

Abstract
Protease activity in inflammation is complex. Proteases released by cells in response to infection, cytokines, or environmental triggers like cigarette smoking cause breakdown of the extracellular matrix (ECM). In chronic inflammatory diseases like chronic obstructive pulmonary disease (COPD), current findings indicate that pathology and morbidity are driven by dysregulation of protease activity, either through hyperactivity of proteases or deficiency or dysfunction their antiprotease regulators. Animal studies demonstrate the accuracy of this hypothesis through genetic and pharmacologic tools. New work shows that ECM destruction generates peptide fragments active on leukocytes via neutrophil or macrophage chemotaxis towards collagen and elastin derived peptides respectively. Such fragments now have been isolated and characterized in vivo in each case. Collectively, this describes a biochemical circuit in which protease activity leads to activation of local immunocytes, which in turn release cytokines and more proteases, leading to further leukocyte infiltration and cyclical disease progression that is chronic. This circuit concept is well known, and is intrinsic to the protease-antiprotease hypothesis; recently analytic techniques have become sensitive enough to establish fundamental mechanisms of this hypothesis, and basic and clinical data now implicate protease activity and peptide signaling as pathologically significant pharmacologic targets. This review discusses targeting protease activity for chronic inflammatory disease with special attention to COPD, covering important basic and clinical findings in the field; novel therapeutic strategies in animal or human studies; and a perspective on the successes and failures of agents with a focus on clinical potential in human disease.
1. INTRODUCTION
In the lungs, chronic inflammatory diseases including COPD, chronic bronchitis, and asthma are increasingly prevalent as humans become more regularly exposed to particulate material in the environment and increased prevalence of cigarette smoking, all of which leads to activation of the immune system(Crystal, 1997; Kobzik, 1999). In the disease asthma, this immune activation manifests as a hypersensitivity response to a particular antigen, which causes airway obstruction by bronchoconstriction in the regions of the tracheobronchial tree possessing smooth muscle. Fortunately, the condition is often self-limited and symptoms are reduced without a ‘trigger’ antigen. For COPD, there is a more sustained and nonspecific response to repeated chemical and particulate exposure (especially to cigarette smoke), with permanent airway remodeling and alveolar space destruction eventually leading to decreased lung elasticity with airflow obstruction at the level of the bronchiole with air retention distal to this collapse.
Cystic fibrosis (CF) is another disease which manifests with chronic pulmonary inflammation as a hallmark, due to genetically determined abnormalities in ion and water transport in the alveolus causing desiccation of the airspace. CF patients are thus more prone to inflammation and infection after environmental exposure to normally innocuous pathogens, leading to permanent tissue remodeling with recurrent infections being common. In each disease, inflammation is the primary culprit for structural changes which result in pulmonary compromise, the progression of symptoms, lifelong disease burden, and in the severely affected, respiratory failure with the possibility of death.
For the purposes of this article, we will deal primarily with chronic obstructive pulmonary disease (COPD), as this disease has a large public health impact and has been the focus of much research in both the clinical and laboratory setting over the last decade(Barnes, Shapiro, & Pauwels, 2003). In the introduction, we will discuss the disease burden of COPD and current treatments for the disease; then in the next section we will shift focus to the scientific developments in our understanding the pathobiology of this complex disease, specifically discussing protease activity and tissue breakdown and the recent developments in this area of investigation. We will then present recent preclinical and clinical data in the use of new pharmacologic agents whose mechanisms of action target protease activity at the enzymatic level with a summary of the various animal and human studies in pulmonary inflammation and COPD. We will then finish with an analysis of the accumulated biochemical and in vivo data and a discussion of the most promising possible therapeutic agents and their spectra of clinical applications that may be explored with such agents.
1.1 Chronic Obstructive Pulmonary Disease (COPD) Diagnosis and Disease Burden
COPD is now the most common cause of death among pulmonary disorders (“From the global strategy for the diagnosis, management and prevention of copd, global initiative for chronic obstructive lung disease (gold)”, 2007); the global burden of disease is increasing with prevalence in the US more than doubling over the past 3 decades and US deaths from the disease also more than doubling between 1980 and 2000(“From the global strategy for the diagnosis, management and prevention of copd, global initiative for chronic obstructive lung disease (gold)”, 2007, “Global surveillance, prevention and control of chronic respiratory diseases: A comprehensive approach “, 2007). The disease is largely considered attributable to cigarette smoking or environmental exposure to smoke or particulate matter which leads to the inflammatory phenotype we will discuss.
Diagnosis of COPD is made by lung spirometry, which measures the dynamics of air movement in the lungs and airway thereby giving data about a patient’s respiratory system mechanics. Obstruction is diagnosed by a reduced forced expiratory volume over the first second of exhalation (FEV1), and can be interpreted as a raw value in liters, as a percentage of predicted based on age and sex matched peers, or in terms of a ratio to total lung volume or forced vital capacity (FVC; giving the FEV1/FVC ratio). Essentially, the lower the FEV1, the greater is the patient’s loss of expiratory force, and the greater is the degree of obstruction.
As a disease, COPD represents a global health problem whose incidence and prevalence is increasing over time. The disease prevalence estimates range from 11 to 17 million people in the US being affected with the disease, with still more undiagnosed cases. The incidence of COPD in the US has therefore been estimated to be on the order of 5% of the population, with most patients diagnosed after age 40. Globally, COPD is projected to affect approximately 340 million people. The costs of disease management are likewise increasing with a recent estimate of ~$1800 per patient year(Miravitlles, Murio, Guerrero, & Gisbert, 2003), which can be extrapolated to a staggering ~$30 billion in the USA annually with its costs expected to rise further as the US population’s elderly proportion increases over the next few decades.
1.2 Current Clinical Management for COPD
As advances in management strategies have been made for these conditions over the last 20 years, our pharmacologic arsenal is still limited to a few classes of agents: bronchodilators or β-adrenergic receptor (β-AR) agonists, anticholinergics, phosphodiesterase inhibitors, glucocorticosteroids, and supplemental oxygen, which are used in various combinations for long term disease management. High potency glucocorticosteroids and antimicrobial therapies are used in combination with these agents during acute COPD exacerbations and respiratory infections. More intensive mechanical interventions such as positive pressure ventilation are used during respiratory failure or severe exacerbations requiring hospitalization.
In general, β-AR agonists such as albuterol (short acting) and salmeterol (long acting) have their effect via bronchiolar smooth muscle, inducing the relaxation and therefore dilation of the airway with an overall increase in airflow. Salmeterol is the standard of care for daily therapy with long term disease modification, while the short acting albuterol is used as a ‘rescue’ therapy via inhaler or nebulizer for patients experiencing acute symptoms or in disease exacerbation(Sutherland & Cherniack, 2004).
Anticholinergic therapies include long acting tiotropium and short acting ipratropium which work synergistically with the β-AR agonists at the level of the airway smooth muscle to maximize muscle relaxation and thus airflow. In identical fashion to the β-AR agonists, the long acting agent is used for daily therapy, and use of ipratropium is reserved for more acute situations or at times when patients need immediate symptomatic relief.
Glucocorticosteroids are agents which act at the nuclear level to suppress the cells of the immune system and thereby reduce inflammation. Locally delivered (i.e. inhaled) corticosteroids like fluticasone are now used as daily therapy and have been shown to benefit patients by reducing disease morbidity and associated hospitalizations(“From the global strategy for the diagnosis, management and prevention of copd, global initiative for chronic obstructive lung disease (gold)”, 2007). These agents are now considered standard for COPD maintenance therapy and are available alone or as a formulation combined with inhaled long acting β-AR agonists for daily or twice daily use. Systemic glucocorticosteroid therapy is not indicated for daily use, but is a mainstay of therapy for COPD exacerbations. The clinical use of these agents is variable and intensity of the steroid regimen is dictated by the severity of the patient’s condition with oral prednisone being commonly used for outpatient treatment of a COPD exacerbation and intravenous agents like methylprednisolone or hydrocortisone used in the hospital setting.
1.3 Protease activity in COPD
Pathologically, COPD is characterized by the destruction of lung parenchyma and enlargement of air spaces with loss of functioning alveoli (“Global surveillance, prevention and control of chronic respiratory diseases: A comprehensive approach “, 2007; Kobzik, 1999). Inflammation of the bronchioles is typically neutrophilic in character, with increased release of pro-inflammatory cytokines. Furthermore, there is accumulating evidence suggesting that excessive proteolytic activity occurs which overwhelms the natural protease regulation of the lung, and results in parenchymal destruction. This model is generally referred to as the ‘protease-antiprotease hypothesis’ of disease. The inflammation associated with the disease elicits release of proteases from a variety of cell types, mainly neutrophils (polymorphonuclear leucocytes, PMN), macrophages, and epithelia resulting in tissue destruction. It is now clear that protease action induces further inflammation by generating protein fragments which can recruit leukocytes into the tissue. Multiple proteases can potentially be involved in this process of degrading the extracellular matrix (ECM) and producing emphysematous lung disease. Recently though, a few specific enzymes have been specifically implicated in the pathophysiology of COPD, including several members of the matrix metalloprotease (MMP) family, as well as some serine proteases which have ECM proteins collagen and elastin as their substrates. In the following sections of this article, we will discuss these proteases and their proposed roles in disease progression as well as the steps made thus far towards interference with this process as a therapy for this medically expensive and intractable disease.
2. PROTEASES IMPLICATED IN COPD
Proteases are enzymes involved in the digestion of a variety of proteins. Although they all catalyze the hydrolysis of the peptide (amide) bond, they have different specificities for the side chains of the scissile peptide bond. Certain proteases break down the connective tissue constituents of the lung parenchyma causing the occurrence of emphysema in the context of an apparent imbalance between proteases and endogenous antiproteases which normally protect from excessive proteolysis.
The extracellular matrix of the lung is predominantly composed of elastin and to a lesser amount collagen. These fibrous proteins primarily provide the structure of the lung architecture. In terms of amino acid composition, elastin contains predominantly glycine (Gly), alanine (Ala), valine (Val), and proline (Pro). Similarly, collagen, and its denatured form termed gelatin, contains more than a third of Gly, and 15 to 30% of Pro or Hyp. The significance of the amino acid composition will be discussed later.
Since emphysema is characterized by the destruction of alveolar walls and obstruction occurs due to destruction of elastic parenchymal tissue surrounding the small airways, initial studies focused on neutrophil elastase (NE), a serine protease which has the capacity to hydrolyze elastin. Since then, many other proteases have been implicated based on their capacity to degrade elastin. These include other serine proteases, cysteine proteases, and macrophage metalloelastase. Furthermore, proteases involved in the degradation of collagen and/or gelatin have also been found to play a major role in COPD. These include certain members of the MMP family, a group of zinc-based proteases whose members are widely expressed by parenchymal and inflammatory cells in the lung. Prolyl endopeptidase (PE) is another serine protease only recently studied in airway research that may also contribute to the pathology of the disease, and whose role will be heavily discussed later.
2.1 Matrix Metalloproteases (MMPs)
The family of proteases currently receiving the most attention in chronic inflammatory lung disease is the matrix metalloproteases. MMPs are one of four subfamilies within the superfamily of zinc-dependent metalloendopeptidases known as metzincins. More than 20 MMPs have been identified and are subdivided into collagenases, gelatinases, stromelysins, matrilysin, macrophage elastase, and membrane-type MMPs, based on structure and substrate specificity (Sternlicht & Werb, 2001). Sequence analysis of various MMP domains demonstrate that MMPs evolved prior to the divergence of vertebrates and invertebrates (Massova, Kotra, Fridman, & Mobashery, 1998). Most of the proteases are released extracellularly as pro-enzymes which are then activated, although 6 members of this family of enzymes are membrane-bound. All MMPs have a catalytic domain (of which, the zinc binding motif of HEXXHXXGXXH is highly conserved) and some have a hemopexin-like domain, which is thought to confer substrate specificity (Stamenkovic, 2003). MMPs perform numerous biologic functions, including remodeling of tissues, modulation of cytokines, growth factors and chemokines, and influences on cell mobility and migration (Kang et al., 2001; Stamenkovic, 2003; Sternlicht & Werb, 2001; Van den Steen, Proost, Wuyts, Van Damme, & Opdenakker, 2000). Known sources of MMPs in the lung include neutrophils, alveolar macrophages, and airway epithelial cells (Greenlee, Werb, & Kheradmand, 2007). Neutrophils are a particularly rich source of MMPs, expressing MMP-8 (neutrophil collagenase) and MMP-9 (gelatinase B). Airway epithelial cells express various MMPs including MMP-8, MMP-9, MMP-2 (gelatinase A), and MMP-7 (matrilysin). MMP-12 (macrophage metalloelastase) is expressed at high levels in monocytes (Greenlee et al., 2007; Stamenkovic, 2003; Sternlicht & Werb, 2001).
Together, MMPs have the capacity to digest multiple extracellular matrix molecules, including types I, II, III and IV collagen in basement membranes, fibronectin, chondroitin sulfate proteoglycans, dermatan sulfate proteoglycans, and laminin (Stamenkovic, 2003). Through degradation of ECM, MMPs can destroy the alveolar epithelium and disrupt reorganization during the repair process (Dunsmore et al., 1998). While MMPs are involved in many normal homeostatic mechanisms, they are commonly elevated in their expression and activities in conditions in which inflammation and tissue destruction is operative. Data suggest that dysregulated cellular production, secretion and activation of MMPs, and/or dysfunction of their inhibitors are involved in pathologic conditions within the lung parenchyma and the airways (Greenlee et al., 2007). Animal studies, in addition to human studies in adults, support a role for an imbalance between MMPs and their inhibitors, tissue inhibitors of metalloproteases (TIMPs), in the pathogenesis of COPD and several other well-recognized pulmonary disorders (Shapiro & Senior, 1999).
2.1.1 MMP-1 (E.C. 3.4.24.7)
MMP-1 (collagenase-1 or fibroblast collagenase) is an interstitial collagenase and was the first described MMP (Pardo & Selman, 2005). Due to its capacity to effectively degrade fibrillar collagen, it is felt to play important roles in conditions with significant matrix remodeling such as COPD. Animal studies examining this protease highlight its potentially important role; guinea-pigs exposed to cigarette smoke demonstrated increased emphysematous changes in the lung with increased MMP-1 expression (D’Armiento, Dalal, Okada, Berg, & Chada, 1992). While mice do not express MMP-1, the human transgene has been overexpressed in mice, demonstrating emphysematous changes (Wright & Churg, 1995).
Indeed, Finlay and colleagues reported that MMP-1 levels were elevated in the bronchoalveolar lavage (BAL) fluid of COPD patients with emphysema (Finlay et al., 1997), although the activity of this enzyme in clinical samples has not been examined except in a single study (Vernooy, Lindeman, Jacobs, Hanemaaijer, & Wouters, 2004). Specific polymorphisms of the MMP-1 gene have been described with increasing frequencies in humans with familial rapidly-progressive emphysema (Joos et al., 2002).
2.1.2 MMP-8 (E.C. 3.4.24.34)
MMP-8 (collagenase-2 or neutrophil collagenase) is a prominent MMP, implicated in a variety of disorders (Van Lint & Libert, 2006). As it has been found to have similar efficacy to MMP-1 in cleaving collagen and is produced at increased levels in conditions with ongoing inflammation, MMP-8 is thought to play an important role in the development of COPD, though few animal studies examining MMP-8 in COPD exist. In humans, levels of MMP-8 protein and activity are elevated in individuals with COPD when compared to healthy smokers (Vernooy et al., 2004). MMP-8 is also detected at increased levels within the neutrophils sequestered in COPD lung tissues compared to control tissues (Segura-Valdez et al., 2000).
2.1.3 MMP-9 (E.C. 3.4.24.35)
MMP-9 (gelatinase B) has been examined a variety of lung disorders (Greenlee et al., 2007). It has several potential substrates (Van den Steen et al., 2002), and its increased activity in COPD may be important in driving multiple modalities of disease. Active MMP-9 can increase the neutrophilic inflammatory response (Malik et al., 2007; Van den Steen et al., 2000), has the capability to increase transcription of specific mucins seen in COPD (Deshmukh et al., 2005), and can cause ongoing matrix remodeling. MMP-9 has been examined in animal models of COPD but has yielded often contradictory results. In a mouse model study, interleukin 13 (IL-13) overexpression demonstrated emphysematous changes and increase in MMP-9 expression (Zheng et al., 2000). Despite this, MMP-9 deficient mice exposed to chronic cigarette smoke develop a similar emphysematous phenotype as compared to wild-type mice (Churg & Wright, 2005).
In humans, MMP-9 protein levels have been shown to be increased in BAL from COPD individuals (Betsuyaku et al., 1999; Culpitt et al., 1999). Cultured alveolar macrophages from individuals with COPD produced increased MMP-9 levels with increased gelatinolytic activity when compared to healthy smokers and nonsmokers (Finlay et al., 1997). Lung parenchyma from patients with COPD demonstrated increased MMP-9 levels (Ohnishi, Takagi, Kurokawa, Satomi, & Konttinen, 1998). The MMP-9/TIMP-1 ratio in sputum from COPD individuals was increased compared to normal and fibrosis controls. Interestingly, MMP-9 levels in this study correlated with the degree of airway obstruction (Beeh, Beier, Kornmann, & Buhl, 2003). A specific polymorphism in the promoter of MMP-9 has been found with increased frequency (−1562C/T) in emphysema individuals (Minematsu, Nakamura, Tateno, Nakajima, & Yamaguchi, 2001), although it role in the specific pathogenesis is unknown.
2.1.4 MMP-12 (E.C. 3.4.24.65)
MMP-12, or macrophage metalloelastase, has attracted much attention recently for its potential role in the cleavage of elastin. In a notable study, knockout mice null for MMP-12 were protected from smoke induced emphysema compared to wild type animals (Hautamaki, Kobayashi, Senior, & Shapiro, 1997). In another study utilizing cigarette smoke challenge, MMP-12 deficient mice demonstrated decreased markers of matrix remodeling and decreased inflammatory cell influx compared to wild type littermates (Churg et al., 2002). In addition, increased MMP-12 levels have been shown in BAL fluid, cultured alveolar macrophages, and tissue sections of patients with COPD when compared to normal controls (Molet et al., 2005). With animal data suggesting an important role of this protease in disease, translational human data implicating its role in COPD pathogenesis may be forthcoming.
2.2 Serine Proteases
The initial proteases implicated in emphysematous changes to the lung include members of the serine protease family, most notably neutrophil elastase. NE and its family members are widely expressed through the body, and NE is present naturally in the lungs as is its natural inhibitor, alpha-1 antitrypsin (α1-AT). Prolyl endopeptidase (PE) has also been detected in the lung and appears to play a role in the degradation of collagen fragments in situ.
The biochemical hallmark of serine proteases is the serine (Ser) residue which exhibits catalytic activity. Although there are slight variations of the last amino acid, sequence alignment of various serine proteases reveals that this Ser residue is located with the following consensus sequence as found in trypsin: Gly-X-Ser-X-Gly (Brenner, 1988). Moreover, the active site of serine proteases is formed by a conformational catalytic triad of aspartic acid-histidine-serine (Asp-His-Ser), His-Asp-Ser or Ser-Asp-His.
2.2.1 Neutrophil Elastase (NE)
Neutrophil elastase is a serine protease which is stored in primary granules of neutrophils (Faurschou & Borregaard, 2003). Due to the clinical manifestation of early-onset emphysema in α-1 antitrypsin deficiency syndrome (Stoller & Aboussouan, 2005), NE has attracted increased attention in COPD and was indeed the first protease implicated in the development of the disease. Beyond the capacity of this protease to cleave elastin, NE has also demonstrated a variety of potent immunologic modulatory effects seen in COPD including the cleavage of neutrophil chemokine receptors(Hartl et al., 2007), activation of matrix metalloproteases (Gaggar et al., 2007; Geraghty et al., 2007), and induction of interleukin 8 (IL-8) expression from airway epithelia (Walsh et al., 2001).
Animal studies have confirmed that intratracheal administration of NE leads to emphysematous changes and increased PMN influx (Senior et al., 1977) while NE-deficient mice were protected from emphysematous changes and had less inflammatory cell recruitment compared to wild type littermates after cigarette smoke exposure (Shapiro et al., 2003). COPD patients with computed tomography (CT) evidence of emphysema demonstrated an increased level of neutrophil elastase relative to amounts of α1-AT (Yoshioka et al., 1995)(9), correlating with decline in lung function (Betsuyaku et al., 2000) and providing a clinical foundation for the protease-antiprotease model of the development of COPD.
2.2.2 Cathepsin G and Proteinase 3
Proteinase 3 and cathepsin G are other neutrophil serine proteases with structural homology to elastase (Kao, Wehner, Skubitz, Gray, & Hoidal, 1988; Rao et al., 1991). Similar to elastase, they digest elastin in vitro, as well as other matrix proteins including fibronectin, laminin, vitronectin, and collagen type IV. Intratracheal administration of each enzyme induces emphysema, implicating relevance in the onset of COPD (Kao et al., 1988). Secretory leukocyte protease inhibitor (SLPI) is a serine antiprotease which inhibits NE and cathepsin G, but only minimally inhibits proteinase 3 (Rao, Marshall, Gray, & Hoidal, 1993), and proteinase 3 has the ability to inactivate SPLI. α1-AT has the capability to inhibit cathepsin-G and proteinase 3 (Duranton & Bieth, 2003). Although these proteases cleave elastin, their presence in disease is not as well documented as that of NE, and their stand-alone clinical impact in COPD is not well established.
2.2.3 Prolyl Endopeptidase (PE)
Oligopeptidases are enzymes that exhibit proteolytic activity on shorter portions of protein (peptides or oligopeptides) but not on full length protein (Barrett & Rawlings, 1992), and their activity is endoproteolytic. Prolyl endopeptidase represents its own family of serine proteases, classified as the S9 family of the SC clan (Garcia-Horsman, Mannisto, & Venalainen, 2007; Venalainen, Juvonen, & Mannisto, 2004). The alternate names of this enzyme found in literature include prolyl oligopeptidase (POP) and post-proline cleaving enzyme (PPCE). Originally, this protease was discovered in the human uterus homogenates as an enzyme which cleaves and deactivates the hormone oxytocin between proline and leucine, resulting in release of a C-terminal leucyl-glycinamide dipeptide (Walter, Shlank, Glass, Schwartz, & Kerenyi, 1971). In the 1970s, it was isolated by three different groups from lamb kidney (Koida & Walter, 1976; Walter, 1976) and rabbit brain (Oliveira, Martins, & Camargo, 1976; Orlowski, Wilk, Pearce, & Wilk, 1979) homogenates. The enzyme was also found to hydrolyze bradykinin in its free form, suggesting selective hydrolysis of low molecular weight polypeptides (Camargo, Caldo, & Reis, 1979; Carvalho & Camargo, 1981). Hereafter, we will refer to this enzyme as prolyl endopeptidase.
Although it has not been found in yeast, PE cDNA has been identified and sequenced from a wide array of organisms including bacteria, protozoa, plants, and animals (Garcia-Horsman et al., 2007; Venalainen et al., 2004). A phylogenetic study in which sequences of PE family members were compared indicated that the genes evolved before the archaea, prokaryota and eukaryota diverged evolutionarily(Venalainen et al., 2004). The ancient nature of the enzyme family is further supported by the fact that the sequence upstream of the PE gene codes for a housekeeping enzyme (Kimura, Yoshida, Takagi, & Takahashi, 1999).
The human PE (hPE) gene has been isolated from human lymphocytes (Vanhoof et al., 1994),(Shirasawa, Osawa, & Hirashima, 1994) and consists of an open reading frame (ORF) of approximately 2130 nucleotides on the 6q22 chromosome(Goossens et al., 1996), corresponding to a 710 amino acid protein which is conserved among mammals(Venalainen et al., 2004). Purification from human lymphocytes shows hPE has a molecular mass of 68 kDa under native conditions, or 76 kDa under reducing and denaturing conditions, and is found in monomeric form (Goossens et al., 1996). Unlike bacterial PE isoforms which are able to digest long polypeptides and proteins (Harwood, Denson, Robinson-Bidle, & Schreier, 1997; Harwood & Schreier, 2001), eukaryotic PE only acts on peptides 30–100 amino acids in length (Fulop, Szeltner, Renner, & Polgar, 2001). In addition to its ability to cleave a variety of naturally occurring molecules (oxytocin, vasopressin, etc.) (Walter, 1976; Walter et al., 1971), PE is able to cleave modeled compounds such as Z-Gly-Pro-pNA and be inhibited by Z-Pro-Prolinal (ZPP) and its analogues (Bakker, Jung, Spencer, Vinick, & Faraci, 1990; Garcia-Horsman et al., 2007). Based on these findings, enzymatic assays have been developed for the detection of PE and PE-like activity not only by the positive identification of enzyme activity, but also its inhibition. Prolyl oligopeptidase activity in humans has been detected in a variety of tissues and cells: lung, prostate, gall bladder, testes, lymphocytes, and small cell lung carcinoma cells (Goossens, De Meester, Vanhoof, & Scharpe, 1996; Kato, Okada, & Nagatsu, 1980). PE enzymatic activity has been detected in a multiple environs including membranes, mitochondria, the extracellular space (Polgar, 2002) and human body fluids including serum and seminal fluid (Goossens, De Meester, Vanhoof, & Scharpe, 1992; Goossens et al., 1996; Yoshimoto, Ogita, Walter, Koida, & Tsuru, 1979). The existence of an 87 kDa membrane bound enzyme with PE-like activity has been reported in bovine brain (O’Leary, Gallagher, & O’Connor, 1996; O’Leary & O’Connor, 1995), with many similarities and differences between the 76 kDa cytosolic PE and this larger enzyme. Therefore it is conceivable that different forms of PE, arising from posttranslational modification and/or different gene products, can account for the variety of functions and cellular locations of the enzyme. PE has been detected in lung samples and is known to be important in processing collagen fragments into smaller molecules with signaling activity(Gaggar et al., 2008), which will be discussed in the next section.
3. THE EXTRACELLULAR MATRIX AND DISEASE
3.1 Constituents of the Extracellular Matrix (ECM) – Collagen and Elastin
As previously discussed, a prominent feature of proteases is the capacity to degrade the extracellular matrix. While many ECM-derived fragments are not involved in cell signaling, some such peptide fragments demonstrate the capacity to induce cellular recruitment and ongoing inflammation. Indeed, fragments of both laminin (Mydel et al., 2008) and fibronectin (Norris et al., 1982) have been shown to be chemotactic for neutrophils and monocytes, respectively, whereas hyaluronan fragments are felt to induce increased alveolar macrophage inflammatory gene expression via Toll-Like Receptor (TLR) dependent mechanism (Jiang et al., 2005). Perhaps the most studied ECM-derived biologically-active fragments are those derived from collagen and elastin which are discussed next.
3.2 Elastin fragmentation and Cell Signaling
Elastin-derived fragments have been implicated in monocyte recruitment in experimental models of disease such as aortic aneurysms (Hance, Tataria, Ziporin, Lee, & Thompson, 2002) and Marfans syndrome (Guo et al., 2006). The monocyte chemotactic activity from elastin degradation was found principally in peptides of 10,000 to 50,000 mol wt (Houghton et al., 2006; Senior, Griffin, & Mecham, 1980). A repeating hexamer Val-Gly-Val-Ala-Pro-Gly was demonstrated to be chemotactic for fibroblasts and monocytes and is likely an important component of the chemotactic activity of elastin (Senior et al., 1984).
The loss of elasticity in the emphysema lung has focused attention to the degradation of elastase in COPD. Interestingly, when macrophage metalloelastase (MMP-12) knockout mice are exposed to long-term cigarette smoke, they do not develop the emphysematous phenotype seen in their wild-type littermates; this difference may be attributable to both the mutants’ lack of structural insult, as well as their relative deficiency in the production of chemotactic elastin peptides (Hautamaki et al., 1997). Elastin fragments, when instilled into the lungs of mice, induce a prominent monocyte influx (Houghton et al., 2006). In addition, BAL fluid and lung homogenates from mice exposed to cigarette smoke induced monocyte migration in vitro, and this activity was blocked by antibody against a specific elastin fragment (Houghton et al., 2006). Elastin fragments isolated from BAL fluids in this study were ~45KD and are thought to be chemotactic through the elastin binding protein expressed on monocytes (Privitera, Prody, Callahan, & Hinek, 1998).
3.3 Collagen Fragmentation and Cell Signaling
It has been known for over 30 years that digested collagen may have the capacity to augment inflammation and injury response. Postlethwaite and colleagues initially described that collagen fragments derived from type I, II, and III collagen have the capacity to cause fibroblasts to migrate in vitro (Postlethwaite, Seyer, & Kang, 1978). This group also demonstrated that type I collagen fragments were chemotactic for monocytes in vitro (Postlethwaite & Kang, 1976). Robert Senior and colleagues have also described in vitro neutrophil chemotaxis to sequences found in type IV collagen (Senior et al., 1989). Laskin et al further demonstrated that collagen sequences with Pro-Pro-Gly or Pro-Hyp-Gly were chemotactic for peripheral blood neutrophils ex vivo and that this molecule was able to induce superoxide production and elastase release from alveolar macrophages (Laskin, Kimura, Sakakibara, Riley, & Berg, 1986). This work was extended when Riley et al demonstrated an increased neutrophilic response in rats after intratracheal administration of collagen fragments (Riley et al., 1988; Riley et al., 1984).
3.3.1 Collagen fragment signaling through the chemokine receptors
The links between proteases, collagen and inflammation are reinforced by older studies in a rabbit model investigating alkali injury to the eye, which identified a peptide, N-acetyl Pro-Gly-Pro (N-a-PGP), as being chemotactic for neutrophils, and was presumed to be derived from collagen (Pfister, Haddox, & Lam, 1993; Pfister, Haddox, Lam, & Lank, 1988; Pfister, Haddox, & Sommers, 1993, 1998; Pfister, Haddox, Sommers, & Lam, 1995). Similarly, collagen fragments administered intratracheally cause the influx of neutrophils (Foronjy, Okada, Cole, & D’Armiento, 2003). Moreover, the overexpression of collagenase-1 causes emphysema, while blocking the activity of MMPs ameliorates the symptoms of COPD (Corbel et al., 2001; Selman et al., 1996).
Further studies have identified N-a-PGP from mouse BAL fluid as a neutrophil chemoattractant both in vivo and in vitro (Weathington et al., 2006). Sequence and structural analyses revealed a similarity between the CXC neutrophil chemokines, collagen fragments and N-a-PGP. Chemotaxis experiments verified that the tripeptide is active on human PMN and acts through the same CXC receptor (CXCR) as is well characterized for CXCR ligands IL-8 in humans and keratinocyte derived chemokine (KC) or macrophage inflammatory protein (MIP-1a) in mice. It was shown that N-a-PGP was produced in vivo following murine airway exposure to lipopolysaccharide (LPS). Most importantly, N-a-PGP was detected in BAL fluid of COPD patients where out of five samples tested, three had significantly higher concentrations of the tripeptide which was undetectable in healthy control samples. Although not all COPD patient BAL fluid was positive for N-a-PGP, the patients who had elevated concentrations had radiographic evidence of emphysema and lower FEV1 scores. In all, these studies linked N-a-PGP with neutrophil influx resulting from ECM degradation during airway inflammation(Weathington et al., 2006). Furthermore, related peptides with homology to N-a-PGP as well as the non-acetylated form of proline-glycine-proline (PGP) have also been shown to possess neutrophil chemoattractant activity (Haddox, Pfister, Sommers, Blalock, & Villain, 2001; Malik et al., 2007). Additional studies have shown that PGP acts via the same CXC receptors as N-a-PGP (Gaggar et al., 2008).
Although the involvement of PE in inflammatory lung diseases is as yet poorly characterized (Gaggar et al., 2008), PE has been shown to have a variety of proline containing peptides as substrates (Garcia-Horsman et al., 2007). The involvement of this enzyme in inflammation represents something of a paradigm shift for its physiologic function, but with multiple proteases implicated in inflammation (Aoyagi et al., 1983; Luscombe, 1963; Smith & Hamerman, 1962; Weissmann, 1972; Weissmann & Spilberg, 1968), it was postulated that PE could also play a role (Aoyagi et al., 1985). In two systemic lupus erythematosus (SLE) mouse models, liver PE enzymatic activity increased with age in diseased animals, compared to control animals, where it decreased (Aoyagi et al., 1987; Aoyagi et al., 1985). In a spontaneous osteoarthritis (OA) model, serum PE activity was again elevated in diseased mice (Fukuoka, Hagihara, Nagatsu, & Kaneda, 1993). In human studies, it was found that patients suffering from rheumatoid arthritis (RA) had higher levels of PE activity directly correlated with the volume of joint fluid suggesting correlation with the severity of inflammation (Kamori, Hagihara, Nagatsu, Iwata, & Miura, 1991).
In cystic fibrosis, which represents another lung disease characterized by chronic neutrophilic inflammation, both N-a-PGP and PGP are elevated in CF patient sputum(Gaggar et al., 2008). Concomitantly, PE activity was five fold higher in the sputum of CF patients as compared to normal control samples and correlated with neutrophil burden. The correlation of PGP production and PE activity in CF sputum samples was very high, implicating PE activity and PGP production in vivo. PE has been identified from literature as the only enzyme which has the capacity to cleave a PGP tripeptide from collagen fragments containing the ‘PPGP’ repeat (Koida & Walter, 1976; Rosenblum & Kozarich, 2003; Walter, 1976; Walter et al., 1971; Yoshimoto et al., 1979). However, in order for PE to cleave the tripeptde, the collagen fragments need to be from 30–100 amino acids in length (Shirasawa et al., 1994).
Based on these observations, it was hypothesized that PGP generation requires a multistep cleavage cascade involving multiple proteases. CF sputum contains increased levels of NE and MMPs 8, 9, 11, and 12(Gaggar et al., 2007), and it has the capacity to produce PGP from intact collagen, indicating that it contains all of the necessary components for the cascade(Gaggar et al., 2008). These data were further verified in an experiment in which PGP generation from collagen was measured in the presence of inhibitors specific for particular enzymes. PE, MMP-8, and MMP-9 inhibitors and doxycycline abrogated PGP production at levels by 80–100%, while MMP-2 and NE inhibitors marginally prevented PGP production, indicating that the latter two proteases likely do not play a role in PGP production. Further, it was shown that PGP production correlates with PMN influx. Based on a compilation of data, a mechanistic cascade has been proposed and is schematically represented in Figure 1: Following injury or infection, PMNs are summoned to the site of the insult. Upon release of MMP-8 and MMP-9 (and potentially other MMPs), the initial cleavage of collagen occurs. When the fragments reach the appropriate size to be utilized by PE as substrates, the enzyme cleaves out the PMN chemoattractant tripeptide PGP. As the figure indicates, it is unclear whether acetylation of PGP occurs before or after PE cleavage. Both molecules can be detected, albeit the acetylation enzyme is yet unknown. N-a-PGP and PGP then act through CXCR1 and CXCR2 to further attract PMNs, and the cycle continues. Additionally, simultaneous fragmentation of elastin signals alveolar macrophages to the site (Houghton et al., 2006), which may in turn release IL-8, further contributing to PMN recruitment. This complex mechanism involves many proteases, however, it is important to note that collagen and elastin turnover is a physiological process. Pathology is observed in inflammatory disease when an imbalance of protease activity causes a dysregulation of the normal physiology.
Figure 1. Proteolytic-Inflammatory changes in the interstitial space drive COPD pathology.

Insult to the airway (far right) by damage from toxic exposure by particulate matter, cigarette smoke, or inflammatory compounds like LPS, or by bacterial or viral infection causes recruitment and activation of inflammatory cells from the bloodstream (far right). In this simplified view, extracellular matrix breakdown is illustrated for collagen (main panel, left side) and elastin (main panel, right). Such substrate breakdown contributes to further inflammation in the tissue by the generation of matrix derived peptides active on neutrophils or macrophages in the cases of collagen or elastin, respectively.
In the case of collagen, Collagenases and elastases (i.e. MMPs-2, 8, or 9) cause the fragmentation of the protein into peptides which are then further digested by Prolyl endopeptidase leading to generation of PGP, which is sometimes acetylated by an unknown mechanism to form N-a-PGP. In the extracellular space, the PGP molecules are then active on neutrophils as chemoattractants and cell activators via the CXCR receptor pathway, causing influx of cells into the interstitium and augmenting the inflammatory phenotype caused by the initial insult.
When elastin is cleaved by neutrophil elastase, matrix metalloelastase, or other peptidases present, many peptides are produced. Among these, a 12 kDa peptide has been identified that can induce chemotaxis of macrophages or cause macrophage activation to release other inflammatory mediators including cytokines or chemokines like IL-8 which is a potent neutrophil chemoattractant.
The end result of these matrix-derived signaling peptides’ activity seems to be the perpetuation of an inflammatory phenotype in the affected tissue. In COPD, chronic insult - by cigarette smoke for example – leads to chronic matrix degradation, which in turn drives chronic inflammation by chemoattraction and cell activation. This model for chronic inflammatory disease connects the reactive inflammation to noxious or infectious stimuli to extracellular matrix breakdown to prolonged leukocyte recruitment and chronic inflammation and remodeling as seen in diseased tissue.
4. PROTEASE INHIBITORS – POTENTIAL TREATMENT FOR COPD
4.1 NE inhibitors
4.1.1 Endogenous Inhibitors
The naturally occurring neutrophil elastase inhibitors are α-1 antitrypsin, SLPI, and skin derived antileukoprotease (SKALP). Of these, one human trial in alpha-1 antitrypsin repletion therapy was performed in Europe with considerable quantity of the enzyme administered (250 mg/kg every 4 weeks for at least 3 years). Ultimately, this study showed no significant benefit in spirometry performance in the treated group(Dirksen et al., 1999). However, in an α1-AT repletion study in mice, the protein was delivered as an inhalant and was reported to abrogate airway enlargement by 73% with moderate effect on cell counts. Despite these benefits, immunization to therapy may severely limit the use of α1-AT, as anti- α1-AT antibodies have been found in treated animals (Pemberton et al., 2006).
Other early trials have been conducted with a small recombinant peptide which is derived from a naturally occurring antiprotease, interalpha trypsin inhibitor. This peptide, Epi-NE-4, has been tested in humans with no major side effects(Bradbury, 2001) and was shown to inhibit NE activity in sputum from CF patients(Attucci et al., 2006). Further experiments into an inhaled formulation of the peptide may be forthcoming.
4.1.2 Synthetic Small Molecule NE Inhibitors
The most well studied NE inhibitors thus far developed belong to two chemical classes, known as acyl-enzyme inhibitors and transition state inhibitors. In each kind, the inhibitor binds the NE molecule and renders it permanently nonfunctional in noncompetitive fashion. Of the ten or so inhibitors that have been significantly developed, we will discuss those most promising and most advanced in their development with clinical data (Ohbayashi, 2002).
4.1.2.1 Sivelestat (ONO-5046)
The development of NE inhibitors was led by this acyl-enzyme inhibitor, which is patented by the Ono Pharmaceutical company of Japan, and has gained approval for experimental use in humans in Japan. In early animal experiments, the compound showed efficacy with intravenous administration in mouse, rat, and hamster models of acute lung injury and COPD. Now that the compound is available for human use, clinical trials have ensued with some initial promising results. In one recent study, septic acute lung injury patients given the drug had reduced serum concentrations of tumor necrosis factor alpha (TNF-α) and IL-8, were more quickly weaned from mechanical ventilation, and had better oxygenation and pulmonary function than untreated patients (Endo et al., 2006). Another Japanese study in critically ill patients shows a significant mortality benefit in patients treated with the drug(Hoshi et al., 2005). Because of its intravenous formulation and relatively short half life, only inpatient studies have been performed on this drug, and it is unknown whether any long term therapeutic strategy exists for this compound. Unfortunately, these promising results have not been reproduced in rigorous studies carried out in the US and Europe by Lilly Pharma corporation(Zeiher et al., 2004) (which had license agreement with Ono to share clinical rights to the compound), and further research outside Japan may be forever abandoned(Pharmaceuticals, 2003).
4.1.2.2 MR-889
MR-889 is a compound whose action is a competitive inhibition of NE, with activity in the micromolar range and has been formulated for oral administration. The drug has been tolerated well in humans, with initial data indicating efficacy at NE inhibition, but no change in the natural course of disease when given to COPD patients in a randomized European study(Luisetti et al., 1996). No other studies regarding therapeutic efficacy have been reported at the time of this writing.
4.1.2.3 ONO-6818
ONO-6818 is a transition-state inhibitor of NE, whose activity appears to be potent and specific with covalent association to the enzyme and permanent inactivation at nanomolar concentrations. This molecule was formulated into an orally active agent and passed phase 1 studies in the US and Japan(Trifilieff, 2002), only to be abandoned in Stage II Japanese trials due to side effects seen in humans which included increased liver enzymes(Pharmaceuticals, 2002). Although no longer in development, there is hope that this drug may serve as a model compound for later generation orally active antiproteases.
4.1.2.4 ICI 200,880
The transition state NE inhibitor ICI-200,880 is a drug that has been under development since 1991, and has shown some promising results in the ICU environment. In similar fashion to the ONO-5046 compound, there was little complication found in administration of the compound intravenously. Also, acute respiratory distress syndrome (ARDS) patients in the treatment group of a study had significant survival benefit and improved pulmonary function compared to untreated patients(GOTTLIEB, ELMER, & STEINER, 1994). Despite these promising early results, further study has not been undertaken on this compound, suggesting that later studied either revealed side effects or proved ineffective.
4.2 MMP Inhibitors
4.2.1 Tissue Inhibitors of Metalloproteases (TIMPs)
The tissue inhibitors of metalloproteases are endogenous proteins which naturally suppress the activity of the MMPs by direct binding and inhibition of enzymatic activity. The TIMPs of interest in COPD are those that are expressed in lung and are active on collagenases MMP-1 and MMP-8, gelatinases MMP-2 and MMP-9, and macrophage metalloelastase MMP-12, and these include TIMPs 1, 2, and 4(Gomez, Alonso, Yoshiji, & Thorgeirsson, 1997). While TIMP binding blocks MMP activity, TIMPs themselves can be inactivated by proteases like neutrophil elastase by direct proteolytic degradation(Gaggar et al., 2007). Though this kind of TIMP/MMP dysregulation has been shown to contribute to the pathology of the disease and to correlate to disease severity in animal and human studies, there have not, to our knowledge, been attempts to replete TIMPs as a treatment for COPD. Though interesting as a concept for intervention and biologically sound, the use of TIMPs for treatment of COPD will probably never be feasible due to the costly, unpredictable, and cumbersome nature of protein therapy, with major hurdles including in vivo degradation and immunization to therapy.
4.2.2 Antibiotics
Macrolides are a frequently prescribed class of antibiotics used to treat a variety of pulmonary infections from community-acquired pneumonias to chronic bronchitis exacerbations. Interestingly, macrolides have been demonstrated to have numerous anti-inflammatory effects that could be important in COPD, including neutrophil survival and activation (Aoshiba, Nagai, & Konno, 1995; Villagrasa et al., 1997). Other studies indicate changes in cytokine/chemokine profiles (Rubin, Druce, Ramirez, & Palmer, 1997; Shimane, Asano, Suzuki, Hisamitsu, & Suzaki, 2001), including reducing IL-8 levels (Takizawa et al., 1997). Recently, the anti-inflammatory properties of macrolide antibiotics have been translated into the usage of these antibiotics as long-term therapeutics in a variety of chronic, neutrophilic-predominant diseases such as bronchiectasis, post-transplant obliterative bronchiolitis, panbronchiolitis and cystic fibrosis (Equi, Balfour-Lynn, Bush, & Rosenthal, 2002; Gerhardt et al., 2003; Kadota et al., 2003; Tsang et al., 1999). Interestingly, these conditions are dominated by increased expression and activity of MMP-9 (Beeh, Beier, Kornmann, Micke, & Buhl, 2001; Gaggar et al., 2007; Sepper, Konttinen, Sorsa, & Koski, 1994), and a study found that use of roxithromycin, another macrolide antibiotic, reduced the expression of MMP-9 from blood neutrophils but did not change levels of TIMP-1 (Kanai, Asano, Hisamitsu, & Suzaki, 2004). The same group also demonstrated that MMP-9 production is significantly reduced from nasal epithelia ex vivo with pretreatment with roxithromycin by either TNF-α or nuclear factor kappa beta (NF-κβ)/activator protein (AP-1) pathways (Kanai, Asano, Hisamitsu, & Suzaki, 2004). Another study has demonstrated that MMP-9 activity and production were reduced in both a macrophage cell line (U937) and splenic macrophages ex vivo when treated with the macrolide erythromycin, and migration of both of these cell types was significantly reduced when treated this drug (Hashimoto et al., 2001).
Tetracyclines are a class of antibiotics used to treat simple upper respiratory tract infections whose use is being reduced in favor of other classes of antibiotics such as fluoroquinolones (Wilson, 2005). New data indicate that tetracyclines have intrinsic anti-inflammatory properties. Doxycycline, a member of the tetracycline family is able to reduce neutrophil chemotaxis ex vivo and in vivo (Elewski, Lamb, Sams, & Gammon, 1983), and has been shown to reduce both interleukin 1 production from LPS-treated corneal epithelium in vitro (Solomon et al., 2000) and nitric oxide production from murine lung epithelial cells (Hoyt, Ballering, Numanami, Hayden, & Robbins, 2006). More pertinently, tetracycline antibiotics have also demonstrated anticollagenase properties both in vitro (Suomalainen et al., 1992) and in vivo (Golub et al., 1983) by inhibiting MMP activity (Golub, Ramamurthy, McNamara, Greenwald, & Rifkin, 1991). In addition to its capacity to inhibit elastin degradation (Boyle et al., 1998) in vivo, doxycycline can also inhibit MMP synthesis from human endothelial cells (Hanemaaijer et al., 1998). As MMPs have been well-regarded as targets for tetracycline antibiotics in disease (Emingil et al., 2004), there is increasing interest for inhibiting MMP-9 by these agents. Another study has shown that MMP-9 inhibition by minocycline reduces hyperresponsiveness and neutrophil counts in a murine model of asthma (Lee, Jin, Kim, & Lee, 2004). Therefore, antibiotic therapy with either tetracyclines or macrolides in COPD may serve not only as an antimicrobial but also as an anti-inflammatory therapy reducing MMP and NE activity and therefore preventing production of signaling peptides derived from elastin or collagen and subsequent leukocyte trafficking.
4.2.3 Small Molecule Inhibitors
To date several broad spectrum MMP inhibitors (MMPIs) have been developed and tested in animal models of cigarette smoke-induced emphysema with generally promising data. Each of the compounds, GM-6001; CP-471,474; Marimastat, and RS113,456 have inhibited airspace enlargement and/or neutrophil recruitment in the context of noxious stimuli known to cause emphysematous or other structural lung tissue changes. However promising the animal data have been, human studies have not come to full fruition, and there are currently no MMP inhibitors approved for use in humans for COPD therapy
Because MMPs have been also implicated in the spread and progression of cancer, a great deal of clinical investigation has been undertaken for the use of MMPIs as antitumor drugs. Though these initial oncologic studies showed no benefit, attention is being turned to the use of these agents against chronic inflammatory diseases(Hu, Van den Steen, Sang, & Opdenakker, 2007).
Early clinical data for broad spectrum MMP inhibitors indicate class wide side effects most commonly involving the musculoskeletal system with arthralgia being the main complaint (Johnson, Dyer, & Hupe, 1998; Pavlaki & Zucker, 2003) described as ‘Musculoskeletal Syndrome’. In one cohort of cancer patients, a majority of patients experienced severe, debilitating muscle and joint pain in response to Marimastat (Steward, 1999). This presumably reflects the natural function of MMPs in the turnover of normal connective tissue and the potential interference that such a biologically tailored therapy may bring about. For the purpose of developing better therapies for chronic inflammatory disease as well as perhaps neoplastic disease, alternate modes of delivery, and more comprehensive side effect description and aversion is an area of great interest(Fingleton, 2008), as it can also help guide the development of new generations of drugs (Overall & Kleifeld, 2006).
4.2.2.1 Ilomastat (GM6001)
The most well studied of the small molecule MMPIs, GM-6001 or ilomastat, is a hydroxamate molecule with a subnanomolar IC50 for MMP-1,2,8,9, and 12, and has been tested in a number of mouse models, including chronic smoke administration(Pemberton et al., 2005) and IL-13 overexpression(Zheng et al., 2000) with reduction of airspace disease and PMN cell counts for all conditions. In the most provocative study, ilomastat was formulated for inhalation and delivered to mice under a cigarette smoke induced emphysema model. The study showed relative decreases in macrophage and neutrophil cell numbers with almost complete blockage of airway enlargement and structural change at 6 months(Pemberton et al., 2005). Use of the MMPI in an inhaled formulation may help to avert some of the systemic side effects, and may in fact enhance therapy by direct airspace delivery.
4.2.2.2 Marimastat
Marimastat is a broad spectrum MMPI developed and tried in cancer therapy as well as pulmonary disease. In one study(Nenan et al., 2007), investigators use airway instillation of recombinant MMP-12 as a model for COPD and evaluate the effects of orally administered marimastat well as the common glucocorticosteroid, dexamethasone on disease progression. While both agents inhibit neutrophil cell trafficking into the lung, marimastat also prevented the later influx of macrophages, and both compounds reduced the quantities of proinflammatory cytokines and MMP-9. Unfortunately, the musculoskeletal side effects (Steward, 1999) preclude further investigation of this drug as a systemic agent.
4.2.2.3 RS-113,456
The broad spectrum MMPI RS-113,456 is active against MMPs-2,8,9, and 12 at subnanomolar concentrations. This compound has been used orally in mice and was shown to prevent neutrophil influx as well as collagen breakdown occurring acutely after exposure to cigarette smoke (Churg et al., 2002). This inhibitor also prevented acute PMN influx in response to quartz dust, and had generally anti-inflammatory properties acutely. This drug has not been used in human trials to date, and it is unclear if further development is planned.
4.2.2.4 CP 471,474
CP 471,474 has inhibitory activity on 6 MMPs with IC50s at the 3–10 ng/ml range, and in guinea pig studies, both neutrophil recruitment and airway remodeling were suppressed when the compound was administered subcutaneously (Selman et al., 2003). Interestingly, this compound is also being tested in models of heart failure(Li & Feldman, 2001), where MMP dysregulation is thought to contribute to myocardial remodeling.
4.3 PE inhibitors
Though inhibition of PE has not been explored for use against lung disease, the recent discovery of PE’s role in the genesis of PGP suggests that it may be of utility in treatment of this disease, and because of these premature but substantial implications, we will discuss compounds that may interrupt its activity. Since the discovery of PE (Koida & Walter, 1976), many bioactive peptides have been identified as substrates (Garcia-Horsman et al., 2007) including several neuropeptides. Because PE is located in the central nervous system and is active on neuropeptides whose dysregulation characterizes neurodegenerative disease, much of the research performed on PE was initiated with attention to neurologic conditions including memory loss, fibromyalgia, depression, schizophrenia, mania, anxiety, bulimia, and anorexia nervosa (Maes et al., 1998; Maes et al., 1995; Maes et al., 1998; Maes et al., 2001; Yoshimoto et al., 1987). Naturally, these studies spurred the research field in the search of specific inhibitors of the enzyme for potential treatment of a variety of neurological disorders in which levels of PE have been altered (Gass & Khosla, 2007; Wallen et al., 2003). Table 1 lists structural formulas of some of the most studied PE inhibitors, a few synthetic compounds used for PE activity detection as well as the targeted region of oxytocin, a natural substrate of the enzyme; these molecules all exhibit structural similarity to each other.
Table 1.
Prolyl Endopeptidase Substrates and Inhibitors
| Compound | Structure/Sequence | Function | Reference |
|---|---|---|---|
| Oxytocin |
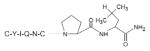
|
Hormone | (Walter, Shlank, Glass, Schwartz, & Kerenyi, 1971) |
| Z-Gly-Pro-Ala-OH |
|
POP Activity Assay | (Garcia- Horsman, Mannisto, & Venalainen, 2007) |
| Z-Gly-Pro-pNA |
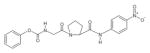
|
POP Activity Assay | (Zolfaghari et al., 1986) |
| Suc-Ala-Pro-pNA |
|
POP Activity Assay | (Garcia- Horsman, Mannisto, & Venalainen, 2007) |
| ZPP |
|
Inhibitor | (Bakker, Jung, Spencer, Vinick, & Faraci, 1990) |
| JTP-4819 |
|
Inhibitor | (Toide, Okamiya, Iwamoto, & Kato, 1995) |
| ONO-1603 |
|
Inhibitor | (Katsube, Sunaga, Chuang, & Ishitani, 1996) |
| SUAM-1221 |
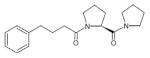
|
Inhibitor | (Atack, Suman- Chauhan, Dawson, & Kulagowski, 1991) |
In scopolamine induced amnesia in rats, PE inhibitor SUAM-1221 had in vivo antiamnesic effects which correlated with inhibition of PE in vitro (Atack, Suman-Chauhan, Dawson, & Kulagowski, 1991; Yoshimoto et al., 1987) with partial reversal of the deficit in memory performance produced by scopolamine. JTP-4819 was tested on a variety of rat models involving age-related memory loss (Toide et al., 1996; Toide, Iwamoto, Fujiwara, & Abe, 1995; Toide, Okamiya, Iwamoto, & Kato, 1995; Toide, Shinoda, Fujiwara, & Iwamoto, 1997), demonstrating that this compound might be useful in treating Alzheimer’s disease since it inhibited PE and therefore the degradation of substance P, arginine-vasopressin, and thyrotropin-releasing hormone (Toide et al., 1997; Toide, Shinoda, & Miyazaki, 1998).
Of the compounds listed in Table 1, ZPP and S 17092 may be most relevant, as the former inhibits mouse PE while the latter inhibits rodent, monkey and human PE (Bakker et al., 1990; Barelli et al., 1999; Morain et al., 2002). S-17092 was shown to be a very specific inhibitor of PE with the capability to inhibit the enzyme within the 293 HEK cell, used in a model for Alzheimer’s disease (Barelli et al., 1999). Moreover, ONO-1603 was shown to have similar effects to the antidementia drug tetrahydroaminoacridine (THA) on neuronal outgrowth in a model system also investigating Alzheimer’s disease. A mouse model examining the age related cognitive decline showed that mice treated with S-17092 showed an improvement in choice accuracy over controls (Marighetto et al., 2000). In a monkey model of Parkinsonism, S17092 was shown to significantly ameliorate cognitive functions when administered orally (Schneider, Giardiniere, & Morain, 2002). In a related study, the effect of S-17092 was identified to be due in part to the increased concentrations of promnesic (good for memory) neuropeptides such as SP and α-MSH which are substrates of PE (Bellemere, Morain, Vaudry, & Jegou, 2003). When S-17092 was administered orally to healthy elderly patients in a Phase I clinical study both α-band electroencephalogram (EEG) and performance in two verbal memory tests improved without serious adverse effects (Morain et al., 2000). In the latest study using quantitative EEG, S-17092 has memory preservative effects and also displayed mood stabilizing properties (Morain, Boeijinga, Demazieres, De Nanteuil, & Luthringer, 2007).
With the exception of ZPP, the PE inhibitory compounds listed have never been tested for the reduction of inflammation along the PE-PGP-PMN axis (Gaggar et al., 2008), but the study provides the proof of principle that other PE inhibitors may reduce PGP production and PMN influx. Moreover, the other inhibitors have been shown to inhibit PE in vivo, since they reduce the serum levels of the enzyme (Barelli et al., 1999; Bellemere et al., 2003; Bellemere, Vaudry, Morain, & Jegou, 2005; Morain et al., 2002; Schneider et al., 2002). Of particular interest is S-17092. In addition to its reduction of PE serum levels, it inhibits the enzyme within HEK293 cells. This indicates that it could potentially be a dual functioning inhibitor by inhibiting PE within PMNs as well as the PE that has been already released. Importantly, this drug has already passed Phase I clinical trials with no major adverse effects in human patients(Morain et al., 2000) indicating that S-17092 could have potential for treating inflammatory diseases with a PMN component such as CF and COPD.
5. CHEMOTAXIS ANTAGONISTS
As detailed above, both inflammatory cell recruitment and proteolytic matrix degradation are required in the pathology of COPD(Churg et al., 2002), and the prevention of either of these mechanisms may effectively suppress disease development. Furthermore, the findings that ECM breakdown and leukocyte recruitment are intrinsically related through the production of chemoattractant peptides(Houghton et al., 2006; Weathington et al., 2006) suggests that by suppressing one of these modalities we might effectively suppress the other. To date, only a few studies have been done to directly antagonize chemotactic activity.
In a phase 2 study evaluating monoclonal antibody against IL-8 (Mahler, Huang, Tabrizi, & Bell, 2004), patients with stable disease received intravenous infusions of the antibody and were evaluated via spirometry and by a dyspnea score. The treatment group had significant improvements in their dyspnea score at all time points measured. In preclinical work, animal studies targeting PGP have shown reductions in cell infiltration and decreased pathology in response to inflammatory insult. Anti-PGP antibody as well as anti-chemokine antibody prevented cell migration into lung in an acute injury model with bacterial LPS exposure(Weathington et al., 2006).
Early development of PGP-blocking peptides has also shown some promise, with the prevention of corneal ulceration in an alkali injury model described earlier. Use of the blocking peptide, arginine-threonine-arginine (RTR), prevented neutrophil infiltration and reduced the severity of injury in response to exposure(Haddox et al., 2001). More recently, the use of RTR has been tested in pulmonary models of inflammation and lung disease in an animal model of LPS- induced emphysema. Pulmonary infiltration of PMN in response to LPS and structural changes in the lung are completely prevented by pretreatment with the RTR drug(van Houwelingen et al., 2008). This study also shows that RTR’s effects include not only inhibition of PGP-mediated neutrophil chemotaxis, but also suppression of IL-8-dependent cell migration, indicating that this agent has two mechanisms of anti-inflammatory activity.
6. SUMMARY
In COPD, the current therapeutic strategies work predominantly towards symptom management with integrated therapy focusing on a few areas of pathology. The only life extending therapies currently employed include supplemental oxygen and smoking cessation. Other approved therapies are disease modifying agents that improve dyspnea and decrease hospitalizations. β-AR agonists and anticholinergics target the airway smooth muscle and improve airflow through the larger airways without action at the level of inflammatory changes in the bronchioles and alveoli. Glucocorticoids act on the level of protein expression to decrease proinflammatory molecules like cytokines and proteases. Given our new understanding of the complex molecular mechanisms of disease, new generations of targeted anti-inflammatory therapy may be on the horizon(Barnes, 2008).
Much of the recent work with collagen and elastin degradation has revealed new signaling activity of these structural proteins that is unmasked upon degradation by protease-mediated cleavage. The presence of collagen and elastin breakdown products in disease and their characterized chemotactic activity for inflammatory cells (PMNs and monocytes, respectively) underscores the hypothesis that interference with these novel pathways of the inflammatory phenotype may help treat the disease. To illustrate the action of these pathways, a discussion of the biochemistry of the small molecule PGP and its derivation from collagen seems appropriate.
As mentioned previously, both collagen and elastin contain a large proportion of proline residues (Voet & Voet, 1995). Proline is a cyclical amino acid in which the side chain R group is bound both to the nitrogen of the amino group and to the α-carbon, making it technically an imino acid (Cunningham & O’Connor, 1997). Due to the cyclical structure, it exhibits two important properties: 1) The angle of rotation between the α carbon and the nitrogen within a peptide bond formed by proline and another amino acid is limited, with a fixed angle introduced into the polypeptide. 2) Since the ring does not contain any oxygen, nitrogen, or sulfur, proline does not participate in the formation of hydrogen bonding without hydroxylation. Therefore, the presence of proline in peptides is incompatible with formation of α-helices and β-sheets. However, when proline is present at regular intervals within the sequence, it can produce a left handed helical structure associated with fibrillar proteins that is reinforced by the presence of hydroxylated proline residues, which participate in interstrand hydrogen bonding (Voet & Voet, 1995).
Therefore, peptides and proteins which possess this amino acid exhibit specific conformational properties which have a direct effect on their biochemical properties. The limited rotational angles of the peptide bonds formed by proline introduce kinks in the peptide and confer rigidity to the overall structure. In general, these conformational properties limit access of the molecule to external enzymatic processes including proteolysis. From an evolutionary standpoint, this makes sense. Collagen is the most abundant protein found in vertebrates and occurs in virtually every tissue where it provides structural support (Bornstein, 1974; Voet & Voet, 1995), therefore it should be generally protected from proteolysis and other enzymatic modification. To maintain healthy tissue turnover, however, specific enzymes have evolved specifically for cleaving proline containing peptides and proteins (MMPs, NE, and PE) (Cunningham & O’Connor, 1997).
Possibly because of its unique structural character, proline containing peptides exhibit the signaling capacity described above only when cleaved into smaller peptides and accessible to the cell receptors they ligate. It is tempting to speculate that this mode of signaling was established even before that of more complex chemokine signaling, with ECM damage acting as a kind of “danger signal” that induces the recruitment of leukocytes into a site of injury. This represents alternate, redundant inflammatory signaling in addition to the more canonical pathways like IL-8 signaling.
Consideration of the pathophysiology presented above gives us a proteolytic cascade of events that is intrinsic to the inflammation of chronic disease like COPD, and presents a series of putative therapeutic targets, as depicted in Figure 2. With new data that better characterizes the biology involved and the advent of inhibitors that might be used, promising avenues have been opened. The proteases PE, MMP-8, MMP-9, NE, and MMP-12 likely play a significant role in disease development.
Figure 2. The steps of the proteolytic cascade present multiple targets for therapeutic intervention.

As seen in Figure 1, extracellular matrix breakdown is a multistep process that drives inflammation. New compounds have been developed to antagonize each of these steps.
Collagen and elastin processing each feature MMPs as active proteases. Nonspecific MMP inhibitors such as GM6001 or CP 471,474 can therefore suppress the proinflammatory signaling caused by collagen or elastin degradation. Furthermore, specific antagonists of either collagen or elastin breakdown have shown promise in preclinical work. Neutrophil elastase inhibitors like ONO-6818 have been developed and have seen mixed results in human studies, while other NE inhibitors have been tested preclinically. Prolyl endopeptidase inhibitors, specifically ZPP have been shown to prevent PGP production as well as neutrophil accumulation in a model of airway inflammation. Finally, chemotaxis inhibitors like anti-IL-8 antibodies and RTR have each shown some promise in reducing the cell number and inflammation associated with pulmonary disease. As development of these compounds continues, Antiproteolytic agents may arise as a completely new class of anti-inflammatory drugs available for treatment of human inflammatory diseases like COPD.
An initial insult by environmental exposure like cigarette smoking results in reactive inflammation and collagen degradation by collagenases and gelatinases, each of which may be targeted by MMP inhibitors. The most promising of these include broad spectrum inhibitors like GM-6001, whose efficacy and tolerability may be enhanced by inhaled delivery. Unfortunately, systemic delivery of these compounds is thus far limited by side effects.
The next step in the collagen cascade is the processing of fragments further by PE, ultimately producing PGP which is chemotactic to neutrophils (Malik et al., 2007; Pfister et al., 1995; Weathington et al., 2006). PE is an evolutionarily conserved enzyme which cleaves proline containing peptides (30–100 amino acids in length) at the C-terminal side of the proline. Model systems in which inflammation has been correlated with an increase of PE levels include RA, alkali injury to the eye, and CF, and targeting PE may prevent signaling through the PGP- CXCR axis. Chemotaxis antagonists such as RTR and anti-IL-8 may be used to inhibit cell trafficking through the CXCRs (Haddox et al., 2001; Mahler et al., 2004). Potentially, CXC receptor antagonists could also be useful, but such agents are beyond the scope of this discussion.
Parallel to collagen degradation, the processing of elastin by proteases including NE and MMP-12 causes tissue breakdown and structural compromise. Elastase degradation also produces protein fragments that contribute to inflammation by chemotaxis of monocytes in similar fashion to collagen fragments’ effects on PMN. The targeting of NE activity is well developed and has spawned a few candidate drugs that show some promise, though no members of this class have developed into approved therapies for inflammatory disease. Antagonism of MMP-12 can be affected by some of the broad spectrum MMPIs already mentioned, and may prove important in the development of new agents, and there is anticipation of new compounds with specific activity for this protease. As with therapy targeting collagen degradation, suppression of elastase has the potential to prevent multiple pathologic mechanisms of disease.
It is clear that the pathophysiology of inflammatory diseases with a neutrophilic component is very complicated and multifaceted. In consideration of COPD where PMN inflammation characterizes the disease, it is clear that the underlying pathophysiologic processes need to be addressed in therapy. While ECM turnover is an ongoing physiological process, an imbalance may result in dysregulation causing pathological, self perpetuating inflammation and tissue destruction, a hallmark of chronic inflammatory disease
COPD is the 4th leading cause of death in the developed world and, due to its increasing prevalence, is expected to be the 3rd leading cause of death in the developed world by 2020. However, with increased research into the mechanisms underlying this inflammatory response, development of adequate disease biomarkers, disease-specific therapeutics (e.g. targeting proteolytic activity), and ongoing public-health efforts regarding smoking cessation, it may be possible to stave off this looming worldwide epidemic.
Acknowledgments
The authors would like to thank Michael P Nelson for assistance with the figures and Dr. J Edwin Blalock for reviewing the manuscript.
UVD is funded by the NIH (T32A107493). AG is funded through the UAB CIFA Award and the Cystic Fibrosis Foundation (GAGGAR07A0). NW is supported by the UAB Department of Medicine, Internal medicine residency program.
Abbreviations
- α1-AT
alpha 1 antitrypsin
- Ala
alanine
- ARDS
acute respiratory distress syndrome
- AP-1
activator protein 1
- Asp
aspartic acid
- BAL
bronchoalveolar lavage
- CF
cystic fibrosis
- COPD
chronic obstructive pulmonary disease
- CXC
referring to cysteine-x-cysteine chemokine family
- CXCR
cysteine-x-cysteine chemokine receptor
- CT
computed tomography scan
- ECM
extracellular matrix
- EEG
electroencephalogram
- FEV1
forced expiratory volume in 1 second
- FVC
forced vital capacity
- Gly
glycine
- hPE
human prolyl endopeptidase
- His
histidine
- Hyp
4-hydoxyproline
- IL-8
interleukin 8
- IL-13
interleukin 13
- KC
keratinocyte-derived chemokine
- LPS
lipopolysaccharide
- MIP
macrophage inflammatory protein
- MMP
matrix metalloproteases
- MMPI
matrix metalloprotease inhibitor
- N-a-PGP
acetylated proline-glycine-proline
- NE
neutrophil elastase
- NF-κβ
nuclear factor kappa beta
- OA
osteoarthritis
- PE
prolyl endopeptidase
- PGP
proline-glycine-proline
- POP
prolyl oligopeptidase
- PMN
polymorphonuclear leukocytes
- pNa
paranitroaniline
- PPCE
post-proline cleaving enzyme
- Pro
proline
- RA
rheumatoid arthritis
- RTR
arginine-thronine-arginine
- SKALP
skin-derived antileukoprotease
- SLE
systemic lupus erythematosus
- SLPI
secretory leukocyte protease inhibitor
- Ser
serine
- TIMP
tissue inhibitor of metalloproteases
- TNF
tumor necrosis factor
- TLR
toll-like receptors
- Val
valine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- From the global strategy for the diagnosis, management and prevention of copd, global initiative for chronic obstructive lung disease (gold) 2007 [Google Scholar]
- Nikolai JBa, Khaltaev, editors. Global surveillance, prevention and control of chronic respiratory diseases: A comprehensive approach. 2007. [Google Scholar]
- Aoshiba K, Nagai A, Konno K. Erythromycin shortens neutrophil survival by accelerating apoptosis. Antimicrob Agents Chemother. 1995;39(4):872–877. doi: 10.1128/aac.39.4.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi T, Wada T, Daskalov HP, Kojima F, Nagai M, Harada S, et al. Dissociation between serine proteinases and proline related enzymes in spleen of mrl mouse as a model of systemic lupus erythematodes. Biochem Int. 1987;14(3):435–441. [PubMed] [Google Scholar]
- Aoyagi T, Wada T, Ishikawa Y, Kojima F, Nagai M, Osanai T, et al. Systemic enzymatic changes in guinea pigs suffering from experimental allergic encephalomyelitis. J Pharmacobiodyn. 1983;6(12):963–973. doi: 10.1248/bpb1978.6.963. [DOI] [PubMed] [Google Scholar]
- Aoyagi T, Wada T, Kojima F, Nagai M, Okubo M, Masaki Y, et al. Abnormality of the post-proline-cleaving enzyme activity in mice with systemic lupus erythematosus-like syndrome. J Appl Biochem. 1985;7(4–5):273–281. [PubMed] [Google Scholar]
- Atack JR, Suman-Chauhan N, Dawson G, Kulagowski JJ. In vitro and in vivo inhibition of prolyl endopeptidase. Eur J Pharmacol. 1991;205(2):157–163. doi: 10.1016/0014-2999(91)90814-7. [DOI] [PubMed] [Google Scholar]
- Attucci S, Gauthier A, Korkmaz B, Delepine P, Martino MF, Saudubray F, et al. Epi-hne4, a proteolysis-resistant inhibitor of human neutrophil elastase and potential anti-inflammatory drug for treating cystic fibrosis. J Pharmacol Exp Ther. 2006;318(2):803–809. doi: 10.1124/jpet.106.103440. [DOI] [PubMed] [Google Scholar]
- Bakker AV, Jung S, Spencer RW, Vinick FJ, Faraci WS. Slow tight-binding inhibition of prolyl endopeptidase by benzyloxycarbonyl-prolyl-prolinal. Biochem J. 1990;271(2):559–562. doi: 10.1042/bj2710559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelli H, Petit A, Hirsch E, Wilk S, De Nanteuil G, Morain P, et al. S 17092-1, a highly potent, specific and cell permeant inhibitor of human proline endopeptidase. Biochem Biophys Res Commun. 1999;257(3):657–661. doi: 10.1006/bbrc.1999.0366. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- Barrett AJ, Rawlings ND. Oligopeptidases, and the emergence of the prolyl oligopeptidase family. Biol Chem Hoppe Seyler. 1992;373(7):353–360. doi: 10.1515/bchm3.1992.373.2.353. [DOI] [PubMed] [Google Scholar]
- Beeh KM, Beier J, Kornmann O, Buhl R. Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjects. Respir Med. 2003;97(6):634–639. doi: 10.1053/rmed.2003.1493. [DOI] [PubMed] [Google Scholar]
- Beeh KM, Beier J, Kornmann O, Micke P, Buhl R. Sputum levels of metalloproteinase-9 and tissue inhibitor of metalloproteinase-1, and their ratio correlate with airway obstruction in lung transplant recipients: Relation to tumor necrosis factor-alpha and interleukin-10. J Heart Lung Transplant. 2001;20(11):1144–1151. doi: 10.1016/s1053-2498(01)00325-4. [DOI] [PubMed] [Google Scholar]
- Bellemere G, Morain P, Vaudry H, Jegou S. Effect of s 17092, a novel prolyl endopeptidase inhibitor, on substance p and alpha-melanocyte-stimulating hormone breakdown in the rat brain. J Neurochem. 2003;84(5):919–929. doi: 10.1046/j.1471-4159.2003.01536.x. [DOI] [PubMed] [Google Scholar]
- Bellemere G, Vaudry H, Morain P, Jegou S. Effect of prolyl endopeptidase inhibition on arginine-vasopressin and thyrotrophin-releasing hormone catabolism in the rat brain. J Neuroendocrinol. 2005;17(5):306–313. doi: 10.1111/j.1365-2826.2005.01308.x. [DOI] [PubMed] [Google Scholar]
- Betsuyaku T, Nishimura M, Takeyabu K, Tanino M, Miyamoto K, Kawakami Y. Decline in fev(1) in community-based older volunteers with higher levels of neutrophil elastase in bronchoalveolar lavage fluid. Respiration. 2000;67(3):261–267. doi: 10.1159/000029508. [DOI] [PubMed] [Google Scholar]
- Betsuyaku T, Nishimura M, Takeyabu K, Tanino M, Venge P, Xu S, et al. Neutrophil granule proteins in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Am J Respir Crit Care Med. 1999;159(6):1985–1991. doi: 10.1164/ajrccm.159.6.9809043. [DOI] [PubMed] [Google Scholar]
- Bornstein P. The biosynthesis of collagen. Annu Rev Biochem. 1974;43(0):567–603. doi: 10.1146/annurev.bi.43.070174.003031. [DOI] [PubMed] [Google Scholar]
- Boyle JR, McDermott E, Crowther M, Wills AD, Bell PR, Thompson MM. Doxycycline inhibits elastin degradation and reduces metalloproteinase activity in a model of aneurysmal disease. J Vasc Surg. 1998;27(2):354–361. doi: 10.1016/s0741-5214(98)70367-2. [DOI] [PubMed] [Google Scholar]
- Bradbury J. Neutrophil elastase inhibitors for cystic fibrosis. Drug Discov Today. 2001;6(9):441–442. doi: 10.1016/s1359-6446(01)01789-5. [DOI] [PubMed] [Google Scholar]
- Brenner S. The molecular evolution of genes and proteins: A tale of two serines. Nature. 1988;334(6182):528–530. doi: 10.1038/334528a0. [DOI] [PubMed] [Google Scholar]
- Camargo AC, Caldo H, Reis ML. Susceptibility of a peptide derived from bradykinin to hydrolysis by brain endo-oligopeptidases and pancreatic proteinases. J Biol Chem. 1979;254(12):5304–5307. [PubMed] [Google Scholar]
- Carvalho KM, Camargo AC. Purification of rabbit brain endooligopeptidases and preparation of anti-enzyme antibodies. Biochemistry. 1981;20(25):7082–7088. doi: 10.1021/bi00528a005. [DOI] [PubMed] [Google Scholar]
- Churg A, Wright JL. Proteases and emphysema. Curr Opin Pulm Med. 2005;11(2):153–159. doi: 10.1097/01.mcp.0000149592.51761.e3. [DOI] [PubMed] [Google Scholar]
- Churg A, Zay K, Shay S, Xie C, Shapiro SD, Hendricks R, et al. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol. 2002;27(3):368–374. doi: 10.1165/rcmb.4791. [DOI] [PubMed] [Google Scholar]
- Corbel M, Theret N, Caulet-Maugendre S, Germain N, Lagente V, Clement B, et al. Repeated endotoxin exposure induces interstitial fibrosis associated with enhanced gelatinase (mmp-2 and mmp-9) activity. Inflamm Res. 2001;50(3):129–135. doi: 10.1007/s000110050736. [DOI] [PubMed] [Google Scholar]
- Crystal RG. The lung: Scientific foundations. 2. Philadelphia: Lippincott-Raven; 1997. [Google Scholar]
- Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthews JL, Barnes PJ. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1635–1639. doi: 10.1164/ajrccm.160.5.9811058. [DOI] [PubMed] [Google Scholar]
- Cunningham DF, O’Connor B. Proline specific peptidases. Biochim Biophys Acta. 1997;1343(2):160–186. doi: 10.1016/s0167-4838(97)00134-9. [DOI] [PubMed] [Google Scholar]
- D’Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell. 1992;71(6):955–961. doi: 10.1016/0092-8674(92)90391-o. [DOI] [PubMed] [Google Scholar]
- Deshmukh HS, Case LM, Wesselkamper SC, Borchers MT, Martin LD, Shertzer HG, et al. Metalloproteinases mediate mucin 5ac expression by epidermal growth factor receptor activation. Am J Respir Crit Care Med. 2005;171(4):305–314. doi: 10.1164/rccm.200408-1003OC. [DOI] [PubMed] [Google Scholar]
- Dirksen A, Dijkman JH, Madsen F, Stoel B, Hutchison DC, Ulrik CS, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1468–1472. doi: 10.1164/ajrccm.160.5.9901055. [DOI] [PubMed] [Google Scholar]
- Dunsmore SE, Saarialho-Kere UK, Roby JD, Wilson CL, Matrisian LM, Welgus HG, et al. Matrilysin expression and function in airway epithelium. J Clin Invest. 1998;102(7):1321–1331. doi: 10.1172/JCI1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duranton J, Bieth JG. Inhibition of proteinase 3 by [alpha]1-antitrypsin in vitro predicts very fast inhibition in vivo. Am J Respir Cell Mol Biol. 2003;29(1):57–61. doi: 10.1165/rcmb.2002-0258OC. [DOI] [PubMed] [Google Scholar]
- Elewski BE, Lamb BA, Sams WM, Jr, Gammon WR. In vivo suppression of neutrophil chemotaxis by systemically and topically administered tetracycline. J Am Acad Dermatol. 1983;8(6):807–812. doi: 10.1016/s0190-9622(83)80010-3. [DOI] [PubMed] [Google Scholar]
- Emingil G, Atilla G, Sorsa T, Luoto H, Kirilmaz L, Baylas H. The effect of adjunctive low-dose doxycycline therapy on clinical parameters and gingival crevicular fluid matrix metalloproteinase-8 levels in chronic periodontitis. J Periodontol. 2004;75(1):106–115. doi: 10.1902/jop.2004.75.1.106. [DOI] [PubMed] [Google Scholar]
- Endo S, Sato N, Yaegashi Y, Suzuki Y, Kojika M, Yamada Y, et al. Sivelestat sodium hydrate improves septic acute lung injury by reducing alveolar dysfunction. Res Commun Mol Pathol Pharmacol. 2006;119(1–6):53–65. [PubMed] [Google Scholar]
- Equi A, Balfour-Lynn IM, Bush A, Rosenthal M. Long term azithromycin in children with cystic fibrosis: A randomised, placebo-controlled crossover trial. Lancet. 2002;360(9338):978–984. doi: 10.1016/s0140-6736(02)11081-6. [DOI] [PubMed] [Google Scholar]
- Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5(14):1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Fingleton B. Mmps as therapeutic targets--still a viable option? Semin Cell Dev Biol. 2008;19(1):61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay GA, O’Driscoll LR, Russell KJ, D’Arcy EM, Masterson JB, FitzGerald MX, et al. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med. 1997;156(1):240–247. doi: 10.1164/ajrccm.156.1.9612018. [DOI] [PubMed] [Google Scholar]
- Finlay GA, Russell KJ, McMahon KJ, D’Arcy EM, Masterson JB, FitzGerald MX, et al. Elevated levels of matrix metalloproteinases in bronchoalveolar lavage fluid of emphysematous patients. Thorax. 1997;52(6):502–506. doi: 10.1136/thx.52.6.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foronjy RF, Okada Y, Cole R, D’Armiento J. Progressive adult-onset emphysema in transgenic mice expressing human mmp-1 in the lung. Am J Physiol Lung Cell Mol Physiol. 2003;284(5):L727–737. doi: 10.1152/ajplung.00349.2002. [DOI] [PubMed] [Google Scholar]
- Fukuoka Y, Hagihara M, Nagatsu T, Kaneda T. The relationship between collagen metabolism and temporomandibular joint osteoarthrosis in mice. J Oral Maxillofac Surg. 1993;51(3):288–291. doi: 10.1016/s0278-2391(10)80177-6. [DOI] [PubMed] [Google Scholar]
- Fulop V, Szeltner Z, Renner V, Polgar L. Structures of prolyl oligopeptidase substrate/inhibitor complexes. Use of inhibitor binding for titration of the catalytic histidine residue. J Biol Chem. 2001;276(2):1262–1266. doi: 10.1074/jbc.M007003200. [DOI] [PubMed] [Google Scholar]
- Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180(8):5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggar A, Li Y, Weathington N, Winkler M, Kong M, Jackson P, et al. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L96–L104. doi: 10.1152/ajplung.00492.2006. [DOI] [PubMed] [Google Scholar]
- Garcia-Horsman JA, Mannisto PT, Venalainen JI. On the role of prolyl oligopeptidase in health and disease. Neuropeptides. 2007;41(1):1–24. doi: 10.1016/j.npep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Gass J, Khosla C. Prolyl endopeptidases. Cell Mol Life Sci. 2007;64(3):345–355. doi: 10.1007/s00018-006-6317-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty P, Rogan MP, Greene CM, Boxio RM, Poiriert T, O’Mahony M, et al. Neutrophil elastase up-regulates cathepsin b and matrix metalloprotease-2 expression. J Immunol. 2007;178(9):5871–5878. doi: 10.4049/jimmunol.178.9.5871. [DOI] [PubMed] [Google Scholar]
- Gerhardt SG, McDyer JF, Girgis RE, Conte JV, Yang SC, Orens JB. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: Results of a pilot study. Am J Respir Crit Care Med. 2003;168(1):121–125. doi: 10.1164/rccm.200212-1424BC. [DOI] [PubMed] [Google Scholar]
- Golub LM, Lee HM, Lehrer G, Nemiroff A, McNamara TF, Kaplan R, et al. Minocycline reduces gingival collagenolytic activity during diabetes. Preliminary observations and a proposed new mechanism of action. J Periodontal Res. 1983;18(5):516–526. doi: 10.1111/j.1600-0765.1983.tb00388.x. [DOI] [PubMed] [Google Scholar]
- Golub LM, Ramamurthy NS, McNamara TF, Greenwald RA, Rifkin BR. Tetracyclines inhibit connective tissue breakdown: New therapeutic implications for an old family of drugs. Crit Rev Oral Biol Med. 1991;2(3):297–321. doi: 10.1177/10454411910020030201. [DOI] [PubMed] [Google Scholar]
- Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur J Cell Biol. 1997;74(2):111–122. [PubMed] [Google Scholar]
- Goossens F, De Meester I, Vanhoof G, Scharpe S. A sensitive method for the assay of serum prolyl endopeptidase. Eur J Clin Chem Clin Biochem. 1992;30(4):235–238. [PubMed] [Google Scholar]
- Goossens F, De Meester I, Vanhoof G, Scharpe S. Distribution of prolyl oligopeptidase in human peripheral tissues and body fluids. Eur J Clin Chem Clin Biochem. 1996;34(1):17–22. doi: 10.1515/cclm.1996.34.1.17. [DOI] [PubMed] [Google Scholar]
- Goossens FJ, Wauters JG, Vanhoof GC, Bossuyt PJ, Schatteman KA, Loens K, et al. Subregional mapping of the human lymphocyte prolyl oligopeptidase gene (prep) to human chromosome 6q22. Cytogenet Cell Genet. 1996;74(1–2):99–101. doi: 10.1159/000134391. [DOI] [PubMed] [Google Scholar]
- GOTTLIEB J, ELMER M, STEINER R. Efficacy of intravenous ici-200,880, a neutrophil elastase inhibitor, in the treatment of adult respiratory distress syndrome. Chest. 1994;107:67. [Google Scholar]
- Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: Multiple, multifarious, and multifaceted. Physiol Rev. 2007;87(1):69–98. doi: 10.1152/physrev.00022.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Booms P, Halushka M, Dietz HC, Ney A, Stricker S, et al. Induction of macrophage chemotaxis by aortic extracts of the mgr marfan mouse model and a gxxpg-containing fibrillin-1 fragment. Circulation. 2006;114(17):1855–1862. doi: 10.1161/CIRCULATIONAHA.105.601674. [DOI] [PubMed] [Google Scholar]
- Haddox JL, Pfister RR, Sommers CI, Blalock JE, Villain M. Inhibitory effect of a complementary peptide on ulceration in the alkali-injured rabbit cornea. Invest Ophthalmol Vis Sci. 2001;42(12):2769–2775. [PubMed] [Google Scholar]
- Hance KA, Tataria M, Ziporin SJ, Lee JK, Thompson RW. Monocyte chemotactic activity in human abdominal aortic aneurysms: Role of elastin degradation peptides and the 67-kd cell surface elastin receptor. J Vasc Surg. 2002;35(2):254–261. doi: 10.1067/mva.2002.120382. [DOI] [PubMed] [Google Scholar]
- Hanemaaijer R, Visser H, Koolwijk P, Sorsa T, Salo T, Golub LM, et al. Inhibition of mmp synthesis by doxycycline and chemically modified tetracyclines (cmts) in human endothelial cells. Adv Dent Res. 1998;12(2):114–118. doi: 10.1177/08959374980120010301. [DOI] [PubMed] [Google Scholar]
- Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, et al. Cleavage of cxcr1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13(12):1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- Harwood VJ, Denson JD, Robinson-Bidle KA, Schreier HJ. Overexpression and characterization of a prolyl endopeptidase from the hyperthermophilic archaeon pyrococcus furiosus. J Bacteriol. 1997;179(11):3613–3618. doi: 10.1128/jb.179.11.3613-3618.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood VJ, Schreier HJ. Prolyl oligopeptidase from pyrococcus furiosus. Methods Enzymol. 2001;330:445–454. doi: 10.1016/s0076-6879(01)30396-8. [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Kawabe T, Hara T, Imaizumi K, Wakayama H, Shimokata K, et al. effects of erythromycin on matrix metalloproteinase-9(mmp-9)and cell migration. Jpn J Antibiot, 54 Suppl A. 2001:137–138. [PubMed] [Google Scholar]
- Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- Hoshi K, Kurosawa S, Kaise A, Kato M, Andoh K, Satoh D. Sivelestat, a neutrophil elastase inhibitor, reduces mortality rate of critically ill patients. Tohoku Journal of Experimental Medicine. 2005;207(2):143–148. doi: 10.1620/tjem.207.143. [DOI] [PubMed] [Google Scholar]
- Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt JC, Ballering J, Numanami H, Hayden JM, Robbins RA. Doxycycline modulates nitric oxide production in murine lung epithelial cells. J Immunol. 2006;176(1):567–572. doi: 10.4049/jimmunol.176.1.567. [DOI] [PubMed] [Google Scholar]
- Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6(6):480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- Johnson LL, Dyer R, Hupe DJ. Matrix metalloproteinases. Curr Opin Chem Biol. 1998;2(4):466–471. doi: 10.1016/s1367-5931(98)80122-1. [DOI] [PubMed] [Google Scholar]
- Joos L, He JQ, Shepherdson MB, Connett JE, Anthonisen NR, Pare PD, et al. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet. 2002;11(5):569–576. doi: 10.1093/hmg/11.5.569. [DOI] [PubMed] [Google Scholar]
- Kadota J, Mukae H, Ishii H, Nagata T, Kaida H, Tomono K, et al. Long-term efficacy and safety of clarithromycin treatment in patients with diffuse panbronchiolitis. Respir Med. 2003;97(7):844–850. doi: 10.1016/s0954-6111(03)00042-8. [DOI] [PubMed] [Google Scholar]
- Kamori M, Hagihara M, Nagatsu T, Iwata H, Miura T. Activities of dipeptidyl peptidase ii, dipeptidyl peptidase iv, prolyl endopeptidase, and collagenase-like peptidase in synovial membrane from patients with rheumatoid arthritis and osteoarthritis. Biochem Med Metab Biol. 1991;45(2):154–160. doi: 10.1016/0885-4505(91)90016-e. [DOI] [PubMed] [Google Scholar]
- Kanai K, Asano K, Hisamitsu T, Suzaki H. Suppression of matrix metalloproteinase-9 production from neutrophils by a macrolide antibiotic, roxithromycin, in vitro. Mediators Inflamm. 2004;13(5–6):313–319. doi: 10.1155/S0962935104000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai K, Asano K, Hisamitsu T, Suzaki H. Suppression of matrix metalloproteinase production from nasal fibroblasts by macrolide antibiotics in vitro. Eur Respir J. 2004;23(5):671–678. doi: 10.1183/09031936.04.00057104. [DOI] [PubMed] [Google Scholar]
- Kang T, Yi J, Guo A, Wang X, Overall CM, Jiang W, et al. Subcellular distribution and cytokine- and chemokine-regulated secretion of leukolysin/mt6-mmp/mmp-25 in neutrophils. J Biol Chem. 2001;276(24):21960–21968. doi: 10.1074/jbc.M007997200. [DOI] [PubMed] [Google Scholar]
- Kao RC, Wehner NG, Skubitz KM, Gray BH, Hoidal JR. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J Clin Invest. 1988;82(6):1963–1973. doi: 10.1172/JCI113816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Okada M, Nagatsu T. Distribution of post-proline cleaving enzyme in human brain and the peripheral tissues. Mol Cell Biochem. 1980;32(3):117–121. doi: 10.1007/BF00227437. [DOI] [PubMed] [Google Scholar]
- Kimura A, Yoshida I, Takagi N, Takahashi T. Structure and localization of the mouse prolyl oligopeptidase gene. J Biol Chem. 1999;274(34):24047–24053. doi: 10.1074/jbc.274.34.24047. [DOI] [PubMed] [Google Scholar]
- Kobzik L. The lung. In: Cotrain VKRS, Collins T, editors. Robbins pathologic basis of disease. Philadelphia: W.B. Saunders Company; 1999. [Google Scholar]
- Koida M, Walter R. Post-proline cleaving enzyme. Purification of this endopeptidase by affinity chromatography. J Biol Chem. 1976;251(23):7593–7599. [PubMed] [Google Scholar]
- Laskin DL, Kimura T, Sakakibara S, Riley DJ, Berg RA. Chemotactic activity of collagen-like polypeptides for human peripheral blood neutrophils. J Leukoc Biol. 1986;39(3):255–266. doi: 10.1002/jlb.39.3.255. [DOI] [PubMed] [Google Scholar]
- Lee KS, Jin SM, Kim SS, Lee YC. Doxycycline reduces airway inflammation and hyperresponsiveness in a murine model of toluene diisocyanate-induced asthma. J Allergy Clin Immunol. 2004;113(5):902–909. doi: 10.1016/j.jaci.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Li YY, Feldman AM. Matrix metalloproteinases in the progression of heart failure: Potential therapeutic implications. Drugs. 2001;61(9):1239–1252. doi: 10.2165/00003495-200161090-00002. [DOI] [PubMed] [Google Scholar]
- Luisetti M, Sturani C, Sella D, Madonini E, Galavotti V, Bruno G, et al. Mr889, a neutrophil elastase inhibitor, in patients with chronic obstructive pulmonary disease: A double-blind, randomized, placebo-controlled clinical trial. Eur Respir J. 1996;9(7):1482–1486. doi: 10.1183/09031936.96.09071482. [DOI] [PubMed] [Google Scholar]
- Luscombe M. Acid phosphatase and catheptic activity in rheumatoid synovial tissue. Nature. 1963;197:1010. doi: 10.1038/1971010a0. [DOI] [PubMed] [Google Scholar]
- Maes M, Goossens F, Lin A, De Meester I, Van Gastel A, Scharpe S. Effects of psychological stress on serum prolyl endopeptidase and dipeptidyl peptidase iv activity in humans: Higher serum prolyl endopeptidase activity is related to stress-induced anxiety. Psychoneuroendocrinology. 1998;23(5):485–495. doi: 10.1016/s0306-4530(98)00020-1. [DOI] [PubMed] [Google Scholar]
- Maes M, Goossens F, Scharpe S, Calabrese J, Desnyder R, Meltzer HY. Alterations in plasma prolyl endopeptidase activity in depression, mania, and schizophrenia: Effects of antidepressants, mood stabilizers, and antipsychotic drugs. Psychiatry Res. 1995;58(3):217–225. doi: 10.1016/0165-1781(95)02698-v. [DOI] [PubMed] [Google Scholar]
- Maes M, Libbrecht I, Van Hunsel F, Lin AH, Bonaccorso S, Goossens F, et al. Lower serum activity of prolyl endopeptidase in fibromyalgia is related to severity of depressive symptoms and pressure hyperalgesia. Psychol Med. 1998;28(4):957–965. doi: 10.1017/s0033291798006801. [DOI] [PubMed] [Google Scholar]
- Maes M, Monteleone P, Bencivenga R, Goossens F, Maj M, van West D, et al. Lower serum activity of prolyl endopeptidase in anorexia and bulimia nervosa. Psychoneuroendocrinology. 2001;26(1):17–26. doi: 10.1016/s0306-4530(00)00032-9. [DOI] [PubMed] [Google Scholar]
- Mahler DA, Huang S, Tabrizi M, Bell GM. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in copd: A pilot study. Chest. 2004;126(3):926–934. doi: 10.1378/chest.126.3.926. [DOI] [PubMed] [Google Scholar]
- Malik M, Bakshi CS, McCabe K, Catlett SV, Shah A, Singh R, et al. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type a and b strains of francisella tularensis. J Immunol. 2007;178(2):1013–1020. doi: 10.4049/jimmunol.178.2.1013. [DOI] [PubMed] [Google Scholar]
- Marighetto A, Touzani K, Etchamendy N, Torrea CC, De Nanteuil G, Guez D, et al. Further evidence for a dissociation between different forms of mnemonic expressions in a mouse model of age-related cognitive decline: Effects of tacrine and s 17092, a novel prolyl endopeptidase inhibitor. Learn Mem. 2000;7(3):159–169. doi: 10.1101/lm.7.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova I, Kotra LP, Fridman R, Mobashery S. Matrix metalloproteinases: Structures, evolution, and diversification. Faseb J. 1998;12(12):1075–1095. [PubMed] [Google Scholar]
- Minematsu N, Nakamura H, Tateno H, Nakajima T, Yamaguchi K. Genetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysema. Biochem Biophys Res Commun. 2001;289(1):116–119. doi: 10.1006/bbrc.2001.5936. [DOI] [PubMed] [Google Scholar]
- Miravitlles M, Murio C, Guerrero T, Gisbert R. Costs of chronic bronchitis and copd: A 1-year follow-up study. Chest. 2003;123(3):784–791. doi: 10.1378/chest.123.3.784. [DOI] [PubMed] [Google Scholar]
- Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, et al. Increase in macrophage elastase (mmp-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54(1):31–36. doi: 10.1007/s00011-004-1319-4. [DOI] [PubMed] [Google Scholar]
- Morain P, Boeijinga PH, Demazieres A, De Nanteuil G, Luthringer R. Psychotropic profile of s 17092, a prolyl endopeptidase inhibitor, using quantitative eeg in young healthy volunteers. Neuropsychobiology. 2007;55(3–4):176–183. doi: 10.1159/000107070. [DOI] [PubMed] [Google Scholar]
- Morain P, Lestage P, De Nanteuil G, Jochemsen R, Robin JL, Guez D, et al. S 17092: A prolyl endopeptidase inhibitor as a potential therapeutic drug for memory impairment. Preclinical and clinical studies. CNS Drug Rev. 2002;8(1):31–52. doi: 10.1111/j.1527-3458.2002.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morain P, Robin JL, De Nanteuil G, Jochemsen R, Heidet V, Guez D. Pharmacodynamic and pharmacokinetic profile of s 17092, a new orally active prolyl endopeptidase inhibitor, in elderly healthy volunteers. A phase i study. Br J Clin Pharmacol. 2000;50(4):350–359. doi: 10.1046/j.1365-2125.2000.00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydel P, Shipley JM, Adair-Kirk TL, Kelley DG, Broekelmann TJ, Mecham RP, et al. Neutrophil elastase cleavesllaminin-332 (laminin-5) generating peptides that are chemotactic for neutrophils. J Biol Chem. 2008 doi: 10.1074/jbc.M706239200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenan S, Lagente V, Planquois JM, Hitier S, Berna P, Bertrand CP, et al. Metalloelastase (mmp-12) induced inflammatory response in mice airways: Effects of dexamethasone, rolipram and marimastat. Eur J Pharmacol. 2007;559(1):75–81. doi: 10.1016/j.ejphar.2006.11.070. [DOI] [PubMed] [Google Scholar]
- Norris DA, Clark RA, Swigart LM, Huff JC, Weston WL, Howell SE. Fibronectin fragment(s) are chemotactic for human peripheral blood monocytes. J Immunol. 1982;129(4):1612–1618. [PubMed] [Google Scholar]
- O’Leary RM, Gallagher SP, O’Connor B. Purification and characterization of a novel membrane-bound form of prolyl endopeptidase from bovine brain. Int J Biochem Cell Biol. 1996;28(4):441–449. doi: 10.1016/1357-2725(95)00154-9. [DOI] [PubMed] [Google Scholar]
- O’Leary RM, O’Connor B. Identification and localisation of a synaptosomal membrane prolyl endopeptidase from bovine brain. Eur J Biochem. 1995;227(1–2):277–283. doi: 10.1111/j.1432-1033.1995.tb20385.x. [DOI] [PubMed] [Google Scholar]
- Ohbayashi H. Neutrophil elastase inhibitors as treatment for copd. Expert Opin Investig Drugs. 2002;11(7):965–980. doi: 10.1517/13543784.11.7.965. [DOI] [PubMed] [Google Scholar]
- Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest. 1998;78(9):1077–1087. [PubMed] [Google Scholar]
- Oliveira EB, Martins AR, Camargo AC. Isolation of brain endopeptidases: Influence of size and sequence of substrates structurally related to bradykinin. Biochemistry. 1976;15(9):1967–1974. doi: 10.1021/bi00654a026. [DOI] [PubMed] [Google Scholar]
- Orlowski M, Wilk E, Pearce S, Wilk S. Purification and properties of a prolyl endopeptidase from rabbit brain. J Neurochem. 1979;33(2):461–469. doi: 10.1111/j.1471-4159.1979.tb05176.x. [DOI] [PubMed] [Google Scholar]
- Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer. 2006;94(7):941–946. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo A, Selman M. Mmp-1: The elder of the family. Int J Biochem Cell Biol. 2005;37(2):283–288. doi: 10.1016/j.biocel.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Pavlaki M, Zucker S. Matrix metalloproteinase inhibitors (mmpis): The beginning of phase i or the termination of phase iii clinical trials. Cancer Metastasis Rev. 2003;22(2–3):177–203. doi: 10.1023/a:1023047431869. [DOI] [PubMed] [Google Scholar]
- Pemberton PA, Cantwell JS, Kim KM, Sundin DJ, Kobayashi D, Fink JB, et al. An inhaled matrix metalloprotease inhibitor prevents cigarette smoke-induced emphysema in the mouse. Copd. 2005;2(3):303–310. doi: 10.1080/15412550500218171. [DOI] [PubMed] [Google Scholar]
- Pemberton PA, Kobayashi D, Wilk BJ, Henstrand JM, Shapiro SD, Barr PJ. Inhaled recombinant alpha 1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Copd. 2006;3(2):101–108. doi: 10.1080/15412550600651248. [DOI] [PubMed] [Google Scholar]
- Pfister RR, Haddox JL, Lam KW. Alkali-degraded cells generate a respiratory burst stimulant for neutrophils. Cornea. 1993;12(2):155–160. doi: 10.1097/00003226-199303000-00011. [DOI] [PubMed] [Google Scholar]
- Pfister RR, Haddox JL, Lam KW, Lank KM. Preliminary characterization of a polymorphonuclear leukocyte stimulant isolated from alkali-treated collagen. Invest Ophthalmol Vis Sci. 1988;29(6):955–962. [PubMed] [Google Scholar]
- Pfister RR, Haddox JL, Sommers CI. Alkali-degraded cornea generates a low molecular weight chemoattractant for polymorphonuclear leukocytes. Invest Ophthalmol Vis Sci. 1993;34(7):2297–2304. [PubMed] [Google Scholar]
- Pfister RR, Haddox JL, Sommers CI. Injection of chemoattractants into normal cornea: A model of inflammation after alkali injury. Invest Ophthalmol Vis Sci. 1998;39(9):1744–1750. [PubMed] [Google Scholar]
- Pfister RR, Haddox JL, Sommers CI, Lam KW. Identification and synthesis of chemotactic tripeptides from alkali-degraded whole cornea. A study of n-acetyl-proline-glycine-proline and n-methyl-proline-glycine-proline. Invest Ophthalmol Vis Sci. 1995;36(7):1306–1316. [PubMed] [Google Scholar]
- Pharmaceuticals O. Announcement of the suspension of clinical study in japan using ono-6818 for chronic obstructive pulmonary disease:BUSINESS WIRE. 2002. [Google Scholar]
- Pharmaceuticals O. Announcement of termination of exclusive license agreement on sivelestat with lilly corporation. 2003. [Google Scholar]
- Polgar L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59(2):349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwaite AE, Kang AH. Collagen-and collagen peptide-induced chemotaxis of human blood monocytes. J Exp Med. 1976;143(6):1299–1307. doi: 10.1084/jem.143.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwaite AE, Seyer JM, Kang AH. Chemotactic attraction of human fibroblasts to type i, ii, and iii collagens and collagen-derived peptides. Proc Natl Acad Sci U S A. 1978;75(2):871–875. doi: 10.1073/pnas.75.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privitera S, Prody CA, Callahan JW, Hinek A. The 67-kda enzymatically inactive alternatively spliced variant of beta-galactosidase is identical to the elastin/laminin-binding protein. J Biol Chem. 1998;273(11):6319–6326. doi: 10.1074/jbc.273.11.6319. [DOI] [PubMed] [Google Scholar]
- Rao NV, Marshall BC, Gray BH, Hoidal JR. Interaction of secretory leukocyte protease inhibitor with proteinase-3. Am J Respir Cell Mol Biol. 1993;8(6):612–616. doi: 10.1165/ajrcmb/8.6.612. [DOI] [PubMed] [Google Scholar]
- Rao NV, Wehner NG, Marshall BC, Gray WR, Gray BH, Hoidal JR. Characterization of proteinase-3 (pr-3), a neutrophil serine proteinase. Structural and functional properties. J Biol Chem. 1991;266(15):9540–9548. [PubMed] [Google Scholar]
- Riley DJ, Berg RA, Soltys RA, Kerr JS, Guss HN, Curran SF, et al. Neutrophil response following intratracheal instillation of collagen peptides into rat lungs. Exp Lung Res. 1988;14(4):549–563. doi: 10.3109/01902148809087827. [DOI] [PubMed] [Google Scholar]
- Riley DJ, Kerr JS, Guss HN, Curran SF, Laskin DL, Berg RA. Intratracheal instillation of collagen peptides induces a neutrophil influx in rat lungs. Trans Assoc Am Physicians. 1984;97:290–295. [PubMed] [Google Scholar]
- Rosenblum JS, Kozarich JW. Prolyl peptidases: A serine protease subfamily with high potential for drug discovery. Curr Opin Chem Biol. 2003;7(4):496–504. doi: 10.1016/s1367-5931(03)00084-x. [DOI] [PubMed] [Google Scholar]
- Rubin BK, Druce H, Ramirez OE, Palmer R. Effect of clarithromycin on nasal mucus properties in healthy subjects and in patients with purulent rhinitis. Am J Respir Crit Care Med. 1997;155(6):2018–2023. doi: 10.1164/ajrccm.155.6.9196110. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Giardiniere M, Morain P. Effects of the prolyl endopeptidase inhibitor s 17092 on cognitive deficits in chronic low dose mptp-treated monkeys. Neuropsychopharmacology. 2002;26(2):176–182. doi: 10.1016/S0893-133X(01)00307-4. [DOI] [PubMed] [Google Scholar]
- Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases a and b, collagenases 1 and 2, and increased parenchymal cell death in copd. Chest. 2000;117(3):684–694. doi: 10.1378/chest.117.3.684. [DOI] [PubMed] [Google Scholar]
- Selman M, Cisneros-Lira J, Gaxiola M, Ramirez R, Kudlacz EM, Mitchell PG, et al. Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in guinea pigs. Chest. 2003;123(5):1633–1641. doi: 10.1378/chest.123.5.1633. [DOI] [PubMed] [Google Scholar]
- Selman M, Montano M, Ramos C, Vanda B, Becerril C, Delgado J, et al. Tobacco smoke-induced lung emphysema in guinea pigs is associated with increased interstitial collagenase. Am J Physiol. 1996;271(5 Pt 1):L734–743. doi: 10.1152/ajplung.1996.271.5.L734. [DOI] [PubMed] [Google Scholar]
- Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. 1980;66(4):859–862. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior RM, Griffin GL, Mecham RP, Wrenn DS, Prasad KU, Urry DW. Val-gly-val-ala-pro-gly, a repeating peptide in elastin, is chemotactic for fibroblasts and monocytes. J Cell Biol. 1984;99(3):870–874. doi: 10.1083/jcb.99.3.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior RM, Hinek A, Griffin GL, Pipoly DJ, Crouch EC, Mecham RP. Neutrophils show chemotaxis to type iv collagen and its 7s domain and contain a 67 kd type iv collagen binding protein with lectin properties. Am J Respir Cell Mol Biol. 1989;1(6):479–487. doi: 10.1165/ajrcmb/1.6.479. [DOI] [PubMed] [Google Scholar]
- Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, Pierce JA. The induction of pulmonary emphysema with human leukocyte elastase. Am Rev Respir Dis. 1977;116(3):469–475. doi: 10.1164/arrd.1977.116.3.469. [DOI] [PubMed] [Google Scholar]
- Sepper R, Konttinen YT, Sorsa T, Koski H. Gelatinolytic and type iv collagenolytic activity in bronchiectasis. Chest. 1994;106(4):1129–1133. doi: 10.1378/chest.106.4.1129. [DOI] [PubMed] [Google Scholar]
- Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163(6):2329–2335. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro SD, Senior RM. Matrix metalloproteinases. Matrix degradation and more. Am J Respir Cell Mol Biol. 1999;20(6):1100–1102. doi: 10.1165/ajrcmb.20.6.f151. [DOI] [PubMed] [Google Scholar]
- Shimane T, Asano K, Suzuki M, Hisamitsu T, Suzaki H. Influence of a macrolide antibiotic, roxithromycin, on mast cell growth and activation in vitro. Mediators Inflamm. 2001;10(6):323–332. doi: 10.1080/09629350120102343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasawa Y, Osawa T, Hirashima A. Molecular cloning and characterization of prolyl endopeptidase from human t cells. J Biochem. 1994;115(4):724–729. doi: 10.1093/oxfordjournals.jbchem.a124402. [DOI] [PubMed] [Google Scholar]
- Smith C, Hamerman D. Aic phosphatase in human synovial fluid. Arthritis Rheum. 1962;5:11–14. doi: 10.1002/art.1780050410. [DOI] [PubMed] [Google Scholar]
- Solomon A, Rosenblatt M, Li D, Monroy D, Ji Z, Lokeshwar BL, et al. Doxycycline inhibition of interleukin-1 in the corneal epithelium. Am J Ophthalmol. 2000;130(5):688. doi: 10.1016/s0002-9394(00)00755-8. [DOI] [PubMed] [Google Scholar]
- Stamenkovic I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J Pathol. 2003;200(4):448–464. doi: 10.1002/path.1400. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward WP. Marimastat (bb2516): Current status of development. Cancer Chemother Pharmacol. 1999;43(Suppl):S56–60. doi: 10.1007/s002800051099. [DOI] [PubMed] [Google Scholar]
- Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet. 2005;365(9478):2225–2236. doi: 10.1016/S0140-6736(05)66781-5. [DOI] [PubMed] [Google Scholar]
- Suomalainen K, Sorsa T, Golub LM, Ramamurthy N, Lee HM, Uitto VJ, et al. Specificity of the anticollagenase action of tetracyclines: Relevance to their anti-inflammatory potential. Antimicrob Agents Chemother. 1992;36(1):227–229. doi: 10.1128/aac.36.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland ER, Cherniack RM. Management of chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2689–2697. doi: 10.1056/NEJMra030415. [DOI] [PubMed] [Google Scholar]
- Takizawa H, Desaki M, Ohtoshi T, Kawasaki S, Kohyama T, Sato M, et al. Erythromycin modulates il-8 expression in normal and inflamed human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;156(1):266–271. doi: 10.1164/ajrccm.156.1.9612065. [DOI] [PubMed] [Google Scholar]
- Toide K, Fujiwara T, Iwamoto Y, Shinoda M, Okamiya K, Kato T. Effect of a novel prolyl endopeptidase inhibitor, jtp-4819, on neuropeptide metabolism in the rat brain. Naunyn Schmiedebergs Arch Pharmacol. 1996;353(3):355–362. doi: 10.1007/BF00168640. [DOI] [PubMed] [Google Scholar]
- Toide K, Iwamoto Y, Fujiwara T, Abe H. Jtp-4819: A novel prolyl endopeptidase inhibitor with potential as a cognitive enhancer. J Pharmacol Exp Ther. 1995;274(3):1370–1378. [PubMed] [Google Scholar]
- Toide K, Okamiya K, Iwamoto Y, Kato T. Effect of a novel prolyl endopeptidase inhibitor, jtp-4819, on prolyl endopeptidase activity and substance p- and arginine-vasopressin-like immunoreactivity in the brains of aged rats. J Neurochem. 1995;65(1):234–240. doi: 10.1046/j.1471-4159.1995.65010234.x. [DOI] [PubMed] [Google Scholar]
- Toide K, Shinoda M, Fujiwara T, Iwamoto Y. Effect of a novel prolyl endopeptidase inhibitor, jtp-4819, on spatial memory and central cholinergic neurons in aged rats. Pharmacol Biochem Behav. 1997;56(3):427–434. doi: 10.1016/s0091-3057(96)00238-9. [DOI] [PubMed] [Google Scholar]
- Toide K, Shinoda M, Iwamoto Y, Fujiwara T, Okamiya K, Uemura A. A novel prolyl endopeptidase inhibitor, jtp-4819, with potential for treating alzheimer’s disease. Behav Brain Res. 1997;83(1–2):147–151. doi: 10.1016/s0166-4328(97)86059-7. [DOI] [PubMed] [Google Scholar]
- Toide K, Shinoda M, Miyazaki A. A novel prolyl endopeptidase inhibitor, jtp-4819--its behavioral and neurochemical properties for the treatment of alzheimer’s disease. Rev Neurosci. 1998;9(1):17–29. doi: 10.1515/revneuro.1998.9.1.17. [DOI] [PubMed] [Google Scholar]
- Trifilieff A. Ono-6818 cortech/ono. Curr Opin Investig Drugs. 2002;3(8):1161–1164. [PubMed] [Google Scholar]
- Tsang KW, Ho PI, Chan KN, Ip MS, Lam WK, Ho CS, et al. A pilot study of low-dose erythromycin in bronchiectasis. Eur Respir J. 1999;13(2):361–364. doi: 10.1183/09031936.99.13236199. [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase b or matrix metalloproteinase-9 (mmp-9) Crit Rev Biochem Mol Biol. 2002;37(6):375–536. doi: 10.1080/10409230290771546. [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase b potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades ctap-iii, pf-4, and gro-alpha and leaves rantes and mcp-2 intact. Blood. 2000;96(8):2673–2681. [PubMed] [Google Scholar]
- van Houwelingen AH, Weathington NM, Verweij V, Blalock JE, Nijkamp FP, Folkerts G. Induction of lung emphysema is prevented by l-arginine-threonine-arginine. Faseb J, Online before print. 2008 doi: 10.1096/fj.1007-096230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint P, Libert C. Matrix metalloproteinase-8: Cleavage can be decisive. Cytokine Growth Factor Rev. 2006;17(4):217–223. doi: 10.1016/j.cytogfr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Vanhoof G, Goossens F, Hendriks L, De Meester I, Hendriks D, Vriend G, et al. Cloning and sequence analysis of the gene encoding human lymphocyte prolyl endopeptidase. Gene. 1994;149(2):363–366. doi: 10.1016/0378-1119(94)90177-5. [DOI] [PubMed] [Google Scholar]
- Venalainen JI, Juvonen RO, Mannisto PT. Evolutionary relationships of the prolyl oligopeptidase family enzymes. Eur J Biochem. 2004;271(13):2705–2715. doi: 10.1111/j.1432-1033.2004.04199.x. [DOI] [PubMed] [Google Scholar]
- Vernooy JH, Lindeman JH, Jacobs JA, Hanemaaijer R, Wouters EF. Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with copd. Chest. 2004;126(6):1802–1810. doi: 10.1378/chest.126.6.1802. [DOI] [PubMed] [Google Scholar]
- Villagrasa V, Berto L, Cortijo J, Perpina M, Sanz C, Morcillo EJ. Effects of erythromycin on chemoattractant-activated human polymorphonuclear leukocytes. Gen Pharmacol. 1997;29(4):605–609. doi: 10.1016/s0306-3623(96)00566-6. [DOI] [PubMed] [Google Scholar]
- Voet D, Voet J. Biochemistry. 2. New York: John Wiley and Sons, Inc; 1995. [Google Scholar]
- Wallen EA, Christiaans JA, Jarho EM, Forsberg MM, Venalainen JI, Mannisto PT, et al. New prolyl oligopeptidase inhibitors developed from dicarboxylic acid bis(l-prolyl-pyrrolidine) amides. J Med Chem. 2003;46(21):4543–4551. doi: 10.1021/jm030811o. [DOI] [PubMed] [Google Scholar]
- Walsh DE, Greene CM, Carroll TP, Taggart CC, Gallagher PM, O’Neill SJ, et al. Interleukin-8 up-regulation by neutrophil elastase is mediated by myd88/irak/traf-6 in human bronchial epithelium. J Biol Chem. 2001;276(38):35494–35499. doi: 10.1074/jbc.M103543200. [DOI] [PubMed] [Google Scholar]
- Walter R. Partial purification and characterization of post-proline cleaving enzyme: Enzymatic inactivation of neurohypophyseal hormones by kidney preparations of various species. Biochim Biophys Acta. 1976;422(1):138–158. doi: 10.1016/0005-2744(76)90015-2. [DOI] [PubMed] [Google Scholar]
- Walter R, Shlank H, Glass JD, Schwartz IL, Kerenyi TD. Leucylglycinamide released from oxytocin by human uterine enzyme. Science. 1971;173(999):827–829. doi: 10.1126/science.173.3999.827. [DOI] [PubMed] [Google Scholar]
- Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, et al. A novel peptide cxcr ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12(3):317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- Weissmann G. Lysosomal mechanisms of tissue injury in arthritis. N Engl J Med. 1972;286(3):141–147. doi: 10.1056/NEJM197201202860307. [DOI] [PubMed] [Google Scholar]
- Weissmann G, Spilberg I. Breakdown of cartilage proteinpolysaccharide by lysosomes. Arthritis Rheum. 1968;11(2):162–169. doi: 10.1002/art.1780110206. [DOI] [PubMed] [Google Scholar]
- Wilson R. Bacteria and airway inflammation in chronic obstructive pulmonary disease: More evidence. Am J Respir Crit Care Med. 2005;172(2):147–148. doi: 10.1164/rccm.2504004. [DOI] [PubMed] [Google Scholar]
- Wright JL, Churg A. Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. Am J Physiol. 1995;268(1 Pt 1):L17–20. doi: 10.1152/ajplung.1995.268.1.L17. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Kado K, Matsubara F, Koriyama N, Kaneto H, Tsura D. Specific inhibitors for prolyl endopeptidase and their anti-amnesic effect. J Pharmacobiodyn. 1987;10(12):730–735. doi: 10.1248/bpb1978.10.730. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Ogita K, Walter R, Koida M, Tsuru D. Post-proline cleaving enzyme. Synthesis of a new fluorogenic substrate and distribution of the endopeptidase in rat tissues and body fluids of man. Biochim Biophys Acta. 1979;569(2):184–192. doi: 10.1016/0005-2744(79)90053-6. [DOI] [PubMed] [Google Scholar]
- Yoshioka A, Betsuyaku T, Nishimura M, Miyamoto K, Kondo T, Kawakami Y. Excessive neutrophil elastase in bronchoalveolar lavage fluid in subclinical emphysema. Am J Respir Crit Care Med. 1995;152(6 Pt 1):2127–2132. doi: 10.1164/ajrccm.152.6.8520785. [DOI] [PubMed] [Google Scholar]
- Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, et al. Neutrophil elastase inhibition in acute lung injury: Results of the strive study. Crit Care Med. 2004;32(8):1695–1702. doi: 10.1097/01.ccm.0000133332.48386.85. [DOI] [PubMed] [Google Scholar]
- Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, et al. Inducible targeting of il-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106(9):1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]