

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, involving a large number of genes, proteins and their complex interactions. Currently, no effective therapeutic agents are available to either stop or reverse the progression of this disease, likely due to its polygenic nature. The complicated pathophysiology of AD remains unresolved. Although it has been hypothesized that the amyloid β cascade and the hyper-phosphorylated tau protein may be primarily involved, other mechanisms, such as oxidative stress, deficiency of central cholinergic neurotransmitter, mitochondrial dysfunction and inflammation have also been implicated. The main focus of this review is to document current therapeutic agents in clinical trials and patented candidate compounds under development based on their main mechanisms of action. It also discusses the relationship between the recent understanding of key targets and the development of potential therapeutic agents for the treatment of AD.
Keywords: Alzheimer’s disease, amyloid β (Aβ), patent, tau, therapeuticagents
Alzheimer’s disease (AD), the most common form of dementia, is a progressive neurodegenerative disease and a complex multi-factorial disorder among the elderly [1]. It is estimated that the morbidity of AD over the age of 65 could reach up to 10–50%. AD has been recognized as one of the most intractable medical problems with heavy social and economic costs [2]. So far, no effective medicines or treatments are available yet to stop or reverse the progression of the disease.
AD is characterized by progressive memory loss and cognitive impairments. The main neuropathological features of AD are extracellular deposits of amyloid β peptide (Aβ) in senile plaques (SP), and the formation of intracellular neurofibrillary tangles (NFTs) [3,4]. Despite extensive research in the pathogenesis of AD, the exact mechanism of AD still remains unknown. During the past decade, several attempts have been made to explain the pathogenesis of the disease. The amyloid β (Aβ) cascade [5] and the hyperphosphorylated tau protein [6] seem to be the primarily involved. However, other mechanisms such as oxidative stress [7], deficiency of central cholinergic neurotransmitter [8], mitochondrial dysfunction [9] and inflammation [10] have also been implicated (Figure 1).
Figure 1. The complicated pathway and promising therapeutics in Alzheimer’s disease.

Aβ peptides are derived from APP. APP can be processed by amyloidogenic and nonamyloidogenic pathways which lead to different outcomes. In a nonamyloidogenic pathway, APP is initially cleaved by α-secretase to release sAPP-α and the left fragment is further processed by γ-secretase complex. In the amyloid pathway, APP is cleaved by β-secretase followed by γ-secretase complex to produce Aβ40/42, sAPP-β and AICD. Aβ42 has a high potential to aggregate to form toxic Aβ oligomers which cause the impairment of synapses and neurons. The Aβ oligomers increase the influx of Ca2+ and other different ions resulting in membrane depolarization. These results affect the function of different receptors and channels such as NAMDR, AMPAR and VDCC. In addition, the elevated Ca2+ can affect the modulation of tau-phosphorylating kinases such as GSK3β and CDK5, and result in the hyperphosphorylation of tau and the subsequent NFTs. Aβ also cause ER stress and mitochondria dysfunction by increase of ROS and Ca2+ dysregulation, which finally lead to dysfunction, degeneration and death of neurons. Based on the mechanisms and underlying targets associated with AD, some promising therapeutics designated by medicine bottles are under development, such as cholinergic drugs, amyloid-targeted therapies as well as drugs to target tau protein, mitochondrial function and neurotrophins.
Aβ: Amyloid β; AD: Alzheimer’s disease; AICD: Amyloid intracellular domain; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; APP: Amyloid precursor protein; CDK5: Cell division protein kinase 5; ER: Endoplasmic reticulum; GSK3β: Glycogen synthase 3β; NAMDR: N-methyl D-aspartate receptors; NFTs: Neurofibrillary tangles; ROS: Reactive oxygen species; sAPP-α: Soluble APP fragment α; sAPP-β: Soluble APP-β; VDCC: Voltage-dependent calcium channels.
In spite of the lack of a clear-cut understanding of the pathology of AD, significant efforts are being directed in developing new agents that are based on potential targets associated with the pathological changes seen in AD. Figure 2 identifies several such processes involved in the pathological changes. Based on this new information, a large number of patents have already been secured. This review is an effort to discuss recent understanding of key targets, and document patented candidate compounds under development and those under preclinical and clinical investigations, hoping that it would promote design of the next generation of therapeutic agents for the treatment of AD.
Figure 2. Advance in research publication reported for Alzheimer’s disease over the past decade.

The green and blue columns represent patents and scientific literature annually published related to AD, respectively.
AD: Alzheimer’s disease.
For color images please see http://www.future-science.com/doi/full/10.4155/ppa.14.22.
Current treatments & cholinergic drugs
Five drugs approved by the US FDA are used to treat the cognitive dysfunction of AD (Figure 3). These drugs are categorized into two major types: cholinergic inhibitors and N-methyl D-aspartate (NMDA) receptor antagonist. Cholinergic inhibitors are developed mainly based on the cholinergic hypothesis [11]. Reduction in the activity of the cholinergic neurons is a well-known feature in AD, leading to the deficiency of the neurotransmitter acetylcholine (ACh). The loss of cholinergic function is closely related to cognitive dysfunction and behavioral disorder. These symptoms can be improved by acetylcholinesterase (AChE) inhibitors or by modulating other cholinergic receptors, such as muscarinic and nicotinic ACh receptors. Since 1993, there have been four AChE inhibitors approved by FDA for AD treatment, including tacrine (1993), donepezil (1996), rivastigmine (2000) and galantamine (2001). These drugs were effective in improving the symptoms, behavioral and cognitive abilities in early-to-moderate stages of AD [12]. Among them, only donepezil is approved for treatment of advanced AD dementia [13]. Recent studies indicated that donepezil, rivastigmine and galantamine can decrease Aβ production and Aβ-induced toxicity, suggesting the cholinergic system may play a role in Aβ generation and aggregation [14]. Apart from the AChE inhibitors approved, Memantine is a novel NMDA receptor antagonist. It acts on the glutamatergic system by blocking NMDA receptors and inhibiting their overstimulation by glutamate. Memantine has been shown to be efficacious in the treatment of moderate-to-severe AD [15].
Figure 3. Five medicines approved by the US FDA for treatment in Alzheimer’s disease.
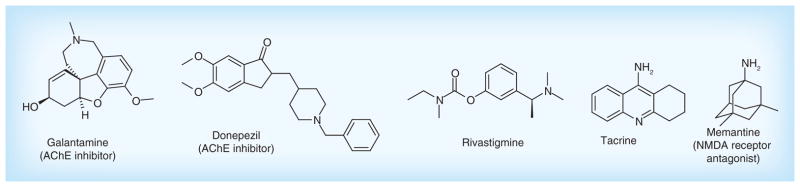
AChE: Acetylcholinesterase; NMDA: N-methyl D-aspartate.
Besides the drugs approved by FDA, there has still been progress in development of cholinergic drugs in clinical trials as well as patented lead compounds (Figure 4 & Table 1).
Figure 4. Cholinergic inhibitors in clinical trials and patented lead compounds.

Table 1.
The development of cholinergic drugs in different stages
| Compound | Target | Assignee | Patent number | Phases | Ref. |
|---|---|---|---|---|---|
| ZT-1 | AChE | Debiopharm SA, Switzerland | WO120774A2 (2009) | Phase II | [16] |
| (-)-Phenserine | AChE | The General Hospital Corporation, USA | WO117727A2 (2010) | Preclinical | [17] |
| GLN-1062/Memogain | AChE | Galantos Pharma GmbH, Germany | WO127218A1 (2009) | Discovery | [18] |
| 1a | AChE | Universidad Autonoma de Madrid UAM, Spain | WO051535A1 (2011) | Discovery | [19] |
| Ladostigil | BChE | Teva Pharmaceutical Industries, Ltd, Israel | US0232691A1 (2007) | Phase II | [20] |
| NGX267 | ACM1 | Solvay Pharmaceuticals BV, The Netherlands | WO128674A2 (2007) | Phase II | [21] |
| EVP-6124 | ACHA7 | EnVivo Pharmaceuticals, Inc., USA | WO169646A1 (2013) | Phase II | [22] |
| GTS-21 | ACHA7 | Niconovum USA, Inc., USA | US0274628A1 (2011) | Phase II | [23] |
AChE: Acetylcholinesterase; ACHA7: Neuronal ACh receptor subunit α-7; ACM1: Muscarinic ACh receptor M1; BChE: Butyrylcholinesterase.
ZT-1, as pro-drug of huperzine A from natural product, is a potent and selective AChE inhibitor. The results from the Phase I clinical trials showed that ZT-1 has an admirable pharmacokinetic with a rapid absorption and a wide distribution in human [24]. In a subsequent Phase IIa study, daily administration of oral ZT-1 delayed cognitive decline for AD patients [25]. Currently, it is undergoing Phase IIb clinical development for the treatment of AD. The (-)-phenserine, a derivative of physostigmine, is also an AChE inhibitor that shows the effects on improving cognition in vivo [26]. In addition to inhibiting AChE, it can significantly reduce Aβ precursor protein (APP) and Aβ concentrations by reducing the translation of APP [26], suggesting (-)-phenserine may be a promising multitarget drug of AD.
Memogain (Gln-1062) developed by Galantos Pharma is an inactive pro-drug of galantamine approved for the treatment of AD. Memogain has more hydrophobic characteristics than galantamine, and therefore has more than 15-fold higher bioavailability in the brain than the same dosage of galantamine. As a cholinergic enhancer, it possibly represents a valuable drug with much lesser gastrointestinal side effects and higher potency in enhancing cognition for AD treatment [27].
Bis(aralkyl)amino-and(hetero)aryl derivatives were designed and patented by Universidad Autonoma de Madrid (UAM). These compounds can increase levels of the neurotransmitter ACh by binding to the catalytic active center of AChE. Furthermore, it possesses the potent neuroprotective activity against mitochondrial oxidative stress. Compound 1a has the significant effect on inhibition of AChE with IC50 level of 900 nM [19], which is a potential lead compound for the treatment of AD.
In addition, ladostigil is a novel multitarget neuroprotective drug with a dual ACh-butyrylcholinesterase and monoamine oxidase A and B inhibitor. It was shown to alleviate scopolamine-induced impairment in spatial memory, and increase in rat brain cholinergic activity. Moreover, it possesses potent neuroprotective and anti-apoptotic activities. These neuroprotective activities are attributed to the regulation of APP processing, activation of protein kinase C and mitogen-activated protein kinase signaling pathways. Currently, the Phase II study of the drug has been completed, and the results have not been published yet [28].
Enhancement of cholinergic transmission with muscarinic receptor agonist and nicotinic receptor agonist has also been investigated. NGX267 (AF267B), as a selective cholinergic M1 muscarinic receptor agonist, can reduce cognitive deficits [29]. In particular, it also decreased Aβ1-42 and tau pathologies in the cortex and hippocampus in transgenic AD mice, suggesting its potential for therapy in AD [30].
EVP-6124 is an α7 nicotinic ACh receptor (nAChR) agonist with highly CNS-penetrant. It can improve memory performance by potentiating the ACh response of α7 nAChRs. The compound has currently successfully completed Phase II trials, supporting a new therapeutic strategy for the treatment of cognitive impairment [31]. Additionally, GTS-21 is selectively agonist of the α7 nicotinic receptor with good safety and tolerability. This drug has displayed promising characteristics during Phase II clinical trial [32].
Amyloid-targeted therapies
The development of AD drugs has been facilitated by the amyloid hypothesis [33,34]. Aβ peptides are derived from amyloid precursor protein (APP) which is an integral glycoprotein expressed in the brain [35]. APP can be processed by amyloidogenic and nonamyloidogenic pathways which lead to different outcomes. In general, APP is cleaved by α-secretase and then γ-secretase, which is nonamyloidogenic. However, in amyloidogenic pathway, APP is initially performed by β-secretase to release the soluble fragment into extracellular region. The remaining section is then processed by γ-secretase, generating amyloidogenic peptides such as Aβ1-40 and Aβ1-42 (Figure 1) [35]. Many evidences have indicated that Aβ is a neurotoxin, and the accumulation of Aβ1-42 in particular induces the formation of toxic Aβ oligomers and fibrils [36], which cause the impairment of synapses and neurons [37]. Based on the amyloid hypothesis, drugs that can reduce the generation of Aβ, prevent the aggregation of Aβ, and promote its clearance are thought to be promising therapeutics for AD.
Decreasing Aβ generation
Since β- and γ-secretases are responsible for the generation of Aβ from the release of the intracellular domain of APP, great efforts have been focused on the inhibition or modulation of activities of β- and γ-secretases, which are recognized as important drug targets of AD.
β-secretase inhibitors
LY2811376 developed by Eli Lilly and Co. is the first orally available nonpeptidic β-secretase inhibitor identified by fragment-based screening. It can reduce Aβ levels in animal models in dose-dependent manner [38]. LY2811376 can also produce long-lasting reductions of Aβ levels in healthy volunteers with safety and good tolerability. However, due to the off-target-based toxicology, it prevented the compound from progressing to clinical development. Another compound LY2886721 is a selective β-secretase inhibitor with agreeable drug properties. The compound lowered cerebral spinal fluid (CSF) Aβ40, Aβ42 and sAPP-β concentrations without safety concerns in Phase I clinical trial [39]. Unfortunately, it also did not undergo subsequent trials due to abnormal liver biochemical tests.
MK-8931 is a β-secretase 1 (BACE1) inhibitor being tested for the treatment of Alzheimer’s type dementia in Phase II clinical trial. Results of Phase I clinical studies demonstrated that MK-8931 resulted in a dose-dependent and sustained reduction in CSF Aβ levels by greater than 90% in healthy volunteers without dose-limiting side effects [40]. Based on these results, global, multicenter Phase II/III clinical trials are conducted to evaluate the safety and efficacy of MK-8931 versus placebo in patients with mild-to-moderate AD.
Several novel lead compounds targeting β-secretase are under development. Lactams derivatives, as β-secretase inhibitors, were patented by Pfizer, Inc. The compound 2a possesses enhanced brain penetration and improved cardiovascular properties. It is a high selective β-secretase inhibitor with IC50 of 32 nM on neuroglioma cell line H4 cells [41]. In addition, Pfizer, Inc. designed the hexahydropyrano [3,4-d] [1,3] thiazin-2-amine compounds that are β-secretase inhibitors with the novel scaffold. The compound 3a showed the strong inhibition effect on β-secretase with IC50 of 23.1 nM with H4 human neuroglioma cells expressing APP695 in vitro [42]. Another novel tricyclic compounds such as compound 4a displayed a stronger inhibition effect with IC50 of 1.1 nM in vitro [43]. These patented compounds could be as a potential candidate drug in the treatment and prevention of AD. Figure 5 & Table 2 list the main β-secretase inhibitors for AD therapies.
Figure 5. β-secretase inhibitors in clinical trials and patented lead compounds.

Table 2.
The development of β-and γ-secretase inhibitors.
| Compounds | Chemical class | Assignee | Patent number | Phases | Ref. |
|---|---|---|---|---|---|
| β-secretase inhibitors | |||||
| LY2886721 | Aminothiazine derivatives | Eli Lilly and Co., USA | WO005738A1(2011) | Phase I/II | [44] |
| LY2811376 | Aminodihydrothiazine derivatives | Eli Lilly and Co., USA | WO134617A1(2009) | Phase I | [45] |
| MK-8931 | Imidazole derivatives | Schering Corp., USA | US0287692A1(2007) | Phase II/III | [46] |
| 2a | Lactams derivatives | Pfizer, Inc., USA | WO172449A1(2012) | Discovery | [41] |
| 3a | Thioamidine derivatives | Pfizer, Inc., USA | WO030713A1(2013) | Discovery | [42] |
| 4a | Tricyclicderivatives | Array Biopharma Inc., USA; Genentech, Inc., USA | WO148851A1(2013) | Discovery | [43] |
| γ-secretase inhibitors | |||||
| LY-450139 | Butanamides | Eli Lilly and Co., USA | US0299053A1(2007) | Phase III | [47] |
| BMS-708163 | Trifluoropentanamides | Bristol-Myers Squibb Co., USA | US0260837A1(2010) | Phase II | [48] |
| NIC5-15 | Pinitol | Waratah Pharmaceuticals, Inc., USA | US0105631A1(2010) | Phase II | [49] |
| GSI-953 | Thiophene sulfonamide | Wyeth, USA | US0181932A1(2009) | Phase I | [50] |
| PF-3084014 | Pentanamide derivatives | Pfizer, Inc., USA | US0215610A1(2005) | Phase I | [51] |
| BMS299897 | Sulfonamide | Elan Pharmaceuticals, Inc., USA | US0045499A1(2008) | Discovery | [52] |
| CHF5074 | Flurbiprofen derivatives | Chiesi Pharmaceuticals, Inc., USA | WO015287A2(2011) | Phase II | [53] |
| E2012 | 2-piperidinone derivatives | Schering Corp., USA | US20110027264A1 | Preclinical | [54] |
| 5a | Bicyclic pyridinones | Pfizer, Inc., USA | WO2012131539 A1 | Discovery | [55] |
| 6a | Tetrasubstituted benzenes | Envivo Pharmaceuticals, Inc., USA | US20130165486 A1 | Discovery | [56] |
Currently, numerous small molecules are designed to target BACE1. Whereas, the pace of research and development on BACE1 as the therapeutic target has been slow. Several concerns have been stated about the potential side effects of BACE1 inhibitors, because BACE1 also cleaves a selection of substrates involved in myelination, neuronal circuits, retinal homeostasis and synaptic function [57]. Inhibition of the enzyme could have toxic consequences. Therefore, the selective BACE1 inhibitors without side effect are expected to design for further evaluation in AD treatment.
γ-secretase inhibitors & modulators
γ-secretase is an intramembrane multisubunit protease complex that is responsible for cleavage of the APP to produce neurotoxic Aβ peptides in the final step. It is also critical in the related processing of several other type of membrane proteins, such as Notch receptor, N-cadherin and ErbB4 [58]. Several γ-secretase inhibitors and modulators have been developed as potential treatments of AD to reduce the formation of Aβ.
Semagacestat (LY-450139) is a γ-secretase inhibitor under development by Eli Lilly and Co. as a treatment for AD. It can reduce Aβ concentrations in the plasma and Aβ production in the CNS. However, the Phase III trial was terminated owing to a high occurrence of adverse effects. Furthermore, patients with AD receiving the drug showed a worsening of cognition function than the placebo group [59]. A possible reason for the highlight of adverse events associated with semagacestat is that inhibiting γ-secretase possibly interferes with the receptor-related nuclear signaling of Notch [60]. Therefore, the developments of γ-secretase inhibitors with severe Notch-related side effects have been discontinued.
The second-generation γ-secretase inhibitors with Notch-sparing effect have been developed. Avagacestat (BMS-708163) is a potent, selective γ-secretase inhibitor of Aβ42 with IC50 of 0.27 nM, demonstrating a 193-fold selectivity against Notch [61]. Phase I clinical trial studies showed that BMS-708163 significantly decreased the level of CSF Aβ40 and Aβ42 approximately 30% with a daily dose of 100 mg in humans [62]. Nevertheless, results from Phase II trials in mild-to-moderate AD showed that the compound did not display obvious efficacy to drive the advancement of Phase III trials [63].
NIC5-15 is a natural product found in soy and several fruits. It can act as a Notch-sparing γ-secretase inhibitor and an insulin sensitizer. This compound modulates γ-secretase to reduce Aβ production, and improves cognitive function and memory deficits in preclinical models of AD [64]. The results from Phase IIa trial in 15 patients with mild-to-moderate AD showed that NIC5-15 is safe and has good tolerability [65]. Additionally, Begacestat (GSI-953) is also a novel γ-secretase inhibitor that selectively inhibits cleavage of APP over Notch. It inhibits Aβ production in a dose-dependent reduction with EC50 value of 7.3 nM [66]. The compound is being tested in Phase I clinical trial.
PF-3084014 is a highly selective γ-secretase inhibitor that reduces Aβ with IC50 of 1.2 nM in vitro. This compound showed dose-dependent reduction in brain, CSF and plasma Aβ in Tg2576 mice. PF-3084014 is currently under clinical development [67]. In addition, BMS-299897 is also a γ-secretase inhibitor. It is shown to be orally available, readily cross blood–brain barrier and effectively suppress plasma and brain Aβ level in human APP-bearing transgenic mice in a time- and dose-dependent manner with ED50 values of 30 mg/kg in vivo, suggesting its potential for therapy in AD [68].
γ-secretase modulators selectively blocking APP proteolysis without Notch-related side effects could offer a more promising strategy [69]. CHF5074, a novel γ-secretase modulator, reduces brain Aβ burden, and attenuates spatial memory deficit in a transgenic mice model of AD [70]. The data from Phase II clinical trial in 96 patients with mild-to-moderate AD showed that CHF5074 is safe and has good efficacy [71]. E2012, a novel compound discovered by Eisai, is also a γ-secretase modulator that inhibits the production of Aβ without affecting Notch cleavage. E2012 significantly decreased the levels of Aβ40 and Aβ42 in rat CSF, brain and plasma in a dose-dependent manner in vivo [72], suggesting the novel γ-secretase modulator could be a promising therapeutic agent for AD.
Besides the drugs in clinical trials, some novel γ-secretase modulators are also under development. Bicyclic pyridinone derivatives patented by Pfizer, Inc. are a novel γ-secretase modulator [55]. Among them, the compound 5a showed a stronger inhibition effect on Aβ42 with IC50 of 4.2 nM in vitro. In addition, tetrasubstituted benzenes compounds designed by Envivo Pharmaceuticals, Inc. are also γ-secretase modulator. The compound 6a can significantly reduce Aβ42 with EC50 of 69 nM in vitro (HEK 293 cell line). The distinct effects on lowering Aβ were also observed in Tg2576 transgenic mice [56]. Figure 6 & Table 2 list the main γ-secretase inhibitors and modulators for AD therapies.
Figure 6. γ-secretase inhibitors and modulators in clinical trials and patented lead compounds.

Preventing Aβ aggregation
Evidence shows that Aβ aggregations induce the formation of toxic Aβ oligomers and fibrils, and cause the impairment of synapses and neurons [73]. Based on this point, some anti-aggregation drugs have been investigated to prevent Aβ fragments from aggregating (Figure 7). The strategy can be implemented by either binding to Aβ monomers to avoid the oligomerisation, or reacting with Aβ oligomers to alleviate toxicity and promoting their clearance.
Figure 7. Drugs in clinical trials and patented lead compounds to prevent Aβ aggregation.

Tramiprosate (3APS) is an orally-administered compound binding to the soluble Aβ and reduces Aβ aggregation. Tramiprosate possesses neuroprotection against Aβ-induced neurotoxicity in vitro, and produces dose-dependent reductions of Aβ in the brain of transgenic mice [74]. Clinical Phase II studies showed that it was safe and tolerable. However, the further Phase III test has been terminated due to its poor clinical efficacy and low CNS bioavailability for mild-to-moderate AD patients [75].
Scyllo-inositol, as a natural product, is another anti-oligomerization compound. It stabilized a small conformer of Aβ42 and neutralized cell-derived Aβ oligomers in vitro, and promoted the generation of low molecular weight Aβ species in vivo. Furthermore, the compound decreased neuronal toxicity and alleviated the cognitive deficits in multiple mouse models of AD [76]. A Phase II clinical trial evaluating efficacy and safety is currently ongoing. Another well-known natural product from green tea, epigallo-catechin-3-gallate (EGCG) also has shown multiple neuroprotective effects. It can prevent the aggregation of Aβ peptides to form toxic oligomers through the direct binding to the unfolded peptide [77]. It is currently being tested in Phase II/III clinical trials for patients with early AD.
PBT1 is a novel metal chelator that inhibits Aβ aggregation by interfering with interactions between Aβ and metal ions. Evidence from Phase II clinical trials suggested that PBT1 could halt cognitive decline in AD [78]. Unfortunately, further Phase II/III studies were halted owing to manufacturing toxicity issues. Subsequently, the second-generation inhibitor, PBT2 was developed as a metal-protein attenuating compound that affects the metal-mediated toxic oligomerisation [79]. It has a better blood–brain barrier permeability than does PBT1. The data from animal experiments showed that PBT2 prevents Aβ oligomerization, reduces soluble and insoluble Aβ in the brain and promotes Aβ oligomer clearance [80]. The positive results from Phase II also showed that PBT2 reduced Aβ42 CSF concentrations and improved cognition function with quality safety and tolerance [81]. The novel metal chaperones could be a promising drug to the treatment of age-related cognitive decline.
Some attractive anti-aggregation compounds have been investigated. Apomorphine derivatives such as compound 7a patented by Cytokine Pharmasciences, Inc. target the nucleation phase of Aβ self-assembly and interfere effectively with aggregation of Aβ1-40 into amyloid fibrils in vitro [82]. Peptidomimetic derivatives are new small molecules for inhibiting Aβ aggregation. Among them, compound 8a displayed distinct inhibition of the Aβ fibril formation with ThT assay in vitro [83]. In addition, Neuropore Therapies, Inc. presented heterocyclic compounds such as compound 9a that specifically target the toxic Aβ oligomers aggregation with a high affinity. The compound is also able to easily cross the blood–brain barrier at high AUC brain/blood ratios [84]. Figure 6 & Table 3 list the main drugs in clinical trials and patented lead compounds to prevent Aβ aggregation.
Table 3.
The development of drugs to prevent Aβ aggregation.
| Compounds | Chemical class | Assignee | Patent number | Phases | Ref. |
|---|---|---|---|---|---|
| Tramiprosate | Amidosulphuric acid | Bellus Health, Inc., Canada | WO054485A1(2010) | Phase III | [85] |
| Scyllo-Inositol | Scyllitol | Elan Pharmaceuticals, USA | WO173808A1(2012) | Phase II | [86] |
| PBT1 | Hydroxyquinoline derivatives | Prana Biotechnology, Ltd, Australia | WO074068A1(2008) | Phase II | [87] |
| PBT2 | Hydroxyquinolines derivatives | Prana Biotechnology, Ltd, Australia | WO071944A1(2010) | Phase II | [88] |
| Epigallocate-chin-3-gallate | Catechin | Max-Delbruck-Centrum FurMolekulare Medizin, Germany | US0117040A1(2009) | Phase I | [89] |
| 7a | Apomorphine derivatives | Cytokine Pharmasciences, Inc., USA | US20080096909 A1 | Discovery | [82] |
| 8a | Peptidomimetic derivatives | Almqvist Fredrik, Sweden | WO 2009134203 A1 | Discovery | [83] |
| 9a | Heterocyclic compounds | Neuropore Therapies, Inc., USA | WO2013134371 A1 | Discovery | [84] |
Promoting Aβ clearance
Anti-amyloid immunotherapy has shown beneficial effects on Aβ clearance in various mice models, which would be a valuable therapeutic strategy. Active or passive Aβ immunization has been developed to prevent Aβ aggregation and promote its clearance.
Active AD immunotherapy
The first active vaccine is AN-1792 using full-length Aβ1-42 tested in clinical trial. However, it was terminated because of severe side effect in some patients, which was attributed to nonspecific immune response [90]. To avoid this point, Novartis designed the second-generation vaccine CAD106 that comprises Aβ1–6 sequence. It can reduce Aβ accumulation and induce a substantial anti-Aβ immune response with quality toleration and safety in Phase II trials [91]. However, no significant changes in Aβ levels were detected in CSF with treatment of CAD106, and some adverse effects on nasopharyngitis and erythema in injection sites were also observed [91].
Subsequently, ACI-24 and UB-311 were designed based on Aβ1–15 and Aβ1–14, respectively. During pre-clinical development, ACI-24 has shown high efficacy on memory restoration and plaque reduction in transgenic mice [92]. The combined Phase I/II clinical trials are currently ongoing for determining its efficacy and tolerability. UB-311 is a novel immunotherapy with the UBITh helper T-cell technology and a particular site-specific epitope to target the Aβ peptide. It has successfully completed clinical Phase I study, demonstrating safety and tolerability [93].
Other on-going active immunization trials include ACC-001 and V-950 as well. ACC-001 was developed by Janssen according to the N-terminal Aβ fragment attached to a carrier protein. It is currently being tested in a Phase II trial. Additionally, Merck designed V-950, as a multivalent Aβ vaccine [94], and just finished the Phase I study in AD patients.
Passive AD immunotherapy
Passive immunotherapy refers to the direct administration of anti-Aβ antibodies, obviating the need for patients to mount an antibody response. Passive immunotherapy is based on specifically designed monoclonal antibodies targeting Aβ to promote its clearance. As an alternative therapy, passive immunization was considered safer and more controllable than active immunization [95].
AAB-001 (Bapineuzumab) is a humanized monoclonal antibody targeting the N-terminal region of Aβ. The antibody was shown to bind to Aβ plaques, lower plaque burden and improve performance on mouse behavioral assays [96]. However, its Phase III trials were halted after completion of two trials owing to a failure to meet primary outcome measures of cognition and activities of daily living [97]. LY-2062430 (Solanezumab) is a monoclonal antibody developed by Eli Lilly. It can bind to the soluble Aβ and lower amyloid pathology in mouse models. The major mechanism of action is thought to be via peripheral Aβ sequestration and a peripheral sink [98]. Nevertheless, the LY-2062430 also failed to meet its primary cognitive and functional end points in Phase III in two clinical trials after AAB-001.
PF-04360365 (Ponezumab) is a humanized IgG2δA monoclonal antibody that binds the free C-terminal amino acids 33–40 of the Aβ1-40 peptide. The results from Phase I trials showed acceptable safety without findings of antibody-induced side effects [99]. Two Phase 2 trials of multiple doses are ongoing.
In addition, GSK-933776, R-1450 and MABT-5102A, which are monoclonal antibodies targeting Aβ, have been entered in the clinical trials. GSK-933776 is a humanized IgG1 monoclonal antibody directed against the N-terminal of Aβ. It can clear the soluble amyloid from the brain, reduce its neurotoxic effect and improve cognition in transgenic mice [100]. GSK-933776 is being tested in Phase II trials for AD. In addition, R-1450 designed by Roche is a novel human anti-Aβ antibody that recognizes the N-terminal and the central region within Aβ. Several preclinical results show that g R-1450 preferentially interacts with aggregated Aβ in the brain and lowers amyloid-β by eliciting effector cell-mediated clearance [101]. MABT5102A was derived by immunization with modified Aβ1–15, possessing a human IgG4 backbone. It is thought to target multiple conformational protofibrillar epitopes of Aβ, including oligomeric forms for inhibiting Aβ aggregation and promoting its disaggregation [102]. A Phase I clinical trial proved its safety and Phase II of MABT5102A is ongoing. Table 4 lists the active and passive immunotherapies for AD.
Table 4.
Active and passive immunotherapy for Alzheimer’s disease in clinical trials.
| Compounds | Class | Assignee | Patent number | Phases | Ref. |
|---|---|---|---|---|---|
| CAD106 | Active immunotherapy | Cytos Biotechnology; Novartis Pharma, Switzerland | WO016282A1(2004) | Phase II | [103] |
| ACC-001 | Active immunotherapy | Janssen Alzheimer Immunotherapy R&D, LLC, Japan | US0276116A1(2012) | Phase II | [104] |
| V-950 | Active immunotherapy | Merck Sharp & Dohme Corp., USA | WO121656 A2(2006) | Phase I | [105] |
| ACI-24 | Active immunotherapy | AC Immune SA, Switzerland; Genentech, Inc., USA | WO156622A1(2008) | Phase I | [106] |
| UB-311 | Active immunotherapy | United Biomedical, USA | US0070255A1(2011) | Phase I | [107] |
| AAB-001 | Passive immunotherapy | Elan Pharma International Ltd, Ireland; Wyeth, John, and Brother Ltd, UK | WO017467A1(2009) | Phase II | [108] |
| LY-2062430 | Passive immunotherapy | Eli Lilly and Co., USA | US0158986A1(2011) | Phase III | [109] |
| PF-04360365 | Passive immunotherapy | Rinat Neuroscience Corp., USA | WO032868A2(2004) | Phase I | [110] |
| GSK-933776 | Passive immunotherapy | GlaxoSmithKline, UK | WO 020722A2(2013) | Phase I | [111] |
| R-1450 | Passive immunotherapy | Hoffmann-La Roche, Switzerland; Morphosys, Germany | US0136747A1(2013) | Phase I | [112] |
| MABT-5102A | Passive immunotherapy | AC Immune SA, Switzerland; Genentech, Inc., USA | WO016173A2(2012) | Phase I | [113] |
Although some clinical trials are still ongoing, the effects of the AD immunotherapy targeting Aβ in pre-clinical studies do not seem to correspond with those observed in clinical trials. The AD immunotherapies failed in clinical trials suggested that even elimination of the Aβ plaques still cannot improve cognition and stop the disease progression in AD patients.
Drugs to target tau protein
Tau pathology is another important hallmark of AD and perhaps the most promising target. Tau is highly enriched within neurons of the central nervous system, in which it appears to play an important role in the formation and stabilization of microtubules (MTs) [114]. In AD, hyperphosphorylated tau protein results in the intracellular NFTs and further disrupts MTs-mediated axonal transport, leading to dysfunction, degeneration and subsequent death for neurons [115]. Several evidences suggest that tau pathology closely correlates with the progressive neuronal loss and cognitive decline in AD patients [116].
Two main therapeutic approaches focused on tau protein can be used to either modulate phosphorylation of tau by inhibitors of tau-phosphorylating kinases, or inhibit the tau aggregation and promote its degradation [117]. Phosphorylation of tau is controlled through different kinases and phosphatases. Among them, the glycogen synthase kinase 3 β (GSK3β) is a key target that regulates tau phosphorylation, which is also involved in amyloid processing and gene transcription [118].
Several GSK3β inhibitors are under development. Tideglusib (NP-031112), as a non-ATP-competitive GSK-3 inhibitor, is a small molecule belonging to the tiadiazolidindiones family. It can decrease tau hyper-phosphorylation, lower brain amyloid plaque levels, improve learning and memory and prevent neuronal loss in a variety of animal models [119]. The results from Phase IIa study demonstrated its valuable safety and efficacy in AD patients [120]. This drug is currently being confirmed in a larger clinical trial. In addition, the indole derivatives designed by Noscira, SA, such as compounds 10a [121], 11a [122] display micro-molar inhibition against GSK3β in vitro. Furthermore, maleimide derivatives inhibit the GSK3β in the micro- and nano-molar ranges. Among them, compound 12a [123] shows higher selective GSK3β inhibiting activity with IC50 of 5.0 nM, suggesting its potential for therapy in AD.
Preventing tau interaction and neurofibrillary tangle accumulation could be a promising treatment for AD. Leuco-methylthioninium (LMTX, TRx0237) is a first-in-class tau aggregation inhibitor. It acts by preventing the formation and spread of NFTs, which comprise abnormal tau protein clusters causing neuronal cell toxicity and death in the brain of AD patients. Additionally, Leuco-methylthioninium has a role in inhibiting Aβ aggregation [124]. It is intended for the treatment of mild-to-moderate AD with a higher bioavailability, and also in Phase III clinical trials for its safety and clinical efficacy. In addition, quinolones derivatives, as potential blockers, prevent the tau aggregation before the formation of NFTs. Compound 13a patented by Universidad De Chile displayed high-binding affinity for tau protein with Kd level of 186 nM [125]. Furthermore, pyrazole derivatives are also novel tau aggregation inhibitors. Among them, compound 14a showed high inhibition against tau aggregation with IC50 of 1.49 μM, and also inhibited β-secretase with IC50 of 2.85 [126]. Myricanol derivatives, such as compound 15a, potently reduces tau protein levels upon treatment of HeLa cells (IC50 <10 μg/ml) [127]. Figure 8 & Table 5 list the main drugs and compounds to target tau protein.
Figure 8. Drugs in clinical trials and patented lead compounds to target tau protein.

Table 5.
The development of drugs and compounds to target tau protein.
| Compounds | Chemical class | Assignee | Patent number | Phases | Ref. |
|---|---|---|---|---|---|
| NP-031112 | Tiadiazolidindiones | Noscira SA, Spain | US0233971A1(2009) | Phase II | [128] |
| Leuco-methylthioninium | Phenothiazine | Wista Laboratories, Ltd, Singapore; TauRx Therapeutics, Ltd, Singapore | WO107706 A1(2012) | Phase III | [129] |
| 10a | Indole-dihydro-imidazole derivatives | Noscira, SA, Spain; The University of Queensland, Australia | WO167635 A1(2013) | Discovery | [121] |
| 11a | Indole-pyrimidine derivatives | Noscira, SA, Spain | WO149976 A1(2013) | Discovery | [122] |
| 12a | Maleimide derivatives | Universidad de Barcelona, Spain | WO113967 A1(2012) | Discovery | [123] |
| 13a | Quinoline derivatives | Universidad de Chile, Chile | WO134098 A1(2011) | Discovery | [125] |
| 14a | Pyrazole derivatives | Pharma Eight Co., Ltd, Japan | WO169576 A1(2013) | Discovery | [126] |
| 15a | Myricanol derivatives | University of South Florida, USA | WO152350 A1(2013) | Discovery | [127] |
Other potential therapeutic strategies in AD
Targeting mitochondrial dysfunction
A large body of evidence suggests that mitochondrial dysfunction and oxidative damage have a significant role in the early development of AD. Mitochondrial dysfunction leads to impaired calcium buffering and generation of reactive oxygen species, promoting synaptic damage and apoptosis [130]. Thus, strategies targeting basic mitochondrial processes, such as energy metabolism or free-radical generation possess great promise in AD treatment.
Latrepirdine, known as Dimebon, is a small-molecule compound developed by Medivation, Inc. and Pfizer for the treatment of AD. Previous research showed that the mechanism of its action was focused on AChE inhibition and NMDA antagonism [131]. While the recent study indicated that the potent neuroprotective effect of latrepirdine is attributed to the enhancement of mitochondrial function under stress conditions [132]. Moreover, it can protect neuronal mitochondria against Aβ-induced toxicity and improve mitochondrial membrane potential and ATP production [133], suggesting the potential for the treatment of neurodegenerative diseases [134].
In addition, several lead compounds to enhance mitochondrial function are under development. Indole and indoline derivatives designed by Bar Ilan University can reduce the production of oxidative stress, and excessive release of NO and pro-inflammatory cytokines. Compound 16a shows significant radical scavenging effect with IC50 of 70 nM, and protection against apoptosis induced by H2O2 with IC50 of 10 nM. Additionally, chroman derivatives such as Compound 17a also prevent damage to neuronal cells caused by mitochondrial dysfunction, oxidative stress in vitro with IC50 around 10 μM, and reduce the MPTP-induced deficit at the doses of 10 mg/kg/day in animal experimental models of mitochondrial dysfunction in vivo. Furthermore, both 4-(p-quinolyl)-2-hydroxybutanamide derivatives (Compound 18a) and catechol derivatives (Compound 19a) patented by Edison Pharmaceuticals, Inc. exhibit protection against oxidative damage in vitro with EC50 of less than 500 nM. Figure 9 & Table 6 list the main compounds to target mitochondrial dysfunction.
Figure 9. Drugs to target mitochondrial dysfunction.
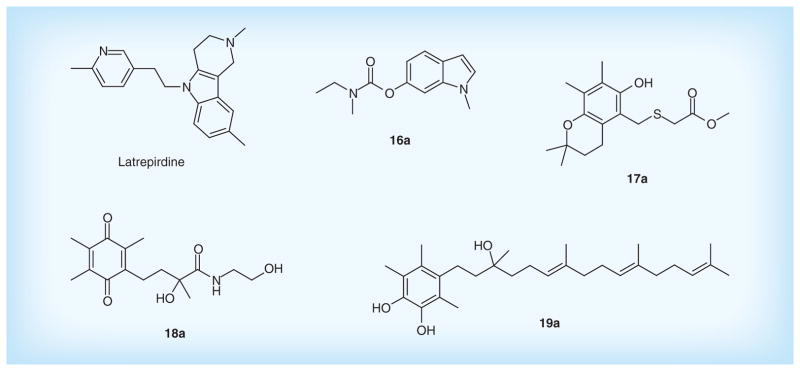
Table 6.
The development of other potential therapeutic strategies in Alzheimer’s disease.
| Compounds | Chemical class | Assignee | Patent number | Phases | Ref. |
|---|---|---|---|---|---|
| Mitochondrial therapy | |||||
| Latrepirdine | Tetracyclic pyrazinoindoles | Pfizer Inc., USA; Medivation Neurology, Inc., USA | WO039675A2 (2011) | Phase I | [135] |
| 16a | Indole, indoline derivatives | BarIlan University, Israel | WO150529 A2(2013) | Discovery | [136] |
| 17a | Chroman derivatives | Ampere Life Sciences, Inc., USA | US0267538 A1(2013) | Discovery | [137] |
| 18a | 4-(p-quinolyl)-2-hydroxybutanamide derivatives | Edison Pharmaceuticals, Inc., USA | US0289034 A1(2013) | Discovery | [138] |
| 19a | Catechol derivatives | Edison Pharmaceuticals, Inc., USA | WO174286 A1(2012) | Discovery | [139] |
| Neurotrophins | |||||
| SR57667 | Tetrahydropyridins | Sanofi-Aventis, France | WO025363 A1(1999) | Phase II | [140] |
| FK962 | Fluorobenzamide | Senju Pharmaceutical, Japan; Astellas Pharma Inc., Japan | WO133198A1(2008) | Phase II | [141] |
| T-817MA | Benzothiophene oxide derivative | Toyama Chemical Co., Ltd, Japan | WO145171A1(2009) | Phase II | [142] |
| 20a | Alkanone derivatives | Sanofi-Aventis, France | US20130303520 A1 | Discovery | [143] |
Neurotrophins
Neurotrophins are dimeric peptide hormones. The first member of the neurotrophin family to be discovered was nerve growth factor (NGF), which plays an important role in development and maintenance of the nervous system, as well as neuronal cell survival and differentiation [144]. Recently, an increasing number of studies have called attention to the correlation between the decreased NGF and AD [145]. Thus, neurotrophins have been acted as an attractive target for treatment of AD.
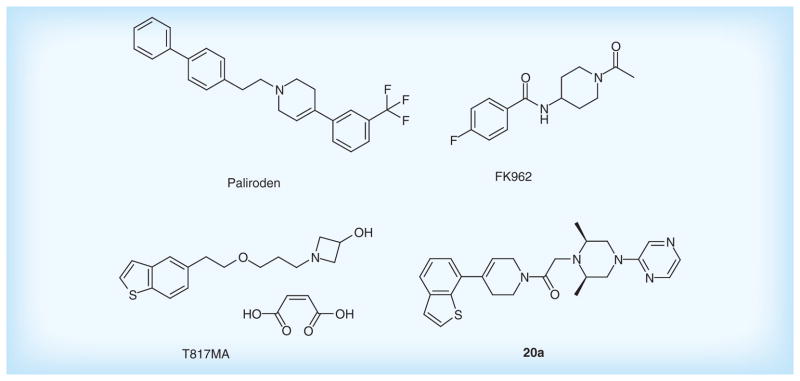
Paliroden (SR57667) developed by Sanofi-Aventis is a nonpeptide compound that activates the synthesis of endogenous neurotrophins [146]. A Phase II study is ongoing to evaluate its safety and tolerability in patients with mild-to-moderate AD. Subsequently, Sanofi also designed several novel compounds with high affinity for the p75NTR receptor of neurotrophins. The p75NTR receptor overexpressed in AD plays a predominant role in mechanisms leading to neuronal death via postischemic apoptosis. Among them, compound 20a shows high inhibitory activity on p75NTR with IC50 of 0.08 nM, suggesting a potential candidate drug for the treatment of AD.
FK962 designed by Astellas Pharma, Inc. is a neurite formation promoter. It can ameliorate cognitive impairment in rats by activation of the somatostatinergic nervous system in the hippocampus [147], and also promote neurite elongation and regeneration of cultured rat trigeminal ganglion cells [148]. However, the Phase II trial has been terminated due to its poor clinical efficacy for mild-to-moderate AD patients.
T-817MA, a neuroprotective agent, prevents Aβ-induced granule cell loss in the dentate gyrus of the hippocampus [149], and improves the motor and cognitive impairments owing to inhibiting neuronal degeneration in P301L tau transgenic mice [150]. The Phase II trial has been completed for its evaluations on safety and tolerability. Figure 10 & Table 6 list the main neurotrophins promoters.
Figure 10. Neurotrophins promoters.
Conclusion & future perspective
So far, the development of AD drugs has achieved some success in aspects of symptomatic improvement, whereas it also had several failures in aspects of disease modifying. Although many clinical and drug design research studies are undergoing, we have to recognize that it is quite difficult to successfully cure AD by a single treatment, which attributes to the complicated pathophysiology of AD. It is thought to be the cause not by defects in single gene, but rather by variations in a large number of genes, proteins and their complex interactions, ultimately leading to this disease [151].
Multitarget drug discovery could be a more promising strategy for AD treatment [152]. It could overcome the deficiency of poor efficacy for one-target-one-compound development. Several multitarget compounds already have been designed, such as dual binding AChE and BACE1 inhibitors [153], AChE inhibitors and antioxidants [154], which provide better therapeutic effects on both symptomatic and disease modifying in AD. In this point, natural products with polypharmacology may serve as good prototypes to design multitarget drugs for AD treatment [155,156]. Of course, it is a challenge to apply such multidrug remedies for AD treatment with clinical rationale. Thus, new approaches such as quantitative system pharmacology with computational system polypharmacology algorithms [157,158] and chemogenomics knowledgebases [159,160] will open up a broad and promising avenue to advance the discovery and development of new-generation drugs for AD in the future.
Supplementary Material
Executive summary.
Background
Current situation and pathological features of Alzheimer’s disease (AD).
Advance in research on AD.
Current treatments & cholinergic drugs
Current treatment: five approved drugs for AD treatments.
Cholinergic drugs: cholinergic hypothesis and main drugs in clinical trials and patented lead compounds.
Amyloid-targeted therapies
Decreasing Aβ generation.
The mechanism of Aβ generation.
The main β-secretase inhibitors in clinical trials and patented compounds.
The main γ-secretase inhibitors and modulators in clinical trials and patented compounds.
Preventing Aβ aggregation.
Drugs in clinical trials and patented lead compounds to prevent Aβ aggregation.
Promoting Aβ clearance.
Active AD immunotherapy and passive AD immunotherapy.
Drugs to target tau protein
The mechanism of tau pathology.
Tau-phosphorylating kinases inhibitors (GSK3β inhibitors).
Inhibit the tau aggregation and promote its degradation (tau aggregation inhibitors).
Other potential therapeutic strategies in AD
Targeting mitochondrial dysfunction.
Neurotrophins.
Future perspective
Multitarget drug design and discovery: a more promising strategy for AD treatment.
Acknowledgments
The authors are grateful to BN Dixit from the Pharmaceutical Science, University of Pittsburgh (Pittsburgh, PA, USA) for proof reading of the manuscript.
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial & competing interests disclosure
The authors would like to acknowledge the funding support for the laboratory at the University of Pittsburgh from the NIH R01DA025612 (Xie) and P30DA035778 (Xie), and Science and Technological Program for Dongguan’s Higher Education, Science and Research, and Health Care Institutions, Guangdong Province, China (Grant number 2012105102002). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Querfurth HW, Laferla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Wimo A, Winblad B, Jonsson L. The worldwide societal costs of dementia: estimates for 2009. Alzheimers Dement. 2010;6(2):98–103. doi: 10.1016/j.jalz.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 4.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 6.Goedert M, Spillantini MG, Crowther RA. Tau proteins and neurofibrillary degeneration. Brain Pathol. 1991;1(4):279–286. doi: 10.1111/j.1750-3639.1991.tb00671.x. [DOI] [PubMed] [Google Scholar]
- 7.Su B, Wang X, Nunomura A, et al. Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res. 2008;5(6):525–532. doi: 10.2174/156720508786898451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66(2):137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Lima F, Barksdale BR, Rojas JC. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochem Pharmacol. 2013;88(4):584–593. doi: 10.1016/j.bcp.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Holmes C. Review: systemic inflammation and Alzheimer’s disease. Neuropathol Appl Neurobiol. 2013;39(1):51–68. doi: 10.1111/j.1365-2990.2012.01307.x. [DOI] [PubMed] [Google Scholar]
- 11.Babic T. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;67(4):558. doi: 10.1136/jnnp.67.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;25(1):CD005593. doi: 10.1002/14651858.CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birks J, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2006;25(1):CD001190. doi: 10.1002/14651858.CD001190.pub2. [DOI] [PubMed] [Google Scholar]
- 14.Nordberg A. Mechanisms behind the neuroprotective actions of cholinesterase inhibitors in Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(2 Suppl 1):S12–S18. doi: 10.1097/01.wad.0000213804.59187.2d. [DOI] [PubMed] [Google Scholar]
- 15.Reisberg B, Doody R, Stoffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 16.Debiopharm SA. Switzerland. WO2009120774A2. 2009
- 17.The General Hospital Corporation, USA; National Institute of Aging Office of Technology Transfer, National Institutes of Health. WO2010117727A2. 2010
- 18.Galantos Pharma GmbH, Germany. WO2009127218A1. 2009
- 19.Consejo Superior de Investigaciones Cientificas CSIC, Spain; Universidad Autonoma de Madrid UAM. WO2011051535A1. 2011
- 20.Teva Pharmaceutical Industries, Ltd, Israel. US20070232691A1. 2007
- 21.Solvay Pharmaceuticals BV, Netherlands. WO2007128674A2. 2007
- 22.Envivo Pharmaceuticals, Inc., USA. WO2013169646A1. 2013
- 23.Niconovum USA, Inc., USA. US20110274628A1. 2011
- 24.Jia JY, Zhao QH, Liu Y, et al. Phase I study on the pharmacokinetics and tolerance of ZT-1, a prodrug of huperzine A, for the treatment of Alzheimer’s disease. Acta Pharmacol Sin. 2013;34(7):976–982. doi: 10.1038/aps.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson D, Cohen S, Inglis F, et al. Results of a double-blind Phase 2b study of monthly mimopezil implants compared with oral donepezil in patients with moderate Alzheimer’s disease (BRAINZ study) Alzheimers Dement. 2009;5(4):P85. [Google Scholar]
- 26.Winblad B, Giacobini E, Frolich L, et al. Phenserine efficacy in Alzheimer’s disease. J Alzheimers Dis. 2010;22(4):1201–1208. doi: 10.3233/JAD-2010-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maelicke A, Hoeffle-Maas A, Ludwig J, et al. Memogain is a galantamine pro-drug having dramatically reduced adverse effects and enhanced efficacy. J Mol Neurosci. 2010;40(1–2):135–137. doi: 10.1007/s12031-009-9269-5. [DOI] [PubMed] [Google Scholar]
- 28.Weinreb O, Amit T, Bar-Am O, Youdim MB. Ladostigil: a novel multimodal neuroprotective drug with cholinesterase and brain-selective monoamine oxidase inhibitory activities for Alzheimer’s disease treatment. Curr Drug Targets. 2012;13(4):483–494. doi: 10.2174/138945012799499794. [DOI] [PubMed] [Google Scholar]
- 29.Fisher A. M1 muscarinic agonists target major hallmarks of Alzheimer’s disease – the pivotal role of brain M1 receptors. Neurodegener Dis. 2008;5(3–4):237–240. doi: 10.1159/000113712. [DOI] [PubMed] [Google Scholar]
- 30.Fisher A, Brandeis R, Bar-Ner RH, et al. AF150(S) and AF267B. M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J Mol Neurosci. 2002;19(1–2):145–153. doi: 10.1007/s12031-002-0025-3. [DOI] [PubMed] [Google Scholar]
- 31.Prickaerts J, Van Goethem NP, Chesworth R, et al. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2012;62(2):1099–1110. doi: 10.1016/j.neuropharm.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 32.Zawieja P, Kornprobst JM, Metais P. 3-(2,4-dimethoxybenzylidene)-anabaseine. a promising candidate drug for Alzheimer’s disease? Geriatr Gerontol Int. 2012;12(3):365–371. doi: 10.1111/j.1447-0594.2011.00827.x. [DOI] [PubMed] [Google Scholar]
- 33.Hardy J. Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006;9(3 Suppl):151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 34.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 35.Hamley IW. The amyloid beta peptide: a chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem Rev. 2012;112(10):5147–5192. doi: 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- 36.Checler F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J Neurochem. 1995;65(4):1431–1444. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- 37.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 38.May PC, Dean RA, Lowe SL, et al. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci. 2011;31(46):16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martenyi F, Dean RA, Lowe S, et al. BACE inhibitor LY2886721 safety and central and peripheral PK and PD in healthy subjects (HSs) Alzheimers Dement. 2012;8(4):P583–P584. [Google Scholar]
- 40.Forman M, Palcza J, Tseng J, et al. The novel BACE inhibitor MK-8931 dramatically lowers cerebrospinal fluid Aβ peptides in healthy subjects following single-and multiple-dose administration. Alzheimers Dement. 2012;8(4):704–705. [Google Scholar]
- 41.Pfizer Inc., USA. WO2012172449A1. 2012
- 42.Rinat Neuroscience Corp., USA. WO2004032868A2
- 43.GlaxoSmithKline Biologicals SA, Belgium, Affiris AG. WO2013148851A1. 2013
- 44.Eli Lilly and Co., USA. WO2011005738A1. 2011
- 45.Eli Lilly and Co., USA. WO2009134617A1. 2009
- 46.Schering Corp., USA. US20070287692A1. 2007
- 47.Eli Lilly and Co., USA. US20070299053A1. 2007
- 48.Bristol-Myers Squibb Co., USA. US20100260837A1. 2010
- 49.Waratah Pharmaceuticals Inc., Can. US20100105631 A1. 2010
- 50.Wyeth, John, and Brother Ltd, USA. WO2009089237A1. 2009
- 51.Pfizer Inc., USA. US20050215610A1. 2005
- 52.Elan Pharmaceuticals, Inc., USA. US20080045499 A1. 2008
- 53.Chiesi Farmaceutici SpA, Italy. WO2011015287A2. 2011
- 54.Schering Corp., USA. US20110027264 A1. 2011
- 55.Pfizer Inc., USA. WO2012131539A1. 2012
- 56.EnVivo Pharmaceuticals, Inc., USA. US20130165486 A1. 2013
- 57.Evin G, Hince C. BACE1 as a therapeutic target in Alzheimer’s disease: rationale and current status. Drugs Aging. 2013;30(10):755–764. doi: 10.1007/s40266-013-0099-3. [DOI] [PubMed] [Google Scholar]
- 58.De Strooper B, Annaert W, Cupers P, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398(6727):518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 59.Blennow K, Zetterberg H, Haass C, Finucane T. Semagacestat’s fall. where next for AD therapies? Nat Med. 2013;19(10):1214–1215. doi: 10.1038/nm.3365. [DOI] [PubMed] [Google Scholar]
- 60.Doody RS, Raman R, Farlow M, et al. A Phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 61.Albright CF, Dockens RC, Meredith JE, Jr, et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J Pharmacol Exp Ther. 2013;344(3):686–695. doi: 10.1124/jpet.112.199356. [DOI] [PubMed] [Google Scholar]
- 62.Dockens R, Wang JS, Castaneda L, et al. A placebo-controlled, multiple ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of avagacestat (BMS-708163) in healthy young and elderly subjects. Clin Pharmacokinet. 2012;51(10):681–693. doi: 10.1007/s40262-012-0005-x. [DOI] [PubMed] [Google Scholar]
- 63.Coric V, Van Dyck CH, Salloway S, et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a Phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012;69(11):1430–1440. doi: 10.1001/archneurol.2012.2194. [DOI] [PubMed] [Google Scholar]
- 64.Pasinetti G, Rosen Z, Grossman H. Nic5–15: a novel natural gamma-secretase inhibitor that attenuates brain beta-amyloid content and improves cognition. Alzheimers Dement. 2009;5(4):e28. [Google Scholar]
- 65.Grossman H, Marzloff G, Luo X, Leroith D, Sano M, Pasinetti G. NIC5–15 as a treatment for Alzheimer’s: safety, pharmacokinetics and clinical variables. Alzheimers Dement. 2009;5(4):P259. [Google Scholar]
- 66.Martone RL, Zhou H, Atchison K, et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein γ-secretase for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther. 2009;331(2):598–608. doi: 10.1124/jpet.109.152975. [DOI] [PubMed] [Google Scholar]
- 67.Lanz TA, Wood KM, Richter KE, et al. Pharmacodynamics and pharmacokinetics of the gamma-secretase inhibitor PF-3084014. J Pharmacol Exp Ther. 2010;334(1):269–277. doi: 10.1124/jpet.110.167379. [DOI] [PubMed] [Google Scholar]
- 68.Anderson JJ, Holtz G, Baskin PP, et al. Reductions in beta-amyloid concentrations in vivo by the gamma-secretase inhibitors BMS-289948 and BMS-299897. Biochem Pharmacol. 2005;69(4):689–698. doi: 10.1016/j.bcp.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 69.Hall A, Patel TR. Gamma-secretase modulators: current status and future directions. Prog Med Chem. 2014;53:101–145. doi: 10.1016/B978-0-444-63380-4.00003-2. [DOI] [PubMed] [Google Scholar]
- 70.Imbimbo BP, Hutter-Paier B, Villetti G, et al. CHF5074, a novel gamma-secretase modulator, attenuates brain beta-amyloid pathology and learning deficit in a mouse model of Alzheimer’s disease. Br J Pharmacol. 2009;156(6):982–993. doi: 10.1111/j.1476-5381.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ross J, Sharma S, Winston J, et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: a 12-week, double-blind, placebo-controlled study. Curr Alzheimer Res. 2013;10(7):742–753. doi: 10.2174/13892037113149990144. [DOI] [PubMed] [Google Scholar]
- 72.Hashimoto T, Ishibashi A, Hagiwara H, Murata Y, Takenaka O, Miyagawa T. E2012: a novel gamma-secretase modulator-pharmacology part. Alzheimers Dement. 2010;6(4):S242. [Google Scholar]
- 73.Savelieff MG, Lee S, Liu Y, Lim MH. Untangling amyloid-beta, tau, and metals in Alzheimer’s disease. ACS Chem Biol. 2013;8(5):856–865. doi: 10.1021/cb400080f. [DOI] [PubMed] [Google Scholar]
- 74.Gervais F, Paquette J, Morissette C, et al. Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28(4):537–547. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 75.Aisen PS, Gauthier S, Ferris SH, et al. Tramiprosate in mild-to-moderate Alzheimer’s disease – a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study) Arch Med Sci. 2011;7(1):102–111. doi: 10.5114/aoms.2011.20612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma K, Thomason LA, Mclaurin J. Scyllo-inositol, preclinical, and clinical data for Alzheimer’s disease. Adv Pharmacol. 2012;64:177–212. doi: 10.1016/B978-0-12-394816-8.00006-4. [DOI] [PubMed] [Google Scholar]
- 77.Mandel SA, Amit T, Kalfon L, Reznichenko L, Weinreb O, Youdim MB. Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: special reference to epigallocatechin gallate (EGCG) J Alzheimers Dis. 2008;15(2):211–222. doi: 10.3233/jad-2008-15207. [DOI] [PubMed] [Google Scholar]
- 78.Adlard PA, Cherny RA, Finkelstein DI, et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59(1):43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 79.Adlard PA, Sedjahtera A, Gunawan L, et al. A novel approach to rapidly prevent age-related cognitive decline. Aging Cell. 2013;13(2):351–359. doi: 10.1111/acel.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adlard PA, Cherny RA, Finkelstein DI, et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron. 2008;59(1):43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 81.Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a Phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7(9):779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 82.Cytokine Pharmasciences, Inc. US20080096909 A1. 2008
- 83.Almqvist Fredrik, Sweden. WO2009134203A1. 2009
- 84.Neuropore Therapies, Inc., USA. WO2013134371A1. 2013
- 85.Bellus Health Limited, Switzerland. WO2010054485A1. 2010
- 86.Elan Pharmaceuticals, Inc., USA. WO2012173808A1. 2012
- 87.Prana Biotechnology Ltd, Australia. WO2008074068A1. 2008
- 88.Prana Biotechnology Ltd, Australia. WO2010071944A1. 2010
- 89.Max-Delbruck-Centrum Fur Molekulare Medizin, Germany. US20090117040 A1. 2009
- 90.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 91.Winblad B, Andreasen N, Minthon L, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease. randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11(7):597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- 92.Muhs A, Hickman DT, Pihlgren M, et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc Natl Acad Sci USA. 2007;104(23):9810–9815. doi: 10.1073/pnas.0703137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Galimberti D, Ghezzi L, Scarpini E. Immunotherapy against amyloid pathology in Alzheimer’s disease. J Neurol Sci. 2013;333(1–2):50–54. doi: 10.1016/j.jns.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 94.Savage MJ, Wu G, Mccampbell A, et al. A novel multivalent Abeta peptide vaccine with preclinical evidence of a central immune response that generates antisera recognizing a wide range of a beta peptide species. Alzheimers Dement. 2010;6(4):S142. [Google Scholar]
- 95.Moreth J, Mavoungou C, Schindowski K. Passive anti-amyloid immunotherapy in Alzheimer’s disease. What are the most promising targets? Immunity Ageing. 2013;10(1):1–9. doi: 10.1186/1742-4933-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kerchner GA, Boxer AL. Bapineuzumab. Expert Opin Biol Ther. 2010;10(7):1121–1130. doi: 10.1517/14712598.2010.493872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grundman M, Dibernardo A, Raghavan N, Krams M, Yuen E. 2012: a watershed year for Alzheimer’s disease research. J Nutr Health Aging. 2013;17(1):51–53. doi: 10.1007/s12603-013-0002-2. [DOI] [PubMed] [Google Scholar]
- 98.Samadi H, Sultzer D. Solanezumab for Alzheimer’s disease. Expert Opin Biol Ther. 2011;11(6):787–798. doi: 10.1517/14712598.2011.578573. [DOI] [PubMed] [Google Scholar]
- 99.Freeman GB, Lin JC, Pons J, Raha NM. 39-week toxicity and toxicokinetic study of ponezumab (PF-04360365) in cynomolgus monkeys with 12-week recovery period. J Alzheimer’s Dis. 2012;28(3):531–541. doi: 10.3233/JAD-2011-110869. [DOI] [PubMed] [Google Scholar]
- 100.McCleery J, Molena EJ, Worrall RE. Passive immunisation with monoclonal anti-Abeta antibodies for the treatment of Alzheimer’s disease. Cochrane Database System Rev. 2012;11:CD010188. [Google Scholar]
- 101.Bohrmann B, Baumann K, Benz J, et al. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimer’s Dis. 2012;28(1):49–69. doi: 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- 102.Adolfsson O, Pihlgren M, Toni N, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;32(28):9677–9689. doi: 10.1523/JNEUROSCI.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cytos Biotechnology AG, Switz.; Novartis Pharma AG. WO2004016282A1. 2004
- 104.Janssen Alzheimer Immunotherapy, Ireland. US20120276116 A1. 2012
- 105.Merck & Co., Inc., USA. WO2006121656A2. 2006
- 106.AC Immune SA, Switzerland; Genentech, Inc. WO2008156622A1. 2008
- 107.United Biomedical, Inc., USA. US20110070255 A1. 2011
- 108.Elan Pharma International Ltd, Ireland; Wyeth, John, and Brother Ltd. O2009017467A1. 2009
- 109.Washington University, USA; Eli Lilly and Co. US20110158986 A1. 2011
- 110.Rinat Neuroscience Corp., USA. WO2004032868A2. 2004
- 111.GlaxoSmithKline Biologicals SA, Belgium; Affiris AG. WO2013020722A2. 2013
- 112.F. Hoffmann-La Roche AG, Switzerland; MorphosysImmune AG. US20130136747 A1. 2013
- 113.AC Immune SA, Switzerland; Genentech, Inc. WO2012016173A2. 2012
- 114.Ballatore C, Brunden KR, Trojanowski JQ, Lee VM, Smith AB, 3rd, Huryn DM. Modulation of protein-protein interactions as a therapeutic strategy for the treatment of neurodegenerative tauopathies. Curr Top Med Chem. 2011;11(3):317–330. doi: 10.2174/156802611794072605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156(6):1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 117.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6(6):464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 118.Rockenstein E, Torrance M, Adame A, et al. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27(8):1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dominguez JM, Fuertes A, Orozco L, Del Monte-Millan M, Delgado E, Medina M. Evidence for irreversible inhibition of glycogen synthase kinase-3beta by tideglusib. J Biol Chem. 2012;287(2):893–904. doi: 10.1074/jbc.M111.306472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Del Ser T, Steinwachs KC, Gertz HJ, et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimer’s Dis. 2013;33(1):205–215. doi: 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- 121.Noscira, SA, Spain; The University of Queensland. WO2013167635A1. 2013
- 122.Noscira, SA, Spain. WO2013149976A1. 2013
- 123.Consejo Superior de Investigaciones Cientificas CSIC, Spain. WO2012113967 A1. 2012
- 124.Rember Wischik C. Issues in design of a Phase 3 disease modifying clinical trial of Tau aggregation inhibitor therapy in Alzheimer’s disease. Alzheimers Dement. 2009;5(4 Suppl):P74. [Google Scholar]
- 125.Universidad de Chile, Chile. WO2011134098 A1. 2011
- 126.Pharma Eight Co., Ltd Japan. WO2012141228A1. 2012
- 127.University of South Florida, USA. WO2013152350A1. 2013
- 128.Neuropharma, SA, Spain; Noscira, SA, USA. 20090233971A1. 2009
- 129.WisTa Laboratories Ltd, Singapore. WO2012107706A1. 2012
- 130.Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2013;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Giorgetti M, Gibbons JA, Bernales S, et al. Cognition-enhancing properties of Dimebon in a rat novel object recognition task are unlikely to be associated with acetylcholinesterase inhibition or N-methyl-D-aspartate receptor antagonism. J Pharmacol Exp Ther. 2010;333(3):748–757. doi: 10.1124/jpet.109.164491. [DOI] [PubMed] [Google Scholar]
- 132.Eckert SH, Eckmann J, Renner K, Eckert GP, Leuner K, Muller WE. Dimebon ameliorates amyloid-beta induced impairments of mitochondrial form and function. J Alzheimers Dis. 2012;31(1):21–32. doi: 10.3233/JAD-2012-120310. [DOI] [PubMed] [Google Scholar]
- 133.Weisova P, Alvarez SP, Kilbride SM, et al. Latrepirdine is a potent activator of AMP-activated protein kinase and reduces neuronal excitability. Transl Psychiatry. 2013;3:e317. doi: 10.1038/tp.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sabbagh MN, Shill HA. Latrepirdine, a potential novel treatment for Alzheimer’s disease and Huntington’s chorea. Curr Opin Investig Drugs. 2010;11(1):80–91. [PMC free article] [PubMed] [Google Scholar]
- 135.Pfizer Inc., USA; Medivation Neurology, Inc. WO2011039675A2. 2011
- 136.Yissum Research Development Company of the Hebrew University of Jerusalem Ltd, Israel; Bar Ilan University. WO2013150529A2. 2013
- 137.Ampere Life Sciences, Inc. US20130267538 A1. 2013
- 138.Edison Pharmaceuticals, Inc., USA. US20130289034 A1. 2013
- 139.Edison Pharmaceuticals, Inc., USA. WO2012174286A1. 2012
- 140.Sanofi, France. WO9925363A1. 1999
- 141.Senju Pharmaceutical Co., Ltd, Japan; Astellas Pharma Inc. WO2008133198A1. 2008
- 142.Toyama Chemical Co., Ltd, Japan. WO2009145171A1. 2009
- 143.Sanofi-Aventis, France. US20130303520 A1. 2013
- 144.Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nagahara AH, Merrill DA, Coppola G, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15(3):331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jacobsen JS, Reinhart P, Pangalos MN. Current concepts in therapeutic strategies targeting cognitive decline and disease modification in Alzheimer’s disease. NeuroRx. 2005;2(4):612–626. doi: 10.1602/neurorx.2.4.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tokita K, Inoue T, Yamazaki S, et al. FK962, a novel enhancer of somatostatin release, exerts cognitive-enhancing actions in rats. Eur J Pharmacol. 2005;527(1–3):111–120. doi: 10.1016/j.ejphar.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 148.Kishimoto Y, Yabuta C, Shearer TR, Azuma M. FK962 promotes neurite elongation and regeneration of cultured rat trigeminal ganglion cells. possible involvement of GDNF. Invest Ophthalmol Vis Sci. 2012;53(9):5312–5319. doi: 10.1167/iovs.11-8957. [DOI] [PubMed] [Google Scholar]
- 149.Nguyen PT, Kimura T, Ho SA, Tran AH, Ono T, Nishijo H. Ameliorative effects of a neuroprotective agent, T-817MA, on place learning deficits induced by continuous infusion of amyloid-beta peptide (1–40) in rats. Hippocampus. 2007;17(6):443–455. doi: 10.1002/hipo.20281. [DOI] [PubMed] [Google Scholar]
- 150.Fukushima T, Nakamura A, Iwakami N, et al. T-817MA, a neuroprotective agent, attenuates the motor and cognitive impairments associated with neuronal degeneration in P301L tau transgenic mice. Biochem Biophys Res Commun. 2011;407(4):730–734. doi: 10.1016/j.bbrc.2011.03.091. [DOI] [PubMed] [Google Scholar]
- 151.Ray M, Ruan J, Zhang W. Variations in the transcriptome of Alzheimer’s disease reveal molecular networks involved in cardiovascular diseases. Genome Biol. 2008;9(10):R148. doi: 10.1186/gb-2008-9-10-r148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Leon R, Garcia AG, Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med Res Rev. 2013;33(1):139–189. doi: 10.1002/med.20248. [DOI] [PubMed] [Google Scholar]
- 153.Zhu Y, Xiao K, Ma L, et al. Design, synthesis and biological evaluation of novel dual inhibitors of acetylcholinesterase and beta-secretase. Bioorg Med Chem. 2009;17(4):1600–1613. doi: 10.1016/j.bmc.2008.12.067. [DOI] [PubMed] [Google Scholar]
- 154.Rosini M, Andrisano V, Bartolini M, et al. Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem. 2005;48(2):360–363. doi: 10.1021/jm049112h. [DOI] [PubMed] [Google Scholar]
- 155.Liu H, Liang F, Su W, et al. Lifespan extension by n-butanol extract from seed of Platycladus orientalis inCaenorhabditis elegans. J Ethnopharmacol. 2013;147(2):366–372. doi: 10.1016/j.jep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 156.Russo P, Frustaci A, Del Bufalo A, Fini M, Cesario A. Multitarget drugs of plants origin acting on Alzheimer’s disease. Curr Med Chem. 2013;20(13):1686–1693. doi: 10.2174/0929867311320130008. [DOI] [PubMed] [Google Scholar]
- 157.Wang L, Ma C, Wipf P, Liu H, Su W, Xie XQ. TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013;15(2):395–406. doi: 10.1208/s12248-012-9449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ma C, Wang L, Xie XQ. Ligand Classifier of Adaptively Boosting Ensemble Decision Stumps (LiCABEDS) and its application on modeling ligand functionality for 5HT-subtype GPCR families. J Chem Inf Model. 2011;51(3):521–531. doi: 10.1021/ci100399j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu H, Wang L, Su W, Xie XQ. AlzPlatform: an Alzheimer’s disease domain-specific chemogenomic knowledgebase for polypharmacology and target identification research. J Chem Inf Model. 2014;54(4):1050–1060. doi: 10.1021/ci500004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xie X, Wang L, Liu H, Ouyang Q, Fang C, Su W. Chemogenomics knowledge based polypharmacology analyses of drug abuse related G-protein coupled receptors and their ligands. Front Pharmacol. 2014;5(3):1–11. doi: 10.3389/fphar.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.