

Abstract
Multiple myeloma (MM) is the second most common hematological malignancy and is characterized by the aberrant proliferation of terminally differentiated plasma B cells with impairment in apoptosis capacity. Particularly, osteolytic bone diseases and renal failure resulting from hyperparaproteinemia and hypercalcemia have been the major serious sequelae that are inextricably linked with MM tumor progression. Despite the introduction of new treatment regimens, problematic neuropathy, thrombocytopenia, drug resistance and high MM relapse rates continue to plague the current therapies. New chemical agents are in development on the basis of understanding several signaling pathways and molecular mechanisms like tumor necrosis factor-α, proteasome, PI3K and MARKs. This review focuses on the most recent patents and clinical trials in the development of new medicine for the treatment of multiple myeloma. Furthermore, the important signaling pathways involved in the proliferation, survival and apoptosis of myeloma cells will be discussed.
Background
Multiple myeloma (MM) is the second most common progressive hematological malignancy in the USA, and is characterized by abnormal monoclonal plasma cells accumulated in the bone marrow and destructive bone lesions [1]. In the USA alone, there were 10,710 deaths related to MM and 21,700 new cases in 2012. By 2013, new MM cases rose to 22,350 [2]. MM comprises 1% of malignant tumors and is the second most common form of blood cancer following lymphomas [3]. It is treated as an elderly disease, as the median age of affected individuals is 70 in the USA and 72 in Europe [4]. MM remains an incurable disease, with a median survival of 3–4 years after conventional treatments [5]. Commonly observed in advanced MM patients are excess bone marrow plasma cells and monoclonal protein, hypercalcemia, anemia, osteolytic bone lesions, renal disease, immunodeficiency and peripheral neuropathy [6,7].
In the 1960s, the chemotherapeutic agent melphalan and corticosteroid prednisone were adopted to prolong survival of MM patients. In the 1980s, it was determined that MM evolves from premalignant stages termed monoclonal gammopathy of undetermined significance and smoldering MM [8,9]. As such, the selection of treatment became dependent on the stage of MM experienced by the patient. By the early 1990s, the standard MM treatment combined high-doses of chemotherapy, followed by autologous hematopoietic stem cell transplantation [10]. Unfortunately, as it is commonly known, such chemotherapies kill both tumor cells and normal cells alike, in this case leading to bone marrow depression and immunosuppression. More recently, however, research has uncovered a new understanding of the bone marrow micro-environment and characteristic molecular mechanisms, resulting in a paradigm shift for the treatment of MM from nonspecific chemotherapy to novel drugs that target bone marrow microenvironments [11]. Since 1998, a combined regimen of thalidomide, bortezomib and lenalidomide has been widely used to treat MM [12].
Among the new medications, bortezomib, approved by the US FDA in 2003, is the first representative synthetic proteasome inhibitor that can inhibit tumor survival pathways and prevent degradation of pro-apoptotic proteins for the treatment of newly diagnosed MM [13,14]. Unfortunately, bortezomib has low oral bioavailability and severe toxic side effects such as diarrhea, fatigue and insomnia that have restricted the dosage [15,16]. Thalidomide is among the first-in-class immunomodulatory drugs (IMiDs) for the treatment of all stages of MM and was approved by the FDA in 2006 to treat newly diagnosed MM [17]. The anticancer mechanisms of IMiDs include inhibition of angiogenesis and the secretion of cytokines, immuno-modulation of regulatory T cells, disruption of interactions between plasma cells and the bone marrow microenvironment, as well as direct antitumor effects [18,19]. Thalidomide, however, is associated with toxicities such as thrombocytopenia and side effects that include constipation and neuropathy [20]. Lenalidomide, a more potent and less toxic drug than thalidomide, was adopted in 2006 as a common treatment in combination with dexamethasone for MM patients who have received one prior therapy [21,22].
Although still limited by unwanted side effects and poor long-term efficacy, the newer agents are designed, for the first time, to modulate pathways that most directly influences the progression of MM. Researchers have been encouraged to develop new treatments that also target the bone microenvironment. Indeed, this is the current trend, as both patents and literature citations from SCI-FINDER related to ‘MM’ have steadily risen in recent years (Figure 1).
Figure 1.

Frequency of patents and literature citations related to ‘multiple myeloma’ from 2005 to 2013.
Pathophysiology of MM
As a tumors form in postgerminal mature B cells, MM is regulated by expression of various cytokines and signal transduction molecules [23]. Released cytokines, chemokines and growth factors from myeloma cells interact with the microenvironment, causing auto-crine and paracrine secretion of insulin-like growth factor 1, interleukin-6 (IL-6), fibroblast growth factor receptor 3 (FGFR3), vascular endothelial growth factor (VEGF), tumor necrosis factor-α (TNF-α) and transforming growth factor-β [24]. The bone marrow microenvironment comprises several cell types such as hematopoietic cells, stromal cells, fibroblasts, bone marrow stromal cells (BMSCs), osteoblasts, osteoclasts, endothelial cells and immune cells, as well as noncellular components that include extracellular matrix, cytokines, growth factors and chemokines [23]. The onset of MM is due to the accumulation of abnormal plasma cells in the bone marrow that adhere to extracellular matrix and BMSCs, which play a key role in the pathogenesis of MM [25]. The adherence of MM cells to BMSCs increases induction of the NF-κB signaling pathway that up-regulates the expression of IL-6, which is itself predominant in the development of MM [26]. IL-6 stimulates proliferation and inhibits apoptosis, which in turn enhances drug resistance in myeloma cells [27]. IL-6 can induce the autocrine and paracrine secretion of VEGF, the presence of which can further up-regulate the induction of IL-6 and trigger the MM cells migration by β1 inte-grin- and phosphatidylinositol 3-kinase-dependent PKC α [28–30].
Over-activation of the Ras/Raf/MEK/ERK/MAPKK pathway significantly contributes to the proliferation and anti-apoptotic characteristics of MM cells [31]. JAK/STAT3 and PI3K/Akt3 signaling cascades further regulate cytokine-induced survival and inhibition of apoptosis in MM cells. IL-6 triggers the phosphorylation of STAT3 through JAK1, and further stimulates anti-apoptotic genes Bcl-2, Bcl-xL and Mcl-1 downstream [32–34]. Accordingly, inhibition of the JAK/STAT3 pathway is correlated with enhanced apoptosis of MM cells [35]. Cytokines like IGF and VEGF can activate PI3K, stimulating Akt, which mediates various biological processes including increased expression of cyclin D and NF-κB that enhance tumor survival [36]. After CDK-cyclin D functions are carried out, cyclins are polyubiquitinated and degraded via the ubiquitin-proteasome signaling pathway, which is responsible for regulating many cellular events including DNA replication, transcription activation, and cell cycle regulation and apoptosis as mentioned [37]. For an illustration, Figure 2 summarizes all MM patent drugs discussed herein and their associated MM targets. With identification of these putative mechanisms, scientists and clinical researchers have developed various novel agents in an effort to improve the standard treatment of MM.
Figure 2. The published patents related to multiple myeloma treatments and their associated targets.

APRIL: A proliferation-inducing ligand; BMSC: Bone marrow stromal cells; CB2: Cannabinoid receptor 2; CD40: TNF receptor superfamily member 5; CXCR4: Chemokine receptor type 4; DKK1: Dickkopf-related protein 1; ERK: Extracellular signal-regulated kinases; FGFR3: Fibroblast growth factor receptor 3; HGF: Hepatocyte growth factor; Hsp90: Heat shock protein 90; JAK: Janus kinase; MAPK: Mitogen-activated protein kinase; MEK: Mitogen-activated protein kinase kinase; MIP-1α: Macrophage inflammatory protein-1α; MM cell: Multiple myeloma cell; mTOR: Mammalian target of rapamycin; PI3K: Phosphatidylinositide 3-kinases; PPARγ: Peroxisome proliferator-activated receptor gamma; PTEN: Phosphatase and tensin homolog; SDF-1α: Stromal cell-derived factor 1; STAT3: Signal transducer and activator of transcription 3; UPR: Unfolded protein response; VCAM: Vascular cell adhesion protein 1; VLA4: Very late antigen-4.
New MM drugs & patents under development or in clinical trials
TNF-αinhibitors
TNF-α is a multifunctional cytokine associated with various biological activities [38]. TNF can up-regulate IL-6 level under tumoral environment and trigger the JAK/STAT pathways, leading to the activation of NF-κB [39]. It was demonstrated that TNF-α acts as a tumor-promoting factor, which is related to transformation, proliferation, angiogenesis and metastasis in tumor cells [40]. Studies have shown that the concentrations of BAFF and APRIL, two members of the TNF-α family, are enhanced in MM patients [41]. In light of this, the TNF receptor is treated as a potential drug-gable target for treatment of MM.
Pomalidomide (Table 1), a derivative of thalidomide with oral bioavailability, was approved by the FDA in February 2013 to treat relapsed and refractory MM patients who had received at least two prior therapies. As a novel IMiD, pomalidomide inhibits the interactions between myeloma cells and bone marrow microenvironment, and decreases the production of TNF-α and other cytokines such as IL-6 [42]. Studies showed that pomalidomide potentiated the inhibitory effects on its target cytokines better than thalidomide, which resulted in improved therapeutic effects [43]. However, pomalidomide can potentially cause severe embryo-fetal toxicity and venous thromboembolism. Several pomalidomide-related clinical studies are under way, including those evaluating the safety of pomalidomide in combination with low dose dexamethasone, with or without bortezomib, in patients with relapsed and refractory MM (Phase 3) [44].
Table 1.
Summary of antimultiple myeloma clinical trial drugs.
| Drug | Structure | Multiple myeloma target | Company | Clinical trial |
|---|---|---|---|---|
| Pomalidomide |
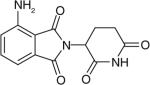
|
Tumor necrosis factor-α | Celgene Corporation | Phase 1–3 |
| Sorafenib |
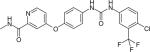
|
Vascular endothelial growth factor R2/ Raf-kinase | Bayer/ONYX pharmaceuticals | Phase 2 |
| Cabozantinib |
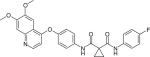
|
Vascular endothelial growth factor R2/MET | Exelixis, Inc. | Phase 1–2 |
| Atiprimod |
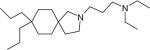
|
Vascular endothelial growth factor | Callisto Pharmaceuticals | Phase 1–2 |
| CHIR-258 |
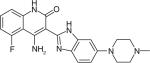
|
Multiple receptor tyrosine kinases | Novartis Pharmaceuticals | Phase 2 |
| Carfilzomib |
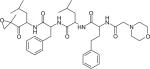
|
Proteasome | ONYX pharmaceuticals | Phase 1–3 |
| Oprozomib (ONX-0912) |
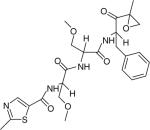
|
Proteasome | ONYX pharmaceuticals | Phase 1–2 |
| MLN9708 |
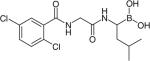
|
Proteasome | Millennium | Phase 1–3 |
| Siltuximab | Monoclonal antibody | Interleukin-6 | Janssen Research & Development, LLC | Phase 2 |
| Daratumumab | Monoclonal antibody | CD38 | Genmab | Phase 1–2 |
| MOR03087 | Monoclonal antibody | CD38 | MorphoSys | Phase 1–2 |
| BI-505 | Monoclonal antibody | CD54 | BioInvent | Phase 2 |
| Panobinostat (LBH589) |
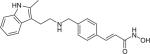
|
Histone deacetylase i | Novartis | Phase 1–2 |
| ACY-1215 |
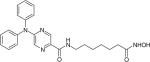
|
Histone deacetylase6 | Acetylon Pharmaceuticals | Phase 1–2 |
| CC-223 | NA | Mammalian target of rapamycin | Celgene Corporation | Phase 1–2 |
| MDX-1338 | Monoclonal antibody | CXCR4 | Mayo Clinic cancer center | Phase 1 |
| BHQ880 | Monoclonal antibody | DKK1 | Novartis | Phase 2 (closed) |
Celgene Corporation patented 3-(4-amino-1-oxo-1,3-dihydro-isoindol-2-yl)-piperidine-2,6-dion (lenalidomide) (P-15, Table 2), which is an orally bio-available immunomodulatory compound. Lenalidomide can promote the degradation of TNF-α mRNA. The IC50 of this compound for inhibiting the production of TNF- α in peripheral blood mononuclear cells under the circumstance of LPS-stimulation was 100 nM, representing a marked improvement over thalidomide (194 μM). In preclinical studies, lenalidomide can efficiently overcome drug resistance in MM cells in vitro and in vivo [45]. Lenalidomide as an oral capsule (Revlimid®) was approved by the FDA in 2006 in combination with dexamethasone for patients with MM who have received one prior therapy [21]. The drug is currently under clinical trials to treat different stages of MM alone or in combination with other drugs [46].
Table 2.
Summary of antimultiple myeloma compounds based on selected patents.
| Number | Patent | Multiple myeloma agents (targets) | Assignee | Year | Ref. |
|---|---|---|---|---|---|
| P-1 | WO061199A1 | Staurosporine derivatives (FGFR3/c-fos/Ras) | Novartis Pharma Gmbh | 2006 | [47] |
| P-2 | WO060676A1 | NPI-0052 (Salinosporamide A) (proteasome) | Dana-Farber Cancer Institute | 2006 | [48] |
| P-3 | WO118953A2 | 17-allylamino- 17-demethoxy-geldanamycin (17-AGG) or 17-amino geldanamycin (17-AG) (Hsp90) | Kosan Biosciences, Inc. | 2006 | [49] |
| P-4 | WO116185A2 | MDX-1338 (CXCR4) | CBR institute for biomedical research, Inc | 2006 | [50] |
| P-5 | US079461A1 | P38 inhibitor (MAPK) | Scios, Inc. | 2006 | [51] |
| P-6 | WO026251A2 | 2-aminoarylthiazoles 2-aminoaryloxazoles (c-Kit/ FGFR3) |
AB SCI- ENCE | 2007 | [52] |
| P-7 | WO059078A1 | Dasatinib (multi-kinase) | Dana-Farber Cancer Institute | 2007 | [53] |
| P-8 | EP-1443927-B1 | Carboline derivatives (NF-κB) | Millennium pharmaceuticals, Inc. | 2007 | [54] |
| P-9 | US7196105 | Curcumin (NF-κB) | Research Development Foundation | 2007 | [55] |
| P-10 | US7211252 | Antagonists for Alpha4 intergrin binding agents (VCAM-1, VLA-4) | University of Texas System | 2007 | [56] |
| P-11 | WO002553 A1 | Erastin analogues (Ras) | Prolexys Pharmaceuticals, Inc. | 2009 | [57] |
| P-12 | WO034414A1 | 2,3-dihydroimidazo[1,2-c] quinazoline (PI3K/Akt) | Bayer Schering PharmaAktiengesellschaft | 2010 | [58] |
| P-13 | WO138141A1 | [(lR)-I-[[(2S,3R)-3-hydroxy-2-[6-phenyl-pyridine-2- carbonyl)amino]-1-oxobutyl]amino]-3-methylbutylboronic acid (proteasome) | Cephalon, Inc. | 2010 | [59] |
| P-14 | EP-1684805-B1 | CHIR-12.12, CHIR-5.9 (CD40 monoclonal antibodies) | Novartis Vaccines and Diagnostics Inc. | 2010 | [60] |
| P-15 | US7968569 | 3-(4-amino-1-oxo-1,3-dihydro-isoindol-2-yl)-piperidine-2,6-dion (TNF- α) | Celgene Corporation | 2011 | [45] |
| P-16 | US8012477 | IL-17 binding molecule (IL-17) | Novartis AG and Novartis Pharma GmbH | 2011 | [61] |
| P-17 | US8268807 | (E)-3-hydroxy-21-[2'-(N,N-dimethylamino)ethoxy]-19-norpregna-1,3,5(10),17(20)-tetraene (IL-6) | SRI International | 2012 | [62] |
| P-18 | WO136732 A1 | Masitinib (c-Kit/ FGFR3/ PDGFR) | AB SCI- ENCE | 2012 | [63] |
| P-19 | WO134915A1 | Ezatiostat (GSTP1-1) | Telik, Inc. | 2012 | [64] |
| P-20 | WO149602A1 | N-(cyanomethyl)-4-[2-[[4-(4-morpholinyl)phenyl]amino]-4-pyrimidinyl]-benzamide (CYT387) (JAK) | YM BioSciences Australia Pty Ltd | 2012 | [65] |
| P-21 | WO022919A1 | 2-((3,4-bis(benzyloxy)benzyl)amino) ethan-1-ol (XRK3) (p62) | Xie, X-Q., et al. University of Pittsburgh | 2013 | [66] |
| P-22 | US0040979 | CCR1 antagonists (CCR1) | Millennium Pharmaceuticals,Inc | 2013 | [67] |
| P-23 | US0172388A1 | phenylacetylamide (PAM) (CB2) | Xie, X-Q., et al. University of Pittsburgh | 2013 | [68] |
CD40 is a member of TNF-α superfamily, having relatively high expression on the surface of MM cell lines and primary MM cells [69]. Inhibition of the CD40/CD40L interaction can have antimyeloma activity by disrupting key signal transduction pathways in MM or BMSCs. Novartis Vaccines and Diagnostics Inc. filed a patent on anti-CD40 monoclonal antibodies CHIR-12.12 and CHIR-5.9 for the treatment of MM (P-14, Table 2) [60]. The agents have high affinity for CD40 and suppress CD40-ligand mediated survival signals in MM cells [60]. Studies indicated that CHIR-12.12 (5 μg/ml) can inhibit the PI3K/Akt, NF-κB and extra-cellular signal-regulated kinase activation induced by CD40L (5 μg/ml) [70]. CHIR-12.12 also decreased the adherence of MM cell to BMSCs induced by CD40L, blocking the induction of IL-6 and VEGF [70].
Receptor tyrosine kinases inhibitors
Receptor tyrosine kinases (RTKs) are transmembrane proteins containing an extracellular lectin binding domain and an intracellular catalytic domain, forming a class of targets for the treatment of MM [71]. Several signaling molecules such as MAPKs and PI3K that are responsible for mediating tumor cell growth, progression and metastasis are downstream of RTKs [72]. Of 91 RTKs, 14 have been found to contain mutations in cases of MM, including insulin-like growth factor 1R, epidermal growth factor receptor, neurotrophic tyro-sine kinase receptor 1 and neurotrophic tyrosine kinase receptor 2 [73]. These mutations in genes of RTKs highlight their importance in the pathogenesis of MM [73].
Among MM tumors, approximately 15 to 20% are characterized by t(4;14)(p16.3;q32) translocation, which is associated with up-regulation of FGFR3 and myeloma SET domain protein [74]. FGFR3 belongs to FGFR, which is a member of a family of highly related transmembrane RTKs [75]. FGFR3 is associated with the intracellular signaling pathways such as Ras/Raf/MAPK and PI3K/Akt/mTOR, and has effects on tumor cell proliferation and migration [74]. Novartis Pharma Gmbh filed a patent for staurosporine derivatives in 2006 (P-1, Table 2) [47]. Staurosporine derivatives including PKC412 are potential agents for the treatment of curative and prophylactic MM, which target the FGFR3 or Ras signaling pathway and c-fos transcription. c-fos proto-oncogene is a member of the AP-1 family of transcription factors, which play a key role in nuclear response to stimulatory signals that regulate the proliferation and differentiation of cells [76]. PKC412 inhibits the activity of Ras and FGFR3 or c-fos in concentrations ranging from 0.01 to 50 μM, showing significant therapeutic effects [47]. Studies suggested that PKC412 also suppresses Akt kinase activation and induces apoptosis in myeloma cell lines (RPMI8226S, U266, MM1S and MMIR) [77].
2-amino aryl thiazoles and 2-amino aryl oxazoles are dual c-Kit/FGFR3 inhibitors (P-6, Table 2), making them candidates for the treatment of relapsed or refractory MM over-expressing FGFR3. Studies showed that these agents inhibited enzymatic activity and the phosphorylation of FGFR3 with an IC50 below 0.1 and 2 μM, respectively, in MM cell lines expressing FGFR3 [52]. Studies indicated that 30% of MM patients have relatively high expression of c-Kit [78]. It was verified that c-Kit expression is connected to survival pathways, which can modulate MM cell death, suggesting that blocking c-Kit is an ideal strategy to accelerate the death of MM cells [78]. The agents inhibited the proliferation of cells, which expressed mutations of c-Kit in juxtamembrane domain with an IC50 of less than 0.1 μM [52].
VEGF, and its receptors VEGFR1, VEGFR2 and VEGF3, have been studied as a target to treat MM, since they are not only essential for angiogenesis but also for triggering growth, survival and migration of MM cells though the MEK/MAPK and PI3K/Akt pathways [79,80]. Elevated levels of VEGF secreted by MM cells will enhance the production of IL-6, promoting the development of MM [81]. Sorafenib (Table 1), a drug initially approved for the treatment of renal and hepatocellular cancer by the FDA in 2007, is a small molecular dual inhibitor of VEGFR 2 and Raf-kinase with oral bioavailability [82]. Administration of sorafenib has been shown to decrease the concentration of plasma VEGF after 8 weeks in patients [83]. Moreover, sorafenib specifically targets Raf kinase by binding to its adenosine triphosphate binding site [84]. A Phase 2 study showed that sorafenib treatment had significant positive effects on two patients among a sample of 11 with relapsed or refractory MM [85].
Cabozantinib (Table 1), developed by Exelixis, Inc., is a small molecule VEGFR2 and MET inhibitor. The signaling pathway of MET is functionally connected to VEGF [86]. The multi-RTK inhibitor cabozantinib is currently under an open label Phase 1–2 clinical trial to study its safety and efficacy for patients with relapsed or refractory myeloma [87].
Atiprimod (Table 1) is a small-molecule drug, belonging to the azaspirane class of cationic amphiphilic drugs with orally bioavailability. It is capable of reducing the production of VEGF and inhibiting the activation of STAT3, which suggests that it may exhibit potent anti-proliferative and anti-angiogenic activities [88]. A Phase 1–2 clinical study is ongoing to identify the maximum tolerated dose and safety of it in refractory or relapsed MM patients by Callisto Pharmaceuticals [89].
CHIR-258 (Dovitinib) (Table 1) is a benzimidazolequinoline that targets multiple RTKs including FGFR1/3 and VEGF receptors VEGFR1/2, PDGFR, c-kit and Flt3 that are associated with the proliferation of solid MM tumors [90,91]. CHIR-258 can be considered a potent small molecular antitumor drug with oral bioavailability [92]. Preclinical studies demonstrated that daily administration of CHIR-258 suppressed the FGFR3-mediated transduction in KMS-11-luc human MM cells in vivo, resulting in significant inhibition of KMS-11-luc tumor growth [92]. The Phase 2 clinical trial to study the safety and efficacy of CHIR-258 for relapsed or refractory MM patients, who are with or without t(4;14) chromosomal translocation, was completed in February 2013 [93].
Dasatinib (P-7, Table 2) is a multi-targeted kinase inhibitor with oral activity, exhibiting inhibitory effects on BCR-ABL, SRC, c-KIT and PDGF-R and ephrin receptor kinases [53]. During preclinical studies, the IC50 of dasatinib was found to be within 100 nM in eight out of 15 MM cell lines [53]. Dasatinib is currently in Phase 2 clinical trials for relapsed or plateau phase MM. The patent suggests a combination use of dasatinib and at least one other anti-MM agent such as dexamethasone, bortezomib and 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor [53].
Masitinib (P-18, Table 2) is a tyrosine kinase inhibitor that selectively targets c-Kit/FGFR3/PDGFR [94]. It shows no kinase-associated cardiotoxicities at therapeutic doses, making it a much safer drug compared with other kinase-targeted drugs [63,95–96]. It was reported that the inhibition effects of masitinib on PDGF-BB-mediated proliferation and PDGFR-α tyrosine phosphorylation were in the range of 300 ± 5 nM (IC50) [63]. It can inhibit the proliferation of two t(4;14) cell lines, LPI and NCI-H929, by 50% at the dose of 2 μM [63]. Data showed that the combination of masitinib and dexamethasone could decrease the proliferation of MM cell lines in vitro [63]. Clinical studies that analyzed the combined administration of masitinib, dexamethasone plus bortezomib demonstrated that the combination therapy is safe and effective for treatment of early stage or first relapsed MM, as evidenced by median progression-free survival and overall survival [63].
Proteasome inhibitors
Proteasomes are multi-enzyme complexes that are responsible for the dysfunctional protein clearance and cell homeostatic maintenance [96]. Tumor cells are sensitive to proteasome inhibitors, which trigger anti-proliferation and pro-apoptotic effects [16]. As mentioned previously, bortezomib is a representative proteasome inhibitor by reversibly blocking the chymotrypsin-like activity of the 20S subunit of the proteasome with limited oral bioavailability and severe side effects [16]. This led to the assessment of other proteasome inhibitors with a more favorable toxicity profile and improved compliance of patients [97]. Carfilzomib (Table 1) is an irreversible, second-in-class proteasome inhibitor for intravenous administration developed by Onyx Pharmaceuticals, Inc. [98]. It was approved by the FDA in July 2012 for the treatment of MM patients who have received at least two prior therapies [99]. The net sales have exceeded US$62M for 2012. Through October 2012, approximately 25% of the estimated 10,000 to 15,000 patients suffering from with third-line or later MM in the USA have been treated with this drug [100]. Compared with bortezomib, carfilzomib targets to chymotrypsin-like proteasome with greater selectivity and less neurotoxicity [101,102]. Clinical trials administering carfilzomib in combination with other agents to cure MM have begun [103].
Oprozomib (ONX-0912) (Table 1), a tripeptide epoxy ketone with oral activity, is now being tested in a Phase 1–2 clinical trial study by Onyx Pharmaceuticals, Inc. [104]. Its anti-MM function is associated with irreversible and specific inhibition of the chymotrypsin-like activities of the proteasome [105]. Oprozomib also induces the activation of caspase-8, caspase-9, caspase-3 and poly (ADP-ribose) polymerase, resulting in a longer duration of effect [16,101].
Another proteasome inhibitor, MLN9708 (Table 1), is an orally bioavailable agent currently in Phase 3 clinical study for the treatment of different stages of MM alone or in combination with other drugs by Millennium Pharmaceuticals, Inc. [106]. Phase 1 and 2 clinical trials of MLN9708 indicated that it was well tolerated and had clinical efficacy with some patients, achieving significant response to the therapy especially at high doses [107]. MLN9708 has greater PDs and PK effects in tissues when compared with bortezomib [108].
Compound [(lR)-I-[[(2S,3R)-3-hydroxy-2-[6-phenyl-pyridine-2- carbonyl)amino]-I-oxobutyl]amino]-3-methylbutylboronic acid (P-13, Table 2), is a reversible proteasome inhibitor in the peptide boronic acid class with oral bioavailability [59]. It has antitumor function in primary MM plasma cells in vitro and in RPMI8226 xenografts in vivo [59]. This compound shares the same mechanisms of action as bortezomib, inhibiting the chymotrypsin-like activity with little inhibition of the trypsin- and caspase-like activity [59]. When used in combination with bortezomib or melphalan, apoptosis was induced in MM cells. The combination can inhibit myeloma tumor growth without cytotoxicity to the normal peripheral blood mono-nuclear cells. The combination can prevent the growth of bortezomib-sensitive LAGκ-1A tumors and significantly delay the progression of bortezomib-resistant LAGκ-1A tumors [59].
NPI-0052 (salinosporamide A) (P-2, Table 2) is a small molecule proteasome inhibitor with oral bio-availability. NPI-0052 can inhibit chymotrypsin-, caspase- and trypsin-like activities of human erythrocyte 20S proteasomes, resulting in apoptosis in various MM cell lines including RMPI-8226, OPM2 and U266 [48]. NPI-0052 can also induce apoptosis in cell lines that are resistant to dexamethasone, bortezomib and thalidomide [48,109]. NPI-0052 is currently under Phase 1 clinical trials by Triphase Research and Development I Corporation [48].
Heat shock protein inhibitor
The molecular chaperone heat shock protein 90 kDa (Hsp90) is an attractive target for the treatment of MM. It can interact with a variety of proteins such as erbB2, raf-1 and Akt, which are involved in several functional signaling pathways that govern cell cycle progression and apoptosis [110]. 17-AAG, a semi-synthetic analog of the naturally occurring compound geldanamycin, and 17-AG, a biologically active geldanamycin derivative (P-3, Table 2), were shown to exhibit inhibitory effects on Hsp90 and further induced cell death [49]. When U266 and primary MM cells were treated with 17-AAG, apoptosis increased compared with control cells [111]. 17-AGG has been studied for its anti-MM activity in vivo using a model of diffuse Green Fluorescent Protein positive MM lesions in SCID/NOD mice [112]. It was shown that the treatment significantly enhanced the median overall survival [112]. 17-AGG is capable of down-regulating Bcl-2, Mcl-1 and Akt when analyzed anti-apoptotic Bcl-2 family proteins and Akt in MM cells incubated with 17-AGG [111].
Surface antigen monoclonal antibodies
IL-6 mediates autocrine and paracrine growth of MM cells [113]. IL-6 triggers tyrosine kinase JAK1, JAK2 and TYK2, leading to the phosphorylation of signal transducers, STAT1 and STAT2 [114]. It also can activate Ras/Raf/MAPK and PI3K/Akt signaling pathways [115]. Therapeutic strategies targeting IL-6 are under development. Siltuximab (Table 1) is a chimeric, human-murine immunoglobulin monoclonal antibody, which can bind to human IL-6 with high affinity and specificity [116]. Developed by Janssen Research & Development, LLC, siltuximab effectively blocks the IL-6/IL-6R/gp130 signal transduction pathway [116,117]. A Phase 2 trial of siltuximab in high-risk smoldering MM has been launched.
(E)-3-hydroxy-21-[2'-(N,N-dimethylamino) ethoxy]-19-norpregna-1,3,5(10),17(20)-tetraene (P-17, Table 2) has an inhibitory effect on IL-6-induced proliferation in RPMI-8226 and U266 cell lines, as claimed in a patent filed by SRI International [62]. It can also down-regulate p-STAT3 induced by IL-6 [62].
Studies have supported that CD38 is expressed at high levels in all malignant MM cells, suggesting that it is a potential therapeutic antibody target for the treatment of MM [118,119]. A human mAb (daratumumab) (Table 1) is currently under clinical trial Phase 1–2 for MM. Daratumumab is a human lgG1 CD38 mAb, which is capable of binding to a unique epitope of CD38 and kills tumor cells via multiple mechanisms, including anti-tumoral antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity [120]. MOR03087 is an anti-CD38 monoclonal antibody, which can bind to CD38 on CD38-positive tumor cells specifically, triggering antibody-dependent cellular cytotoxicity and ultimately cell death [121]. MOR03087 is under an open-label Phase 1–2 clinical trial for its safety and dosing study.
The intercellular adhesion molecule-1 (CD54) is known to be involved in multiple adhesion-dependent leukocyte interactions and immune functions [122]. BI-505 is a humanized immunoglobulin G1 monoclonal antibody that directly targets CD54 (Table 1). It can bind to CD54 with high selectively, leading to the hyper-cross-linking-induced apoptosis and decreased proliferation of tumor cells expressing ICAM-1 [123]. It is currently under Phase 2 clinical trial study to explore its effects on smoldering MM.
Interleukin-17 (IL-17) is a family member of new cytokines that is known to induce expression of several chemokines and cytokines such as IL-6 and ICAM-1 [124]. It was suggested that IL-17 is an important target for the treatment of MM, since molecules that bind to IL-17 are useful in inhibiting the growth of certain solid and hematological tumors [125]. In 2011, Novartis AG and Novartis Pharma GmbH put a patent on record to illustrate a IL-17 binding molecule (P-16, Table 2) [61].
Histone deacetylase inhibitors
Inhibitors of histone deacetylase (HDAC) enzymes can be classified as novel anticancer agents. It has been shown that HDAC inhibitors are responsible for regulating gene expression, which can influence apoptosis and cell cycle progression in cancer cells [126]. Panobinostat (LBH589) (Table 1) is a type of HDAC inhibitor with oral bioavailability that induced apoptosis in cancer cells [127]. It further demonstrated antitumor activity in solid and hematological malignancies in preclinical studies and is currently in Phase 1–2 clinical trials for MM [85,126]. Some researches have reported, however, that panobinostat is nonselective, targeting classes I, II, and IV HDACs with significant toxicity and negative side effects such as gastrointestinal issues and fatigue [84].
ACY-1215 is an orally bioavailable HDAC6 inhibitor (Table 1). Since ACY-1215 selectively inhibit class II HDACs, it has reduced toxic effects on normal cells [128]. Panobinostat and ACY-1215 are currently under clinical trials alone, or in combination with other drugs such as bortezomib and dexamethasone, for MM patients.
PI3K/Akt inhibitor
Enhanced upstream PI3K activation of Akt commonly occurs in late-stage MM [129]. Evidence has shown that inhibition of the PI3K-Akt pathway has potential in preclinical studies of MM, making it a novel target for treatment [36]. 2,3-dihydroimidazo [1,2-c] quinazoline compounds (P-12, Table 2) have potential anti-MM activity via the PI3K/Akt pathway, both as a sole agent or in combination with other active ingredients. Bayer Schering Pharma Aktiengesellschaft filed a patent for the compound, noting that its IC50 ranged from 3–100 nM upon inhibition of nine MM cell lines [58].
Mammalian target of rapamycin inhibitor
The mammalian target of rapamycin (mTOR) kinase is responsible for tumor cell proliferation, anti-apoptosis and pro-angiogenesis through regulating the expression of multiple proteins [130]. mTOR is downstream of the PI3K/Akt pathway and has been evaluated for anti-MM drug discovery [131]. CC-223 (Table 1) is an orally available mTOR inhibitor, resulting in the induction of tumor cell apoptosis and decreased proliferation of tumor cells. A Phase 1–2 study began in July 2010 to assess its safety, PKs and efficacy for MM [132].
Ras inhibitor
Mutations of Ras occur in nearly 50% of MM cases and it is a potent activator of multiple downstream signaling pathways like MEK/ERK [133]. Erastin analogues (P-19, Table 2) patented by Prolexys pharmaceuticals Inc. can selectively kill cancer cells that have elevated Ras pathway activity and have no effect on normal cells without elevated Ras activity [57]. Studies showed that 34 of 46 MM cell lines were sensitive to the agent with IC50 values of <300 nM for 24 of them [57]. The agent was even effective for the treatment of MM cells resistant to conventional and novel anti-MM agents, and further overcame the protective effect of IL-6 [57].
MAPK inhibitor
A patent for a p38 mitogen-activated protein kinase (MAPK) inhibitors has been put on record (P-5, Table 2) [51]. p38/MAPK is a member of the MAPK family and plays a crucial role in MM as an important regulator of the release of IL-6 and VEGF in BMSCs induced by cytokines and inflammation in the bone marrow microenvironment, enhancing MM cell survival [25]. A potential treatment for MM could thus utilize p38 MAPK inhibitors alone or in combination with other agents. The structures of the p38 MAPK inhibitors indicate that they are derivatives of indole-type compounds containing a mandatory substituent [51,134]. The agents can fully suppress p38 MAPK activity shown by immunodetection of p38 MAPK kinase target HSP-27 [51]. Activated p38 phosphorylates can trigger heat shock protein 27 (HSP-27), resulting in anti-apoptotic effects [135]. The agents blocked phosphorylation of HSP-27 completely in MM cells and in BMSCs [51].
JAK/STAT3 inhibitor
The JAK/STAT3 signaling pathway plays a key role in the development of MM by promoting cell accumulation through its anti-apoptotic activity [136]. CYT387 is a phenylaminopyrimidine compound and a novel JAK-targeted drug that can inhibit JAK1, JAK2, JAK3 and TYK2 kinase activity (P-20, Table 2) [65]. Studies have shown that CYT387 can modulate signaling downstream of IL-6, inhibiting proliferation and disrupting the cycle of MM cells [65]. It is currently in Phase 1–2 evaluation, showing no relevant hematological toxicity. It can be considered as a potential anti-MM drug alone or in combination with other agents [65,137].
NF-κB inhibitors
Transcription factor NF-κB is crucial for the survival and proliferation of MM cells. Numerous tumor cells with enhanced expression of NF-κB are resistant to apoptosis induced by chemotherapy and radiation [138]. NF-κB binds to its inhibitory protein IκB and maintains it in the inactive form in the cytosol [139]. Once IκB is phosphorylated, NF-κB is translocated into the nucleus and stimulates the transcription of specific NF-κB-regulated genes, which control the production of cytokines and chemokines [140]. All MM cell lines showed active IκB kinase and IκBα phosphorylation [55]. Therefore, IκB is a potential target for several diseases. Carboline derivatives (P-8, Table 2) are specific IκB kinase inhibitors, which can inhibit IκBα phosphorylation in MM.1S cells and patient MM cells [139]. The agents can inhibit the up-regulation of adhesion molecule mediated by NF-κB in cancer cells and block the protective effect of IL-6 against drug-induced apoptosis [54].
Curcumin (P-9, Table 2) is a chemopreventive agent that suppresses IκBα phosphorylation through the inhibition of IκB kinase activity and down-regulation of NF-κB. Curcumin is capable of suppressing constitutive NF-κB activation in all four MM cell lines including U266, MM.1, MM.1R and RPMI 8226 through blocking constitutively active IκB kinase present in MM cells [55]. Curcumin was shown to suppress MM cell proliferation and arrested cells at the G1/S cell cycle phase [55].
Alpha4 intergrin antagonists
Very late antigen-4 (VLA-4) is over-expressed in MM cells, serving as a key adhesion molecule that acts as a receptor for the extracellular matrix protein fibronectin and the cellular counter-receptor VCAM-1 [141]. It is responsible for the adherence of myeloma cells to bone marrow, promoting the development of MM and drug resistance [142]. Studies performed at the University of Texas demonstrated that the VCAM-1/VLA-4 integrin interaction is crucial for the cell–cell interaction between marrow stromal cells and 5TGM1 myeloma cells, leading to increased production of osteoclastogenic and bone resorption activity [56]. A published patent has indicated that antagonist agents that disrupt VCAM-1/VLA-4 binding may serve as potential treatments of MM and myeloma-induced bone resorption (P-10, Table 2) [56].
P62/sequestosome-1 inhibitor
Sequestosome-1 (p62) is a multifunctional protein that facilitates the formation of complexes involved in multiple signaling pathways such as NF-κβ, p38 MAPK and PI3K/Akt [143]. As a key regulator of these signaling pathways, p62 has been shown to play an important role in triggering cell autophagy and apoptosis in tumorgenesis [144]. Studies identified that the ZZ domain of p62 is crucial for BMSC support of MM cells and Osteoclast (OCL) formation. The ZZ domain of p62 can bind to RIP1 protein, interacting with TNF receptor, and activating NF-κB and p38/MAPK signaling [145]. Binding of innate defense regulator peptide (IDR-1) can promote RIP1 complex formation, inhibiting inflammation via p38 and IL6 signaling by interacting with RIP1 [145]. Study of a p62-knockout stromal cell-line showed that the ZZ domain of p62 is required for stromal cell support of MM cell growth, increased IL-6, VCAM-1 expression and OCL formation [66]. Xie et al. filed a patent in 2013 for the first p62-ZZ chemical inhibitor compounds (P-21, Table 2) [66]. The small molecule p62-ZZ inhibitor, 2-((3,4-bis(benzyloxy)benzyl)amino)ethan-1-ol (XRK3) had an IC50 of <5 μM when tested with MM cell lines. Furthermore, one patented compound demonstrated that it can suppress the expression of VCAM-1 and IL-6 induced by TNF- α in stromal cells by 30 and 90%, compared with the control vehicle [146]. The compounds can block the phosphorylation of PKC-zeta since Phospho-PKK¾ is a unique downstream signal activated by interactions with p62-ZZ domain. Moreover, p62-ZZ inhibitors can block human OCL formation in a dose-dependent way without blocking the differentiation of normal hematopoietic precursors. The compounds involved in the study can treat drug-resistant MM, that is, resistant to one or more of dexamethasone, alkylating agents, anthracyclines, thalidomide, lenalidomide, CC-4047, bortezomib and multi-targeted kinase inhibitors [66].
Other agents
MM patients are known to have elevated levels of bone marrow plasma protein Dickkopf-1 (DKK1) [147]. Evidence further suggests that concentrations of DKK1 are associated with myeloma stage and osteolytic lesions in MM patients [148–150]. The Wnt/β pathway is crucial in osteoblastogenesis and the regulation of bone metabolism [151]. DKK1 can inhibit Wnt/β signaling by binding to LRP5/6, resulting in myeloma bone diseases [152]. Thus, DKK1 can be treated as a potential therapeutic target for MM in the bone marrow microenvironment. Preclinical studies have shown that an anti-DKK1 neutralizing antibody (BHQ880) (Table 1) promoted bone formation and inhibited osteolytic diseases induced by myeloma [153]. Furthermore, BHQ 880 can decrease the level of IL-6 production by BMSCs [154]. Phase 2 clinical study of BHQ880 for the treatment of high risk smoldering MM was complete in October 2013 by Novartis Pharmaceuticals.
Nearly all MM patients suffer from osteolytic bone lesions, as the inherent balance between osteoclast and osteoblast function is disrupted in MM, leading to net bone resorption [155]. Osteoclast-activating factors such as macrophage inflammatory protein-1α and RANK induced by myeloma cells enhance the formation of osteoclasts, and ultimately bone destruction [156]. It has been reported that chemokine receptor CCR1 is employed by macrophage inflammatory protein-1α to form osteoclasts [157]. It is thus suggested that a CCR1 inhibitor could serve as a potential treatment for MM (P-22, Table 2). Studies showed that a CCR1 antagonist could delay the progression of monoclonal gammopathy of undetermined significance or smoldering MM in MM and further demonstrated that MM cell adhesion to osteoclasts was inhibited [67].
The CXCR4 receptor, a chemokine GPCR receptor, is expressed in human MM cells, influencing their migration to the bone marrow [158]. CXCR4 inhibitors can interfere with the CXCR4/SDF signaling pathway by disrupting CXCR4 binding to chemokine stromal derived factor-1/CXCL12 [159]. Studies demonstrated that the attenuation of CXCR4 expression in 5T33 MM cells completely disrupted the development of MM in recipient mice; thus, a CXCR4 inhibitor such as AMD3100 could serve as a potential therapeutic method to treat MM [50]. MDX-1338 (P-4, Table 2) is an orally bioavailable CXCR4 inhibitor that displayed decreased tumor proliferation and migration [50]. MDX-1338 is currently under Phase 1 clinical study to analyze its safety and toxicity [160].
A recent patent (P-23, Table 2) reported the important role of cannabinoid receptor CB2 in MM cell lines [68]. CB2, of the rhodopsin-family GPCR class, is predominantly expressed in the immune system, particularly in plasma B cells, whose dystregulation is the primary characteristic of MM. This was the first report about CB2 as new anti-MM drug target. In their studies, CB2 was found to be highly expressed in primary CD138+ MM cells and other human MM cells by Western blot and RT-PCR experiments. The discovered CB2 ligands, phenylacetylamide as one example, resulted in significant inhibition of MM growth through cell cycle modulation, mitotic death and cyto-skeleton disruption [68]. Importantly, this inhibition was rescued by CB2 gene silencing in the treated MM cells. Furthermore, the reported novel CB2 compounds also exhibit great inhibition of osteoclastogenesis [161,162]. Of course, extensive research studies are still needed to characterize the role of the CB2 receptor in MM cell-signaling pathways in order to facilitate new anti-MM drug targeting CB2.
Ezatiostat (TLK199 or TER 199) (P-11, Table 2) is a potent agent for inhibiting MM cell proliferation and treating MM, either alone or in combination with other anti-myeloma drugs such as bortezomib and cyclophosphamide. Ezatiostat hydrochloride (Telintra), a pharmaceutically acceptable salt of ezatiostat, is a glutathione-analog and a reversible inhibitor of the enzyme glutathione S-transferase P1–1 (GSTP1–1) as well as a therapeutic agent for the treatment of myelodysplastic syndrome [163]. It was shown that Telintra inhibited RPMI8226 MM cell proliferation with an IC50 of 33.0 μM [64].
Conclusion & future perspective
Bortezomib, thalidomide and lenalidomide are the representative therapeutic agents to treat MM with specific targets. Extensive research is in progress that utilizes a new understanding of MM cell interactions with the microenvironment and the key signaling pathways. Several anti-MM drugs are undergoing different stages of clinical trials to analyze their efficiency and safety. IMiDs, proteasome inhibitors, p38 inhibitors and other new anti-MM drugs with novel targets have displayed good therapeutic indexes and acceptable toxicity. These new agents will gradually transform the treatment of MM away from administrated high-doses of chemotherapy and towards more rational, selective therapies. It is anticipated that the novel anti-MM agents recently patented will ultimately be groundbreaking in MM research and will lead to future target-specific anti-MM drugs that will benefit millions of patients who suffer from this immune system-related cancer for which there is currently no cure.
Key terms.
Monoclonal gammopathy of undetermined significance: Condition in which an abnormal protein (monoclonal protein or monoclonal immunoglobulin) is in the blood with no clinical symptoms. It can progress over years to other disorders such as multiple myeloma.
Smoldering multiple myeloma: Asymptomatic plasma cell disorder that is characterized by the presence of a serum monoclonal protein at a concentration of ≥30 g/l and/or clonal bone marrow plasma cells ≥10% and the absence of end-organ damage. It has a high risk of progression to multiple myeloma.
Immunomodulatory drugs: Therapeutic agents that are structural and functional analogs of thalidomide that can suppress the immune system, inhibiting lymphocyte functions, especially T and NK cells.
Key terms.
Hsp90: Chaperone protein that assists other proteins in folding properly, stabilizing proteins against heat stress and aiding in protein degradation
Histone deacetylase: Class of enzymes that remove acetyl groups from histone, allowing the histones to bind DNA more tightly and inhibit gene transcription.
Key term.
Sequestosome-1 (p62): Multidomain and multifunctional protein that acts as a signaling hub for multiple signaling complexes in bone marrow stromal cells.
Executive summary.
Background
Multiple myeloma (MM) is the second most prevalent blood cancer and remains an incurable disease.
Novel medications have been approved for the treatment of MM.
Pathophysiology of multiple
Interaction between MM cells and bone marrow microenvironment play a critical role in promoting the growth of tumor cells and therefore related drug targets have been identified.
New MM drugs & patents under development or in clinical trials
With understanding of the bone marrow microenvironment and molecular mechanisms, many clinical trial drugs and new patents were developed based on different targets: tumor necrosis factor-α inhibitor, receptor tyrosine kinases inhibitors, heat shock protein inhibitor, surface antigen monoclonal antibodies, histone deacetylase inhibitors, mammalian target of rapamycin inhibitor, Ras inhibitor, MARK inhibitor, JAK/STAT3 inhibitor, NF-κB inhibitors and alpha4 intergrin antagonists.
Other agents such as DKK1 inhibitors, CCR1 inhibitors and GSTP1–1 inhibitors have been identified, and can be potential anti-MM drugs.
Conclusion & future perspective
Improved anti-MM drugs will be designed based on new research on MM-associated targets.
Acknowledgments
The authors would like to acknowledge financial support for the laboratory at the University of Pittsburgh from NIH grants DA025612 and HL109654 (X-Q Xie).
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
- 1.Abe M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int. J. Hematol. 2011;94(4):334–343. doi: 10.1007/s12185-011-0949-x. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J. Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Dimopoulos MA, Terpos E. Multiple myeloma. Ann. Oncol. 2010;21(Suppl. 7):vii143–vii150. doi: 10.1093/annonc/mdq370. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig H, Bolejack V, Crowley J, et al. Survival and years of life lost in different age cohorts of patients with multiple myeloma. J. Clin. Oncol. 2010;28(9):1599–1605. doi: 10.1200/JCO.2009.25.2114. [DOI] [PubMed] [Google Scholar]
- 5.Nolan KD, Mone MC, Nelson EW. Plasma cell neoplasms. Review of disease progression and report of a new variant. Surg. Oncol. 2005;14(2):85–90. doi: 10.1016/j.suronc.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma – 2009. Lancet. 2009;374(9686):25. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- 7.Laubach J, Richardson P, Anderson K. Multiple myeloma 2011. Annu. Rev. Med. 2011;62:249–264. doi: 10.1146/annurev-med-070209-175325. [DOI] [PubMed] [Google Scholar]
- 8.Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2006;354(13):1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 9.Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood. 2011;117(21):5573–5581. doi: 10.1182/blood-2011-01-270140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bates SE. Multiple myeloma. Clin. Cancer Res. 2011;17(6):1224. doi: 10.1158/1078-0432.CCR-11-0294. [DOI] [PubMed] [Google Scholar]
- 11.Ocio EM, Richardson PG, Rajkumar SV, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the international myeloma working group (IMWG). Leukemia. 2013;28(3) doi: 10.1038/leu.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singhal S, Mehta J. Multiple myeloma. Clin. J. Am. Soc. Nephrol. 2006;1(6):1322–1330. doi: 10.2215/CJN.03060906. [DOI] [PubMed] [Google Scholar]
- 13.Boccadoro M, Morgan G, Cavenagh J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int. 2005;5(1):18. doi: 10.1186/1475-2867-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsukamoto S, Yokosawa H. Inhibition of the ubiquitin-proteasome system by natural products for cancer therapy. Planta Med. 2010;76(11):1064–1074. doi: 10.1055/s-0029-1240901. [DOI] [PubMed] [Google Scholar]
- 15.Reece D, Imrie K, Stevens A, Smith CA. Bortezomib in multiple myeloma and lymphoma: a systematic review and clinical practice guideline. Curr. Oncol. 2006;13(5):160–172. [PMC free article] [PubMed] [Google Scholar]
- 16.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011;10(11):2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knight R. IMiDs: a novel class of immunomodulators. Semin. Oncol. 2005;32:24–30. doi: 10.1053/j.seminoncol.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 18.McCurdy AR, Lacy MQ. Pomalidomide and its clinical potential for relapsed or refractory multiple myeloma-an update for the hematologist. Ther. Adv. Hematol. 2013;4(3):211–216. doi: 10.1177/2040620713480155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22–32. doi: 10.1038/leu.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latif T, Chauhan N, Khan R, Moran A, Usmani S. Thalidomide and its analogues in the treatment of multiple myeloma. Exp. Hematol. Oncol. 2012;1(1):27. doi: 10.1186/2162-3619-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US FDA Approval for Lenalidomide. National Cancer Institute; www.cancer.gov/cancertopics/druginfo/fdalenalidomide. [Google Scholar]
- 22.Zou Y, Sheng Z, Niu S, Wang H, Yu J, Xu J. Lenalidomide versus thalidomide based regimens as first-line therapy for patients with multiple myeloma. Leuk. Lymphoma. 2013;54(10):2219–2225. doi: 10.3109/10428194.2013.774393. [DOI] [PubMed] [Google Scholar]
- 23.Chng WJ, Lau LG, Yusof N, Mow BMF. Targeted therapy in multiple myeloma. Cancer Control. 2005;12(2):13. doi: 10.1177/107327480501200204. [DOI] [PubMed] [Google Scholar]
- 24.Roodman GD. Role of the bone marrow microenvironment in multiple myeloma. J. Bone Miner. Res. 2002;17(11):1921–1925. doi: 10.1359/jbmr.2002.17.11.1921. [DOI] [PubMed] [Google Scholar]
- 25.Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012;2012:157496. doi: 10.1155/2012/157496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bommert K, Bargou RC, Stuhmer T. Signalling and survival pathways in multiple myeloma. Eur. J. Cancer. 2006;42(11):1574–1580. doi: 10.1016/j.ejca.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 27.Klein B, Zhang XG, Lu ZY, Bataille R. IL-6 in human multiple myeloma. Blood. 1995;85(4):9. [PubMed] [Google Scholar]
- 28.Le Gouill S, Podar K, Amiot M, et al. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood. 2004;104(9):2886–2892. doi: 10.1182/blood-2004-05-1760. [DOI] [PubMed] [Google Scholar]
- 29.Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat. Rev. Cancer. 2002;2(12):927–937. doi: 10.1038/nrc952. [DOI] [PubMed] [Google Scholar]
- 30.Podar K, Tai YT, Lin BK, et al. Vascular endothelial growth factor-induced migration of multiple myeloma cells is associated with beta 1 integrin- and phosphatidylinositol 3-kinase-dependent PKC alpha activation. J. Biol. Chem. 2002;277(10):7875–7881. doi: 10.1074/jbc.M109068200. [DOI] [PubMed] [Google Scholar]
- 31.Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009;23(1):10–24. doi: 10.1038/leu.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatterjee M, Stuhmer T, Herrmann P, Bommert K, Dorken B, Bargou RC. Combined disruption of both the MEK/ERK and the IL-6R/STAT3 pathways is required to induce apoptosis of multiple myeloma cells in the presence of bone marrow stromal cells. Blood. 2004;104(12):3712–3721. doi: 10.1182/blood-2004-04-1670. [DOI] [PubMed] [Google Scholar]
- 33.Oancea M, Mani A, Hussein MA, Almasan A. Apoptosis of multiple myeloma. Int. J. Hematol. 2004;80(3):224–231. doi: 10.1532/IJH97.04107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puthier D, Derenne S, Barille S, et al. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br. J. Haematol. 1999;107(2):392–395. doi: 10.1046/j.1365-2141.1999.01705.x. [DOI] [PubMed] [Google Scholar]
- 35.Chatterjee M, Honemann D, Lentzsch S, et al. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood. 2002;100(9):3311–3318. doi: 10.1182/blood-2002-01-0102. [DOI] [PubMed] [Google Scholar]
- 36.Younes H, Leleu X, Hatjiharissi E, et al. Targeting the phosphatidylinositol 3-kinase pathway in multiple myeloma. Clin. Cancer Res. 2007;13(13):3771–3775. doi: 10.1158/1078-0432.CCR-06-2921. [DOI] [PubMed] [Google Scholar]
- 37.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002;82(2):373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 38.Van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11(4):397–408. doi: 10.1634/theoncologist.11-4-397. [DOI] [PubMed] [Google Scholar]
- 39.M Jourdan KT, Legouffe E, Brochier J, Rossi JF, Klein B. Tumor necrosis factor is a survival and proliferation factor for human myeloma cells. Eur. Cytokine Netw. 1999;10(1):65–70. [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer. 2010;102(4):639–644. doi: 10.1038/sj.bjc.6605530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolkun L, Lemancewicz D, Jablonska E, et al. BAFF and APRIL as TNF superfamily molecules and angiogenesis parallel progression of human multiple myeloma. Ann. Hematol. 2013;93(4):635–644. doi: 10.1007/s00277-013-1924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terpos E, Kanellias N, Christoulas D, Kastritis E, Dimopoulos MA. Pomalidomide: a novel drug to treat relapsed and refractory multiple myeloma. Onco Targets Ther. 2013;6:531–538. doi: 10.2147/OTT.S34498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chanan-Khan AA, Swaika A, Paulus A, et al. Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer. 2013;3:e143. doi: 10.1038/bcj.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clinical Trials of Pomalidomide. National Cancer Institute; www.cancer.gov/clinicaltrials/search/results?protocolsearchid=7443751. [Google Scholar]
- 45.Celgene Corporation 2011 US7968569.
- 46.Clinical Trials of Lenalidomide. National Cancer Institute; www.cancer.gov/clinicaltrials/search/results?protocolsearchid=6467615. [Google Scholar]
- 47.Novartis Pharma Gmbh 2006 WO2006061199.
- 48.Dana-Farber Cancer Institute 2006 WO2006060676.
- 49.Kosan Biosciences, Inc. 2006 WO2006118953.
- 50.CBR institute for biomedical research, Inc. 2006 WO2006116185.
- 51.Scios, Inc. 2006 US2006079461.
- 52.AB Science 2007 WO2007026251.
- 53.Dana-Farber Cancer Institute 2007 WO2007059078.
- 54.Millennium pharmaceuticals, Inc. 2007 EP1443927-B.
- 55.Research Development Foundation 2007 US20077196105.
- 56.Board of Regents, the University of Texas System 2007 US20077211252.
- 57.Prolexys Pharmaceuticals, Inc. 2009 WO2009002553.
- 58.Bayer schering pharma aktiengesellschaft 2010 WO2010034414.
- 59.Cephalon, Inc. 2010 WO2010138141.
- 60.Novartis Vaccines and Diagnostics Inc. 2010 EP1684805B.
- 61.Novartis AG, Novartis Pharma 2011 US8012477.
- 62.SRI International 2012 US8268807.
- 63.Ab Science 2012 WO2012136732.
- 64.Telik, Inc. 2012 WO2012134915.
- 65.YM BioSciences Australia Pty Ltd 2012 WO2012149602.
- 66.Xie X-Q, Myint K, Kurihara N, Roodman D. 2013 WO2013022919.
- 67.Millennium Pharmaceuticals, Inc. 2013 US20130040979.
- 68.Xie X-Q, Feng RT, Yang P. 2013 US20130172388.
- 69.Pellat-Deceunynck C, Bataille R, Robillard N, et al. Expression of CD28 and CD40 in human myeloma cells: a comparative study with normal plasma cells. Blood. 1994;84(8):2597–2603. [PubMed] [Google Scholar]
- 70.Tai YT, Li X, Tong X, et al. Human anti-CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res. 2005;65(13):5898–5906. doi: 10.1158/0008-5472.CAN-04-4125. [DOI] [PubMed] [Google Scholar]
- 71.Ribatti D. Tyrosine kinase inhibitors as antiangiogenic drugs in multiple myeloma. Pharmaceuticals. 2010;3(4):1225–1231. doi: 10.3390/ph3041125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trudel S, Stewart AK, Rom E, et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma cells. Blood. 2006;107(10):4039–4046. doi: 10.1182/blood-2005-10-4179. [DOI] [PubMed] [Google Scholar]
- 73.Leich E, Weissbach S, Klein HU, et al. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion- and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013;3:e102. doi: 10.1038/bcj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalff A, Spencer A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: prognostic implications and current clinical strategies. Blood Cancer J. 2012;2:e89. doi: 10.1038/bcj.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petit V, Nussbaumer U, Dossenbach C, Affolter M. Downstream-of-FGFR Is a fibroblast growth factor-specific scaffolding protein and recruits corkscrew upon receptor activation. Mol. Cell. Biol. 2004;24(9):3769–3781. doi: 10.1128/MCB.24.9.3769-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Orlandini M, Marconcini L, Ferruzzi R, Oliviero S. Identification of a c-fos-induced gene that is related to the platelet-derived growth factor:vascular endothelial growth factor family. Proc. Natl Acad. Sci. USA. 1996;93(21) doi: 10.1073/pnas.93.21.11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bahlis NJ, Miao Y, Koc ON, Lee K, Boise LH, Gerson SL. N-Benzoylstaurosporine (PKC412) inhibits Akt kinase inducing apoptosis in multiple myeloma cells. Leuk. Lymphoma. 2005;46(6):899–908. doi: 10.1080/10428190500080595. [DOI] [PubMed] [Google Scholar]
- 78.Montero JC, Lopez-Perez R, San Miguel JF, Pandiella A. Expression of c-Kit isoforms in multiple myeloma: differences in signaling and drug sensitivity. Haematologica. 2008;93(6):851–859. doi: 10.3324/haematol.12171. [DOI] [PubMed] [Google Scholar]
- 79.Podar K, Anderson KC. The pathophysiologic role of VEGF in hematologic malignancies: therapeutic implications. Blood. 2005;105(4):1383–1395. doi: 10.1182/blood-2004-07-2909. [DOI] [PubMed] [Google Scholar]
- 80.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006;7(5):359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 81.Podar K, Anderson KC. Inhibition of VEGF signaling pathways in multiple myeloma and other malignancies. Cell Cycle. 2007;6(5):538–542. doi: 10.4161/cc.6.5.3922. [DOI] [PubMed] [Google Scholar]
- 82.Kumar SK, Jett J, Marks R, et al. Phase 1 study of sorafenib in combination with bortezomib in patients with advanced malignancies. Invest. New Drugs. 2013;31(5):1201–1206. doi: 10.1007/s10637-013-0004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsuchiya K, Asahina Y, Matsuda S, et al. Changes in plasma vascular endothelial growth factor at 8 weeks after sorafenib administration as predictors of survival for advanced hepatocellular carcinoma. Cancer. 2013 doi: 10.1002/cncr.28384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramakrishnan V, Timm M, Haug JL, et al. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene. 2010;29(8):1190–1202. doi: 10.1038/onc.2009.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yordanova A, Hose D, Neben K, et al. Sorafenib in patients with refractory or recurrent multiple myeloma. Hematol. Oncol. 2013;31(4) doi: 10.1002/hon.2043. [DOI] [PubMed] [Google Scholar]
- 86.Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011;10(12):2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 87.Clinical Trials of Cabozantinib. National Cancer Institute; www.cancer.gov/clinicaltrials/search/view?cdrid=750134&version=HealthProfessional&protocolsearchid=6574837. [Google Scholar]
- 88.Shailubhai KDS, Picker D, Kaur G, Sausville EA, Jacob GS. Atiprimod is an inhibitor of cancer cell proliferation and angiogenesis. J. Exp. Ther. Oncol. 2004;4(4):267–279. [PubMed] [Google Scholar]
- 89.Clinical trials of Atiprimod. National Cancer Institute; www.cancer.gov/clinicaltrials/search/view?cdrid=378176&version=HealthProfessional&protocolsearchid=12341971. [Google Scholar]
- 90.Trudel S, Li ZH, Wei E, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105(7):2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 91.Lee SH, Lopes De Menezes D, Vora J, et al. In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin. Cancer Res. 2005;11(10):3633–3641. doi: 10.1158/1078-0432.CCR-04-2129. [DOI] [PubMed] [Google Scholar]
- 92.Abrams TJ, Hollenbach PW, Rendahl KG, et al. CHIR-258 is efficacious in a newly developed fibroblast growth factor receptor 3-expressing orthotopic multiple myeloma model in mice. Clin. Cancer Res. 2006;12(16):4908–4915. doi: 10.1158/1078-0432.CCR-06-0957. [DOI] [PubMed] [Google Scholar]
- 93.Clinical Trials of TKI258. National Cancer Institute; www.cancer.gov/clinicaltrials/search/view?cdrid=665482&version=HealthProfessional&protocolsearchid=6706026. [Google Scholar]
- 94.Humbert M, Casteran N, Letard S, et al. Masitinib combined with standard gemcitabine chemotherapy: in vitro and in vivo studies in human pancreatic tumour cell lines and ectopic mouse model. PLoS ONE. 2010;5(3):e9430. doi: 10.1371/journal.pone.0009430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mellor HR, Bell AR, Valentin JP, Roberts RR. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol. Sci. 2011;120(1):14–32. doi: 10.1093/toxsci/kfq378. [DOI] [PubMed] [Google Scholar]
- 96.Adams J. The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer. 2004;4(5):349–360. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 97.Ocio EM, Mateos MV, Maiso P, Pandiella A, San-Miguel JF. New drugs in multiple myeloma: mechanisms of action and phase I/II clinical findings. Lancet Oncol. 2008;9(12):1157–1165. doi: 10.1016/S1470-2045(08)70304-8. [DOI] [PubMed] [Google Scholar]
- 98.Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171–003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120(14):2817–2825. doi: 10.1182/blood-2012-05-425934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carfilzomib. US FDA; www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm312945.htm. [Google Scholar]
- 100.Onyx Pharmaceuticals Announces Updates and Reviews 2012 Achievements at J.P. Morgan Healthcare Conference. 2013 www.onyx.com/view.cfm/651/onyx-pharmaceuticals-announces-2013-updates-and-reviews-2012-achievements-at-jp-morgan-healthcare-conference.
- 101.Zang Y, Thomas SM, Chan ET, et al. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012;18(20):5639–5649. doi: 10.1158/1078-0432.CCR-12-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arastu-Kapur S, Anderl JL, Kraus M, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin. Cancer Res. 2011;17(9):2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 103.Clinical trials of Carfilzomib. National Cancer Institute; www.cancer.gov/clinicaltrials/search/results?protocolsearchid=6526214. [Google Scholar]
- 104.Chauhan D, Singh AV, Aujay M, et al. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood. 2010;116(23):4906–4915. doi: 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zang Y, Thomas SM, Chan ET, et al. The next generation proteasome inhibitors carfilzomib and oprozomib activate prosurvival autophagy via induction of the unfolded protein response and ATF4. Autophagy. 2012;8(12):1873–1874. doi: 10.4161/auto.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Clinical trials of MLN9708. National Cancer Institute; www.cancer.gov/clinicaltrials/search/results?protocolsearchid=6786386. [Google Scholar]
- 107.Gupta V, Gonsalves WI, Kumar SK. Novel proteasome inhibitors for multiple myeloma. www.targetedonc.com/publications/targeted-therapies-cancer/2013/june-2013/novel-proteasome-inhibitors-for-multiple-myeloma/1.
- 108.Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70(5):1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 109.Chauhan D, Singh A, Brahmandam M, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111(3):1654–1664. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Allegra A, Sant'antonio E, Penna G, et al. Novel therapeutic strategies in multiple myeloma: role of the heat shock protein inhibitors. Eur. J. Haematol. 2011;86(2):93–110. doi: 10.1111/j.1600-0609.2010.01558.x. [DOI] [PubMed] [Google Scholar]
- 111.Duus J, Bahar HI, Venkataraman G, et al. Analysis of expression of heat shock protein-90 (HSP90) and the effects of HSP90 inhibitor (17-AAG) in multiple myeloma. Leuk. Lymphoma. 2006;47(7):1369–1378. doi: 10.1080/10428190500472123. [DOI] [PubMed] [Google Scholar]
- 112.Mitsiades CS, Mitsiades NS, Mcmullan CJ, et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood. 2006;107(3):1092–1100. doi: 10.1182/blood-2005-03-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Urashima M, Ogata A, Chauhan D, et al. Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood. 1996;88(6):2219–2227. [PubMed] [Google Scholar]
- 114.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ara T, Declerck YA. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer. 2010;46(7):1223–1231. doi: 10.1016/j.ejca.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman's disease. J. Clin. Oncol. 2010;28(23):3701–3708. doi: 10.1200/JCO.2009.27.2377. [DOI] [PubMed] [Google Scholar]
- 117.Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014;141(2):125–139. doi: 10.1016/j.pharmthera.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 118.Lin P, Owens R, Tricot G, Wilson CS. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am. J. Clin. Pathol. 2004;121(4):482–488. doi: 10.1309/74R4-TB90-BUWH-27JX. [DOI] [PubMed] [Google Scholar]
- 119.Stevenson GT. CD38 as a therapeutic target. Mol. Med. 2006;12(11–12):345–346. doi: 10.2119/2006-00082.Stevenson. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De Weers M, Tai YT, Van Der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011;186(3):1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 121.Danylesko I, Beider K, Shimoni A, Nagler A. Monoclonal antibody-based immunotherapy for multiple myeloma. Immunotherapy. 2012;4(9):919–938. doi: 10.2217/imt.12.82. [DOI] [PubMed] [Google Scholar]
- 122.Huang YW, Richardson JA, Vitetta ES. Anti-CD54 (ICAM-1) has antitumor activity in SCID mice with human myeloma cells. Cancer Res. 1995;55(3):610–616. [PubMed] [Google Scholar]
- 123.Veitonmäki NHM, Zhan F, Sundberg A, et al. ICAM1-targeted immunotherapy is effective in multiple myeloma. Cancer Discov. 2013;3(6):602. [Google Scholar]
- 124.Lemancewicz D, Bolkun L, Jablonska E, et al. The role of interleukin-17A and interleukin-17E in multiple myeloma patients. Med. Sci. Monit. 2012;18(1):BR54–BR59. doi: 10.12659/MSM.882204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Anderson KC. New insights into therapeutic targets in myeloma. ASH Education Program Book. 2011;2011(1):184–190. doi: 10.1182/asheducation-2011.1.184. [DOI] [PubMed] [Google Scholar]
- 126.Neri P, Bahlis NJ, Lonial S. Panobinostat for the treatment of multiple myeloma. Expert Opin. Investig. Drugs. 2012;21(5):733–747. doi: 10.1517/13543784.2012.668883. [DOI] [PubMed] [Google Scholar]
- 127.Anne M, Sammartino D, Barginear MF, Budman D. Profile of panobinostat and its potential for treatment in solid tumors: an update. Onco Targets Ther. 2013;6:1623–1624. doi: 10.2147/OTT.S30773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ikeda H, Hideshima T, Fulciniti M, et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood. 2010;116(9):1460–1468. doi: 10.1182/blood-2009-06-222943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gera J, Lichtenstein A. The mTOR Pathway in multiple myeloma. In: Munshi NC, Anderson KC, editors. Advances in Biology and Therapy of Multiple Myeloma. Springer; New York, NY, USA: 2013. pp. 97–116. [Google Scholar]
- 131.Cirstea D, Hideshima T, Rodig S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol. Cancer Ther. 2010;9(4):963–975. doi: 10.1158/1535-7163.MCT-09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Clinical trials of CC-223. National Cancer Institute; www.cancer.gov/clinicaltrials/search/results?protocolsearchid=9204255. [Google Scholar]
- 133.Hu L, Shi Y, Hsu JH, Gera J, Van Ness B, Lichtenstein A. Downstream effectors of oncogenic ras in multiple myeloma cells. Blood. 2003;101(8):3126–3135. doi: 10.1182/blood-2002-08-2640. [DOI] [PubMed] [Google Scholar]
- 134.Nguyen AN, Stebbins EG, Henson M, et al. Normalizing the bone marrow microenvironment with p38 inhibitor reduces multiple myeloma cell proliferation and adhesion and suppresses osteoclast formation. Exp. Cell Res. 2006;312(10):1909–1923. doi: 10.1016/j.yexcr.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 135.Xu L, Chen S, Bergan RC. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene. 2006;25(21):2987–2998. doi: 10.1038/sj.onc.1209337. [DOI] [PubMed] [Google Scholar]
- 136.Yang HH, Ma MH, Vescio RA, Berenson JR. Overcoming drug resistance in multiple myeloma: the emergence of therapeutic approaches to induce apoptosis. J. Clin. Oncol. 2003;21(22):4239–4247. doi: 10.1200/JCO.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 137.Monaghan KA, Khong T, Burns CJ, Spencer A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia. 2011;25(12):1891–1899. doi: 10.1038/leu.2011.175. [DOI] [PubMed] [Google Scholar]
- 138.Demchenko YN, Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010;1(1):59–68. doi: 10.18632/oncotarget.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009;1(4):a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002;277(19):16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 141.Kiziltepe T, Ashley JD, Stefanick JF, et al. Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacy in vivo. Blood Cancer J. 2012;2(4):e64. doi: 10.1038/bcj.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Soodgupta D, Hurchla MA, Jiang M, et al. Very late antigen-4 (α4β1 Integrin) targeted PET imaging of multiple myeloma. PLoS ONE. 2013;8(2):e55841. doi: 10.1371/journal.pone.0055841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hiruma Y, Honjo T, Jelinek DF, et al. Increased signaling through p62 in the marrow microenvironment increases myeloma cell growth and osteoclast formation. Blood. 2009;113(20):4894–4902. doi: 10.1182/blood-2008-08-173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol. Cancer Ther. 2011;10(9):1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yu HB, Kielczewska A, Rozek A, et al. Sequestosome-1/p62 is the key intracellular target of innate defense regulator peptide. J. Biol. Chem. 2009;284(52):36007–36011. doi: 10.1074/jbc.C109.073627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Teramachi JMK, Feng Rt, Xie Xq, Windle Jj, Roodman D, Kurihara N. Blocking the ZZ Domain of Sequestosome 1/p62 suppress the enhancement of myeloma cell growth and osteoclast formation by marrow stromal cells. Blood. 2011;118(21) [Google Scholar]
- 147.Politou MC, Heath DJ, Rahemtulla A, et al. Serum concentrations of Dickkopf-1 protein are increased in patients with multiple myeloma and reduced after autologous stem cell transplantation. Int. J. Cancer. 2006;119(7):1728–1731. doi: 10.1002/ijc.22033. [DOI] [PubMed] [Google Scholar]
- 148.Tian E, Zhan F, Walker R, et al. The role of the wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 2003;349(26):2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 149.Dun Xy Jiang H, Hou J. Bone marrow plasma concentrations of Dickkopf1 in patients with multiple myeloma. Zhejiang Da Xue Xue Bao Ban. 2009;38(5):453–458. doi: 10.3785/j.issn.1008-9292.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 150.Sato N, Yamabuki T, Takano A, et al. Wnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapy. Cancer Res. 2010;70(13):5326–5336. doi: 10.1158/0008-5472.CAN-09-3879. [DOI] [PubMed] [Google Scholar]
- 151.Yamashita T, Takahashi N, Udagawa N. New roles of osteoblasts involved in osteoclast differentiation. World J. Orthop. 2012;3(11):175–181. doi: 10.5312/wjo.v3.i11.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Heath DJ, Chantry AD, Buckle CH, et al. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J. Bone Miner. Res. 2009;24(3):425–436. doi: 10.1359/jbmr.081104. [DOI] [PubMed] [Google Scholar]
- 153.Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109(5):2106–2111. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fulciniti M, Tassone P, Hideshima T, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. 2009;114(2):371–379. doi: 10.1182/blood-2008-11-191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Giuliani N, Rizzoli V, Roodman GD. Multiple myeloma bone disease: pathophysiology of osteoblast inhibition. Blood. 2006;108(13):3992–3996. doi: 10.1182/blood-2006-05-026112. [DOI] [PubMed] [Google Scholar]
- 156.Roodman GD. Biology of osteoclast activation in cancer. J. Clin. Oncol. 2001;19(15):3562–3571. doi: 10.1200/JCO.2001.19.15.3562. [DOI] [PubMed] [Google Scholar]
- 157.Oba Y, Lee JW, Ehrlich LA, et al. MIP-1α utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp. Hematol. 2005;33(3):272–278. doi: 10.1016/j.exphem.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 158.Cojoc M, Peitzsch C, Trautmann F, Polishchuk L, Telegeev GD, Dubrovska A. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther. 2013;6:1347–1361. doi: 10.2147/OTT.S36109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010;16(11):2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 160.Clinical trial of MDX-1338. National Cancer Institute; www.cancer.gov/clinicaltrials/search/results?protocolsearchid=12342358. [Google Scholar]
- 161.Yang P, Wang L, Feng R, et al. Novel triaryl sulfonamide derivatives as selective cannabinoid receptor 2 inverse agonists and osteoclast inhibitors: discovery, optimization, and biological evaluation. J. Med. Chem. 2013;56(5):2045–2058. doi: 10.1021/jm3017464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yang P, Myint KZ, Tong Q, et al. Lead discovery, chemistry optimization, and biological evaluation studies of novel biamide derivatives as CB2 receptor inverse agonists and osteoclast inhibitors. J. Med. Chem. 2012;55(22):9973–9987. doi: 10.1021/jm301212u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Raza A, Galili N, Mulford D, et al. Phase 1 dose-ranging study of ezatiostat hydrochloride in combination with lenalidomide in patients with non-deletion (5q) low to intermediate-1 risk myelodysplastic syndrome (MDS). J. Hematol. Oncol. 2012;5:18. doi: 10.1186/1756-8722-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]