins, and antimicrobial and antiapoptotic peptides.100 The outpouring of inflammatory cytokines and other inflammatory mediators after LPS exposure contributes to generalized inflammation, procoagulant activity, tissue injury, and septic shock.101
In macrophages, Lipid A activation of TLR4 triggers the biosynthesis of diverse mediators of inflammation and activates the production of costimulatory molecules required for the adaptive immune response.4 Once activated, macrophages are the fundamental secretory cells of the immune system.102 To date, more than 100 macrophage products have been identified—with molecular weights ranging from 32 (superoxide anion) to 440,000 Da (fibronectin).103 Among these, inflammatory cytokines such as TNFα, IL-1β, and IL-6 are the most studied.
1. The Cytokine Networks
The cytokine TNFα, an endogenous monocyte/macrophage-derived protein, is one of the most important soluble mediators of inflammation. It is mainly synthesized by activated monocytes/macrophages and is responsible for a wide range of signaling events within cells. In response to an LPS challenge, TNFα is synthesized very quickly and the production peaks in a matter of 1.5 h.104 Secretion of this molecule triggers a proinflammatory response in neutrophils and endothelial cells and leads to cell damage.105 TNFα exerts most of its effects by binding, as a trimer, to either a 55-kDa cell membrane receptor called TNFR-1 or the 75 kDa cell membrane receptor TNFR-2; both are members of the TNF receptor superfamily.106 In animal studies, the administration of TNFα has been shown to have lethal consequences.107 In human volunteers, dramatic hemodynamic, metabolic, and hematologic changes are observed after administration of TNFα. Perhaps the most dramatic demonstration of the pathophysiologic significance of systemic cytokine release was observed recently in a phase I study of an experimental anti-CD28 monoclonal antibody.101a The antibody was well tolerated in animal studies, but was found to be markedly toxic to humans. Within a few hours of receiving the antibody, all six healthy human volunteers developed shock, disseminated intravascular coagulation (DIC), and multiorgan failure. The “cytokine storm” that often accompanies septic shock was clearly demonstrated by the striking elevations in IL-1, TNF, IL-8, IFN-γ, and other cytokines and chemokines that were released almost immediately into the patient’s bloodstream.
Another important cytokine in host defense during sepsis is the IL-1 gene family.108 This family consists of three members: IL-1α, IL-1β (both agonists with proinflammatory character), and the IL-1 receptor antagonist (IL-1ra, anti-inflammatory counterpart). While IL-1β is solely active in its processed and secreted form, IL-1α is active in its intracellular precursor, membrane-associated, or secreted form.109 The activation of numerous cell types by IL-α and IL-β leads to diverse proinflammatory events.110
Both IL-1 and TNFα act synergistically in the initiation of the inflammatory cascade in sepsis, leading to the expression of further factors.111 These factors include other proinflammatory cytokines (IL-12, IL-18),71b,112 and chemokines (IL-8, monocyte chemoattractant protein-1/MCP-1).113 The chemokines IL-8 and MCP-1 are key factors in chemotaxis—IL-8 is involved in neutrophil chemotaxis, while MCP-1 is involved in the chemotaxis of monocytes. IL-8 also causes neutrophils to degranulate and cause tissue damage.114
2. The Coagulation Cascade
Activation of the coagulation cascade has traditionally been synonymous with the need for hemostasis (stoppage of bleeding) at sites of injury. Over the past several decades, however, it has been increasingly recognized that initiation of coagulation is an integral and consistent element of the local and systemic response to inflammatory stimuli. The precise mechanism whereby coagulation contributes to the full expression of inflammation is an area of active study.
Tissue factor (TF) expression on the surface of endothelial cells and monocytes—induced by the presence of endotoxins or inflammatory cytokines—initiates the coagulation process.115 Thus, TF on the cell surface activates factor VII, and the resulting complex of factor VIIa and TF converts factor X into factor Xa. In concert with factor Va, factor Xa converts prothrombin into thrombin, which in turn results in the cleavage of fibrinogen to fibrin. Deposition of fibrin plays a critical role in hemostasis and in the localization of microorganisms within an abscess cavity. This process, however, can impede delivery of oxygen to tissues and can induce further inflammatory injury indirectly through the response to hypoxia (lack of oxygen) and directly through signals delivered to the thrombin receptor. Engagement of the thrombin receptor activates the nuclear transcription factor NF-κB,116 causing the transcription of a broad array of proinflammatory gene products and resulting in release of nitric oxide.117 The thrombin receptor is not a unique mechanism through which an inflammatory response is amplified. Clustering of TF has also been shown to initiate gene expression for proinflammatory cytokines, including TNF.118
In general, the activation of the coagulation pathway induces anticoagulant mechanisms that function to limit progression of the coagulation cascade. During sepsis, however, an imbalance of the procoagulant and anticoagulant systems occurs, resulting in a sustained hypercoagulable state. The specific abnormalities of the coagulation system that occur following endotoxemia and cytokinemia have been documented in detailed studies involving human volunteers and septic patients. Thus, in human volunteers injected with small doses of TNFα or LPS, there is gradual activation of coagulation, as shown by increases in thrombin–antithrombin (TAT) complexes, prothrombin activation fragments, and fibrinopeptide A.119 This process begins by the second hour, peaks at 4–5 h, and persists for 6–12 h. There is also an early increase in plasma fibrinolytic activity (1–2 h) due to the presence of plasminogen activators, which leads to plasmin generation. The anticoagulant effect of the latter, however, is rapidly neutralized by an increase in the amount of antifibrinolytic plasminogen activator inhibitor-1 protein (PAI-1) released into the bloodstream. Both ATIII and protein C are also rapidly consumed during the septic state.120 Additionally, the downregulation of thrombomodulin due to both local and systemic release of such cytokines as TNF and IL-1 results in impaired activation of the anticoagulant APC–protein S complex.121
The increased procoagulant activity, decreased anticoagulant activity, and impaired fibrinolysis in septic patients lead to the development of the clinical syndrome called DIC. DIC is clinically defined as an overexuberant systemic clotting that depletes coagulation proteins and platelets from the blood and leads to bleeding complications.122 It becomes increasingly common as patients advance from sepsis (SIRS) to septic shock.123 The microvascular thrombosis that develops concomitantly results in organ injury, partly on an ischemic basis. As such, while DIC is considered in terms of bleeding complications, the clinical outcome is ultimately decided by the accompanying microvascular thrombosis and end-organ damage.
In summary, multiple and diverse pathways lead to activation of the cytokine networks and the coagulation cascade. To alleviate the fatal outcome of sepsis and septic shock, many therapeutic interventions have been targeted toward the later stages of endotoxin response: (a) blocking cytokine synthesis/release by interfering with the transduction of cell surface signals,124 (b) neutralizing released cytokines by passive immunization125 and soluble receptors,126 and (c) blocking cytokine cell surface receptors with a specific receptor antagonist.127 The majority of these approaches have demonstrated efficacy in both in vitro and animal models, but none has proven to be effective at treating human sepsis. Due to the large diversity and quantity of cytokines released by activated cells, it is likely that the approach of blocking only a single cytokine may be inadequate. Since no single therapeutic agent has proven to be unequivocally beneficial for managing the abnormalities of sepsis, it has become increasingly clear that the therapeutic path to sepsis does not lie on the treatment of the downstream events. Under the assumption that end-organ damage ensues from an exuberant or hyperactivated immunological response that becomes unresponsive to supervening counter-regulatory mechanisms, the approach to treatment of sepsis lies in interrupting the cascading inflammatory response by blocking the initial signaling events. Current progress towards this goal is discussed next.
V. Modulation of the LPS Receptor Complex by Lipid A Analogues
It is understood, thus far, that endotoxin is essentially a signaling molecule that alerts the vertebrate host to the presence of a Gram-negative pathogen. While the endotoxin molecule itself is not intrinsically toxic, the exaggerated host response to endotoxin accounts for septic shock from Gram-negative bacterial organisms. To summarize from the foregoing, endotoxin mediates its injurious effects through systemic activation of host-derived inflammatory mediators.
While the evolution of organ dysfunction in septic shock is a complex, highly variable and multifactorial process involving many mediators, the past few decades have seen enormous advances toward understanding the cellular and molecular basis of the initial events in this process. The thorough characterization of these events has allowed researchers to design rational therapies directed at blocking the initial signaling events in LPS-induced sepsis.
A successful approach to downregulating LPS signaling involves the use of compounds that compete with LPS binding to MD-2 and inhibit the induction of the signal transduction pathway by impairing LPS-initiated receptor dimerization. To date, several Lipid A variants that specifically block the LPS-binding site on human (h) MD-2 have been identified: lipid IVA (a biosynthetic precursor of the Lipid A of E. coli, 1, Fig. 3)128 and a nonpathogenic Lipid A from Rhodopseudomonas sphaeroides (2),129 which served as the structural basis for the synthetic antisepsis drug candidate Eritoran (E5564, 3).130
Fig. 3.

Structure of Lipid IVA (1), Lipid A of R. sphaeroides (2), Eritoran (3), and Lipid A of E. coli (4).
The central structure of Lipid A, as exemplified by the E. coli Lipid A structure (4), is a highly conserved glycosidically β-(1→6)-linked di-D-glucosamine backbone bisphosphorylated at the 1-O- and 4′-O-positions.131 As mentioned earlier, LPS and Lipid A trigger innate immune responses through the TLR4/MD-2 complex, the activation of which leads to two distinct signaling pathways—the Myd88-dependent pathway and the TRIF-dependent pathway.132 The Myd88-dependent pathway results in the production of proinflammatory cytokines such as TNFα, IL-1β, and IL-6. The TRIF-dependent pathway, on the other hand, results in interferon-β and nitric oxide production.
Despite considerable data on the activity of both isolated133 and synthetic Lipid A derivatives,134 there is no universal correlation between the chemical structure of Lipid A and its activity in the TLR4/MD-2 complex. An important approach to understanding how a receptor system functions is to define its pharmacology. Recent work on the crystal structure of the TLR4/MD-2 complex, bound to either agonistic LPS135 or antagonistic Lipid IVA128 (1), provided a deeper understanding of the structural requirements of the LPS receptor complex. Thus, a large hydrophobic cavity is noted in MD-2, whereby all four lipid chains of compound 1 are contained, as opposed to five of the six lipid chains of LPS. The remaining chain of LPS is exposed to the surface of MD-2 and forms hydrophobic interactions with conserved phenylalanine residues in TLR4. Consequently, it is speculated that structural properties of MD-2 play a critical role in differentiating among varying Lipid A structures and potentiating a biological response.
Subtle differences in the length and distribution pattern of acyl chains, the phosphorylation status of the di-glucosamine backbone, and changes in the di-glucosamine backbone of Lipid A are known to profoundly affect its biological activity.134b–d The following sections summarize the effect of these variables on the agonistic or antagonistic activity of Lipid A derivatives.
1. Length and Distribution of Acyl Chains
The acyl chains of Lipid A, particularly its number, length, symmetry, and saturation, have been shown to be a major determinant of the potency of LPS in eliciting TLR4-dependent host responses.136 The relationship between acyl-chain length and bioactivity has been investigated to some extent with tetraacyl disaccharide analogues137 of lipid IVA (1). Recently, having demonstrated that the toxic effects of Lipid A of Salmonella minnesota R595 (5, Fig. 4) could be ameliorated by selective hydrolysis of the 1-O-phosphono and (R)-3-hydroxytetradecanoyl groups (6),138 Johnson and coworkers proceeded to clarify the importance of normal fatty acid chain length by preparing and evaluating chain-length homologues of 5 (also known as monophosphoryl Lipid A, MPL). Thus, derivatives 7a–f were synthesized and evaluated against MPL 6 for induction of nitric oxide synthase (iNOS) in murine macrophages and production of cytokine in human peripheral monocytes.139 It was found that the induction of both iNOS and proinflammatory cytokines exhibit a profound bimodal dependence on the length of the normal fatty acid chains, reaching a maximum when n=8 (7d) in each case. The iNOS response is more sensitive than cytokine induction to variations in chain length, showing a 100-fold difference in potency between 7d, which possesses a greater macrophage-stimulating ability, and 7f. However, both models show a similar threshold chain length for activity—iNOS and cytokine responses are abolished when n=4 (7b) and n=6 (7c), respectively, and for shorter chain derivatives.
Fig. 4.
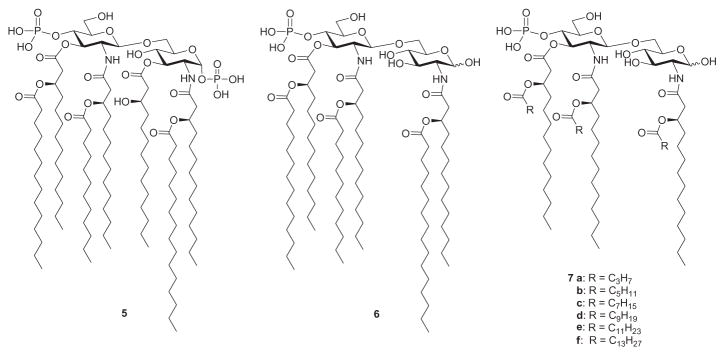
Monophosphoryl Lipid A from S. minnesota 5, and its chain-length homologues 6 and 7a–f.
In general, increasing or decreasing the number of acyl residues present in disaccharide Lipid A derivatives from the optimum of six diminishes endotoxic activity.140 The known141 monosaccharide GLA-47, corresponding to the (tetraacylated) non-reducing sugar portion of compound 7f, is devoid of activity in both the iNOS and cytokine models.142 These observations are consistent with the hypothesis that subtle modifications to the hydrophobic side chains of Lipid A derivatives induce conformations that dramatically affect cellular activation and the expression of endotoxic activities.143 Indeed, Lipid A having six lipid chains has optimal inflammatory activity, Lipid A molecules with five lipid chains are 100-fold less active, and those with four lipid chains, such as Eritoran, lack agonistic activity completely.144
The contribution of acyl chains on ligand specificity and receptor activation mechanism of the TLR4–MD-2–LPS complex is evident in the crystal structure.135 Thus, Park and coworkers demonstrated that binding of LPS induced the formation of an m-shaped receptor multimer composed of two copies of the TLR4–MD-2–LPS complex arranged symmetrically. LPS interacts with a large hydrophobic pocket in MD-2, whereby five of the six lipid chains of LPS are buried deep inside the pocket, and the remaining chain is exposed to the surface of MD-2. The latter forms a hydrophobic interaction with the conserved phenylalanine residues of TLR4. The F126 loop of MD-2 undergoes localized structural change and supports this core hydrophobic interface by making hydrophilic interactions with TLR4. Comparison with the structures of Eritoran (3, Fig. 3) and Lipid IVA (1) bound to MD-2 indicates that two lipid chains in LPS displace the phosphorylated glucosamine backbone by 5 Å towards the solvent area.128,145 This structural shift allows phosphate groups of LPS to contribute to receptor multimerization by forming ionic interactions with a cluster of positively charged residues in TLR4 and MD-2. Thus, the bioactivity of Lipid A is mainly influenced by the length, number, and symmetry of acyl chains, as well as the number and distribution of negative charges.146
Lipid A from the LPS of Rhizobium sin-1 (8, Fig. 5), a nitrogen-fixing bacterial species, is structurally unusual and differs in almost every aspect from endotoxic Lipid A molecules.147 R. sin-1 Lipid A is devoid of phosphates, and a 2-amino-2-deoxy-D-gluconolactone moiety is present at the reducing end. In particular, it contains a very long-chain fatty acid, 27-hydroxyoctacosanoic acid, which can be esterified by 2-hydroxybutanoate. Since Lipid A of R. sin-1 (8) prevents the induction of TNFα by Lipid A of E. coli, Boons and coworkers decided to investigate the contribution of the unusual acyl chain to the antagonistic activity of 8, and they synthesized derivatives 9a and 9b.148 Their data show that the hydroxyl moiety of the 27-hydroxyoctacosanoic acid moiety of R. sin-1 Lipid A is not important for antagonistic properties, whereas shortening the octacosanoic acid moiety (9b) decreases the inhibitory potential. Derivative 9a, which contains the octacosanoic acid moiety without the hydroxyl group, exhibits the same LPS-antagonistic activity as Lipid A of R. sin-1 (8). It should also be noted that while performing detailed biological evaluations on synthetic R. sin-1 Lipid A derivatives, Boons and coworkers149 observed that the ester group at C-3 of the reducing-end residue in 9a is prone to elimination and readily produces enone derivative 10. Also obtained was compound 11, wherein the β-hydroxy ester at C-3 of the proximal sugar unit has been replaced by an ether-linked moiety. Interestingly, compound 11, which has a much improved chemical stability in comparison to that of 9a, was found to be as potent as 9a in antagonizing LPS-induced cytokine production by a human monocytic cell line. Moreover, compound 11 was found to inhibit both MyD88- and TRIF-dependent cell-signaling events.
Fig. 5.
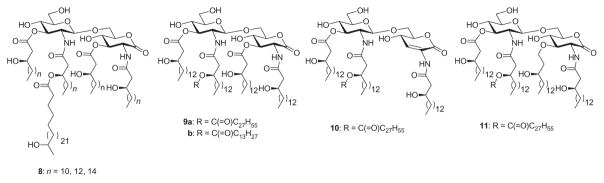
Rhizobium sin -1 Lipid A 8 and its derivatives 9–11.
The chemical and biological properties of Porphyromonas gingivalis LPS and its Lipid A are different from those of enterobacterial LPSs and their Lipid A structures.150 The Lipid A moiety of the LPS of P. gingivalis also displays considerable heterogeneity. The elucidated structures of four Lipid A molecules from P. gingivalis (12–15, Fig. 6) differ in their fatty acid substitution pattern. A common structural feature of these derivatives is the presence of unusual branched fatty acids, such as (R)-3-hydroxy-13-methyltetradecanoic acid and (R)-3-hydroxy-15-methyl hexadecanoic acid. In a recent report, innate host responses to Lipid A species from P. gingivalis LPS were found to be unusual in that these Lipid A molecules were able to function as an agonist for TLR2 and also as an antagonist or agonist for TLR4.151 In order to identify the P. gingivalis Lipid A capable of antagonizing E. coli Lipid A, compounds 12–15 were chemically synthesized.152 Thus, Ogawa and coworkers152a demonstrated that compounds 12 and 13 were agonists for TLR4 but not TLR2, and that both compounds were antagonists for E. coli Lipid A. On the other hand, Boons et al. showed that while compound 14 was a potent LPS antagonist, compound 15 showed significantly diminished activity.152b This prompted their conclusion that the acylation pattern of 14 is critical for optimal antagonistic activity.
Fig. 6.

Lipid A structures from Porphyromonas gingivalis 12–15.
2. Degree of Phosphorylation of the Di-Glucosamine Backbone
As mentioned earlier, the phosphorylation status of the di-glucosamine backbone is known to profoundly affects the biological activity of Lipid A.134b–d At the same time, it has been identified that Lipid A from R. sin-1, which lacks phosphate groups, does not stimulate production of TNFα by human monocytes153 and prevents the induction of TNFα by E. coli LPS. The Lipid A of R. sin-1 is perhaps the most structurally unusual Lipid A reported to date; its structure (8, Fig. 5) differs in almost every aspect from those known to contribute to the toxicity of enteric Lipid A.147 In particular, the disaccharide moiety of rhizobial Lipid A is devoid of phosphate groups and the glucosamine phosphate is replaced by 2-amino-2-deoxy-D-glucono-1,5-lactone. The microheterogeneity of rhizobial Lipid A limits the identification of specific structural features that make it an antagonist rather than an agonist. To study the contribution of the reducing-end phosphate group to the antagonistic activity of R. sin-1 Lipid A, Boons and coworkers153a synthesized compounds 16 and 17 (Fig. 7) and compared their biological activity. Thus, they found that 17 was able to antagonize E. coli LPS, while 16 was devoid of this activity. These results suggest that the gluconolactone moiety is important for this property. Compound 17 is the first example of a synthetic Lipid A derivative that lacks phosphate groups and inhibits production of cytokine initiated by E. coli LPS.
Fig. 7.
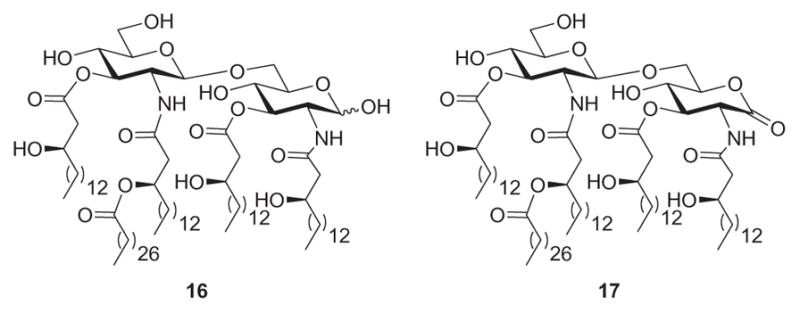
R. sin-1 Lipid A derivatives.
On a similar note, Ribi and coworkers154 found an LPS-mimetic compound that exhibits potent adjuvant activity, but is 100- to 10,000-fold less toxic than LPS.154,155 This compound, MPL,156 is a Lipid A derivative that lacks the phosphate moiety at the reducing-end glucosamine residue. To understand the molecular mechanism underlying the low toxicity of MPL, Nishijima and coworkers157 examined the effects of E. coli Lipid A and MPL on the production of IL-1β and the activation of caspase-1 in mouse peritoneal macrophages. They found that MPL is defective in the induction of IL-1β secretion and is incapable of activating caspase-1. Since caspase-1 has been shown to be essential for the induction of endotoxin shock (via caspase-1-deficient mice),158 these results suggest that the lack of caspase-1 activation in MPL-stimulated macrophages contributes to the low toxicity of MPL. Similarly, Mitchell and coworkers159 reported that the low toxicity of MPL in mice is associated with a bias toward TRIF signaling. To determine whether alteration of a single phosphate group can cause TRIF-biased signaling, Mitchell and coworkers160 performed extensive comparisons of the signaling activities of synthetic MPL (sMPL) and diphosphate Lipid A (sDPL), in the context of an E. coli-type Lipid A structure. They found that sMPL largely retained their TRIF bias as compared to sDPL of E. coli, indicating that the loss of a single phosphoryl group is sufficient to bring about TRIF-biased activation of TLR4.
3. Changes in the β-(1→6)-Linked-Di-Glucosamine Backbone
Partial structures of Lipid A have been useful in investigating the mechanism of LPS binding and cell activation. These include LPS antagonists such as deacylated LPS, lipid IVA, and R. sphaeroides Lipid A,161 as well as several unnatural synthetic Lipid A-like structures.162 An example for the latter are derivatives synthesized by Shiozaki and coworkers,163 whereby they altered the β-(1→6)-linked di-glucosamine backbone common to natural Lipid A structures with a β-(1→6)-linked glucosamine-glucose disaccharide. They found that these novel derivatives, based on the structure of Eritoran (3, Fig. 3), had almost the same (or stronger) LPS-antagonistic activities toward both human blood cells and murine macrophages, as compared to classic Lipid A-type disaccharides having the glucosamine–glucosamine moiety. Following their initial success, Shiozaki’s group decided to reverse their aforementioned backbone design and created Lipid A derivatives featuring β-(1→6)-linked glucose-glucosamine backbones (18–23, Fig. 8).164 Thus, they found that, except for compound 21, these synthetic derivatives exhibited LPS-antagonistic activity comparable to R. sphaeroides Lipid A. Compounds 20 and 23 were even more potent than E5564 (3) in inhibiting production of TNFα in LPS-challenged mice, while compound 19 was more potent than 3 in protecting mice from lethal LPS challenge.
Fig. 8.

Unnatural Lipid A derivatives 18–23 based on Eritoran (3).
Peri and coworkers165 developed synthetic Lipid A derivatives that, on the other hand, largely depart from natural Lipid A structural motifs. Thus, compound 24 (Fig. 9) features a β-(1→6)-N(OMe)-linked di-glucose backbone, four linear C-14 hydrophobic alkyl chains, and no phosphate groups, while compound 25 is the β-O-linked analogue of 24. Compounds 24 and 25 antagonized the inflammatory effect of E. coli Lipid A on MT2 macrophages and did not exhibit proinflammatory effects on the same cell lines. Their most significant result, however, is the observation that N- and O-linked disaccharides have very similar activities. It indicates that the chemical nature of the interglycosidic bridge does not influence LPS antagonist activity.
Fig. 9.
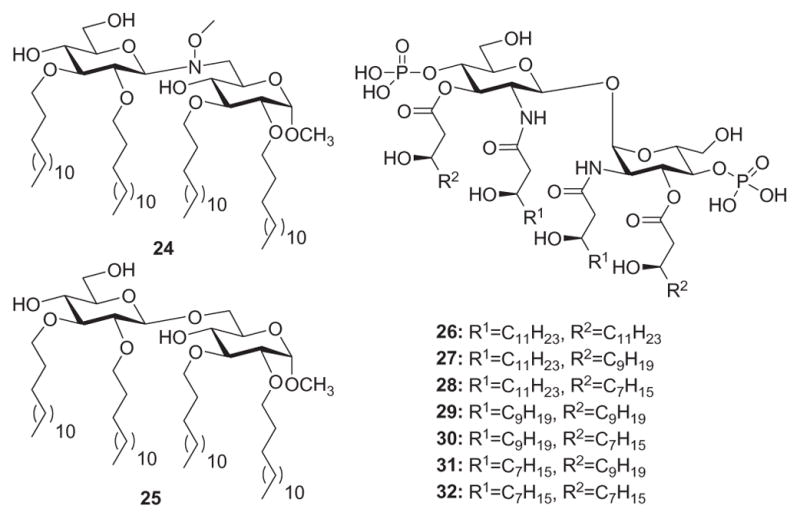
Nonnatural Lipid A derivatives.
In another example of exploring structure–function relationships in the LPS receptor complex with nonnatural Lipid A derivatives, Zamyatina and coworkers166 synthesized Lipid A mimetics featuring a β,α−(1→1′)-linked di-glucosamine scaffold (26–32, Fig. 9). With the restricted internal flexibility imposed on the di-glucosamine backbone, these trehalose-type derivatives aim to elucidate the parameters whereby the three-dimensional molecular shape of MD-2-bound Lipid A/Lipid IVA determines endotoxicity. Thus, they found that the variably acylated Lipid A mimetics 26–32 lacked proinflammatory activities in hTLR4/MD-2-transfected HEK293 cells. Except for compound 26, which is a conformationally constrained counterpart of Lipid IVA, the compounds were shown to potently inhibit proinflammatory responses in TLR4-transfected HEK293 cells stimulated with E. coli Lipid A (4, Fig. 3). The failure of 26 to compete with 4 for the binding site on MD-2 could be due to an increase in the hydrophobic volume of the lipid chains, which would prevent the ligand from entering the binding groove of MD-2. These Lipid A mimics were further examined for their capacity to inhibit E. coli O111 LPS-induced activation of the TLR4/MD-2 complex in TLR4-transfected HEK293 cells. Upon application of 27 and 29 at a concentration of 500 ng/mL, the inflammatory responses to E. coli LPS were entirely abrogated. The shorter chain Lipid A mimetics 31, 29, and 30 allowed for 50% inhibition at the submolar concentration of 5 ng/mL. Compound 32, which contains four (R)-3-hydroxydecanoic acid groups, was shown to suppress cell activation elicited by 4, but did not antagonize E. coli LPS. Thus, with its smaller hydrophobic volume, compound 32 is capable of competing with Lipid A for the binding site on hMD-2, but is incapable of displacing LPS, which possesses a higher affinity for the MD-2/TLR4 complex than the corresponding Lipid A.167 Overall, these results define a crucial role of the inherent plasticity of the carbohydrate backbone of Lipid A—it decides the relocation of a single lipid chain onto the surface of MD-2 in the ligand–receptor structure–function relationships. Replacement of the flexible (1→6)-linked Lipid A backbone by a conformationally constrained trehalose-type scaffold resulted in abrogation of species-specific agonistic activity of lipid IVA. Consequently, manipulating conformational flexibility of the carbohydrate backbone of Lipid A is a useful tool in the rational design of immunomodulating therapeutics targeting the LPS receptor complex.
Lipid A analogues that lack the disaccharide backbone, that is to say monosaccharide Lipid A mimetics, have also been used to study the structure–activity relationship of the LPS receptor complex. The structural simplicity of these truncated molecules allows for abridged synthetic routes and easier access to a broad library of analogues for chemical genetics. GLA-60 (33, Fig. 10)—a synthetic monosaccharide Lipid A analogue having an ester-branched acyl group—has been used extensively as a model structure for structure–activity studies of this type on account of its broad endotoxic activities. To this end, Matsuura and coworkers168 altered the ester-branched acyl group of GLA-60 into alkyl-branched types (34–40, Fig. 10) and evaluated the biological activities of the corresponding analogues to determine the role of a branched side-chain in the expression of endotoxic activities. In terms of ability to induce TNFα production, compounds 34 and 35 were found to be more potent than 33, but not as potent as E. coli Lipid A. Compound 36 was found to be less potent than 33 in inducing production of TNFα, and 37 was even weaker. Similar results were obtained when compounds 33–37 were tested for (1) ability to activate macrophages from LPS-nonresponsive C3H/HeJ mice and (2) tolerance-inducing potency against LPS-stimulated macrophage activation. Compounds 38–40, on the other hand, were found to be more potent than 33 in terms of induction of TNFα expression, mitogenic activity, and LPS lethality. Since the results were comparable to that of 36 and 37, it can be concluded that branching position has a very slight contribution in the expression of endotoxic activities. These studies demonstrate that the usual ester-branched acyl groups in Lipid A analogues can be replaced by alkyl-branched acyl groups with no great consequence to endotoxic activities.
Fig. 10.

Monosaccharide Lipid A analogues 34–45 based on GLA-60 (33).
Subsequently, Matsuura and coworkers proceeded to clarify the structure–activity relationship of monosaccharide Lipid A analogues having different acylation patterns in terms of production of nitric oxide (NO) and cytokine (TNFα, IL-6) in murine macrophages. Briefly, the stimulatory effects of analogues 41–45 (Fig. 10) on the production of NO varied from strongly positive to negative, depending on their structure, and the intensity of activity correlated well with cytokine production. These results strongly suggest the existence of closely related regulatory mechanisms and signaling pathways for the three inflammatory mediators studied. Compound 43 was previously reported169 to possess the ability to protect mice from LPS lethality under D-galactosamine-sensitized conditions (in vivo protective activity) and the potency to induce a hyporesponsive state to LPS stimulation in macrophages. In this study, compound 43 also demonstrated antagonistic activity against LPS, E. coli Lipid A, and 33 for induction of NO, TNFα, and IL-6 when it coexisted with the stimulants.
Following the lead of compound 43,170 as well as their previous work with nonphosphorylated N(OMe)-linked disaccharides,165 Peri, Nicotra, and coworkers synthesized monosaccharide Lipid A analogues that featured no phosphate groups, a glucose backbone, and an amine (46), ammonium species (47), and hydroxylamine (48) moiety (Fig. 11).171 In terms of inhibiting Lipid A-induced TNFα expression in bone-marrow derived macrophages and dendritic cells, compounds 46 and 47 interfered with Lipid A activity in a dose-dependent manner, while compound 48 showed consistently lower activity. Compound 47 was more potent than 46, which in turn was more potent than 48. The activity of the most promising lead, compound 47, was then analyzed in more detail by monitoring the production of another inflammatory cytokine, IL-1β. Thus, 47 inhibited IL-1β and TNFα expression in macrophages and dendritic cells, and the inhibitory effect was proportional to the concentration of the inhibitor. To evaluate the selectivity for TLR4, compound 47 was investigated for TNFα expression in response to stimulation by the CpG motif of bacterial DNA (TLR9 ligand) and trihexadecanoyl cysteine (Pam3Cys-SK4, TLR2 ligand). Production of cytokine was not inhibited by 47 in either the TLR9- or the TLR2-mediated inflammatory cascade, indicating a relevant level of selectivity. Further direct evidence that the activity of compound 47 is selective for the TLR4 receptor was obtained with experiments on a TLR4- and TLR9-transfected HEK 293 cell system. Thus, 47 was able to counteract significantly the effect of Lipid A in TLR4-transfected cells, whereas it was inactive at the maximal dose of 50 mm in influencing the effect of CpG on TLR9-transfected HEK 293.
Fig. 11.
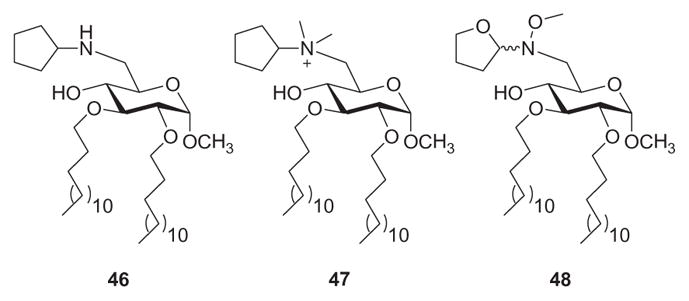
Methyl α-D-glucopyranoside-derived monosaccharide Lipid A analogues 46–48.
In another example of rational designs for monosaccharide Lipid A analogues, Fukase and coworkers134a synthesized a series of Lipid A analogues to investigate the structure–activity relationships governing the biological activity of triacyl-type Lipid A analogue 49, which was found to be an LPS antagonist (Fig. 12). In analogues 50–55, the nonreducing end of Lipid A (3-phosphorylated glucosamine) was substituted with an acidic amino acid, such as aspartic acid or phosphoserine, and the acylation arrangement was varied. These analogues were evaluated for induction of human cytokines and inhibitory activities against LPS. In terms of cytokine induction, most analogues exhibited no induction of IL-6, whereas tetraacylated 54a exhibited concentration-dependent induction at ≥100 ng/mL. Compound 54b also exhibited IL-6 induction at 10 μg/mL concentration, although the activity was about 100-fold weaker than that of 54a. As for inhibition of LPS-induced cytokine production, only analogues 53a and 54a failed to exhibit inhibitory activities. The phosphoserine-containing analogues showed stronger inhibitory activities than the corresponding aspartic acid-containing analogues that feature the same acylation pattern.
Fig. 12.

Triacylated Lipid A 49 and its monosaccharide analogues 50–55 containing acidic N-acyl amino acids.
Obvious trends were also observed between the inhibitory activities and the acylation patterns. In either enantiomeric form (D- and L-phosphoserine), the acylation pattern of compound 51 gave the highest inhibitory activity. The acylation pattern in 50 allowed for slightly weaker activity than that of 51, but it was slightly stronger than that of 52. For the aspartic acid analogues, the L-form showed stronger inhibition than the D-form, whereas the D- and L-phosphoserine analogues showed no significant differences. In the Limulus test, the expression of Limulus activity was dramatically influenced by the acylation patterns. This result clearly showed that structural requirements for expression of the Limulus activity are different from cytokine-inducing activities or LPS-antagonistic activities.
Following Peri’s171 and Fukase’s lead,134a Demchenko and coworkers172 proceeded to synthesize Lipid A analogues containing a glucopyranoside core, hydrophobic ether substituents, and an amino acid moiety to provide ionic character to the constructs. The inhibitory activity of compounds 56 and 57 (Fig. 13) on LPS-induced TNFα expression was investigated in vitro using THP-1 macrophages. Compounds 56a, 57a, and 57b exhibited no inhibitory activity against LPS-induced TNFα expression in the concentration range of 0.1–10 mM. Compound 56b, which has a free carboxylic group, was able to significantly inhibit LPS-induced expression of TNFα at concentrations greater than 10 μM. Unfortunately, cell viability measurements indicated that compound 56b was toxic to the cells in the 30–100 mM range. Indeed, the similarities between the inhibition and toxicity curves suggested that much of the antagonistic activity by 56b was related to toxicity. With compound 47 as a positive control, lipidated compound 58 displayed a marked enhancement in LPS-antagonistic ability (550 nM range). It was able to inhibit 80% of the LPS response at a concentration of 5 mM, with no observable toxicity. Cell viability began to be compromised at 10 mM, and some agonist activity was found in the 10–30 mM range. Alkylated compound 59, on the other hand, demonstrated 70% inhibition of LPS-induced TNFα expression at a concentration of 1 mM and reached 90% inhibition at 40 mM. Compound 59 exhibited no toxicity or agonist activity within the 0.2–40 mM range. These results were better than those obtained with compound 47, which had an inhibition range of 3–10 mM and began to show agonist activity at concentrations >10 mM. Overall, these studies validate the conclusion that Lipid A analogues that lack the di-glucosamine backbone, phosphate moieties, and typical acylation patters can still demonstrate significant antagonistic activity toward LPS-induced cytokine production.
Fig. 13.
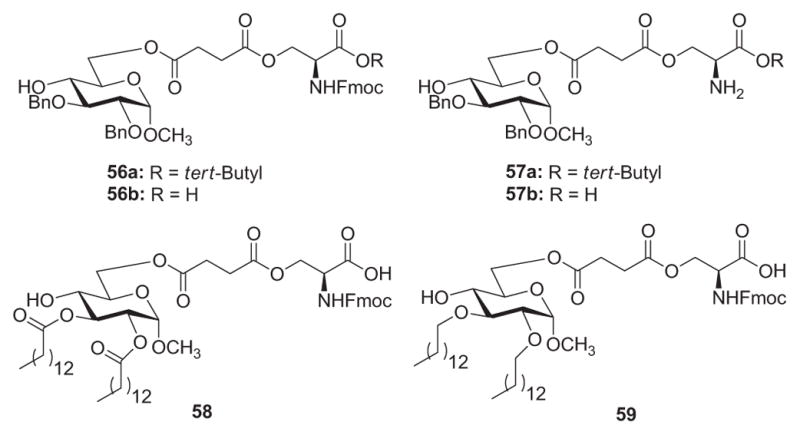
Monosaccharide Lipid A analogues 56–59 with glucopyranoside-spacer–amino acid motifs.
In a similar effort to define the parameters of antagonistic activity by monosaccharide Lipid A analogues, Johnson and coworkers134d identified a new class of potent monosaccharide immunomodulators called aminoalkyl glucosaminide 4-phosphates (AGPs), wherein the less-conserved reducing-sugar unit of Lipid A is substituted with a flexible N-acyloxyacyl aglycone unit.142 The flexible AGP motif permits energetically favored close packing173 of fatty acid moieties, facilitating intercalation of the lipids into the hydrophobic pocket of MD-2. In addition, the carboxyl group of seryl-based AGPs serves as a stable bioisostere of the labile anomeric phosphate of Lipid A, which, along with phosphate groups, presumably binds electrostatically to lysine residues along the edge of the hydrophobic pocket of MD-2.95 Among seryl-based AGPs, compounds 60 and 61 (Fig. 14) containing 10-carbon and 6-carbon secondary acyl residues, respectively, have been found to exhibit potent TLR4 agonist and antagonist activity, respectively, in both murine and human models.174 In order to overcome the inherent chemical and metabolic instability of the ester-linked secondary fatty acids present in 60 and 61, and to further evaluate structural modifications in the AGP series, the corresponding ether-linked lipid analogues 62 and 63 (Fig. 14) were synthesized.134d Thus, it was found that 60 and its ether analogue 62 exhibited similar abilities in inducing TNFα expression in human monocytes, diminishing splenic bacteria following a Listeria challenge, and providing protection from a lethal influenza challenge. In contrast, neither 61 nor its ether analogue 63 induced detectable cytokines in human cell assays. However, both compounds 61 and 63 were able to inhibit LPS-induced expression of TNFα in human monocytes. Compound 61 effectively inhibited expression of serum TNFα when coadministered intravenously with LPS, but 63 did not demonstrate any antagonist activity. The weak TLR4 agonist activity of ether analogue 63 in vivo is further exemplified by its ability to induce TNFα when administered intravenously to mice—a dose–response comparison of serum TNFα levels induced by 60 and 63 showed that 63 was between 10 and 100 times less active than 61. Overall, ether lipids 62 and 63 exhibited similar TLR4 agonist and antagonist activities, respectively, as compared to their ester analogs 60 and 61. Unlike the potent TLR4 antagonist 61, the ether lipid 63 was a weak agonist in murine models, suggesting that one or more of the ester carbonyl groups in 61 play a pivotal role in binding to murine MD-2 and preventing TLR4 activation.
Fig. 14.
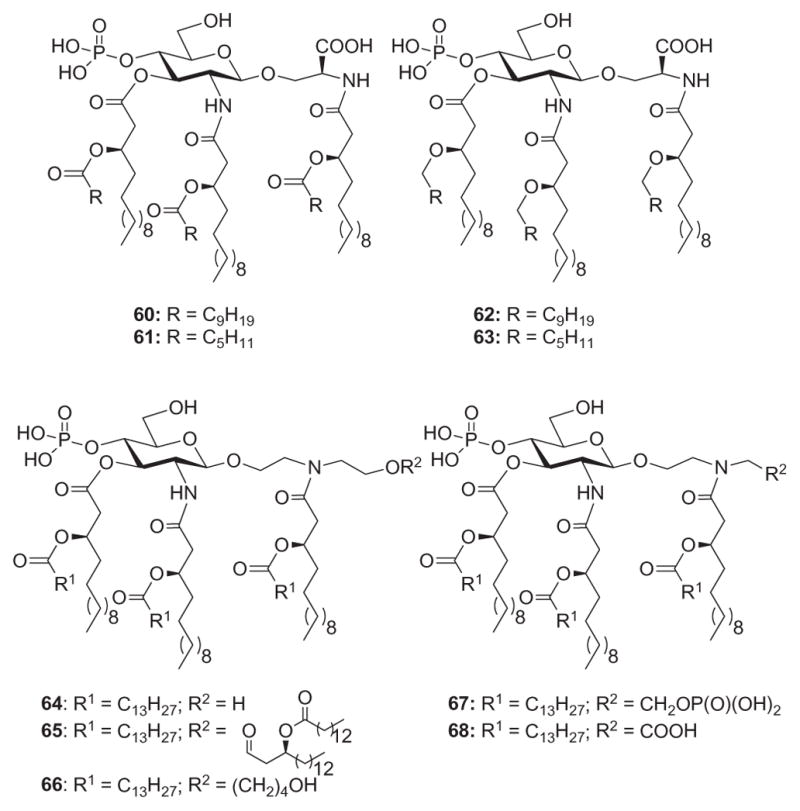
AGP class of Lipid A mimetics 60–68.
Following Johnson’s lead,134d Jiang and coworkers132 synthesized Lipid A mimics (64–66, Fig. 14) wherein the reducing glucosamine residue of the archetypal disaccharide scaffold in Lipid A has been replaced by an acylated diethanolamine moiety. The design of mimics 64–66 was based on the following considerations: (a) conservation of essential functional groups involved in TLR4/MD-2 ligand binding, namely, the phosphate and fatty acid chains; (b) conservation of the glycosidic linkage; and (c) the appropriate location of each functional group through the diethanolamine moiety. Thus, it was found that all three compounds (64–66) showed no cell toxicity at the highest concentrations tested for each, as measured via Trypan Blue exclusion. Compound 64 and 66 increased ICAM-1 expression in human THP-1 cells, but 66 was found to be less potent than 64. The maximum ICAM-1 expression level was achieved at a concentration of 2.0 mM for both. In contrast, no significant increase in ICAM-1 expression level is noted for the octa-acylated analogue 65 at the highest concentration tested (4.0 mM). To further characterize the immunostimulatory properties of 64–66, production of TNFα, IL-6, and IL-1β cytokines was also measured. In general, the responses measured for all three cytokines mirror that of the response for ICAM-1 expression—compound 64 showed the greatest potency, compound 66 showed a slightly decreased potency, and compound 65 induced very little to no response. While the octa-acylated analogue 65 is inactive in the induction of ICAM-1 and other cytokines tested, it induces a significant level of IL-1β at the concentration of 9 mM. Building on the results of 64, Jiang and coworkers175 synthesized compounds 67 and 68 (Fig. 14) with the view of improving the potency of 64 as a TLR4 agonist. Thus, compounds 67 and 68 showed increased potency over 64; mimic 68 showed the greatest potency for stimulating production of all three cytokines (TNFα, IL-6, and IL-1β). In comparison with the terminal free hydroxyl group in 64, the terminal phosphate group in 67 and the terminal acidic moiety in 68 increased the potency of the immunostimulatory response, with the terminal acidic moiety contributing more toward enhancing the potency. Competitive-inhibition studies with lipid IVa, a known human TLR4 antagonist, confirm TLR4 as the target of diethanolamine-containing Lipid A mimics.
To further define structure–activity relationships that influence the LPS-antagonistic activity of compounds 46 and 47 (Fig. 11), Peri and coworkers176 synthesized compounds 69–73 (Fig. 15). The activity of compounds 46, 47, and 69–73 as inhibitors of the TLR4 signal pathway was tested in vitro using a HEK-Blue LPS Detection Kit. They found that compounds 47, 71, 72, and 73 exhibited significant inhibition of Lipid A activity in a concentration-dependent manner. The cytotoxic potentials of 47, 71, 72, and 73 were also measured and it was found that 47, 71 and 72 showed no inhibitory effects on cell viability. Compound 73, in contrast, showed a diminution of the cell viability of about 17%. Finally, they tested compounds 46, 47, 71, and 73 for protective capacity against LPS-induced lethality in vivo. The administration of 10 mg/kg of 47, 71, and 73 significantly increased survival of mice against LPS-induced lethality. In particular, compound 47 increased the survival rate from 0% to 25%, compound 73 from 0% to 67%, and all mice that received compound 71 survived. Indeed, at a smaller dose of 3 mg/kg, compound 71 evoked a 100% survival of mice treated with LPS. Compound 46, which has the weakest TLR4 antagonistic activity in vitro, was found to be ineffective in the lethal endotoxin-shock model. In order to clarify the mode of action of 47 and 71, Peri and coworkers176b analyzed possible interactions with the extracellular components that bind and shuttle endotoxin to TLR4, namely, LBP, CD14, and MD-2. Briefly, their data strongly suggest that compounds 47 and 71 inhibit TLR4 activation by competitively occupying CD14, thereby inhibiting the delivery of active endotoxin to the MD-2/TLR4 complex.
Fig. 15.
Glycolipid-based (69–72) and benzylammonium lipid-based (73) Lipid A mimics.
VI. Summary and Conclusions
Mortality from Gram-negative sepsis remains a serious problem and the concomitant challenges continue to be intimidating. After several decades of vigorous research in the field, several carefully designed pharmaceutical approaches to modifying the clinical outcome of sepsis have failed. As with any illness, a true understanding of the pathophysiology of disease is a critical step in designing effective remedies. The recent determination of crystal structures of the dimeric TLR4–MD-2 complex with bound endotoxic E. coli Lipid A or LPS antagonists (Eritoran, Lipid IVA) clarified important aspects of the structure–activity relationship in bacterial Lipid A, the endotoxic principle of LPS. Investigations on compounds that can modulate the LPS–receptor complex, such as the synthetic Lipid A analogues outlined in this chapter, not only offer novel pharmacological targets but also contribute to the clarification of basic structural and mechanistic aspects of TLR4 signaling, including the role of LBP, CD14, and MD-2 coreceptors. The identification of new pharmacological targets for LPS-mediated diseases should lead to renewed optimism that effective therapies against Gram-negative sepsis will ultimately be achieved.
Acknowledgments
AVD is thankful to the National Institute of General Medical Sciences (Award GM111835) for providing generous support for synthetic and biomedical studies in his group.
Abbreviations
- AGPs
aminoalkyl glucosaminide 4-phosphates
- BPI
bactericidal/permeability-increasing protein
- CD
cluster of differentiation
- CETP
cholesteryl ester transfer protein
- CHO
Chinese hamster ovary
- DIC
disseminated intravascular coagulation
- EGFP
enhanced green fluorescent protein
- GPI
glycosyl phosphatidylinositol
- HDL
high-density lipoprotein
- iNOS
nitric oxide synthase
- IRAK
interleukin-1 receptor-associated kinase
- LBP
lipopolysaccharide-binding protein
- MCP
monocyte chemoattractant protein
- MD
myeloid differentiation antigen
- MPL
monophosphoryl Lipid A
- MPs
mononuclear phagocytes
- MyD88
myeloid differentiation factor 88
- NF-κB
nuclear factor-kappa B
- PAI-1
plasminogen activator inhibitor-1 protein
- PAMPs
pathogen-associated molecular patterns
- PLTP
phospholipid transfer protein
- PMNs
polymorphonuclear leukocytes
- PRRs
pattern-recognition receptors
- SIRS
systemic inflammatory response syndrome
- TAT
thrombin–antithrombin
- TF
tissue factor
- TLR
Toll-like receptor
- TRIF
TLR-domain-containing adapter-inducing interferon-β
References
- 1.Marshall JC. Lipopolysaccharide: An endotoxin or an exogenous hormone? Clin Infect Dis. 2005;41:S470–S480. doi: 10.1086/432000. [DOI] [PubMed] [Google Scholar]
- 2.Dyall SD, Brown MT, Johnson PJ. Ancient invasions: From endosymbionts to organelles. Science. 2004;304:253–257. doi: 10.1126/science.1094884. [DOI] [PubMed] [Google Scholar]
- 3.(a) Veldhuyzen van Zanten SJO, Sherman PM. Helicobacter pylori infection as a cause of gastritis, duodenal ulcer, gastric cancer and nonulcer dyspepsia: A systematic overview. CMAJ. 1994;150:177–185. [PMC free article] [PubMed] [Google Scholar]; (b) Kalayoglu MV, Libby P, Byrne GI. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA. 2002;288:2724–2731. doi: 10.1001/jama.288.21.2724. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway CAJ. Innate immunity. New Engl J Med. 2000;434:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 5.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 6.Pinsky MR. Sepsis: A pro-and anti-inflammatory disequilibrium syndrome. Contrib Nephrol. 2001;132:354–366. doi: 10.1159/000060100. [DOI] [PubMed] [Google Scholar]
- 7.Bone RC, Sprung CL, Sibbald WJ. Definitions for sepsis and organ failure. Crit Care Med. 1992;20:724–726. doi: 10.1097/00003246-199206000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epdemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Lemaitre B, Nicolas E, Michaut L, Reichhart J-M, Hoffmann JA. The dorsoventral regulatory gene cassette spaetzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 10.Michel T, Reichhart J-M, Hoffmann JA, Royet J. Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature. 2001;414:756–759. doi: 10.1038/414756a. [DOI] [PubMed] [Google Scholar]
- 11.Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/20ScCr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 12.(a) Pfeiffer R. Untersuchungen über das Choleragift. Z Hyg. 1892;11:393–411. [Google Scholar]; (b) Centanni E. Untersuchungen über das Infektionsfieber – das Fiebergift der Bakterien. Dtsch Med Wochenschr. 1894;20:148–153. [Google Scholar]; (c) Rietschel ET, Brade H. Bacterial endotoxins. Sci Am. 1992;267:54–61. doi: 10.1038/scientificamerican0892-54. [DOI] [PubMed] [Google Scholar]; (d) Beutler B, Th Rietschel E. Innate immune sensing and its roots: the story of endotoxin. Nature Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 13.Boivin A, Izard Y. Method for the purification of diphtheria, tetanus and staphylococcus toxins and anatoxins by trichloroacetic acid. C R Soc Biol. 1937;124:25–28. [Google Scholar]
- 14.Westphal O, Lüderitz O. Chemical and biological analysis of highly purified bacterial polysaccharides. Dtsch Med Wochenschr. 1946;78:17–19. [PubMed] [Google Scholar]
- 15.Van Amersfoort ES, Van Berkel TJC, Kuiper J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin Microbiol Rev. 2003;16:379–414. doi: 10.1128/CMR.16.3.379-414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacken A, Rietschel ET, Brade H. Methylation analysis of the heptose/3-deoxy-D-manno-2-octulosonic acid region (inner core) of the lipopolysaccharide from Salmonella minnesota rough mutants. Carbohydr Res. 1986;149:279–291. doi: 10.1016/s0008-6215(00)90051-x. [DOI] [PubMed] [Google Scholar]
- 17.Heinrichs DE, Yethon JA, Whitfield C. Molecular basis for structural diversity in the core regions of the lipopolysaccharides of Escherichia coli and Salmonella enterica. Mol Microbiol. 1998;30:221–232. doi: 10.1046/j.1365-2958.1998.01063.x. [DOI] [PubMed] [Google Scholar]
- 18.Imoto M, Kusumoto S, Shiba T, Naoki H, Zähringer U, Rietschel ET, Unger FM. Structural and synthetic study on lipopolysaccharide of Escherichia coli Re mutant. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu. 1985;27:585–592. [Google Scholar]
- 19.Weckesser J, Mayer H, Drews G, Fromme I. Lipophilic O-antigens containing D-glycero-D-mannoheptose as the sole neutral sugar in Rhodopseudomonas gelatinosa. J Bacteriol. 1975;123:449–455. doi: 10.1128/jb.123.2.449-455.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Schweda EKH, Richards JC, Hood DW, Moxon ER. Expression and structural diversity of the lipopolysaccharide of Haemophilus influenzae: Implication in virulence. Int J Med Microbiol. 2007;297:297–306. doi: 10.1016/j.ijmm.2007.03.007. [DOI] [PubMed] [Google Scholar]; (b) Sadovskaya I, Brisson JR, Thibault P, Richards JC, Lam JS, Altman E. Structural characterization of the outer core and the O-chain linkage region of lipopolysaccharide from Pseudomonas aeruginosa serotype O5. Eur J Biochem. 2000;267:1640–1650. doi: 10.1046/j.1432-1327.2000.01156.x. [DOI] [PubMed] [Google Scholar]; (c) Cox AD, Brisson J, Varma V, Perry MB. Structural analysis of the lipopolysaccharide from Vibrio cholerae O139. Carbohydr Res. 1996;290:43–58. doi: 10.1016/0008-6215(96)00135-8. [DOI] [PubMed] [Google Scholar]; (d) Muller-Loennies S, Linder B, Brade H. Structural analysis of oligosaccharides from lipopolysaccharide (LPS) of Escherichia coli K12 strain W3100 reveals a link between inner and outer core LPS biosynthesis. J Biol Chem. 2003;278:34090–34101. doi: 10.1074/jbc.M303985200. [DOI] [PubMed] [Google Scholar]
- 21.(a) Stanislavskii ES, Mashilova GM, Dmitriev BA, Knirel YA, Vinogradov EV. The structure and immunochemical specificity of O-antigens of 03 serogroup Pseudomonas aeruginosa. J Hyg Epidemiol Microbiol Immunol. 1984;29:289–295. [PubMed] [Google Scholar]; (b) Richards JC, Leitch RA. Elucidation of the structure of the Pasteurella haemolytica serotype T10 lipopolysaccharide O-antigen by n.m.r spectroscopy. Carbohydr Res. 1989;186:275–286. doi: 10.1016/0008-6215(89)84041-8. [DOI] [PubMed] [Google Scholar]; (c) Stanislavsky ES, Kholodkova EV, Knirel YA, Kocharova NA. Saccharides of seven Pseudomonas aeruginosa immunotypes. FEMS Microbiol Immunol. 1989;1:245–251. doi: 10.1111/j.1574-6968.1989.tb02389.x. [DOI] [PubMed] [Google Scholar]; (d) Whitfield C, Richards JC, Perry MB, Clarke BR, MacLean LL. Expression of two structurally distinct D-galactan O antigens in the lipopolysaccharide of Klebsiella pneumoniae serotype O1. J Bacteriol. 1991;173:1420–1431. doi: 10.1128/jb.173.4.1420-1431.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lerouge I, Vanderleyden J. O-antigen structural variation: Mechanisms and possible roles in animal/plant microbe interactions. FEMS Microbiol Rev. 2002;26:17–47. doi: 10.1111/j.1574-6976.2002.tb00597.x. [DOI] [PubMed] [Google Scholar]
- 23.Wiese A, Gutsmann T, Seydel U. Towards antibacterial strategies: Studies on the mechanisms of interactions between antibacterial peptides and model membranes. J Endotoxin Res. 2003;9:67–84. doi: 10.1179/096805103125001441. [DOI] [PubMed] [Google Scholar]
- 24.Lüderitz O, Freudenberg MA, Galanos C, Lehmann V, Rietschel ET, Shaw DH. Lipopolysaccharides of gram-negative bacteria. Curr Top Membr Transp. 1982;17:79–151. [Google Scholar]
- 25.Westphal O, Lüderitz O. Chemical research on lipopolysaccharides of gram-negative bacteria. Angew Chem. 1954;66:407–417. [Google Scholar]
- 26.Imoto M, Yoshimura H, Sakaguchi N, Kusumoto S, Shiba T. Total synthesis of Escherichia coli Lipid A. Tetrahedron Lett. 1985;26:1545–1548. [Google Scholar]
- 27.(a) Galanos C, Lüderitz O, Rietschel ET, Westphal O, Brade H, Brade L, Freudenberg M, Schade U, Imoto M, Yoshimura H, Kusumoto S, Shiba T. Synthetic and natural Escherichia coli free Lipid A express identical endotoxic activities. Eur J Biochem. 1985;148:1–5. doi: 10.1111/j.1432-1033.1985.tb08798.x. [DOI] [PubMed] [Google Scholar]; (b) Kotani S, Takada H, Tsujimoto M, Ogawa T, Takashi I, Ikeda T, Otsuka K, Shimauchi H, Kasai N. Synthetic Lipid A with endotoxic and related biological activities comparable to those of a natural Lipid A from an Escherichia coli Re-mutant. Infect Immun. 1985;49:225–237. doi: 10.1128/iai.49.1.225-237.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takada H, Kotani S. Structural requirements of Lipid A for endotoxicity and other biological activities. Crit Rev Microbiol. 1989;16:477–523. doi: 10.3109/10408418909104475. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol. 2007;193:323–330. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- 29.Freudenberg MA, Freudenberg N, Galanos C. Time course and cellular distribution of endotoxin in liver, lungs, and kidney of rats. Br J Exp Pathol. 1982;63:56–65. [PMC free article] [PubMed] [Google Scholar]
- 30.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 31.Tobias PS, Soldau K, Ulevitch RJ. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med. 1986;164:777–793. doi: 10.1084/jem.164.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramadori G, Meyer zum Buschenfelde KH, Tobias PS, Mathison JC, Ulevitch RJ. Biosynthesis of lipopolysaccharide-binding protein in rabbit hepatocytes. Pathobiology. 1990;58:90–94. doi: 10.1159/000163569. [DOI] [PubMed] [Google Scholar]
- 33.Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 34.(a) Zweigner J, Schumann RR, Weber JR. The role of lipopolysaccharide-binding protein in modulating the innate immune response. Microbes Infect. 2006;8:946–952. doi: 10.1016/j.micinf.2005.10.006. [DOI] [PubMed] [Google Scholar]; (b) Su GL, Frees-wick PD, Geller DA, Wang Q, Shapiro RA, Wan Y-H, Billiar TR, Tweardy DJ, Simmons RL, Wang SC. Molecular cloning, characterization, and tissue distribution of rat lipopolysaccharide binding protein. Evidence of extraheptic expression. J Immunol. 1994;153:743–752. [PubMed] [Google Scholar]
- 35.Beamer LJ, Carroll SF, Eisenberg D. Crystal structure of human BPI and two bound phospholipids at 2.4 Å ngstrom resolution. Science. 1997;276:1861–1864. doi: 10.1126/science.276.5320.1861. [DOI] [PubMed] [Google Scholar]
- 36.Beamer LJ, Carroll SF, Eisenberg D. The BPI/LBP family of proteins: A structural analysis of conserved regions. Protein Sci. 1998;7:906–914. doi: 10.1002/pro.5560070408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamping N, Hoess A, Yu B, Park TC, Kirschning CJ, Pfeil D, Reuter D, Wright SD, Herrmann F, Schumann RR. Effects of site-directed mutagenesis of basic residues (Arg 94, Lys 95, Lys 99) of lipopolysaccharide (LPS)-binding protein on binding and transfer of LPS and subsequent immune cell activation. J Immunol. 1996;157:4648–4656. [PubMed] [Google Scholar]
- 38.(a) Hailman E, Lichenstein HS, Wurfel MM, Miller DS, Johnson DA, Kelley M, Busse LA, Zukowski MM, Wright SD. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179:269–277. doi: 10.1084/jem.179.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu B, Wright SD. Catalytic properties of lipopolysaccharide (LPS) binding protein. Transfer of LPS to soluble CD14. J Biol Chem. 1996;271:4100–4105. doi: 10.1074/jbc.271.8.4100. [DOI] [PubMed] [Google Scholar]
- 39.(a) Wurfel MM, Hailman E, Wright SD. Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J Exp Med. 1995;181:1743–1754. doi: 10.1084/jem.181.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutsmann T, Muller M, Carroll SF, MacKenzie RC, Wiese A, Seydel U. Dual role of lipopolysaccharide (LPS)-binding protein in neutralization of LPS and enhancement of LPS-induced activation of mononuclear cells. Infect Immun. 2001;69:6942–6950. doi: 10.1128/IAI.69.11.6942-6950.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Theofan G, Horwitz AH, Williams RE, Liu PS, Chan I, Birr C, Carroll SF, Meszaros K, Parent JB. An amino-terminal fragment of human lipopolysaccharide-binding protein retains lipid A binding but not CD14-stimulatory activity. J Immunol. 1994;152:3623–3629. [PubMed] [Google Scholar]
- 41.Schumann RR, Lamping N, Hoess A. Interchangeable endotoxin-binding domains in proteins with opposite lipopolysaccharide-dependent activities. J Immunol. 1997;159:5599–5605. [PubMed] [Google Scholar]
- 42.Martin TR, Mathison JC, Tobias PS, Leturcq DJ, Moriarty AM, Maunder RJ, Ulevitch RJ. Lipopolysaccharide binding protein enhances the responsiveness of alveolar macrophages to bacterial lipopolysaccharide. Implications for cytokine production in normal and injured lungs. J Clin Invest. 1992;90:2209–2219. doi: 10.1172/JCI116106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tobias PS, Soldau K, Gegner JA, Mintz D, Ulevitch RJ. Lipopolysaccharide binding protein-mediated complexation of lipopolysaccharide with soluble CD14. J Biol Chem. 1995;270:10482–10488. doi: 10.1074/jbc.270.18.10482. [DOI] [PubMed] [Google Scholar]
- 44.Gegner JA, Ulevitch RJ, Tobias PS. Lipopolysaccharide (LPS) signal transduction and clearance. Dual roles for LPS binding protein and membrane CD14. J Biol Chem. 1995;270:5320–5325. doi: 10.1074/jbc.270.10.5320. [DOI] [PubMed] [Google Scholar]
- 45.Ulevitch RJ. Recognition of bacterial endotoxins by receptor-dependent mechanisms. Adv Immunol. 1993;53:267–289. doi: 10.1016/s0065-2776(08)60502-7. [DOI] [PubMed] [Google Scholar]
- 46.(a) Wright SD, Detmers PA, Aida Y, Adamowski R, Anderson DC, Chad Z, Kabbash LG, Pabst MJ. CD18-deficient cells respond to lipopolysaccharide in vitro. J Immunol. 1990;144:2566–2571. [PubMed] [Google Scholar]; (b) Wright SD, Tobias PS, Ulevitch RJ, Ramos RA. Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel receptor on macrophages. J Exp Med. 1989;170:1231–1241. doi: 10.1084/jem.170.4.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 48.Golenbock DT, Liu Y, Millham FH, Freeman MW, Zoeller RA. Surface expression of human CD14 in Chinese hamster ovary fibroblasts imparts macrophage-like responsiveness to bacterial endotoxin. J Biol Chem. 1993;268:22055–22059. [PubMed] [Google Scholar]
- 49.Lee JD, Kato K, Tobias PS, Kirkland TN, Ulevitch RJ. Transfection of CD14 into 70Z/3 cells dramatically enhances the sensitivity to complexes of lipopolysaccharide (LPS) and LPS binding protein. J Exp Med. 1992;175:1697–1705. doi: 10.1084/jem.175.6.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirkland TN, Finley F, Leturcq D, Moriarty A, Lee JD, Ulevitch RJ, Tobias PS. Analysis of lipopolysaccharide binding by CD14. J Biol Chem. 1993;268:24818–24823. [PubMed] [Google Scholar]
- 51.(a) Bazil V, Horejsi V, Baudys M, Kristofova H, Strominger JL, Kostka W, Hilgert I. Biochemical characterization of a soluble form of the 53-kDa monocyte surface antigen. Eur J Immunol. 1986;16:1583–1589. doi: 10.1002/eji.1830161218. [DOI] [PubMed] [Google Scholar]; (b) Bazil V, Baudys M, Hilgert I, Stefanova I, Low MG, Zbrozek J, Horejsi V. Structural relationship between soluble and membrane-bound forms of human monocyte surface glycoprotein CD14. Mol Immunol. 1989;26:657–662. doi: 10.1016/0161-5890(89)90048-5. [DOI] [PubMed] [Google Scholar]
- 52.(a) Simmons DL, Tan S, Tenen DG, Nicholson-Weller A, Seed B. Monocyte antigen CD14 is a phospholipid anchored membrane protein. Blood. 1989;73:284–289. [PubMed] [Google Scholar]; (b) Landmann R, Wesp M, Dukor P. Modulation of interferon-gamma-induced major histocompatibility (MHC) and CD14 antigen changes by lipophilic muramyltripeptide MTP-PE in human monocytes. Cell Immunol. 1988;117:45–55. doi: 10.1016/0008-8749(88)90075-5. [DOI] [PubMed] [Google Scholar]
- 53.Ferrero E, Goyert SM. Nucleotide sequence of the gene encoding the monocyte differentiation antigen, CD14. Nucleic Acids Res. 1988;16:4173. doi: 10.1093/nar/16.9.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Setoguchi M, Nasu N, Yoshida S, Higuchi Y, Akizuki Si, Yamamoto S. Mouse and human CD14 (myeloid cell-specific leucine-rich glycoprotein) primary structure deduced from cDNA clones. Biochim Biophys Acta. 1989;1008:213–222. doi: 10.1016/0167-4781(80)90012-3. [DOI] [PubMed] [Google Scholar]
- 55.(a) Juan TSC, Kelley MJ, Johnson DA, Busse LA, Hailman E, Wright SD, Lichtenstein HS. Soluble CD14 truncated at amino acid 152 binds lipopolysaccharide (LPS) and enables cellular response to LPS. J Biol Chem. 1995;270:1382–1387. doi: 10.1074/jbc.270.3.1382. [DOI] [PubMed] [Google Scholar]; (b) Yu W, Soprana E, Cosentino G, Volta M, Lichenstein HS, Viale G, Vercelli D. Soluble CD141-152 confers responsiveness to both lipoarabinomannan and lipopolysaccharide in a novel HL-60 cell bioassay. J Immunol. 1998;161:4244–4251. [PubMed] [Google Scholar]
- 56.Stelter F, Bernheiden M, Menzel R, Jack RS, Witt S, Fan XL, Pfister M, Schuett C. Mutation of amino acids 39-44 of human CD14 abrogates binding of lipopolysaccharide and Escherichia coli. Eur J Biochem. 1997;243:100–109. doi: 10.1111/j.1432-1033.1997.00100.x. [DOI] [PubMed] [Google Scholar]
- 57.(a) Juan TSC, Hailman E, Kelley MJ, Wright SD, Lichenstein HS. Identification of a domain in soluble CD14 essential for lipopolysaccharide (LPS) signaling but not LPS binding. J Biol Chem. 1995;270:17237–17242. doi: 10.1074/jbc.270.29.17237. [DOI] [PubMed] [Google Scholar]; (b) Stelter F, Loppnow H, Menzel R, Grunwald U, Bernheiden M, Jack RS, Ulmer AJ, Schutt C. Differential impact of substitution of amino acids 9-13 and 91-101 of human CD14 on soluble CD14-dependent activation of cells by lipopolysaccharide. J Immunol. 1999;163:6035–6044. [PubMed] [Google Scholar]
- 58.(a) Antal-Szalmas P, Van Strijp JAG, Weersink AJL, Verhoef J, Van Kessel KPM. Quantitation of surface CD14 on human monocytes and neutrophils. J Leukoc Biol. 1997;61:721–728. doi: 10.1002/jlb.61.6.721. [DOI] [PubMed] [Google Scholar]; (b) Liu S, Khemlani LS, Shapiro RA, Johnson ML, Liu K, Geller DA, Watkins SC, Goyert SM, Billiar TR. Expression of CD14 by hepatocytes: Upregulation by cytokines during endotoxemia. Infect Immun. 1998;66:5089–5098. doi: 10.1128/iai.66.11.5089-5098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nanbo A, Nishimura H, Muta T, Nagasawa S. Lipopolysaccharide stimulates HepG2 human hepatoma cells in the presence of lipopolysaccharide-binding protein via CD14. Eur J Biochem. 1999;260:183–191. doi: 10.1046/j.1432-1327.1999.00141.x. [DOI] [PubMed] [Google Scholar]; (d) Peterson PK, Gekker G, Hu S, Sheng WS, Anderson WR, Ulevitch RJ, Tobias PS, Gustafson KV, Molitor TW. CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect Immun. 1995;63:1598–1602. doi: 10.1128/iai.63.4.1598-1602.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sugawara S, Sugiyama A, Nemoto E, Rikiishi H, Takada H. Heterogeneous expression and release of CD14 by human gingival fibroblasts: Characterization and CD14-mediated interleukin-8 secretion in response to lipopolysaccharide. Infect Immun. 1998;66:3043–3049. doi: 10.1128/iai.66.7.3043-3049.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ziegler-Heitbrock HWL, Pechumer H, Petersmann I, Durieux JJ, Vita N, Labeta MO, Stroebel M. CD14 is expressed and functional in human B cells. Eur J Immunol. 1994;24:1937–1940. doi: 10.1002/eji.1830240835. [DOI] [PubMed] [Google Scholar]
- 59.(a) Marchant A, Duchow J, Delville JP, Goldman M. Lipopolysaccharide induces up-regulation of CD14 molecule on monocytes in human whole blood. Eur J Immunol. 1992;22:1663–1665. doi: 10.1002/eji.1830220650. [DOI] [PubMed] [Google Scholar]; (b) Matsuura K, Ishida T, Setoguchi M, Higuchi Y, Akizuki Si, Yamamoto S. Upregulation of mouse cd14 expression in Kupffer cells by lipopolysaccharide. J Exp Med. 1994;179:1671–1676. doi: 10.1084/jem.179.5.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ziegler-Heitbrock HWL, Ulevitch RJ. CD14: Cell surface receptor and differentiation marker. Immunol Today. 1993;14:121–125. doi: 10.1016/0167-5699(93)90212-4. [DOI] [PubMed] [Google Scholar]
- 60.(a) Landmann R, Fisscher AE, Obrecht JP. Interferon-.gamma. and interleukin-4 down-regulate soluble CD14 release in human monocytes and macrophages. J Leukoc Biol. 1992;52:323–330. doi: 10.1002/jlb.52.3.323. [DOI] [PubMed] [Google Scholar]; (b) Schuett C, Schilling T, Grunwald U, Schoenfeld W, Krueger C. Endotoxin-neutralizing capacity of soluble CD14. Res Immunol. 1992;143:71–78. doi: 10.1016/0923-2494(92)80082-v. [DOI] [PubMed] [Google Scholar]
- 61.Tobias PS, Ulevitch RJ. Lipopolysaccharide binding protein and CD14 in LPS dependent macrophage. Immunobiology. 1993;187:227–232. doi: 10.1016/S0171-2985(11)80341-4. [DOI] [PubMed] [Google Scholar]
- 62.Landmann R, Zimmerli W, Sansano S, Link S, Hahn A, Glauser MP, Calandra T. Increased circulating soluble CD14 is associated with high mortality in gram-negative septic shock. J Infect Dis. 1995;171:639–644. doi: 10.1093/infdis/171.3.639. [DOI] [PubMed] [Google Scholar]
- 63.Labeta MO, Vidal K, Nores JER, Arias M, Vita N, Morgan BP, Guillemot JC, Loyaux D, Ferrara P, Schmid D, Affolter M, Borysiewicz LK, Donnet-Hughes A, Schiffrin EJ. Innate recognition of bacteria in human milk is mediated by a milk-derived highly expressed pattern recognition receptor, soluble CD14. J Exp Med. 2000;191:1807–1812. doi: 10.1084/jem.191.10.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.(a) Detmers PA, Thieblemont N, Vasselon T, Pironkova R, Miller DS, Wright SD. Potential role of membrane internalization and vesicle fusion in adhesion of neutrophils in response to lipopolysaccharide and TNF. J Immunol. 1996;157:5589–5596. [PubMed] [Google Scholar]; (b) Lichtman SN, Wang J, Lemasters JJ. Lipopolysaccharide-stimulated TNF-alpha release from cultured rat Kupffer cells: Sequence of intracellular signaling pathways. J Leukoc Biol. 1998;64:368–372. doi: 10.1002/jlb.64.3.368. [DOI] [PubMed] [Google Scholar]
- 65.(a) Pollack M, Ohl CA, Golenbock DT, Di Padova F, Wahl LM, Koles NL, Guelde G, Monks BG. Dual effects of LPS antibodies on cellular uptake of LPS and LPS-induced proinflammatory functions. J Immunol. 1997;159:3519–3530. [PubMed] [Google Scholar]; (b) Poussin C, Foti M, Carpentier JL, Pugin J. CD14-dependent endotoxin internalization via a macropinocytic pathway. J Biol Chem. 1998;273:20285–20291. doi: 10.1074/jbc.273.32.20285. [DOI] [PubMed] [Google Scholar]
- 66.Thieblemont N, Wright SD. Transport of bacterial lipopolysaccharide to the Golgi apparatus. J Exp Med. 1999;190:523–534. doi: 10.1084/jem.190.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vasselon T, Hailman E, Thieringer R, Detmers PA. Internalization of monomeric lipopolysaccharide occurs after transfer out of cell surface CD14. J Exp Med. 1999;190:509–521. doi: 10.1084/jem.190.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 1991;254:1016–1019. doi: 10.1126/science.1719635. [DOI] [PubMed] [Google Scholar]
- 69.Lee JD, Kravchenko V, Kirkland TN, Han J, Mackman M, Moriarty A, Leturcq D, Tobias PS, Ulevitch RJ. Glycosyl-phosphatidylinositol-anchored or integral membrane forms of CD14 mediate identical cellular responses to endotoxin. Proc Natl Acad Sci U S A. 1993;90:9930–9934. doi: 10.1073/pnas.90.21.9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williams MJ, Rodriguez A, Kimbrell DA, Eldon ED. The 18-wheeler mutation reveals complex antibacterial gene regulation in Drosophila host defense. EMBO J. 1997;16:6120–6130. doi: 10.1093/emboj/16.20.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.(a) Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoshino K, Tsutsui H, Kawai T, Takeda K, Nakanishi K, Takeda Y, Akira S. Cuting edge: Generation of IL-18 receptor-deficient mice: Evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol. 1999;162:5041–5044. [PubMed] [Google Scholar]
- 72.Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: The induction of polarity by the Toll gene product. Cell. 1985;42:791–798. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 73.Imler J-L, Hoffmann JA. Signaling mechanisms in the antimicrobial host defense of Drosophila. Curr Opin Microbiol. 2000;3:16–22. doi: 10.1016/s1369-5274(99)00045-4. [DOI] [PubMed] [Google Scholar]
- 74.Gay NJ, Keith FJ. Drosophila toll and IL-1 receptor. Nature. 1991;351:355–356. doi: 10.1038/351355b0. [DOI] [PubMed] [Google Scholar]
- 75.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homolog of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–396. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 76.Jenkin C, Palmer DL. Changes in the titre of serum opsonins and phagocytic properties of mouse peritoneal macrophages following injection of endotoxin. J Exp Med. 1960;112:419–429. doi: 10.1084/jem.112.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skidmore BJ, Chiller JM, Morrison DC, Weigle WO. Immunologic properties of bacterial lipopolysaccharide (LPS). Correlation between the mitogenic, adjuvant, and immunogenic activities. J Immunol. 1975;114:770–775. [PubMed] [Google Scholar]
- 78.Underhill DM, Ozinsky A, Haijar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 79.Heine H, Kirschning CJ, Lien E, Monks BG, Rothe M, Golenbock DT. Cutting edge: Cells that carry a null allele for toll-like receptor 2 are capable of responding to endotoxin. J Immunol. 1999;162:6971–6975. [PubMed] [Google Scholar]
- 80.(a) Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signaling. Nature. 1998;395:284–288. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]; (b) Da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. Transfer from CD14 to TLR4 and MD-2. J Biol Chem. 2001;276:21129–21135. doi: 10.1074/jbc.M009164200. [DOI] [PubMed] [Google Scholar]
- 81.(a) Kirschning CJ, Wesche H, Ayres TM, Rothe M. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med. 1998;188:2091–2097. doi: 10.1084/jem.188.11.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. Cutting edge: Cell surface expression and lipopolysaccharide signaling via the Toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–3475. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- 83.Gangloff M, Gay NJ. MD-2: The Toll “gate-keeper” in endotoxin signaling. Trends Biochem Sci. 2004;29:294–300. doi: 10.1016/j.tibs.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 84.Visintin A, Mazzoni A, Spitzer JA, Segal DM. Secreted MD-2 is a large polymeric protein that efficiently confers lipopolysaccharide sensitivity to Toll-like receptor 4. Proc Natl Acad Sci U S A. 2001;98:12156–12161. doi: 10.1073/pnas.211445098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.(a) Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]; (b) Ohnishi T, Muroi M, Tanamoto K-i. MD-2 is necessary for the Toll-like receptor 4 protein to undergo glycosylation essential for its translocation to the cell surface. Clin Diagn Lab Immunol. 2003;10:405–410. doi: 10.1128/CDLI.10.3.405-410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Viriyakosol S, Tobias PS, Kitchens RL, Kirkland TN. MD-2 binds to bacterial lipopolysaccharides. J Biol Chem. 2001;276:38044–38051. doi: 10.1074/jbc.M105228200. [DOI] [PubMed] [Google Scholar]
- 87.Jia HP, Kline JN, Penisten A, Apicella MA, Gioannini TL, Weiss J, McCray PB. Endotoxin responsiveness of human airway epithelia is limited by low expression of MD-2. Am J Physiol. 2004;287:L428–L437. doi: 10.1152/ajplung.00377.2003. [DOI] [PubMed] [Google Scholar]
- 88.Viriyakosol S, Tobias PS, Kirkland TN. Mutational analysis of membrane and soluble forms of human MD-2. J Biol Chem. 2006;281:11955–11964. doi: 10.1074/jbc.M511627200. [DOI] [PubMed] [Google Scholar]
- 89.Mancek M, Pristovsek P, Jerala R. Identification of LPS-binding peptide fragment of MD-2, a toll-receptor accessory protein. Biochem Biophys Res Commun. 2002;292:880–885. doi: 10.1006/bbrc.2002.6748. [DOI] [PubMed] [Google Scholar]
- 90.(a) Visintin A, Latz E, Monks BG, Espevik T, Golenbock DT. Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J Biol Chem. 2003;278:48313–48320. doi: 10.1074/jbc.M306802200. [DOI] [PubMed] [Google Scholar]; (b) Kawasaki K, Nogawa H, Nishijima M. Identification of mouse MD-2 residues important for forming the cell surface TLR4-MD-2 complex recognized by anti-TLR4-MD-2 antibodies, and for conferring LPS and taxol responsiveness on mouse TLR4 by alanine-scanning mutagenesis. J Immunol. 2003;170:413–420. doi: 10.4049/jimmunol.170.1.413. [DOI] [PubMed] [Google Scholar]; (c) Schromm AB, Lien E, Henneke P, Chow JC, Yoshimura A, Heine H, Latz E, Monks BG, Schwartz DA, Miyake K, Golenbock DT. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: A point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J Exp Med. 2001;194:79–88. doi: 10.1084/jem.194.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Re F, Strominger JL. Separate functional domains of human MD-2 mediate toll-like receptor 4-binding and lipopolysaccharide responsiveness. J Immunol. 2003;171:5272–5276. doi: 10.4049/jimmunol.171.10.5272. [DOI] [PubMed] [Google Scholar]; (e) Da Silva Correia J, Ulevitch RJ. MD-2 and TLR4 N-linked glycosylation are important for a functional lipopolysaccharide receptor. J Biol Chem. 2002;277:1845–1854. doi: 10.1074/jbc.M109910200. [DOI] [PubMed] [Google Scholar]; (f) Ohnishi T, Muroi M, Tanamoto K-i. N-linked glycosylations at Asn26 and Asn114 of human MD-2 are required for toll-like receptor 4-mediated activation of NF-kappaB by lipopolysaccharide. J Immunol. 2001;167:3354–3359. doi: 10.4049/jimmunol.167.6.3354. [DOI] [PubMed] [Google Scholar]
- 91.Gruber A, Mancek M, Wagner H, Kirschning CJ, Jerala R. Structural model of MD-2 and functional role of its basic amino acid clusters involved in cellular lipopolysaccharide recognition. J Biol Chem. 2004;279:28475–28482. doi: 10.1074/jbc.M400993200. [DOI] [PubMed] [Google Scholar]
- 92.Mullen GED, Kennedy MN, Visintin A, Mazzoni A, Leifer CA, Davies DR, Segal DM. The role of disulfide bonds in the assembly and function of MD-2. Proc Natl Acad Sci U S A. 2003;100:3919–3924. doi: 10.1073/pnas.0630495100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J Immunol. 2005;175:6465–6472. doi: 10.4049/jimmunol.175.10.6465. [DOI] [PubMed] [Google Scholar]
- 94.(a) Latz E, Visintin A, Lien E, Fitzgerald KA, Espevik T, Golenbock DT. The LPS receptor generates inflammatory signals from the cell surface. J Endotoxin Res. 2003;9:375–380. doi: 10.1179/096805103225003303. [DOI] [PubMed] [Google Scholar]; (b) Re F, Strominger JL. Monomeric recombinant MD-2 binds Toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem. 2002;277:23427–23432. doi: 10.1074/jbc.M202554200. [DOI] [PubMed] [Google Scholar]
- 95.Gioannini TL, Teghanemt A, Zhang D, Coussens NP, Dockstader W, Ramaswamy S, Weiss JP. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci U S A. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kennedy MN, Mullen GED, Leifer CA, Lee CW, Mazzoni A, Dileepan KN, Segal DM. A complex of soluble MD-2 and lipopolysaccharide serves as an activating ligand for toll-like receptor 4. J Biol Chem. 2004;279:34698–34704. doi: 10.1074/jbc.M405444200. [DOI] [PubMed] [Google Scholar]
- 97.Jack RS, Fan X, Bernheiden M, Rune G, Ehlers M, Weber A, Kirsch G, Mentel R, Furll B, Freudenberg M, Schmitz G, Stelter F, Schutt C. Lipopolysaccharide-binding protein is required to combat a murine Gram-negative bacterial infection. Nature. 1997;389:742–745. doi: 10.1038/39622. [DOI] [PubMed] [Google Scholar]
- 98.(a) Saitoh S-i, Akashi S, Yamada T, Tanimura N, Kobayashi M, Konno K, Matsumoto F, Fukase K, Kusumoto S, Nagai Y, Kusumoto Y, Kosugi A, Miyake K. Lipid A antagonist, Lipid IVA, is distinct from Lipid A in interaction with Toll-like receptor 4 (TLR4)-MD2 and ligand-induced TLR4 oligomerization. Int Immunol. 2004;16:961–969. doi: 10.1093/intimm/dxh097. [DOI] [PubMed] [Google Scholar]; (b) Inohara N, Nunez G. ML—A conserved domain involved in innate immunity and lipid metabolism. Trends Biochem Sci. 2002;27:219–221. doi: 10.1016/s0968-0004(02)02084-4. [DOI] [PubMed] [Google Scholar]
- 99.(a) Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]; (b) Beutler B, Rietschel ET. Timeline: Innate immune sensing and its roots: The story of endotoxins. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 100.Karin M, Lin A. NF-kB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 101.(a) Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]; (b) Reitsma PH, Branger J, Van Den Blink B, Weijer S, Van Der Poll T, Meijers JCM. Procoagulant protein levels are differently increased during human endotoxemia. J Thromb Haemost. 2003;1:1019–1023. doi: 10.1046/j.1538-7836.2003.00237.x. [DOI] [PubMed] [Google Scholar]; (c) Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 102.Lopez-Bojorquez LN, Dehesa AZ, Reyes-Teran G. Molecular mechanisms involved in the pathogenesis of septic shock. Arch Med Res. 2004;35:465–479. doi: 10.1016/j.arcmed.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 103.Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taveira da Silva AM, Kaulbach HC, Chuidian FS, Lambert DR, Suffredini AF, Danner RL. Brief report: Shock and multiple-organ dysfunction after self-administration of Salmonella endotoxin. New Engl J Med. 1993;328:1457–1460. doi: 10.1056/NEJM199305203282005. [DOI] [PubMed] [Google Scholar]
- 105.(a) Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]; (b) Schlayer HJ, Karck U, Ganter U, Hermann R, Decker K. Enhancement of neutrophil adherence to isolated rat liver sinusoidal endothelial cells by supernatants of lipopolysaccharide-activated monocytes. Role of tumor necrosis factor. J Hepatol. 1987;5:311–321. doi: 10.1016/s0168-8278(87)80037-5. [DOI] [PubMed] [Google Scholar]; (c) Varani J, Ward PA. Mechanisms of endothelial cell injury in acute inflammation. Shock. 1994;2:311–319. doi: 10.1097/00024382-199411000-00001. [DOI] [PubMed] [Google Scholar]
- 106.Baud V, Karin M. Signal transduction by tumor necrosis factor and it relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 107.Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteremia. Nature. 1987;330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 108.Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60:49–57. doi: 10.1016/s0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 109.(a) Rajalingam D, Kacer D, Prudovsky I, Kumar TKS. Molecular cloning, overexpression and characterization of human interleukin 1alpha. Biochem Biophys Res Commun. 2007;360:604–608. doi: 10.1016/j.bbrc.2007.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brody DT, Durum SK. A plasma membrane anchoring mechanism for IL-1. Prog Leukoc Biol. 1988;8:101–107. [Google Scholar]; (c) Wingfield P, Payton M, Graber P, Rose K, Dayer JM, Shaw AR, Schmeissner U. Purification and characterization of human interleukin-1.alpha produced in Escherichia coli. Eur J Biochem. 1987;165:537–541. doi: 10.1111/j.1432-1033.1987.tb11472.x. [DOI] [PubMed] [Google Scholar]; (d) Gubler U, Chua AO, Stern AS, Hellmann CP, Vitek MP, Dechiara TM, Benjamin WR, Collier KJ, Dukovich M. Recombinant human interleukin 1alpha: Purification and biological characterization. J Immunol. 1986;136:2492–2497. [PubMed] [Google Scholar]
- 110.Dinarello CA. Biological basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 111.Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hazelzet JA, Kornelisse RF, van der Pouw Kraan TCTM, Joosten KFM, van der Voort E, van Mierlo G, Suur MH, Hop WCJ, de Groot R, Hack CE. Interleukin 12 levels during the initial phase of septic shock with purpura in children: Relation to severity of disease. Cytokine. 1997;9:711–716. doi: 10.1006/cyto.1997.0215. [DOI] [PubMed] [Google Scholar]
- 113.(a) Van Zee KJ, DeForge LE, Fischer E, Marano MA, Kenney JS, Remick DG, Lowry SF, Moldawer LL. IL-8 in septic shock, endotoxemia, and after IL-1 administration. J Immunol. 1991;146:3478–3482. [PubMed] [Google Scholar]; (b) Bossink AWJ, Paemen L, Jansen PM, Hack CE, Thijs LG, Van Damme J. Plasma levels of the chemokines monocyte chemotactic proteins-1 and -2 are elevated in human sepsis. Blood. 1995;86:3841–3847. [PubMed] [Google Scholar]
- 114.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 115.(a) Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA., Jr Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med. 1984;160:618–623. doi: 10.1084/jem.160.2.618. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gregory SA, Morrissey JH, Edgington TS. Regulation of tissue factor gene expression in the monocyte procoagulant response to endotoxin. Mol Cell Biol. 1989;9:2752–2755. doi: 10.1128/mcb.9.6.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maruyama I. Biology of endothelium. Lupus. 1998;7:S41–S43. doi: 10.1177/096120339800700210. [DOI] [PubMed] [Google Scholar]
- 117.Ryu J, Pyo H, Jou I, Joe E. Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-kappaB. J Biol Chem. 2000;275:29955–29959. doi: 10.1074/jbc.M001220200. [DOI] [PubMed] [Google Scholar]
- 118.McGilvray ID, Rotstein OD. Signaling pathways of tissue factor expression in monocytes and macrophages. Sepsis. 1999;3:93–101. [Google Scholar]
- 119.(a) Suffredini AF, Harpel PC, Parrillo JE. Promotion and subsequent inhibition of plasminogen activation after administration of intravenous endotoxin to normal subjects. New Engl J Med. 1989;320:1165–1172. doi: 10.1056/NEJM198905043201802. [DOI] [PubMed] [Google Scholar]; (b) Van Deventer SJH, Buller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: Analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990;76:2520–2526. [PubMed] [Google Scholar]; (c) Van der Poll T, Bueller HR, ten Cate H, Wortel CH, Bauer KA, Van Deventer SJH, Hack CE, Sauerwein HP, Rosenberg RD, ten Cate JW. Activation of coagulation after administration of tumor necrosis factor to normal subjects. New Engl J Med. 1990;322:1622–1627. doi: 10.1056/NEJM199006073222302. [DOI] [PubMed] [Google Scholar]; (d) Van Der Poll T, Levi M, Buller HR, Van Deventer SJH, De Boer JP, Hack CE, ten Cate JW. Fibrinolytic response to tumor necrosis factor in healthy subjects. J Exp Med. 1991;174:729–732. doi: 10.1084/jem.174.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.(a) Fourrier F, Chopin C, Goudemand J, Hendrycx S, Caron C, Rime A, Marey A, Lestave P. Septic shock, multiple organ failure, and disseminated intravascular coagulation. Compared patterns of antithrombin III, protein C, and protein S deficiencies. Chest. 1992;101:816–823. doi: 10.1378/chest.101.3.816. [DOI] [PubMed] [Google Scholar]; (b) Mesters RM, Mannucci PM, Coppola R, Keller T, Ostermann H, Kienast J. Factor VIIa and antithrombin III activity during severe sepsis and septic shock in neutropenic patients. Blood. 1996;88:881–886. [PubMed] [Google Scholar]
- 121.Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163:740–755. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.(a) Senior RM, Skogen WF, Griffin GL, Wilner GD. Effects of fibrinogen derivatives upon the inflammatory response. Studies with human fibrinopeptide B. J Clin Invest. 1986;77:1014–1019. doi: 10.1172/JCI112353. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bick RL. Disseminated intravascular coagulation. Hematol Oncol Clin North Am. 1992;6:1259–1285. [PubMed] [Google Scholar]; (c) Bick RL. Disseminated intravascular coagulation and related syndromes: A clinical review. Semin Thromb Hemost. 1988;14:299–338. doi: 10.1055/s-2007-1002793. [DOI] [PubMed] [Google Scholar]; (d) ten Cate H, Brandjes DP, Wolters HJ, van Deventer SJ. Disseminated intravascular coagulation: Pathophysiology, diagnosis, and treatment. New Horiz. 1993;1:312–323. [PubMed] [Google Scholar]; (e) Taylor FB., Jr The inflammatory-coagulant axis in the host response to gram-negative sepsis: Regulatory roles of proteins and inhibitors of tissue factor. New Horiz. 1994;2:555–565. [PubMed] [Google Scholar]; (f) Carrico CJ, Meakins JL, Marshall JC, Fry D, Maier RV. Multiple-organ-failure syndrome. Arch Surg. 1986;121:196–208. doi: 10.1001/archsurg.1986.01400020082010. [DOI] [PubMed] [Google Scholar]; (g) Shibayama Y. Sinusoidal circulatory disturbance by microthrombosis as a cause of endotoxin-induced hepatic injury. J Pathol. 1987;151:315–321. doi: 10.1002/path.1711510412. [DOI] [PubMed] [Google Scholar]; (h) Schlag G, Redl H. Morphology of the microvascular system in shock: Lung, liver, and skeletal muscles. Crit Care Med. 1985;13:1045–1049. [PubMed] [Google Scholar]
- 123.Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. 1995;273:117–123. [PubMed] [Google Scholar]
- 124.(a) Bulusu M, Hildebrandt J, Lam C, Liehl E, Loibner H, Macher I, Scholz D, Schutze E, Stutz P, Vyplel H, Unger F. Enzymic synthesis of analogs of bacterial lipid A and design of biologically active LPS-antagonists and -mimetics. Pure Appl Chem. 1994;66:2171–2174. [Google Scholar]; (b) Cohen PS, Nakshatri H, Dennis J, Caragine T, Bianchi M, Cerami A, Tracey KJ. CNI-1493 inhibits monocyte/macrophage tumor necrosis factor by suppression of translation efficiency. Proc Natl Acad Sci U S A. 1996;93:3967–3971. doi: 10.1073/pnas.93.9.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rice GC, Brown PA, Nelson RJ, Bianco JA, Singer JW, Burstein S. Protection from endotoxic shock in mice by pharmacologic inhibition of phosphatidic acid. Proc Natl Acad Sci U S A. 1994;91:3857–3861. doi: 10.1073/pnas.91.9.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heyes JR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]; (e) Bianchi M, Ulrich P, Bloom O, Meistrell MI, Zimmerman GA, Schmidtmayerova H, Bukrinsky M, Donnelley T, Bucala R. An inhibitor of macrophage arginine transport and nitric oxide production (CNI-1493) prevents acute inflammation and endotoxin lethality. Mol Med. 1995;1:254–266. [PMC free article] [PubMed] [Google Scholar]
- 125.(a) McGeehan GM, Becherer JD, Bast RCJ, Boyer CM, Champion B, Connolly KM, Conway JG, Furdon P, Karp S, Kidao S, McElroy AB, Nichols J, Pryzxansky KM, Schoenen F, Sekut L, Truesdale A, Verghese M, Warner J, Ways JP. Regulation of tumor necrosis factor-.alpha. processing by a metalloproteinase inhibitor. Nature. 1994;370:558–561. doi: 10.1038/370558a0. [DOI] [PubMed] [Google Scholar]; (b) Galloway CJ, Madanat MS, Mitra G. Monoclonal anti-tumor necrosis factor (TNF) antibodies protect mouse and human cells from TNF cytotoxicity. J Immunol Methods. 1991;140:37–43. doi: 10.1016/0022-1759(91)90124-x. [DOI] [PubMed] [Google Scholar]; (c) Pauli U, Bertoni G, Duerr M, Peterhans E. A bioassay for the detection of tumor necrosis factor from eight different species: Evaluation of neutralization rates of a monoclonal antibody against human TNF-.alpha. J Immunol Methods. 1994;171:263–265. doi: 10.1016/0022-1759(94)90047-7. [DOI] [PubMed] [Google Scholar]; (d) Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 126.Hale KK, Smith CG, Baker SL, Vanderslice RW, Squires CH, Gleason TM, Tucker KK, Kohno T, Russell DA. Multifunctional regulation of the biological effects of TNF-.alpha. by the soluble type I and type II TNF receptors. Cytokine. 1995;7:26–38. doi: 10.1006/cyto.1995.1004. [DOI] [PubMed] [Google Scholar]
- 127.(a) Dinarello CA, Gelfand JA, Wolff SM. Anticytokine strategies in the treatment of the systemic inflammatory response syndrome. JAMA. 1993;269:1829–1835. [PubMed] [Google Scholar]; (b) Redmond HP, Chavin KD, Bromberg JS, Daly JM. Inhibition of macrophage-activating cytokines is beneficial in the acute septic response. Ann Surg. 1991;214:502–508. doi: 10.1097/00000658-199110000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Natanson C, Hoffman WD, Suffredini AF, Eichacker PQ, Danner RL. Selected treatment strategies for septic shock based on proposed mechanisms of pathogenesis. Ann Intern Med. 1994;120:771–783. doi: 10.7326/0003-4819-120-9-199405010-00009. [DOI] [PubMed] [Google Scholar]
- 128.Ohto U, Fukase K, Miyake K, Satow Y. Crystal structures of human MD-2 and its complex with antiendotoxic Lipid IVa. Science. 2007;316:1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 129.(a) Takayama K, Qureshi N, Beutler B, Kirkland TN. Diphosphoryl lipid A from Rhodopseudomonas sphaeroides ATCC 17023 blocks induction of cachectin in macrophages by lipopolysaccharide. Infect Immun. 1989;57:1336–1338. doi: 10.1128/iai.57.4.1336-1338.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Christ WJ, McGuinness PD, Asano O, Wang Y, Mullarkey MA, Perez M, Hawkins LD, Blythe TA, Dubuc GR, Robidoux AL. Total synthesis of the proposed structure of Rhodobacter sphaeroides Lipid A resulting in the synthesis of new potent lipopolysaccharide antagonists. J Am Chem Soc. 1994;116:3637–3638. [Google Scholar]
- 130.(a) Hawkins LD, Christ WJ, Rossignol DP. Inhibition of endotoxin response by synthetic TLR4 antagonists. Curr Top Med Chem. 2004;4:1147–1171. doi: 10.2174/1568026043388123. [DOI] [PubMed] [Google Scholar]; (b) Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J, Chen WH, Ernst RK, Rossignol DP, Gusovsky F, Blanco JCG, Vogel SN. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 2013;497:498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.(a) Gmeiner J, Lüderitz O, Westphal O. Biochemical studies on lipopolysaccharides of Salmonella R mutants. Eur J Biochem. 1969;7:370–379. doi: 10.1111/j.1432-1033.1969.tb19618.x. [DOI] [PubMed] [Google Scholar]; (b) Hase S, Rietschel ET. Lipid A structure of lipopolysaccharides from various bacterial groups. Eur J Biochem. 1976;63:101–107. doi: 10.1111/j.1432-1033.1976.tb10212.x. [DOI] [PubMed] [Google Scholar]
- 132.Lewicky JD, Ulanova M, Jiang Z-H. Synthesis and immunostimulatory activity of diethanolamine-containing lipid A mimics. R Soc Chem Adv. 2012;2:1917–1926. doi: 10.1016/j.bmc.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 133.(a) Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS. Differential induction of the Toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun. 2005;73:2940–2950. doi: 10.1128/IAI.73.5.2940-2950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trent MS, Stead CM, Tran AX, Hankins JV. Invited review: Diversity of endotoxin and its impact on pathogenesis. J Endotoxin Res. 2006;12:205–223. doi: 10.1179/096805106X118825. [DOI] [PubMed] [Google Scholar]
- 134.(a) Akamatsu M, Fujimoto Y, Kataoka M, Suda Y, Kusumoto S, Fukase K. Synthesis of lipid A monosaccharide analogs containing acidic amino acid: Exploring the structural basis for the endotoxic and antagonistic activities. Bioorg Med Chem. 2006;14:6759–6777. doi: 10.1016/j.bmc.2006.05.051. [DOI] [PubMed] [Google Scholar]; (b) Fujimoto Y, Adachi Y, Akamatsu Masao, Fukase Yoshiyuki, Kataoka Mikayo, Suda Yasuo, Fukase Koichi, Kusumoto S. Synthesis of lipid A and its analogues for investigation of the structural basis for their bioactivity. J Endotoxin Res. 2005;11:341–347. doi: 10.1179/096805105X76841. [DOI] [PubMed] [Google Scholar]; (c) Zhang Y, Gaekwad J, Wolfert MA, Boons GJ. Modulation of innate immune responses with synthetic lipid a derivatives. J Am Chem Soc. 2007;129:5200–5216. doi: 10.1021/ja068922a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bazin HG, Murray TJ, Bowen WS, Mozaffarian A, Fling SP, Bess LS, Livesay MT, Arnold JS, Johnson CL, Ryter KT, Cluff CW, Evans JT, Johnson DA. The ‘Ethereal’ nature of TLR4 agonism and antagonism in the AGP class of lipid A mimetics. Bioorg Med Chem Lett. 2008;18:5350–5354. doi: 10.1016/j.bmcl.2008.09.060. [DOI] [PubMed] [Google Scholar]
- 135.Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 136.(a) Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat Immunol. 2002;3:354–359. doi: 10.1038/ni777. [DOI] [PubMed] [Google Scholar]; (b) Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Poltorak A, Ricciardi-Castagnoli P, Citterio S, Beutler B. Physical contact between lipopolysaccharide and Toll-like receptor 4 revealed by genetic complementation. Proc Natl Acad Sci U S A. 2000;97:2163–2167. doi: 10.1073/pnas.040565397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fukase K, Fukase Y, Oikawa M, Liu W-C, Suda Y, Kusumoto S. Divergent synthesis and biological activities of Lipid A analogues of shorter acyl chains. Tetrahedron. 1998;54:4033–4050. [Google Scholar]
- 138.Qureshi N, Mascagni P, Ribi E, Takayama K. Monophosphoryl lipid A obtained from lipopolysaccharides of Salmonella minnesota R595. Purification of the dimethyl derivative by high performance liquid chromatography and complete structural determination. J Biol Chem. 1985;260:5271–5278. [PubMed] [Google Scholar]
- 139.Johnson DA, Keegan DS, Sowell CG, Livesay MT, Johnson CL, Taubner LM, Harris A, Myers KR, Thompson JD, Gustafson GL, Rhodes MJ, Ulrich JT, Ward JR, Yorgensen YM, Cantrell JL, Brookshire VG. 3-O-desacyl monophosphoryl lipid a derivatives: Synthesis and immunostimulant activities. J Med Chem. 1999;42:4640–4649. doi: 10.1021/jm990222b. [DOI] [PubMed] [Google Scholar]
- 140.Funatogawa K, Matsuura M, Nakano M, Kiso M, Hasegawa A. Relationship of structure and biological activity of monosaccharide lipid A analogs to induction of nitric oxide production by murine macrophage RAW264.7 cells. Infect Immun. 1998;66:5792–5798. doi: 10.1128/iai.66.12.5792-5798.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kiso M, Tanaka S, Fujita M, Fujishima Y, Ogawa Y, Ishida H, Hasegawa A. Synthesis of the optically active 4-O-phosphono-D-glucosamine derivatives related to the nonreducing-sugar subunit of bacterial lipid A. Carbohydr Res. 1987;162:127–140. doi: 10.1016/0008-6215(87)80207-0. [DOI] [PubMed] [Google Scholar]
- 142.Johnson DA, Sowell CG, Johnson CL, Livesay MT, Keegan DS, Rhodes MJ, Ulrich JT, Ward JR, Cantrell JL, Brookshire VG. Synthesis and biological evaluation of a new class of vaccine adjuvants: Aminoalkyl glucosaminide 4-phosphates (AGPs) Bioorg Med Chem Lett. 1999;9:2273–2278. doi: 10.1016/s0960-894x(99)00374-1. [DOI] [PubMed] [Google Scholar]
- 143.Rietschel ET, Kirikae T, Schade FU, Ulmer AJ, Holst O, Brade H, Schmidt G, Mamat U, Grimmecke HD, Kusumoto S, Zähringer U. The chemical structure of bacterial endotoxin in relation to bioactivity. Immunobiology. 1993;187:169–190. doi: 10.1016/S0171-2985(11)80338-4. [DOI] [PubMed] [Google Scholar]
- 144.(a) Teghanemt A, Zhang D, Levis EN, Weiss JP, Gioannini TL. Molecular basis of reduced potency of underacylated endotoxins. J Immunol. 2005;175:4669–4676. doi: 10.4049/jimmunol.175.7.4669. [DOI] [PubMed] [Google Scholar]; (b) Rossignol DP, Lynn M. TLR4 antagonists for endotoxemia and beyond. Curr Opin Investig Drugs. 2005;6:496–502. [PubMed] [Google Scholar]
- 145.Kim HM, Park BS, Kim J-I, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 146.Netea MG, Deuren Mv, Kullberg BJ, Cavaillon J-M, Meer JWMVd. Does the shape of lipid A determine the interaction of LPS with Toll-like receptors? J Immunol. 2002;23:135–139. doi: 10.1016/s1471-4906(01)02169-x. [DOI] [PubMed] [Google Scholar]
- 147.Jeyaretnam B, Glushka J, Kolli VSK, Carlson RW. Characterization of a novel lipid-A from Rhizobium species sin-1: A unique lipid-A structure that is devoid of phosphate and has a glycosyl backbone consisting of glucosamine and 2-aminogluconic acid. J Biol Chem. 2002;277:41802–41810. doi: 10.1074/jbc.M112140200. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Y, Wolfert MA, Boons G-J. The influence of the long chain fatty acid on the antagonistic activities of Rhizobium sin-1 lipid A. Bioorg Med Chem. 2007;15:4800–4812. doi: 10.1016/j.bmc.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vasan M, Wolfert MA, Boons G-J. Agonistic and antagonistic properties of a Rhizobium sin-1 lipid A modified by an ether-linked lipid. Org Biomol Chem. 2007;5:2087–2097. doi: 10.1039/b704427e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.(a) Ogawa T. Chemical structure of lipid A from Porphyromonas (Bacteroides) gingivalis lipopolysaccharide. FEBS Lett. 1993;332:197–201. doi: 10.1016/0014-5793(93)80512-s. [DOI] [PubMed] [Google Scholar]; (b) Ogawa T. Immunobiological properties of chemically defined lipid A from lipopolysaccharide of Porphyromonas (Bacteroides) gingivalis. Eur J Biochem. 1994;219:737–742. doi: 10.1111/j.1432-1033.1994.tb18552.x. [DOI] [PubMed] [Google Scholar]
- 151.(a) Darveau RP, Pham TTT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. Porphyromonas gingivalis lipopolysaccharide contains multiple Lipid A species that functionally interact with both Toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reife RA, Coats SR, Al-Qutub M, Dixon DM, Braham PA, Billharz RJ, Howald WN, Darveau RP. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: Differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol. 2006;8:857–868. doi: 10.1111/j.1462-5822.2005.00672.x. [DOI] [PubMed] [Google Scholar]
- 152.(a) Sawada N, Ogawa T, Asai Y, Makimura Y, Sugiyama A. Toll-like receptor 4-dependent recognition of structurally different forms of chemically synthesized lipid As of Porphyromonas gingivalis. Clin Exp Immunol. 2007;148:529–536. doi: 10.1111/j.1365-2249.2007.03346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Y, Gaekwad J, Wolfert MA, Boons GJ. Synthetic tetra-acylated derivatives of lipid A from Porphyromonas gingivalis are antagonists of human TLR4. Org Biomol Chem. 2008;6:3371–3381. doi: 10.1039/b809090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.(a) Demchenko AV, Wolfert MA, Santhanam B, Moore JN, Boons GJ. Synthesis and biological evaluation of Rhizobium sin-1 Lipid A derivatives. J Am Chem Soc. 2003;125:6103–6112. doi: 10.1021/ja029316s. [DOI] [PubMed] [Google Scholar]; (b) Vandenplas ML, Carlson RW, Jeyaretnam BS, McNeill B, Barton MH, Norton N, Murray TF, Moore JN. Rhizobium Sin-1 lipopolysaccharide (LPS) prevents enteric LPS-induced cytokine production. J Biol Chem. 2002;277:41811–41816. doi: 10.1074/jbc.M205252200. [DOI] [PubMed] [Google Scholar]
- 154.Takayama K, Ribi E, Cantrell JL. Isolation of a nontoxic Lipid A fraction containing tumor regression activity. Cancer Res. 1981;41:2654–2657. [PubMed] [Google Scholar]
- 155.Vuopio-Varkila J, Nurminen M, Pyhälä L, Mäkelä PH. Lipopolysaccharide-induced non-specific resistance to systemic Escherichia coli infection in mice. J Med Microbiol. 1988;25:197–203. doi: 10.1099/00222615-25-3-197. [DOI] [PubMed] [Google Scholar]
- 156.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 157.Okemoto K, Kawasaki K, Hanada K, Miura M, Nishijima M. A potent adjuvant monophosphoryl lipid A triggers various immune responses, but not secretion of IL-1 beta or activation of caspase-1. J Immunol. 2006;176:1203–1208. doi: 10.4049/jimmunol.176.2.1203. [DOI] [PubMed] [Google Scholar]
- 158.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 159.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 160.Cekic C, Casella CR, Eaves CA, Matsuzawa A, Ichijo H, Mitchell TC. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl Lipid A. J Biol Chem. 2009;284:31982–31991. doi: 10.1074/jbc.M109.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.(a) Kitchens RL, Ulevitch RJ, Munford RS. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J Exp Med. 1992;176:485–494. doi: 10.1084/jem.176.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Golenbock DT, Hampton RY, Qureshi N, Takayama K, Raetz CRH. Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. J Biol Chem. 1991;266:19490–19498. [PubMed] [Google Scholar]; (c) Kovach NL, Yee E, Munford RS, Raetz CR, Harlan JM. Lipid IVA inhibits synthesis and release of tumor necrosis factor induced by lipopolysaccharide in human whole blood ex vivo. J Exp Med. 1990;172:77–84. doi: 10.1084/jem.172.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.(a) Christ WJ, Asano O, Robidoux ALC, Perez M, Wang Y, Dubuc GR, Gavin WE, Hawkins LD, McGuinness PD. E5531—A pure endotoxin antagonist of high potency. Science. 1995;268:80–83. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]; (b) Ingalls RR, Monks BG, Savedra R, Jr, Christ WJ, Delude RL, Medvedev AE, Espevik T, Golenbock DT. CD11/CD18 and CD14 share a common Lipid A signaling pathway. J Immunol. 1998;161:5413–5420. [PubMed] [Google Scholar]; (c) Lien E, Chow JC, Hawkins LD, McGuinness PD, Miyake K, Espevik T, Gusovsky F, Golenbock DT. A novel synthetic acyclic Lipid A-like agonist activates cells via the lipopolysaccharide/Toll-like receptor 4 signaling pathway. J Biol Chem. 2001;276:1873–1880. doi: 10.1074/jbc.M009040200. [DOI] [PubMed] [Google Scholar]
- 163.(a) Shiozaki M, Watanabe Y, Iwano Y, Kaneko T, Doi H, Tanaka D, Shimozato T, Kurakata Si. Synthesis of lipid A analogues containing glucose instead of glucosamine and their LPS-antagonistic activities. Tetrahedron. 2005;61:5101–5122. [Google Scholar]; (b) Shiozaki M, Doi H, Tanaka D, Shimozato T, Kurakata S-i. Syntheses of glucose-containing Lipid A analogues and their LPS-antagonistic activities. Bull Chem Soc Jpn. 2005;78:1091–1104. [Google Scholar]
- 164.Shiozaki M, Doi H, Tanaka D, Shimozato T, Kurakata S-i. Syntheses of glucose-containing E5564 analogues and their LPS-antagonistic activities. Tetrahedron. 2006;62:205–225. [Google Scholar]
- 165.Peri F, Marinzi C, Barath M, Granucci F, Urbano M, Nicotra F. Synthesis and biological evaluation of novel lipid A antagonists. Bioorg Med Chem. 2006;14:190–199. doi: 10.1016/j.bmc.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 166.Artner D, Oblak A, Ittig S, Garate JA, Horvat S, Arrieumerlou C, Hofinger A, Oostenbrink C, Jerala R, Kosma P, Zamyatina A. Conformationally constrained Lipid A mimetics for exploration of structural basis of TLR4/MD-2 activation by lipopolysaccharide. ACS Chem Biol. 2013;8:2423–2432. doi: 10.1021/cb4003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gaekwad J, Zhang Y, Zhang W, Reeves J, Wolfert MA, Boons G-J. Differential induction of innate immune responses by synthetic Lipid A derivatives. J Biol Chem. 2010;285:29375–29386. doi: 10.1074/jbc.M110.115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Matsuura M, Shimada S-I, Kiso M, Hasegawa A, Nakano M. Expression of endotoxic activities by synthetic monosaccharide lipid A analogs with alkyl-branched acyl substituents. Infect Immun. 1995;63:1446–1451. doi: 10.1128/iai.63.4.1446-1451.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Matsuura M, Kiso M, Hasegawa A, Nakano M. Multistep regulation mechanisms for tolerance induction to lipopolysaccharide lethality in the tumor necrosis factor-.alpha.-mediated pathway. Application of non-toxic monosaccharide lipid A analogs for elucidation of mechanisms. Eur J Biochem. 1994;221:335–341. doi: 10.1111/j.1432-1033.1994.tb18745.x. [DOI] [PubMed] [Google Scholar]
- 170.Matsuura M, Kiso M, Hasegawa A. Activity of monosaccharide lipid A analogues in human monocytic cells as agonists or antagonists of bacterial lipopolysaccharide. Infect Immun. 1999;67:6286–6292. doi: 10.1128/iai.67.12.6286-6292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Peri F, Granucci F, Costa B, Zanoni I, Marinzi C, Nicotra F. Inhibition of lipid A stimulated activation of human dendritic cells and macrophages by amino and hydroxylamino monosaccharides. Angew Chem Int Ed Engl. 2007;46:3308–3312. doi: 10.1002/anie.200604932. [DOI] [PubMed] [Google Scholar]
- 172.Kaeothip S, Paranjape G, Terrill SE, Bongat AFG, Udan MLD, Kamkhachorn T, Johnson HL, Nichols MR, Demchenko AV. Development of LPS antagonistic therapeutics: Synthesis and evaluation of glucopyranoside-spacer-amino acid motifs. R Soc Chem Adv. 2011;1:83–92. [Google Scholar]
- 173.Seydel U, Labischinski H, Kastowsky M, Brandenburg K. Phase behavior, supramolecular structure, and molecular conformation of lipopolysaccharide. Immunobiology. 1993;187:191–211. doi: 10.1016/S0171-2985(11)80339-6. [DOI] [PubMed] [Google Scholar]
- 174.(a) Cluff CW, Baldridge JR, Stöver AG, Evans JT, Johnson DA, Lacy MJ, Clawson VG, Yorgensen VM, Johnson CL, Livesay MT, Hershberg RM, Persing DH. Synthetic Toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect Immun. 2005;73:3044–3052. doi: 10.1128/IAI.73.5.3044-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stöver AG, Correia JDS, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, Ulevitch RJ, Persing DH, Hershberg RM. Structure-activity relationship of synthetic Toll-like receptor 4 agonists. J Biol Chem. 2004;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]; (c) Fort MM, Mozaffarian A, Stöver AG, Correia JdS, Johnson DA, Crane RT, Ulevitch RJ, Persing DH, Bielefeldt-Ohmann H, Probst P, Jeffery E, Fling SP, Hershberg RM. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J Immunol. 2005;174:6416–6423. doi: 10.4049/jimmunol.174.10.6416. [DOI] [PubMed] [Google Scholar]
- 175.Lewicky JD, Ulanova M, Jiang Z-H. Improving the immunostimulatory potency of diethanolamine-containing lipid A mimics. Bioorg Med Chem. 2013;21:2199–2209. doi: 10.1016/j.bmc.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 176.(a) Piazza M, Rossini C, Della Fiorentina S, Pozzi C, Comelli F, Bettoni I, Fusi P, Costa B, Peri F. Glycolipids and benzylammonium lipids as novel antisepsis agents: Synthesis and biological characterization. J Med Chem. 2009;52:1209–1213. doi: 10.1021/jm801333m. [DOI] [PubMed] [Google Scholar]; (b) Piazza M, Yu L, Teghanemt A, Gioannini T, Weiss J, Peri F. Evidence of a specific interaction between new synthetic antisepsis agents and CD14. Biochemistry. 2009;48:12337–12344. doi: 10.1021/bi901601b. [DOI] [PMC free article] [PubMed] [Google Scholar]