

Abstract
Sponge diseases have been widely reported, yet the causal factors and major pathogenic microbes remain elusive. In this study, two individuals of the sponge Crella cyathophora in total that showed similar disease-like characteristics were collected from two different locations along the Red Sea coast separated by more than 30 kilometers. The disease-like parts of the two individuals were both covered by green surfaces, and the body size was much smaller compared with adjacent healthy regions. Here, using high-throughput pyrosequencing technology, we investigated the prokaryotic communities in healthy and disease-like sponge tissues as well as adjacent seawater. Microbes in healthy tissues belonged mainly to the Proteobacteria, Cyanobacteria and Bacteroidetes, and were much more diverse at the phylum level than reported previously. Interestingly, the disease-like tissues from the two sponge individuals underwent shifts of prokaryotic communities and were both enriched with a novel clade affiliated with the phylum Verrucomicrobia, implying its intimate connection with the disease-like Red Sea sponge C. cyathophora. Enrichment of the phylum Verrucomicrobia was also considered to be correlated with the presence of algae assemblages forming the green surface of the disease-like sponge tissues. This finding represents an interesting case of sponge disease and is valuable for further study.
Keywords: Low microbial abundance, Verrucomicrobia, Sponge symbiont, Disease-like sponge
Introduction
Sponges (phylum Porifera) are one of the oldest and most primitive metazoans (Hedges et al., 2004). They are distributed globally and are widely known to be associated with dense and diverse microbial communities of ecological and biotechnological importance (Taylor et al., 2007). Reports have shown that microbes can constitute up to 60% of the sponge tissue volume (Hentschel et al., 2003). In recent years, pyrosequencing technology has facilitated environmental microbial research and increased our knowledge concerning sponge-associated microbial diversity (Webster & Taylor, 2012). According to a recent study, up to 32 bacterial phyla and candidate phyla were documented in sponges (Schmitt et al., 2012). The sponge-species and sampling location specificity of sponge-associated microbes were also described (Lee et al., 2011; Webster et al., 2013). In addition, sponges can be grouped into high microbial abundance (HMA; previously called bacteriosponges) and low microbial abundance (LMA) sponges based on the density of microbes in the host (Hentschel et al., 2003).
An increasing number of reports have demonstrated that sponges are being threatened by disease and mortality events, most of which are accounted for increases of seawater temperature, global climate change and other independent prevailing environmental conditions (Wulff, 2006; Webster et al., 2008; Simister et al., 2012). Shifts of microbial community structures have been observed in diseased sponges (Angermeier et al., 2012; Gao et al., 2014). Microorganisms including Cyanobacteria, fungi, viruses, Alphaproteobacteria, and representatives of the genera Bacillus and Pseudomonas are considered potential pathogens that cause sponge disease (Webster, 2007). However, determining the causative agent of sponge diseases is difficult. Only one pathogen belonging to the Alphaproteobacteria and infecting the Great Barrier Reef sponge Rhopaloeides odorabile has been identified to date (Webster et al., 2002). Other studies have failed to establish a disease-like syndrome in healthy sponges using infection assays failed (Angermeier et al., 2011), and the roles of microbes on sponge disease remain inconclusive.
Crella cyathophora is a LMA sponge species, and its bacterial communities have been reported to be dominated by Proteobacteria (Giles et al., 2013). Here, two individuals of the LMA sponge C. cyathophora with similar disease-like characteristics were found at two different locations in Saudi Arabia along the Red Sea coast. The disease-like parts of the two individuals were both covered by green surfaces, and the body size was much smaller than adjacent healthy parts of the sponge (Fig. 1). Healthy and disease-like tissues of the two sponge individuals as well as adjacent seawater were collected and 16S rRNA genes were amplified. Using pyrosequencing of tagged 16S rRNA amplicons, shifts of prokaryotic communities in these disease-like individuals were investigated and enriched microbes in the disease-like tissues were elucidated.
Figure 1. Healthy and abnormal tissues of the sponge Crella cyathophora.

Sample IDs are referred to Table 1.
Material and methods
Sample collection and DNA extraction
Sponge specimens and seawater were collected from two sites in Saudi Arabia along the Red Sea coast in April 2012 (Table 1 and Fig. 1). RB3 is the southern open water region off the Rabigh Bay and approximates an outfall. KL2 is located near the mouth of the Al Kharar Lagoon to the north of the Rabigh Bay and is more than 30 kilometers away from the former site. At each site, the sponge C. cyathophora was checked by scuba divers and disease-like individuals were targeted. The sponge species was identified with the help of Professor Rob von Soest from the Zoological Museum, University of Amsterdam. Disease-like and healthy tissues from the ectoderm of each individual were collected as well as 4 liters of seawater around each sampling site. Samples were stored in separate sterile plastic bags and transported immediately back to the laboratory. Next, sponge tissues were flushed using 0.22 µm-membrane-filtered seawater to remove loosely attached microbes and debris. The flushed tissues from each sample were separated into two parts: 10 mL for identification of sponge species was preserved in 70% of ethanol; approximately 2 × 0.5 mL for DNA extraction, which was used as two technical replicates, was dissected, cut into small pieces with a sterile razor blade, and frozen in 0.8 mL of extraction buffer (100 mM Tris–HCl, 100 mM EDTA, 100 mM Na2HPO4, 1.5 M NaCl, 1% CTAB, pH 8.0). 2 × 1 L of each seawater sample, used as two replicates, were filtered sequentially through two membranes: the first was a 1.6-µm-pore-sized glass fiber membrane (GF/A, diameter 125 mm; Whatman, Little Chalfont, UK) to remove suspended particles and eukaryotes; the second was a 0.22-µm polycarbonate membrane to capture microbial cells (polycarbonate, 47 mm in diameter; Millipore, Billerca, Massachusetts, USA). The latter polycarbonate membranes were then frozen in 0.8 mL of extraction buffer for DNA extraction. Total genomic DNA was extracted using the modified sodium dodecyl sulfate-based method described previously (Lee et al., 2011), and further purified with the Mo Bio Soil DNA Isolation Kit (Mo Bio Laboratories, Carlsbad, California, USA). The purified DNA samples were subjected to a NanoDrop ND-100 device for quantification (Thermo Fisher, Waltham, Massachusetts, USA), and then were stored at −20 °C until use.
Table 1. Tissue samples of the sponge Crella cyathophora and adjacent seawater.
Samples were collected from the Rabigh Bay (RB) and Al Kharar Lagoon (KL) along the Red Sea coast.
| Site | Coordinate | Depth (m) | Sample ID | Description |
|---|---|---|---|---|
| RB3 | 22°42′38″N | ∼9.5 | D1 | Abnormal tissue (green surface) from individual 1 |
| 38°59′47″E | H1 | Healthy tissue (white surface) from individual 1 | ||
| SW3 | Sea water from the site RB3 | |||
| KL2 | 22°57′39″N | ∼2.75 | D2 | Abnormal tissue (green surface) from individual 2 |
| 38°49′30″E | H2 | Healthy tissue (white surface) from individual 2 | ||
| SW5 | Sea water from the site KL2 |
PCR amplification of 16S rRNA genes and 454 pyrosequencing
The V5-V9 regions of bacterial and archaeal 16S rRNA genes were amplified by PCR using the universal primers U905F (5’-TGAAACTYAAAGGAATTG-3’) and U1492R (5’-GGTTACCTTGTTACGACTT-3’) (Leser et al., 2002; Wang & Qian, 2009). Eight-nucleotide barcodes for distinguishing sponge samples were added at the terminal of primers. A 50-µl PCR reaction volume consisted of 2.5 U of Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA, USA), 1 × Pfu reaction buffer, 0.2 mM of dNTPs (TaKaRa, Dalian, China), 0.1 µM of each pair of barcoded primers and 5–10 ng of genomic DNA template. PCR was performed using a thermal cycler (Bio-Rad, Hercules, California, USA) with the following conditions: initial denaturation at 94 °C for 5 min; 30 cycles of denaturation at 94 °C for 40 s, annealing at 50 °C for 40 s and extension at 72 °C for 60 s; and a final extension at 72 °C for 5 min. PCR products were quantified using the NanoDrop device, mixed equally and subjected to the ROCHE 454 FLX Titanium platform (Roche, Basel, Switzerland) for 454 pyrosequencing. 18S rRNA genes of kinds of Eukaryotes were also amplified using the same primer pair and following the above procedure.
Processing of sequencing data
Raw pyrosequencing data were submitted to the NCBI Sequence Read Archive under accession number SRA065210. QIIME 1.5.0 pipelines (Caporaso et al., 2010b) were used in the downstream bioinformatics analysis. First, low quality reads, including those shorter than 150 bp, those containing ambiguous bases and homopolymers of 6 bp or more and those with an average flowgram score lower than 25 in a quality windows of 50 bp, were removed. The remaining reads were assigned to respective samples according to their barcodes and subjected to Denoiser for the second round of quality control (Reeder & Knight, 2010). Singletons from Denoiser were included in the data set for the following analysis. Operational taxonomic units (OTUs) were picked at a 97% similarity from the qualified reads using UCLUST (Edgar, 2010). The most abundant read in each OTU was selected as the representative sequence. Representative reads were aligned against the SILVA 111 database with PyNAST (Caporaso et al., 2010a). The aligned sequences were searched for chimeras using ChimeraSlayer (Haas et al., 2011). Non-chimeric representatives were assigned taxonomically using the Ribosomal Database Project (RDP) classifier version 2.2 (Wang et al., 2007) against the SILVA111 database (Pruesse et al., 2007) with a confidence threshold of 0.5. Sequences that were annotated as chloroplast, mitochondria and eukaryotes were filtered out. Taxonomic abundance of reads in each sample was summarized at the phylum, class, order, family and genus levels. Species diversity, Shannon index, richness and rarefaction curves were calculated using the QIIME alpha diversity pipeline with a step size of 100 and 100 repetitions per step. Using the above mentioned primer pair, partial eukaryotic 18S rRNA genes were also successfully amplified and included in the pyrosequencing reads. They were separately summarized to reveal the relative abundance of Eukaryotes in the sponge samples.
16S rRNA phylogenetic tree construction
A predominant OTU assigned to the phylum Verrucomicrobia was imported into “The All-Species Living Tree” Project LTPs 115 tree (Yarza et al., 2008) and 16S rRNA sequences of the closest representative species were obtained. The OTU was further searched against the NCBI GenBank database using the BLASTn program to find closely related sequences. A neighbour-joining tree was constructed using MEGA5.1 software (Tamura et al., 2011). Multiple alignment was performed using ClustalW (Thompson, Higgins & Gibson, 1994). Distance matrices were calculated using Kimura’s two-parameter correction model (Kimura, 1980). Bootstrap values were determined with 1000 replications.
Results and Discussion
Diversity of sponge-associated prokaryotic communities
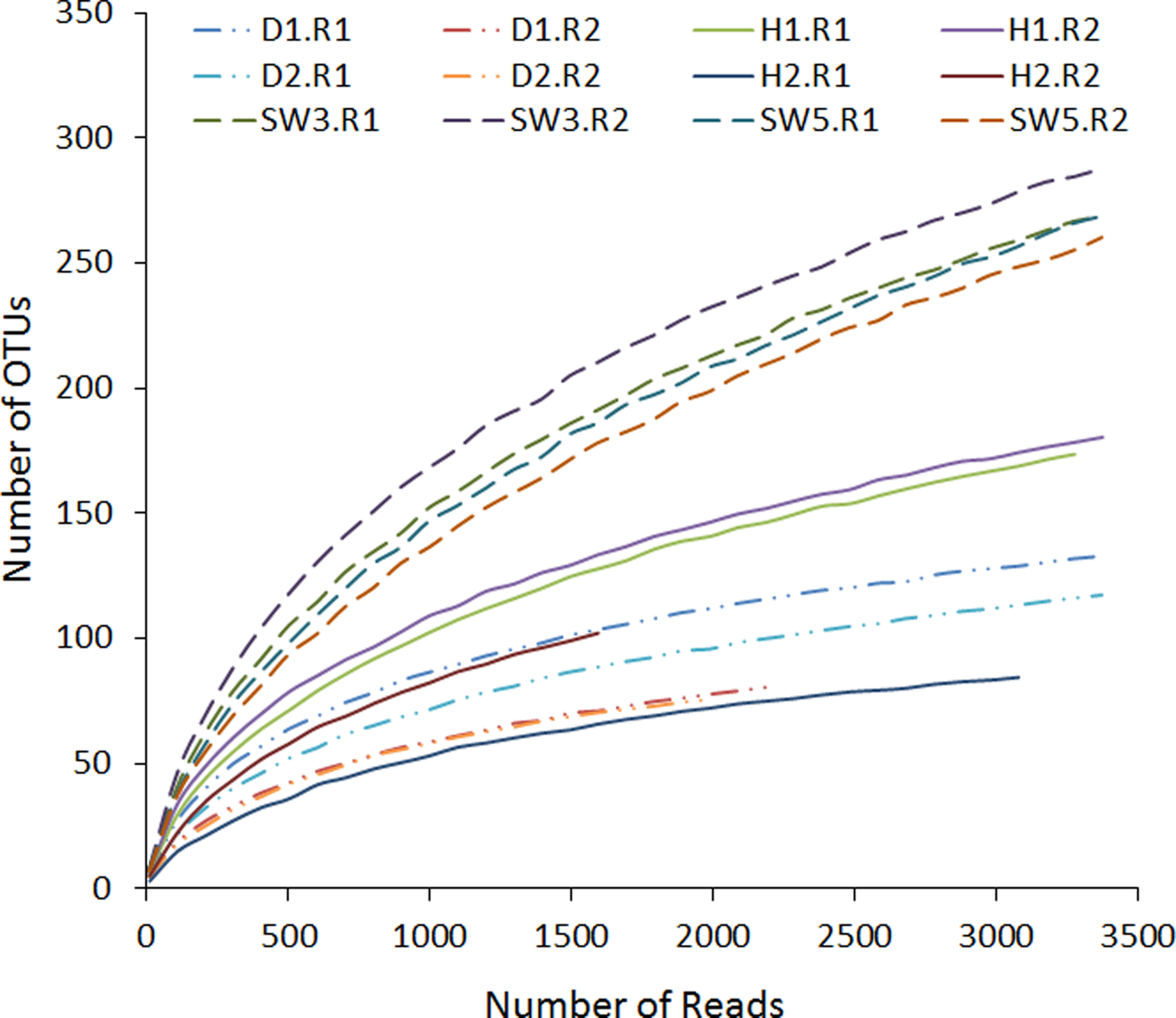
Two sponge individuals of the sponge C. cyathophora were collected, and two technical replicates of healthy and disease-like tissues were cut off from each individuals. Together with the surrounding seawater of each sampling site, a total of 12 DNA samples were obtained (Table 2). Their 16S rRNA gene amplicons were subjected to pyrosequencing. After quality filtering, 56,473 reads with an average length of 549 bp were obtained. Total number of prokaryotic OTUs in healthy sponge tissues ranged from 85 to 182, and that in disease-like tissues ranged from 76 to 142. The total number of prokaryotic OTUs was close to the number of Chao-based estimations of species richness (Table 2). Together with rarefaction curves based on observed species (OTUs, drawn using the QIIME alpha diversity pipeline at a 3% dissimilarity), the sequencing depth for all of the sponge samples was indicated to be representative, but the number of reads in seawater samples was still insufficient (Fig. S1).
Table 2. Pyrosequencing summary of microbial communities in sponges and seawater.
OTUs, Shannon index and Chao1 were determined at 3% dissimilarity. Refer to Table 1 for sample IDs. ‘R1’ and ‘R2’ denote technical replicates 1 and 2, respectively.
| Sample ID | Number of qualified reads | Total OTUs | Normalized reads (n = 1594) | ||
|---|---|---|---|---|---|
| OTUs | Shannon index | Chao1 | |||
| D1.R1 | 4187 | 142 | 103 | 3.71 | 142 |
| D1.R2 | 2212 | 81 | 71 | 2.98 | 105 |
| H1.R1 | 3344 | 175 | 128 | 4.27 | 210 |
| H1.R2 | 3449 | 182 | 134 | 4.71 | 208 |
| D2.R1 | 3426 | 118 | 89 | 3.04 | 135 |
| D2.R2 | 2028 | 76 | 70 | 3.00 | 92 |
| H2.R1 | 3149 | 85 | 66 | 1.89 | 93 |
| H2.R2 | 1662 | 104 | 102 | 3.22 | 157 |
| SW3.R1 | 7552 | 368 | 191 | 4.95 | 330 |
| SW3.R2 | 4707 | 325 | 210 | 5.41 | 335 |
| SW5.R1 | 12598 | 480 | 186 | 4.99 | 348 |
| SW5.R2 | 8159 | 378 | 178 | 4.75 | 338 |
The number of OTUs, Shannon index, and Chao1 estimation of species richness based on normalized reads (n = 1594) showed that the microbial diversity of healthy sponge samples was lower than that of seawater samples (Table 2). This finding is consistent with the low microbial diversity of the sponge C. cyathophora (Giles et al., 2013). The microbial diversity of disease-like sponge samples was also lower than that of seawater samples, yet no obvious divergence of microbial diversity was observed between healthy and disease-like sponge tissues.
Taxonomic assignment of prokaryotic pyrosequencing reads at the phylum level
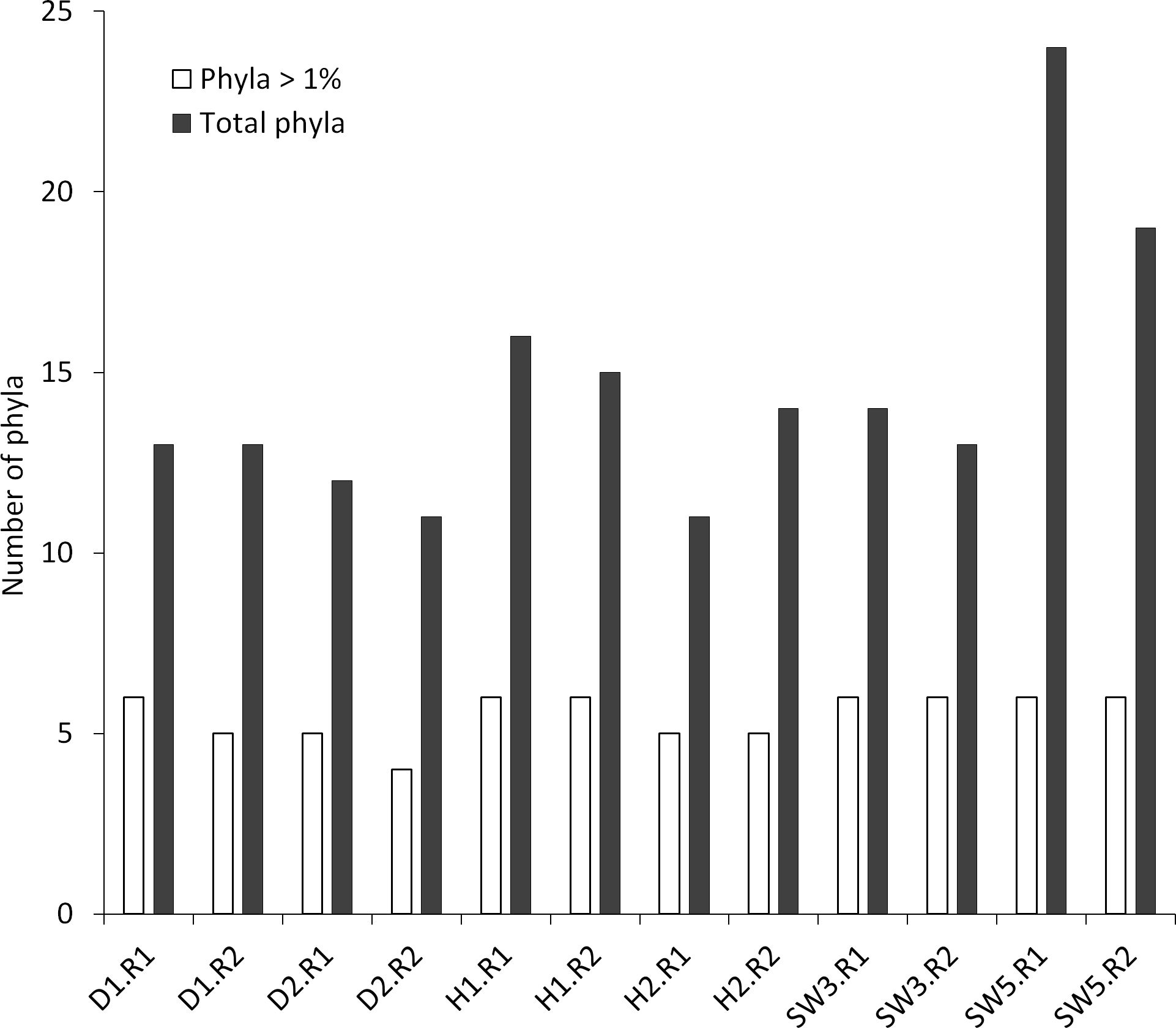
Taxonomic assignment of pyrosequencing reads at the phylum level provided insights into the shifts of prokaryotic communities in disease-like sponge tissues (Fig. 2). Healthy tissues of the sponge C. cyathophora were mainly composed of Proteobacteria, Cyanobacteria and Bacteroidetes. Archaea that accounted for a considerable proportion of the prokaryotic community of the surrounding seawater were rarely detected in the sponge samples, which was consistent with a previous report by Giles et al. (2013). The report also demonstrated a low phylum-level microbial diversity in the sponge C. cyathophora with up to only five bacterial phyla could be detected. However, using high-throughput pyrosequencing technology, we showed herein that phylum-level microbial diversity in healthy sponge tissues was higher than that reported previously, although the number of phyla that accounted for an abundance of higher than 1% of each sample was no more five (Fig. S2). In all of the disease-like sponge tissues, an additional phylum Verrucomicrobia was enriched and became predominant. The predominance of the same novel phylum in two disease-like sponge individuals collected from different locations (Table 1) should not be separated occasional events, and there should be an identical factor influencing both the two individuals.
Figure 2. Taxonomic classification of microbial reads in sponges and seawater at the phylum level.

Microbial reads were assigned taxonomically using the RDP classifier against the SILVA 111 database with a confidence threshold of 50%. Sample IDs are referred to Tables 1 and 2.
Taxonomic assignment of prokaryotic pyrosequencing reads at the genus level
Taxonomic assignment of pyrosequencing reads at the genus level was further summarized and the abundance of the genera that showed more than 5% abundance in at least one sample was depicted in a heatmap (Fig. 3). Unclassified Alphaproteobacteria and Gammaproteobacteria showed a high abundance in healthy sponge tissues as well as in several disease-like tissues, yet they were rarely found in surrounding seawater. Unclassified Alphaproteobacteria and Gammaproteobacteria also have been reported to be dominant in the sponge C. cyathophora according to a previous study (Giles et al., 2013), which implied that these uncultured bacteria are key sponge-associated components. LMA sponges were reported to share more microbes with surrounding seawater than HMA sponges (Moitinho-Silva et al., 2014). The genus Synechococcus, which was highly abundant in surrounding water, could also be found in several healthy and disease-like sponge samples. Alternatively, there was a random abundance pattern of the genus Ralstonia among samples. Although the genus Ralstonia could not be detected in adjacent seawater, it was only highly abundant in healthy tissues of sponge individual 2 but not in individual 1. It was present as a significant proportion of the disease-like samples D1.R2 and D2.R2, but was missing in the other two disease-like samples. Previous studies have reported the spatial distribution of sponge-associated prokaryotic communities, and some microbes were only found in the endosome of the sponge Tethya aurantium (Thiel et al., 2007). Our study focused on the ectoderm of the sponge C. cyathophora, but some samples have also included endosome tissues, which maybe the reason causing the large difference between disease-like technical replicates. Ralstonia is usually heterotrophic and rarely reported in sponge-associated microbial communities. Detection of the genus Ralstonia in the sponge C. cyathophora is curious, but it may represent a novel sponge-associated bacterium that was overlooked in other sponge-related studies.
Figure 3. Heatmap showing the abundance of microbial reads in disease-like and healthy sponge tissues and in adjacent seawater at the genus level.

Genera that showed less than 5% abundance in all the samples were filtered out. Sample IDs are referred to Tables 1 and 2. Abbreviations: α, Alphaproteobacteria; β, Betaproteobacteria; γ, Gammaproteobacteria; C, Cyanobacteria; B, Bacteroidetes; V, Verrucomicrobia.
Interestingly, the phylum Verrucomicrobia, which was specifically enriched in disease-like sponge tissues fell into a single unclassified cluster at the genus level (Fig. 3). An in-depth analysis showed that the verrucomicrobial bacteria belonged mainly to a single OTU (OTU_1064) that became dominant in the disease-like sponge tissues and occupied up to 53% of the prokaryotic communities (Fig. 4). OTU_1064 formed a deep clade in the phylum Verrucomicrobia and showed a high dissimilarity with the representative identified species. A BLASTn search further showed that OTU_1064 shared less than 90% identity with all of the sequences stored in the GenBank NR database. This finding suggested the presence of a novel verrucomicrobial clade with a function that cannot be predicted based on its close relatives. Enrichment of this novel clade implied its intimate connection with the disease-like sponge tissues and likely represents a pathogen infecting the disease-like sponge tissues. However, there is also the possibility that the potential disease attracted this novel bacterium into the sponge tissues. The two disease-like sponge individuals were both covered with a green surface, which might be assemblages of algae. There is a large possibility that this novel Verrucomicrobia clade correlated with the assumed algal assemblages. We also identified 18S rRNA gene sequences of green algae affiliated with the class Prasinophyceae (Table S1), although it still could not be sure whether these are the assumed algal assemblages.
Figure 4. The abundance of OTUs affiliated with the phylum Verrucomicrobia and the phylogenetic relationship of OTU_1064.

(A), proportions of the OTUs in disease-like tissues of the sponge Crella cyathophora; (B), the phylogenetic relationship of OTU_1064 with representative species in the LTPs 115 tree and uncultured clones in the NCBI GenBank NR database. The tree was constructed based on the neighbor-joining method. Values are expressed based on 1000 replications. Bar, 0.02% estimated sequences divergence.
The primer pair we used as above could also successfully amplify eukaryotic 18S rRNA genes, and the relative abundance of eukaryotic communities was also summarized at phylum, order, class and genus level. Green algae affiliated with the class Prasinophyceae dominated the eukaryotic communities of the sponge samples (Table S1), and were potentially related to the green assemblages. Yet, it still could not be sure whether these are the assumed algal assemblages because Prasinophyceae appeared both in healthy and diseased-like sponges tissues.
Conclusions
Here, using high throughput pyrosequencing technology, we re-analyzed the prokaryotic communities of this sponge species and their shifts in disease-like tissues. Healthy tissues showed a low microbial diversity and were dominated by Proteobacteria, as reported previously (Giles et al., 2013). Disease-like tissues from two sponge individuals underwent shifts of prokaryotic communities, and both showed enrichment for a novel verrucomicrobial clade. Because there were only two individuals, the extent of the disease spreading could not be evaluated. However, the finding of disease-like sponges at two distinct locations was interesting. The occurrence of a novel verrucomicrobial clade in the disease-like sponge tissues with a small body size and covered by green surfaces certainly indicated an intimate relationship between this novel clade and the disease-like Red Sea sponge C. cyathophora. Collecting more biological sponge replicates that display this disease-like phenomenon from different locations of the Red Sea is required to re-assess the prevalence of this novel verrucomicrobial clade. In addition, further studies are also needed to uncover the source of the novel verrucomicrobial clade and its ascertained roles in disease-like sponges.
Supplemental Information
The abundance was calculated as the proportions of reads of each eukaryotic clade against all reads of Eukaryotes in each sample.
The curves were drawn based on the number of OTUs at a 3% dissimilarity level. Sample IDs were referred to Tables 1 and 2.
{kind=link}
The black bar indicated total number of microbial phyla in each sample, and the blank bar indicated the number of phyla of which the abundance in microbial communities is higher than 1%. Sample IDs were referred to Tables 1 and 2.
{kind=link}
Acknowledgments
We thank WP Zhang and G Zhang from Hong Kong University of Science and Technology (HKUST), and the technical team from King Abdullah University of Science and Technology (KAUST) for their assistance during sample collection. We also thank Professor Rob von Soest, Zoological Museum, University of Amsterdam, for identification of sponges.
Funding Statement
This study was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB06010102 and XDB06010201), SKLMP Seed Collaborative Research fund (CITYU12SC01), and an award (SA-C0040/UK-C0016) granted to PY Qian from the King Abdullah University of Science and Technology. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Zhao-Ming Gao conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables.
Yong Wang wrote the paper, reviewed drafts of the paper.
Ren-Mao Tian performed the experiments, contributed reagents/materials/analysis tools.
On On Lee, Yue Him Wong and Zenon B. Batang contributed reagents/materials/analysis tools.
Abdulaziz Al-Suwailem and Feras F. Lafi reviewed drafts of the paper.
Vladimir B. Bajic and Pei-Yuan Qian conceived and designed the experiments, reviewed drafts of the paper.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
Raw pyrosequencing data were submitted to the NCBI Sequence Read Archive under accession number SRA065210.
References
- Angermeier et al. (2012).Angermeier H, Glockner V, Pawlik JR, Lindquist NL, Hentschel U. Sponge white patch disease affecting the Caribbean sponge Amphimedon compressa. Diseases of Aquatic Organisms. 2012;99:95–102. doi: 10.3354/dao02460. [DOI] [PubMed] [Google Scholar]
- Angermeier et al. (2011).Angermeier H, Kamke J, Abdelmohsen UR, Krohne G, Pawlik JR, Lindquist NL, Hentschel U. The pathology of sponge orange band disease affecting the Caribbean barrel sponge Xestospongia muta. FEMS Microbiology Ecology. 2011;75:218–230. doi: 10.1111/j.1574-6941.2010.01001.x. [DOI] [PubMed] [Google Scholar]
- Caporaso et al. (2010a).Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010a;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso et al. (2010b).Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 2010b;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar (2010).Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Gao et al. (2014).Gao ZM, Wang Y, Lee OO, Tian RM, Wong YH, Bougouffa S, Batang Z, Al-Suwailem A, Lafi FF, Bajic VB, Qian PY. Pyrosequencing reveals the microbial communities in the red sea sponge carteriospongia foliascens and their impressive shifts in abnormal tissues. Microbial Ecology. 2014;68:621–632. doi: 10.1007/s00248-014-0419-0. [DOI] [PubMed] [Google Scholar]
- Giles et al. (2013).Giles EC, Kamke J, Moitinho-Silva L, Taylor MW, Hentschel U, Ravasi T, Schmitt S. Bacterial community profiles in low microbial abundance sponges. FEMS Microbiology Ecology. 2013;83:232–241. doi: 10.1111/j.1574-6941.2012.01467.x. [DOI] [PubMed] [Google Scholar]
- Haas et al. (2011).Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ, Human Microbiome Consortium. Petrosino JF, Knight R, Birren BW. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges et al. (2004).Hedges SB, Blair JE, Venturi ML, Shoe JL. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evolutionary Biology. 2004;4:2. doi: 10.1186/1471-2148-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentschel et al. (2003).Hentschel U, Fieseler L, Wehrl M, Gernert C, Steinert M, Hacker J, Horn M. Microbial diversity of marine sponges. In: Müller WEG, editor. Sponges (Porifera) Heidelberg: Springer; 2003. pp. 59–83. [DOI] [PubMed] [Google Scholar]
- Kimura (1980).Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Lee et al. (2011).Lee OO, Wang Y, Yang JK, Lafi FF, Al-Suwailem A, Qian PY. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. The ISME Journal: Multidisciplinary Journal of Microbial Ecology. 2011;5:650–664. doi: 10.1038/ismej.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leser et al. (2002).Leser TD, Amenuvor JZ, Jensen TK, Lindecrona RH, Boye M, Moller K. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Applied and Environmental Microbiology. 2002;68:673–690. doi: 10.1128/AEM.68.2.673-690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitinho-Silva et al. (2014).Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, Ravasi T, Hentschel U. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Molecular Ecology. 2014;23:1348–1363. doi: 10.1111/mec.12365. [DOI] [PubMed] [Google Scholar]
- Pruesse et al. (2007).Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder & Knight (2010).Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nature Methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt et al. (2012).Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, Perez T, Rodrigo A, Schupp PJ, Vacelet J, Webster N, Hentschel U, Taylor MW. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. The ISME Journal: Multidisciplinary Journal of Microbial Ecology. 2012;6:564–576. doi: 10.1038/ismej.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simister et al. (2012).Simister R, Taylo MW, Tsai P, Fan L, Bruxner TJ, Crowe ML, Webster N. Thermal stress responses in the bacterial biosphere of the Great Barrier Reef sponge, Rhopaloeides odorabile. Environmental Microbiology. 2012;14:3232–3246. doi: 10.1111/1462-2920.12010. [DOI] [PubMed] [Google Scholar]
- Tamura et al. (2011).Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor et al. (2007).Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiology and Molecular Biology Reviews. 2007;71:295–347. doi: 10.1128/MMBR.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel et al. (2007).Thiel V, Neulinger SC, Staufenberger T, Schmaljohann R, Imhoff JF. Spatial distribution of sponge-associated bacteria in the Mediterranean sponge Tethya aurantium. FEMS Microbiology Ecology. 2007;59:47–63. doi: 10.1111/j.1574-6941.2006.00217.x. [DOI] [PubMed] [Google Scholar]
- Thompson, Higgins & Gibson (1994).Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al. (2007).Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang & Qian (2009).Wang Y, Qian PY. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE. 2009;4:e890. doi: 10.1371/journal.pone.0007401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster (2007).Webster NS. Sponge disease: a global threat? Environmental Microbiology. 2007;9:1363–1375. doi: 10.1111/j.1462-2920.2007.01303.x. [DOI] [PubMed] [Google Scholar]
- Webster et al. (2013).Webster NS, Luter HM, Soo RM, Botté ES, Simister RL, Abdo D, Whalan S. Same, same but different: symbiotic bacterial associations in GBR sponges. Frontiers in Microbiology. 2013;3:444. doi: 10.3389/fmicb.2012.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster et al. (2002).Webster NS, Negri AP, Webb RI, Hill RT. A spongin-boring alpha-proteobacterium is the etiological agent of disease in the Great Barrier Reef sponge Rhopaloeides odorabile. Marine Ecology Progress Series. 2002;232:305–309. doi: 10.3354/meps232305. [DOI] [Google Scholar]
- Webster & Taylor (2012).Webster NS, Taylor MW. Marine sponges and their microbial symbionts: love and other relationships. Environmental Microbiology. 2012;14:335–346. doi: 10.1111/j.1462-2920.2011.02460.x. [DOI] [PubMed] [Google Scholar]
- Webster et al. (2008).Webster NS, Xavier JR, Freckelton M, Motti CA, Cobb R. Shifts in microbial and chemical patterns within the marine sponge Aplysina aerophoba during a disease outbreak. Environmental Microbiology. 2008;10:3366–3376. doi: 10.1111/j.1462-2920.2008.01734.x. [DOI] [PubMed] [Google Scholar]
- Wulff (2006).Wulff JL. Sponge systematics by starfish: predators distinguish cryptic sympatric species of Caribbean fire sponges, Tedania ignis and Tedania klausi n. sp. (Demospongiae, Poecilosclerida) Biological Bulletin. 2006;211:83–94. doi: 10.2307/4134581. [DOI] [PubMed] [Google Scholar]
- Yarza et al. (2008).Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH, Ludwig W, Glockner FO, Rossello-Mora R. The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Systematic and Applied Microbiology. 2008;31:241–250. doi: 10.1016/j.syapm.2008.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The abundance was calculated as the proportions of reads of each eukaryotic clade against all reads of Eukaryotes in each sample.
The curves were drawn based on the number of OTUs at a 3% dissimilarity level. Sample IDs were referred to Tables 1 and 2.
The black bar indicated total number of microbial phyla in each sample, and the blank bar indicated the number of phyla of which the abundance in microbial communities is higher than 1%. Sample IDs were referred to Tables 1 and 2.