

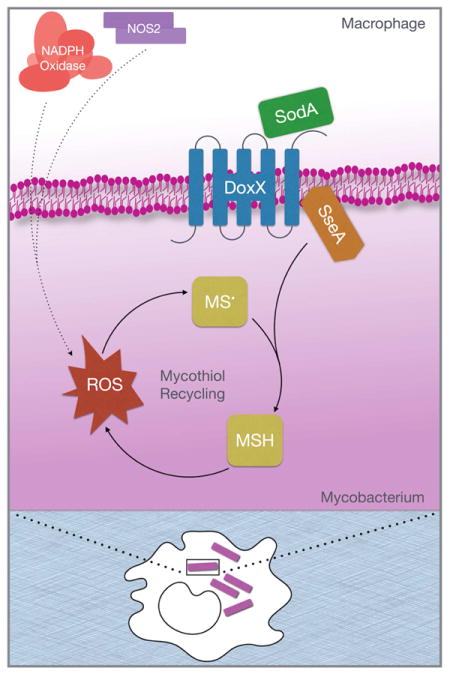
SUMMARY
M. tuberculosis (Mtb) survives a hostile environment within the host that is shaped in part by oxidative stress. The mechanisms used by Mtb to resist these stresses remain ill-defined because the complex combination of oxidants generated by host immunity is difficult to accurately recapitulate in vitro. We performed a genome-wide genetic interaction screen to comprehensively delineate oxidative stress resistance pathways necessary for Mtb to resist oxidation during infection. Our analysis predicted functional relationships between the superoxide-detoxifying enzyme (SodA), an integral membrane protein (DoxX), and a predicted thiol-oxidoreductase (SseA). Consistent with that, SodA, DoxX and SseA form a membrane-associated oxidoreductase complex (MRC) that physically links radical detoxification with cytosolic thiol homeostasis. Loss of any MRC component correlated with defective recycling of mycothiol and accumulation of cellular oxidative damage. This previously uncharacterized coordination between oxygen radical detoxification and thiol homeostasis is required to overcome the oxidative environment Mtb encounters in the host.
Graphical Abstract
Introduction
A cornerstone of metazoan immunity is the production of anti-microbial oxygen and nitrogen radicals by phagocytes. In mammals, superoxide (O2−) is generated by the phagocyte NADPH oxidase and xanthine oxidase systems (Halliwell and Gutteridge, 2007). While this reactive species can interact directly with its targets, the superoxide radical is also converted into a number of chemically-distinct oxidants, such as peroxide (H2O2), hypochlorite (HClO), hydroxyl radicals (OH•), and peroxynitrite ONOO−. Together, these species damage microbial DNA, lipids and proteins, as well as particularly susceptible cellular constituents such as iron-sulfur (4Fe-4S) cluster proteins.
The complexity of the phagocyte oxidative burst is matched by the numerous strategies used by bacterial pathogens, such as M. tuberculosis (Mtb), to resist these insults. Virtually all cells protect themselves from oxidative stress using a cytosolic thiol redox buffer, such as the tripeptide, glutathione. In Mtb, the functional analog of glutathione is the cysteine glycoconjugate, mycothiol (Newton et al., 1996). In addition to this common redox buffering system, Mtb stress defense mechanisms also include dedicated antioxidant enzymes such as superoxide dismutase (SOD), catalase/peroxidase (KatG), thioredoxin reductase (Tpx), alkylhydroperoxide reductase (AhpC) and peroxiredoxin (AhpE) (Bryk et al., 2002; Edwards et al., 2001; Jaeger et al., 2004; Wilson and Collins, 1996).
Despite the identification of several enzymes that could protect Mtb from defined oxidative stresses, it remains unclear how the activities of these pathways are coordinated. Genetic interaction (GI) studies have the capacity to systematically define functional relationships between genes or pathways. A GI is defined by two mutations that modify the phenotype of the other. Aggravating interactions often result from loss-of-function mutations in redundant genes that produce a greater than additive effect. Alleviating interactions occur between genes in the same pathway that depend upon one another for their function and therefore produce a less than additive effect when simultaneously mutated. In order to understand the functional network that Mtb employs to resist the oxidative stresses produced during infection, we delineated a comprehensive genetic interaction network centered on superoxide dismutase activity.
Results
Delineating the oxidative stress network during infection
The primary oxidant produced by the phagocyte oxidative burst is superoxide. Defining a comprehensive oxidative stress interaction network required an Mtb mutant that is sensitive to this radical, as well as the array of additional superoxide-derived oxidants produced in vivo. The major SOD of Mtb, SodA, is essential for bacterial viability, and therefore not useful for this GI study. However, the P-type ATPase (CtpC) responsible for metalating SodA with Mn2+ is known, and cells lacking ctpC are both viable and sensitive to superoxide (Padilla-Benavides et al., 2013). We leveraged this SodA hypomorph to generate a global genetic interaction map of oxidative stress resistance during infection in mice.
Saturated transposon libraries were generated in wild type (WT) Mtb and the ctpC::hyg background. As described previously (Sassetti and Rubin, 2003; Joshi et al., 2006) both libraries were subjected to a period of selection in the mouse spleen, an environment in which the bacteria encounter the full complexity of host-derived oxidants. Surviving mutants were recovered from these animals and the relative representation of each transposon mutant was compared between the WT and ctpC::hyg libraries to generate a map of 181 aggravating or alleviating mutations (Figure 1A–C). This number of interacting genes was consistent with that observed for highly-connected “hub” genes in S. cerevisiae and E. coli genetic interaction maps, and our previous GI studies in Mtb (Babu et al., 2014; Costanzo et al., 2010; Griffin et al., 2011; Joshi et al., 2006). Among the previously recognized ROS detoxification systems, the peroxiredoxin, ahpE, showed the strongest interaction with ctpC (Table S1), whereas weaker and statistically non-significant interactions were found with katG and ahpCD. In addition, a much more complex network emerged that was composed of several distinct functional classes of genes, discussed below (Figure 1C).
Figure 1. Genetic screen to define oxidative stress network.

A] Scheme for identifying genes that are involved in oxidative stress defense during infection. Saturated transposon libraries were generated in WT or ctpC::hyg strains and the respective libraries were used to infect mice. The relative fitness of each mutant was estimated by TnSeq. B] The aggravating and alleviating interactions identified by TnSeq analysis described in A]. Genes that are similarly required for in vivo growth in both genetic backgrounds will produce ratios near to 1. Alleviating or aggravating transposon insertions will produce numerically larger or smaller ratios in the mutant background, respectively. Dotted lines indicate significance thresholds (p < 0.05). C] Network map of mutants with predictable functions. Shorter edge lengths indicate stronger interactions. Genes of related functions are grouped in clusters. Red indicates aggravating interactions and black indicates alleviating interactions, throughout the figure.
Alleviating interactions were detected between ctpC and iron homeostatic genes, such as those in operons involved in siderophore synthesis (mbtA-N), the irtAB-encoded siderophore importer (Chavadi et al., 2011; Farhana et al., 2008), an iron-sulfur cluster assembly protein (rv1465), and the ferric uptake regulation protein (furA). This is consistent with the known role of Fe2+ in potentiating oxidative stress via the Fenton reactions (Winterbourn, 1995) and suggests that cytosolic ferric iron concentration is a significant determinant of oxidative damage during infection.
Several other interacting pathways indicate a major role for redox cofactor homeostasis in oxidative stress resistance in vivo. The reduction of both nicotinamide- (NAD+) and flavin-derived (F420) cofactors via succinate dehydrogenase (rv0247c-rv0249c) and the F420-dependent glucose dehydrogenase (fgd) (Hasan et al., 2010) were found to promote oxidative stress resistance. Mutations in central carbon metabolism and respiration produced both alleviating and aggravating effects, which are also likely related to the maintenance of the redox state of cofactors.
The aggravating effect of mutations in genes necessary for the synthesis or modification of the major outer membrane lipids, phthiocerol dimycocerosate and mycolic acid, could also be related to NADP+/NADPH homeostasis as mycobacteria regulate lipid anabolism as a mechanism of redox control (Singh et al., 2009). However, genes involved in subtle chemical modifications of mycolates, such as methylation (mmaA1) or cyclopropanation (mmaA2), also potentiated oxidative damage, suggesting a distinct role for the cell envelope in resistance. The observed function for ESX (Type VII) secretion systems in cell envelope biogenesis (Garces et al., 2010) may similarly explain the presence of these genes in the network.
Finally, mutations in cysteine synthetic and catabolic genes had opposing effects on oxidative stress resistance. Blocking cysteine synthesis via mutation of cysM aggravated the ctpC phenotype. Conversely, inhibition of cysteine catabolism to alanine via mutation of iscS alleviated this phenotype. These complementary genetic interactions suggested that the availability of cysteine, or small molecular weight thiols derived from cysteine, might be a critical determinant of oxidative stress resistance during infection. In sum, the architecture of the Mtb oxidative stress network affords a quantitative assessment of the cellular functions that are important for resisting the complex mixture of oxidants encountered during infection, and illustrates functional interactions between the canonical oxidative defense mechanism imparted by SOD activity and a variety of other cellular functions including thiol metabolism (Tables S1 and S2, and Figure 1C).
SodA exists in a membrane-associated thiol oxidoreductase complex
To understand the biochemical basis for the identified GI, we used a mass spectrometry-based approach to determine if members of the GI network formed stable protein complexes. When a FLAG-tagged allele of Rv3005c, one of the strong alleviating interactors of ctpC, was purified from detergent extracts of Mtb, two additional proteins were co-purified (Figure 2A and S1). One was SseA (Rv3283), another strong alleviating interactor of ctpC. The second was SodA, which depends on CtpC for its metalation. This complex of functionally related proteins was evolutionarily conserved, as we found that these three proteins also co-purified from lysates of M. smegmatis (Msm), a saprophytic relative of Mtb (Figure 2B). In addition to this conserved complex, a small number of additional Rv3005c-interacting partners were found in Mtb, which function in either sulfur metabolism or oxidative defense (Figure 2A).
Figure 2. DoxX, SseA, and SodA form an oxidoreductase complex.

A] FLAG-tagged DoxX was purified from detergent extracts of M. tuberculosis and interacting proteins were identified by MS/MS. B] A similar analysis was performed as [A] but from Msm expressing FLAG-DoxX. C] Recombinant FLAG-tagged DoxX or SseA was expressed in the indicated strain background, and SNAP-tagged DoxX, SseA or SodA was simultaneously expressed. FLAG-tagged proteins were purified and western blot analysis was performed using anti-tag antibody. D] Kinetics of DoxX and SseA enzyme activity using potassium ferricyanide as the electron acceptor and measuring absorbance at 420 nm. Enzyme complexes were affinity purified using FLAG-DoxX expressed in WT Msm and the enzyme assay was carried out at different pH values in the presence or absence of sulfite (left). Enzyme complexes were purified using FLAG-DoxX from the indicated mutant strains and activity was determined at pH 6 (middle). Enzyme complexes were purified using FLAG-SseA from the indicated mutant strains and kinetics was determined at pH 6 (right). All samples were normalized by relative protein content. Data are representative of three independent experiments.
Homology indicated that the two SodA-interacting proteins of Mtb could be involved in sulfur redox reactions. The hypothetical protein, Rv3005c, is predicted to contain 4–5 transmembrane domains and is weakly homologous to “Dox” proteins of chemotrophic archea that contribute to sulfur oxidation (Müller et al., 2004). SseA is predicted to be a member of a ubiquitous family of thiosulfate sulfurtransferases (TST) that are most commonly thought to alleviate cyanide toxicity by converting cyanide to thiocyanate (Cipollone et al., 2007). However, TST-like enzymes play diverse roles, and can also act as thiol oxidoreductases (Nandi et al., 2000). In particular, their ability to regenerate glutathione thiyl radicals has been proposed to explain the oxidative stress sensitivity of A. vinelandii TST mutants (Remelli et al., 2012). The possible roles for Rv3005c and SseA in thiol redox reactions, and their physical and genetic interactions with SodA led us to hypothesize that this three-protein complex might coordinate multiple enzymatic functions necessary for oxidative stress resistance during Mtb infection. Henceforth, we will refer to Rv3005c as “DoxX”.
To directly investigate if DoxX and SseA possess biochemical functions consistent with oxidative stress resistance, we examined the thiosulfate oxidizing activity of the complex using potassium ferricyanide as an electron acceptor. We generated mutant strains of both Msm and Mtb lacking SseA (ΔMsmeg_1809, Δrv3283) or DoxX (ΔMsmeg_2370, Δrv3005c). SodA mutants of Mtb are not viable, so an Msm mutant lacking the orthologous gene was generated (ΔMsmeg_6427). The physical interaction of DoxX, SseA and SodA in the different mutant backgrounds was confirmed by alternatively tagging either with SNAP- or FLAG-tags. DoxX interacts with SseA even in the absence of SodA. However, the interaction of SseA with SodA was dependent on DoxX, indicating that the latter protein nucleated the complex (Figure 2C). The native enzyme complex was purified from WT or mutant strains lacking each gene using either FLAG-tagged DoxX or FLAG-tagged SseA. The complex purified from WT cells possessed thiosulfate oxidation activity (Figure 2D). This reaction was inhibited by sulfite, similar to the inhibition observed for cyanide detoxification by other SseA homologs. Thiosulfate oxidation activity required both DoxX and SseA to be present in the complex, whereas SodA was dispensable (Figure 2D). Thus, DoxX is necessary for SseA activity, and these proteins form a membrane oxidoreductase complex (MRC) that is physically coupled to SodA.
The coordinated activities of MRC components relieves oxidative stress by maintaining thiol homeostasis
To investigate the interdependency between SseA, DoxX and SodA in the intact cell, we quantified the sensitivity of each mutant strain to oxidative stressors. Consistent with the apparent biochemical dependency between SseA and DoxX, mutation of these genes in Mtb (Figure 3B, S2B and S2D) or Msm (Figure 3A, S2C and S2E) increased sensitivity to tert-butyl-hydroperoxide (tBHP), cumene hydroperoxide (CHP), and the nitric oxide donor, DETA-NO. In contrast, only the ΔMsSodA mutant, but neither the ΔMsSseA or ΔMsDoxX mutants, was sensitive to the superoxide generators, plumbagin and menadione (Figure S2A). Hydrogen peroxide and xanthine oxidase-generated superoxide were not toxic to either WT or mutant strains at the concentrations tested (Figure S2).
Figure 3. Sensitivity of sodA, sseA and doxX mutant strains to redox stressors.

Effect of tBHP on viability of Msm WT and mutant strains A] and Mtb WT and mutant strains B]. Log phase cultures of the indicated strains were exposed to 1 mM tBHP for 1h and CFU were enumerated. Values represent the mean ± S.E. of duplicate determinations of experiments repeated thrice. **indicates p < 0.01 by unpaired student’s t test. The effect of diamide on Msm WT and mutant strains C] and Mtb WT and mutant strains D] was determined. CFUs were measured after 12 h of treatment with 5 mM diamide. Values represent the mean ± S.E. of duplicate determinations of experiments repeated thrice. *indicates p < 0.05 by unpaired student’s t test. E] Conversion of diamide to hydrazine was measured spectrophotometrically at 390 nM at the indicated time points. Values represent the mean ± S.E. (error bars) of three independent experiments. The rate of diamide conversion for Msm WT and ΔMsSodA was 0.24±0.08 and 0.21±0.07 μmoles/mL/h, respectively, and ΔMsDoxX and ΔMsSseA was 0.10±0.05 and 0.10±0.04 μmoles/mL/h, respectively. Ratiometric sensor response of Mrx1-roGFP2 was measured from the indicated Msm strains F] and Mtb strains H] by determining the fluorescence emission at 510 nm after excitation at 390 nm and 490 nm, which indicates the relative abundance of the oxidized and reduced roGFP respectively. Data are representative of three independent experiments. **indicates p < 0.01 by unpaired student’s t test. G] The redox state of the mycothiol pool was assessed during oxidative stress using the Mrx1-roGFP2 sensor. The indicated strains were exposed to 100 μM of cysteine, sodium sulfate or NaHS for 1 h and the ratiometric sensor response was measured. Lipid hydroperoxides were measured after incubation with FOX2 reagent, in Msm WT and mutant strains I] and Mtb WT and mutant strains J]. Data are normalized relative to the cell mass. “Log” and “Stat” represent exponential and stationary phases of growth. Data represent the mean of three independent replicates ± S. E. (error bars). **indicates p < 0.01 by unpaired student’s t test. The effect of pretreatment with 100 μM NaHS for 1 h on tBHP K] and diamide L] sensitivity of Mtb WT and mutant strains was determined. CFUs were enumerated. Values shown represent the mean ± S.E. of duplicate determinations of experiments repeated twice. **indicates p < 0.01 and *indicates p < 0.05 by unpaired student’s t test.
Unlike the superoxide generated by plumbagin and menadione, tBHP and CHP react with cytosolic thiols (Fernandes et al., 2010; Kučera et al., 2014). The specific sensitization of ΔsseA and ΔdoxX strains to tBHP and CHP could reflect a role for the encoded proteins in cytosolic thiol recycling. Consistent with this hypothesis, we observed that mutation of ΔsseA and ΔdoxX in either Mtb or Msm increased sensitivity to the thiol-specific oxidant, diamide (Figure 3C and 3D). To estimate the rate of thiol recycling in each mutant, we quantified the rate of diamide reduction to colorless hydrazine, which occurs as a consequence of thiol oxidation. Diamide reduction was significantly retarded in ΔDoxX and ΔSseA mutants, relative to WT (Figure 3E and S2J), indicating that thiol recycling was impaired. While the MsSodA mutant was equally sensitive to diamide as mutants lacking SseA or DoxX, diamide reduction was unimpaired. These results suggest that SseA and DoxX act as a complex to promote resistance to agents that disrupt thiol homeostasis. SodA does not directly influence thiol recycling, however the unanticipated requirement of SodA for resisting thiol-specific oxidation indicates that this protein plays an essential role in thiol homeostasis as a component of the MRC.
The most abundant small molecular weight thiol in mycobacteria is mycothiol (MSH). To determine if MRC influences MSH cycling, we used the Mrx1-roGFP reporter protein to assess the ratio of reduced:oxidized mycothiol (MSH:MSSM) (Bhaskar et al., 2014). In ΔMsSseA and ΔMsDoxX mutant strains, we found that the MSH:MSSM ratio was significantly lower than WT and ΔMsSodA strains (Figure 3F). A similar decrease in MSH:MSSM ratio was observed in the mutant Mtb strains, ΔMtSseA and ΔMtDoxX (Figure 3H), directly implicating the MRC in MSH recycling. The relative oxidation of the MSH/MSSH pool in the Mtb mutants was further confirmed by mass spectrometry (Figure S3).
The defect in thiol homeostasis expressed by MRC mutants under unperturbed axenic culture conditions suggested that ROS produced endogenously via respiration might be inefficiently detoxified in these bacteria. Indeed, we found that a stable product of cellular oxidation, lipid hydroperoxides, accumulated in ΔMsSseA, ΔMsDoxX, ΔMtSseA and ΔMtDoxX bacteria compared to WT controls (Figure 3I and 3J). The degree of lipid peroxidation observed in these mutants was significantly greater than even the ΔMsSodA mutant. This observation likely explains the presence of phospholipid synthetic genes in the oxidative stress network (Figure 1C). As these cultures contained catalase to reduce exogenous ROS, these data indicate that the MRC plays a significant role in detoxifying endogenously produced radicals.
We used a chemical complementation strategy to investigate whether the observed mycothiol homeostasis defect of MRC mutants was related to their oxidative stress sensitivity. We found that supplementation with 100 μM sulfur, in the form of cysteine, sulfate (SO42−) or hydrogen sulfide (H2S) could restore the MSH:MSSM ratio of ΔsseA and ΔdoxX mutants to WT levels in vitro (Figure 3G and 3H). The restoration of MSH:MSSH ratio also reversed the sensitivity of these mutants to diamide and tBHP (Figure 3K and 3L), demonstrating functional complementation. The differing redox potentials of the sulfur in these compounds did not alter this activity. In particular, the ability of sulfate, which has a lower reduction potential than MSH, to restore MSH:MSSM ratio indicated that these supplements were not directly reducing MSSM. Instead, sulfur supplementation likely complements the recycling defect by increasing MSH synthesis, as has been shown for glutathione synthesis in E. coli (Loewen, 1979).
MRC maintains thiol homeostasis during infection
Our initial genetic interaction screen was performed using a competitive infection of mouse spleen. To confirm the role of the MRC, we assessed the growth dynamics of the ΔMtDoxX and ΔMtSseA mutants in a variety of infection models. Bone marrow-derived macrophages were used to quantify the ability of these mutants to grow intracellularly. In resting macrophages, all mutants behaved similarly to WT bacteria. However, in macrophages that were prestimulated with interferon gamma (IFNγ), both ΔMtSseA and ΔMtDoxX mutants were unable to replicate (Figure 4A). As the antimicrobial effect of IFNγ is at least partially mediated by augmenting the production of ROS and RNI (Podinovskaia et al., 2013), the attenuation of these mutants was consistent with their sensitivity to oxidative stress. This intracellular growth defect was also apparent after low dose aerosol infection of both resistant (C57BL/6) and susceptible (C3HeB/FeJ) mice (Kramnik et al., 2000) with the ΔMtDoxX mutant (Figure 4B and 4C). All growth defects were specific to infection conditions, as both ΔMtDoxX and ΔMtSseA mutants grew at WT rates in vitro (Figure 4E and 4F).
Figure 4. MRC maintains thiol homeostasis in vitro and during infection.

A] Immortalized C57BL/6 bone marrow derived macrophages were infected with Mtb mutants in the presence of the indicated compounds. CFU were enumerated at the indicted time points. Groups of C57BL/6 mice B] or C3HeB/FeJ mice C] were infected with the indicated Mtb strains. Bacterial burdens in the lungs were determined at 4 weeks postinfection. Mice were inoculated with 202 ± 32 CFU of WT, 245 ± 55 CFU of ΔMtDoxX and 173 ± 47 CFU of the complemented mutant (errors indicate SD of duplicate measurements). D] In vivo chemical complementation. Groups of C57BL/6 mice were infected with the indicated strains. After 4 weeks, mice were administered saline (n = 3) or NaHS (n = 3) for 6 weeks by osmotic minipump. Resulting bacterial burdens in the lungs are shown. Growth measured by optical density (OD600) of Msm and mutant strains E] and Mtb and mutant strains F] grown in 7H9 media.
H2S is an endogenous metabolite of mammals, and is broadly distributed through tissue after administration of its sodium salt (NaHS) (Wagner et al., 2009). We leveraged the ability of this compound to chemically complement the in vitro defects of MRC mutants to test whether similar mechanisms were likely to underlie the attenuation of MRC mutants during infection. The addition of NaHS completely complemented the intracellular growth defects of ΔMtSseA and ΔMtDoxX mutants in IFNγ-stimulated macrophages (Figure 4A). To extend these findings to the mouse model, NaHS was delivered continually to animals for six weeks, beginning at 4 weeks postinfection. As we observed in macrophage infections, NaHS supplementation significantly increased the growth of ΔMtDoxX bacteria to near WT levels (Figure 4D). In both the macrophage and mouse infection models, NaHS supplementation had no discernable effect on the growth or replication of WT Mtb, indicating specific complementation of the MRC mutant phenotype. The ability of sulfur supplementation to complement the in vitro MSH redox defect, oxidative stress sensitivity, and growth during infection supports a primary role for the MRC in maintenance of thiol homeostasis in the oxidative environments encountered during infection.
DISCUSSION
This study describes a systematic genetic approach to decipher the functional network responsible for oxidative defense in Mtb during infection. The major SOD of Mtb, SodA, was found to form a stable complex with two-previously uncharacterized proteins, DoxX and SseA, which coordinately act to maintain the redox state of the MSH pool. DoxX and SseA were both required for thiol oxidoreductase activity of the MRC and resistance to specific oxidative stresses that affect the thiol pool. In contrast, SodA was dispensable for thiol recycling, but required for resisting thiol-specific oxidants. These observations imply a biochemical model in which the SseA/DoxX portion of the complex is responsible for recycling thiyl radicals that are formed upon cytosolic thiol oxidation either by peroxides or diamide (Figure S4). This mechanism is analogous to the canonical glutathione (GSH) system, which dissipates free radicals through the formation of a GSH thiyl radical that is recycled through conversion to GSSG by glutaredoxin and then GSH by glutathione reductase (Starke et al., 2003). However, SseA belongs to a large family of “rhodanese” enzymes that form a transient sulfane sulfur during catalysis (Toohey, 1989) and thereby use a wide variety of substrates that could include other low molecular weight thiols in addition to MSH. In mycobacteria, this promiscuity may be advantageous, as alternative thiol redox buffering systems such as those using ergothionine have been described (Kumar et al., 2011). The requirement for SodA in the MRC is likely due to its ability to detoxify radicals that are known to be generated during the SseA reaction (Remelli et al., 2012). Thus, the physical association between SOD and thiol oxidoreductase activities provides a biochemical explanation for the unexpected diamide sensitivity of SodA-deficient mutants.
While genome-wide GI networks have been described in a number of organisms, these interactions are generally determined under defined in vitro conditions. The plasticity of cellular metabolism implies that the structure of these networks will change in different environments. Indeed, the unanticipated coordination between superoxide detoxification and thiol homeostasis described in this study was discovered because the Mtb GI network was defined under relevant environmental conditions that included the complex mixture of host-derived oxidants. These considerations suggest that GI networks are most valuable when they are examined under the correct conditions. In addition to defining functional pathways, GI networks are also useful for identifying genetic synergies that could be exploited therapeutically. Novel treatments that inhibit these synergistic defense mechanisms could be used to accelerate tuberculosis therapy.
EXPERIMENTAL PROCEDURES
Strains and plasmids used in the study
M. tuberculosis H37Rv and M. smegmatis mc2155 were grown at 37°C in 7H9 and 7H10 media (BD Biosciences) supplemented with 10% OADC enrichment plus hygromycin (50 μg/mL) and kanamycin (25 μg/mL) as needed. ΔleuD ΔpanCD auxotrophic Mtb was grown in the presence of lysine and pantothenate (Sampson et al., 2004).
Genetic analysis
Msm and Mtb mutants were constructed following by allelic exchange, as described previously (Murphy et al., 2015; van Kessel and Hatfull, 2007). Transposon libraries were made using the pMycoMarT7 transposon in WT and ctpC::hyg strains (Sassetti et al., 2001). 106 colony-forming units (cfu) of WT or ctpC mutant libraries were introduced intravenously by tail vein injection into four groups of three C57BL/6 mice. At days 0 and 32, the mice were sacrificed and the surviving bacteria isolated by plating spleen homogenates. The composition of these libraries was compared by deep sequencing amplicons derived from the transposon-chromosome junctions, as described in the Supplemental section.
Infection models
Immortalized C57BL/6 bone marrow derived macrophages seeded at 1 ×105 cells per well in 24-well plates were infected with Mtb at an MOI of 3. Extracellular bacteria were washed away 4 h later. At day 7, infected macrophages were washed, lysed, and CFU were enumerated. IFN-γ was added at a concentration of 25 ng/mL. C57BL/6 and C3HeB/FeJ were infected using an aerosol generation chamber (Glas-Col) standardized to deliver ~200 cfu of Mtb per mouse. At the indicated time points, animals were sacrificed and CFU enumerated from tissue homogenates.
Detailed procedures can be found in the Supplemental information.
Supplementary Material
Highlights.
In vivo Mtb screen identifies the membrane-associated oxidoreductase complex (MRC)
The MRC coordinates ROS detoxification and thiol homeostasis during infection
Loss of the MRC reduces thiol recycling and increases sensitivity to oxidative damage
Mtb mutants of MRC component are highly attenuated
In Brief.
Host-derived oxidants impose stress on Mtb and limit its growth. How Mtb coordinates resistance to oxidative stress remains unclear. Nambi et al. define the Mtb oxidative stress network during infection and identify the membrane-associated oxidoreductase complex, a three-protein complex that coordinates ROS detoxification with thiol homeostasis as required for infection.
Acknowledgments
We thank J. Lezsyk, K.G. Papavinasasundaram, C. Smith and X. Meniche for technical assistance, A. Brass, K. Rhee and M. Volkert for valuable suggestions and reagents, and upport from NIH awards F32A1093049 (JEL) and U19 AI107774 (CMS), and the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: SN JEL CS. Performed the experiments: SN JEL BBM KM AJO HPN. Analyzed the data: SN JEL RB HPN SAS CS. Wrote the paper: SN CS.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babu M, Arnold R, Bundalovic-Torma C, Gagarinova A, Wong KS, Kumar A, Stewart G, Samanfar B, Aoki H, Wagih O, et al. Quantitative genome-wide genetic interaction screens reveal global epistatic relationships of protein complexes in Escherichia coli. PLoS Genet. 2014;10:e1004120. doi: 10.1371/journal.pgen.1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar A, Chawla M, Mehta M, Parikh P, Chandra P, Bhave D, Kumar D, Carroll KS, Singh A. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog. 2014;10:e1003902. doi: 10.1371/journal.ppat.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- Chavadi SS, Stirrett KL, Edupuganti UR, Vergnolle O, Sadhanandan G, Marchiano E, Martin C, Qiu WG, Soll CE, Quadri LEN. Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. Journal of Bacteriology. 2011;193:5905–5913. doi: 10.1128/JB.05811-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipollone R, Ascenzi P, Visca P. Common themes and variations in the rhodanese superfamily. IUBMB Life. 2007;59:51–59. doi: 10.1080/15216540701206859. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards KM, Cynamon MH, Voladri RK, Hager CC, DeStefano MS, Tham KT, Lakey DL, Bochan MR, Kernodle DS. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2001;164:2213–2219. doi: 10.1164/ajrccm.164.12.2106093. [DOI] [PubMed] [Google Scholar]
- Farhana A, Kumar S, Rathore SS, Ghosh PC, Ehtesham NZ, Tyagi AK, Hasnain SE. Mechanistic insights into a novel exporter-importer system of Mycobacterium tuberculosis unravel its role in trafficking of iron. PLoS ONE. 2008;3:e2087. doi: 10.1371/journal.pone.0002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes AS, Gaspar J, Cabral MF, Rueff J, Castro M, Batinic-Haberle I, Costa J, Oliveira NG. Protective role of ortho-substituted Mn(III) N-alkylpyridylporphyrins against the oxidative injury induced by tert-butylhydroperoxide. Free Radic Res. 2010;44:430–440. doi: 10.3109/10715760903555844. [DOI] [PubMed] [Google Scholar]
- Garces A, Atmakuri K, Chase MR, Woodworth JS, Krastins B, Rothchild AC, Ramsdell TL, Lopez MF, Behar SM, Sarracino DA, et al. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog. 2010;6:e1000957. doi: 10.1371/journal.ppat.1000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine (OUP Oxford) 2007 [Google Scholar]
- Hasan MR, Rahman M, Jaques S, Purwantini E, Daniels L. Glucose 6-phosphate accumulation in mycobacteria: implications for a novel F420-dependent anti-oxidant defense system. J Biol Chem. 2010;285:19135–19144. doi: 10.1074/jbc.M109.074310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger T, Budde H, Flohé L, Menge U, Singh M, Trujillo M, Radi R. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch Biochem Biophys. 2004;423:182–191. doi: 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc Natl Acad Sci USa. 2006;103:11760–11765. doi: 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramnik I, Dietrich WF, Demant P, Bloom BR. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci USa. 2000;97:8560–8565. doi: 10.1073/pnas.150227197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kučera O, Endlicher R, Roušar T, Lotková H, Garnol T, Drahota Z, Cervinková Z. The effect of tert-butyl hydroperoxide-induced oxidative stress on lean and steatotic rat hepatocytes in vitro. Oxid Med Cell Longev. 2014;2014:752506–752512. doi: 10.1155/2014/752506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Farhana A, Guidry L, Saini V, Hondalus M, Steyn AJC. Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med. 2011;13:e39. doi: 10.1017/S1462399411002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen PC. Levels of glutathione in Escherichia coli. Can J Biochem. 1979;57:107–111. doi: 10.1139/o79-013. [DOI] [PubMed] [Google Scholar]
- Murphy KC, Papavinasasundaram K, Sassetti CM. Mycobacteria Protocols, Methods in Molecular Biology. New York: Springer Science+business media; 2015. [Google Scholar]
- Müller FH, Bandeiras TM, Urich T, Teixeira M, Gomes CM, Kletzin A. Coupling of the pathway of sulphur oxidation to dioxygen reduction: characterization of a novel membrane-bound thiosulphate:quinone oxidoreductase. Molecular Microbiology. 2004;53:1147–1160. doi: 10.1111/j.1365-2958.2004.04193.x. [DOI] [PubMed] [Google Scholar]
- Nandi DL, Horowitz PM, Westley J. Rhodanese as a thioredoxin oxidase. Int J Biochem Cell Biol. 2000;32:465–473. doi: 10.1016/s1357-2725(99)00035-7. [DOI] [PubMed] [Google Scholar]
- Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. Journal of Bacteriology. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Benavides T, Long JE, Raimunda D, Sassetti CM, Argüello JM. A novel P(1B)-type Mn2+-transporting ATPase is required for secreted protein metallation in mycobacteria. J Biol Chem. 2013;288:11334–11347. doi: 10.1074/jbc.M112.448175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podinovskaia M, Lee W, Caldwell S, Russell DG. Infection of macrophages with Mycobacterium tuberculosis induces global modifications to phagosomal function. Cell Microbiol. 2013;15:843–859. doi: 10.1111/cmi.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remelli W, Guerrieri N, Klodmann J, Papenbrock J, Pagani S, Forlani F. Involvement of the Azotobacter vinelandii rhodanese-like protein RhdA in the glutathione regeneration pathway. PLoS ONE. 2012;7:e45193. doi: 10.1371/journal.pone.0045193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson SL, Dascher CC, Sambandamurthy VK, Russell RG, Jacobs WR, Bloom BR, Hondalus MK. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect Immun. 2004;72:3031–3037. doi: 10.1128/IAI.72.5.3031-3037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USa. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USa. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJC. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 2009;5:e1000545. doi: 10.1371/journal.ppat.1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke DW, Chock PB, Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem. 2003;278:14607–14613. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- Toohey JI. Sulphane sulphur in biological systems: a possible regulatory role. Biochem J. 1989;264:625–632. doi: 10.1042/bj2640625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- Wagner F, Asfar P, Calzia E, Radermacher P, Szabó C. Bench-to-bedside review: Hydrogen sulfide--the third gaseous transmitter: applications for critical care. Crit Care. 2009;13:213. doi: 10.1186/cc7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TM, Collins DM. ahpC, a gene involved in isoniazid resistance of the Mycobacterium tuberculosis complex. Molecular Microbiology. 1996;19:1025–1034. doi: 10.1046/j.1365-2958.1996.449980.x. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82–83:969–974. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.