

Abstract
Neurotensin (NT) serves as a neuromodulator in the brain where it is involved in modulating a variety of physiological functions including nociception, temperature, blood pressure and cognition, and many neurological diseases such as Alzheimer’s disease, schizophrenia and Parkinson’s disease. Whereas there is compelling evidence demonstrating that NT facilitates cognitive processes, the underlying cellular and molecular mechanisms have not been fully determined. Because the dentate gyrus expresses high densities of NT and NT receptors, we examined the effects of NT on the synaptic transmission at the synapse formed between the perforant path (PP) and granule cells (GC) in the rats. Our results demonstrate that NT persistently increased the amplitude of the AMPA receptor-mediated EPSCs at the PP-GC synapse. NT-induced increases in AMPA EPSCs were mediated by presynaptic NTS1 receptors. NT reduced the coefficient of variation and paired-pulse ratio of AMPA EPSCs suggesting that NT facilitates presynaptic glutamate release. NT increased the release probability and the number of readily releasable vesicles with no effects on the rate of recovery from vesicle depletion. NT-mediated augmentation of glutamate release required the influx of Ca2+ via L-type Ca2+ channels and the functions of calmodulin and myosin light chain kinase. Our results provide a cellular and molecular mechanism to explain the roles of NT in the hippocampus.
Keywords: synapse, Ca2+ channels, Ca2+, hippocampus, G proteins, synaptic transmission, glutamate, Alzheimer’s disease, myosin light chain kinase
1. Introduction
Neurotensin (NT) is a tridecapeptide that is widely distributed in both the periphery such as the gastrointestinal tract, liver, pancreas, heart, lung and spleen, and the central nervous system including the hypothalamus, entorhinal cortex (EC), hippocampus, amygdala, septum, ventral tegmental area and substantia nigra (Boules et al., 2013; Caceda et al., 2006; Vincent et al., 1999). NT interacts with 3 types of receptors: NTS1, NTS2 and NTS3 (Hermans and Maloteaux, 1998; Mazella, 2001; Navarro et al., 2001; Nouel et al., 1999; St-Gelais et al., 2006; Vincent et al., 1999). NTS1 and NTS2 are G-protein-coupled receptors with seven-transmembrane domains and are distinguished based on their affinity for NT and sensitivity to the nonpeptide antagonist, SR48692, and the histamine receptor antagonist, levocabastine. NTS1 displays high affinity for NT and its effects are usually blocked by SR48692, whereas NTS2 shares only around 40% amino acid identities with NTS1, shows low affinity for NT and is sensitive to levocabastine (Pelaprat, 2006; St-Gelais et al., 2006; Vincent et al., 1999). NTS1 receptors are usually coupled to Gq proteins resulting in activation of phospholipase C (PLC) and the inositol phosphate signaling cascade (Watson et al., 1992), although other signaling molecules including cyclic GMP (Mistry and Vijayan, 1987; Slusher et al., 1994), cyclic AMP (Yamada et al., 1993), MAP kinase (Ehlers et al., 2000; Poinot-Chazel et al., 1996) and Akt (Liu et al., 2004) have been implicated. Unlike NTS1 receptors, the pharmacological and signaling properties of NTS2 receptors are still controversial and it is still uncertain as to whether NT is an agonist, inverse agonist or antagonist for this receptor type (Hwang et al., 2009; Mazella et al., 1996; Sarret et al., 2002). NTS3 receptors are originally identified as the intracellular sorting protein, sortilin, and they bind NT with high affinity once converted to the mature form upon cleavage by the protein convertase furin. This non G protein-coupled receptor appears to be involved in molecule sorting between the cell surface and intracellular compartments (Mazella, 2001; Navarro et al., 2001).
NT has been implicated in modulating many physiological functions including nociception, temperature, blood pressure and cognition, and a variety of neurological diseases such as Alzheimer’s disease (AD), schizophrenia and Parkinson’s disease (Boules et al., 2013; Tyler-McMahon et al., 2000). Among these physiological functions and pathological disorders, there is compelling evidence demonstrating that NT modifies the cognitive processes and functional alterations of NT may undergo the pathology of AD that is characterized as declination of memory. For example, NTS1 polymorphisms are significantly associated with variation in working memory performance among healthy adults (Li et al., 2011); NT binding sites are negatively correlated with age and cognitive status (Rowe et al., 2006); application of NT receptor agonists in vivo enhances (Azmi et al., 2006; Laszlo et al., 2010; Ohinata et al., 2007; Xiao et al., 2014) whereas administration of NT receptor antagonists decreases (Laszlo et al., 2010; Tirado-Santiago et al., 2006) cognitive functions; whereas NTS2 knockout (KO) mice showed reduced fear memory (Yamauchi et al., 2007), NTS1 receptors are usually considered to be responsible for the effects of NT on memory (Azmi et al., 2006; Laszlo et al., 2010; Tirado-Santiago et al., 2006). Paradoxically, the cellular and molecular mechanisms whereby NT modifies the processes of cognition have not been fully determined. In the previous study, we have demonstrated that activation of NTS1 receptors in layer II principal neurons in the EC persistently increases the excitability of these neurons (Xiao et al., 2014). Because the axons of layer II neurons form the perforant path (PP) that synapses onto the dentate gyrus granule cells (GC) of the hippocampus, we examined the effects of NT on synaptic transmission at the PP-GC synapse. Our results demonstrate that NT persistently increases glutamate release at the PP-GC synapse via activation of NTS1 receptors. NT-induced persistent glutamate release involves increases in the readily releasable pool size and release probability without altering quantal size. The effects of NT require an increase in intracellular Ca2+ mediated by the influx of Ca2+ via L-type Ca2+ channels and the functions of calmodulin and myosin light chain kinas (MLCK). Our results further the understanding of the cellular and molecular mechanisms whereby NT facilitates learning and memory in the hippocampal formation.
2. Materials and Methods
2.1. Slice preparation
The ages of the animals used for electrophysiological recordings were postnatal 3–5 weeks for Sprague-Dawley rats and mice in which NTS1 gene or NTS2 gene were deleted as described previously (Xiao et al., 2014). Pairs of homozygous NTS1 or NTS2 knockout (KO) mice were initially provided by Dr. Etsuko Wada from the Department of Degenerative Neurological Diseases, National Institute of Neuroscience, Tokyo, Japan. The mice used for the experiments in this study were bred in the animal facility of the University of North Dakota. Detailed experimental procedures for generating and genotyping the KO mice were described previously (Maeno et al., 2004). Horizontal brain slices (400 μm) including the hippocampus, subiculum and EC were cut using a vibrating blade microtome (VT1000S; Leica, Wetzlar, Germany) as described previously (Deng and Lei, 2008; Wang et al., 2011; Xiao et al., 2009) with the following modifications (Xiao et al., 2014). After being deeply anesthetized with isoflurane, animals were decapitated and their brains were dissected out in ice-cold saline solution that contained (in mM) 130 N-methyl-D-glucamine (NMDG)-Cl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 5.0 MgCl2, and 10 glucose, saturated with 95% O2 and 5% CO2 (pH 7.4, adjusted with HCl). Slices were then incubated in the above solution except NMDG-Cl was replaced with NaCl at 35°C for 1 h for recovery and then kept at room temperature (~24°C) until use. All animal procedures conformed to the guidelines approved by the University of North Dakota Animal Care and Use Committee.
2.2. Recordings of synaptic currents
Whole-cell patch-clamp recordings using a Multiclamp 700B amplifier in voltage-clamp mode were made from dentate gyrus GCs in hippocampal slices. Cells in the slices were visually identified with infrared video microscopy and differential interference contrast optics (Ramanathan et al., 2012; Wang et al., 2012; Wang et al., 2013). Recording electrodes were filled with the solution containing (in mM) 100 Cs-gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 2 ATP2Na, 0.3 GTPNa, 40 HEPES and 1 QX-314 (pH 7.3). The extracellular solution comprised (in mM) 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.5 MgCl2 and 10 glucose, saturated with 95% O2 and 5% CO2 (pH 7.4). Bicuculline (10 μM) was included in the extracellular solution to block GABAA receptors. The holding potential was at −60 mV. AMPA receptor-mediated EPSCs were evoked by placing a stimulation electrode in the middle to the inner one third of molecular layer of dentate gyrus to stimulate the medial PP. Under these conditions, the recorded currents were completely blocked by application of DNQX (10 μM) or GYKI 52466 (100 μM) at the end of experiments confirming that they were mediated by AMPA receptors. Series resistance was rigorously monitored by delivering a 5 mV voltage step after each evoked current. Experiments were discontinued if the series resistance changed by >15%. Data were filtered at 2 kHz, digitized at 10 kHz, acquired on-line and analyzed after-line using pCLAMP 9 or 10 software (Molecular Devices, Sunnyvale, CA). To avoid potential desensitization induced by repeated bath applications of NT, one slice was limited to only one application of NT and only one cell was recorded from each slice.
2.3. Data analysis
Data are presented as the means ± S.E.M. Concentration-response curve of NT was fit by Hill equation: , where Imax is the maximum response, EC50 is the concentration of ligand producing a half-maximal response, and n is the Hill coefficient. Student’s paired or unpaired t test or analysis of variance (ANOVA) was used for statistical analysis as appropriate; P values are reported throughout the text and significance was set as P<0.05. N numbers in the text represent the number of cells examined unless stated otherwise.
3. Results
3.1. NT increases glutamatergic transmission at the PP-GC synapses via activation of NTS1 receptors
We examined the effects of NT on glutamatergic transmission by recording, from the dentate gyrus GCs, AMPA EPSCs evoked by stimulating the PP. Bath application of NT (0.5 μM) for 5 min enhanced AMPA EPSCs (Fig. 1A1–A2). The amplitude of AMPA EPSCs began to increase gradually and reached maximal in ~20 min after the beginning of NT application (150±12% of control, n=10, P=0.002 vs. baseline, Figs. 1A1–A2). NT-induced increases in AMPA EPSCs persisted for at least 1 h in our whole-cell recording configuration (136±6% of control, n=10, P<0.001 vs. baseline, Figs. 1A1–A2). This phenomenon resembles long-term potentiation (LTP) suggesting that it may be related to learning and memory. However, bath application of the heat-inactive NT (0.5 μM, inactivated by heating at 100°C for 20 min) for 5 min failed to significantly increase the amplitude of AMPA EPSCs (101±2% of control, n=5, P=0.78 vs. baseline, Figs. 1B1–B2) suggesting that it is unlikely that NT-induced increases in AMPA EPSCs are due to the nonspecific action of the NT preparation. Furthermore, bath application of the active fragment of NT (NT8-13, 0.5 μM), also enhanced AMPA EPSCs (171±18% of control, n=5, P=0.016 vs. baseline, Fig. 1C) whereas application of the inactive fragment of NT (NT1-8, 0.5 μM) did not alter AMPA EPSCs (101±7% of control, n=6, P=0.89 vs. baseline, Fig. 1C) suggesting that NT-induced facilitation of AMPA EPSCs is mediated by activation of NT receptors. The EC50 of NT was measured to be 94 nM (Fig. 1D).
Fig. 1.

Application of NT facilitates AMPA EPSCs at the PP-GC synapses. A1–A2, Time course of NT-induced increases in the amplitude of AMPA EPSCs recorded from the dentate gyrus GCs evoked by stimulation of the PP. A1, Amplitude of AMPA EPSCs recorded from a dentate gyrus GC evoked by stimulation of PP in response to bath application of NT (0.5 μM). Upper panel shows the traces averaged from 10 EPSCs taken from the time points indicated in the figure. A2, Pooled time course of NT-induced facilitation of AMPA EPSC amplitudes. B1–B2, Bath application of heat-inactivated NT (0.5 μM) failed to augment the amplitudes of AMPA EPSCs. The figures are assembled in the same fashion. C, Application of the active fragment of NT (NT8-13, 0.5 μM) enhanced the amplitudes of AMPA EPSCs at the PP-GC synapses, whereas application of the inactive fragment of NT (NT1-8, 0.5 μM) did not alter AMPA EPSC amplitudes. D, Concentration-response relationship of NT-mediated augmentation of AMPA EPSC amplitudes. Numbers in the parentheses are the numbers of cells recorded.
We next tested the roles of NT receptors in NT-mediated facilitation of AMPA EPSCs. Pretreatment of slices with and continuous bath application of NTS1 antagonist, SR48692 (1 μM), blocked NT-induced increases in AMPA EPSCs (109±7% of control, n=5, P=0.24 vs. baseline, Fig. 2A). However, application of the NTS2 antagonist, levocabastine (30 μM), failed to alter the effect of NT significantly (148±16% of control, n=6, P=0.034 vs. baseline; P=0.89 vs. NT alone, Fig. 2A). Furthermore, application of PD149163 (0.5 μM), a NTS1 agonist, also significantly augmented AMPA EPSCs (136±6% of control, n=6, P=0.002 vs. baseline, Fig. 2B). These results demonstrate that NT increases AMPA EPSCs by activation of NTS1 receptors. We further probed the involvement of NT receptors by utilizing NTS1 and NTS2 KO mice. Application of NT (0.5 μM) to slices cut from NTS1 KO mice failed to significantly alter AMPA EPSCs (99±4% of control, n=12 cells from 3 mice, P=0.89 vs. baseline, Fig. 2C) whereas application of the same concentration of NT to slices cut from NTS2 KO mice still significantly enhanced AMPA EPSCs (156±7% of control, n=10 cells from 3 mice, P<0.001 vs. baseline, Fig. 2C). These results together demonstrate that NT increases AMPA EPSCs by activating NTS1 receptors.
Fig. 2.
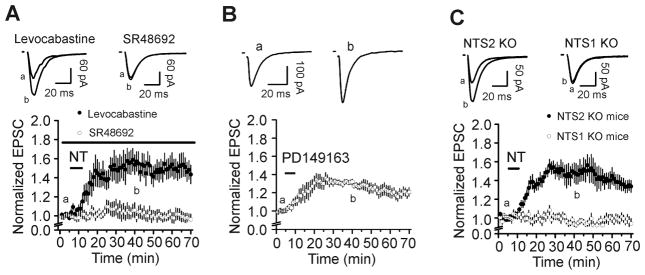
NT facilitates AMPA EPSCs by activation of NTS1 receptors. A, Pretreatment of slices with and continuous bath application of the NTS1 antagonist, SR48692 (1 μM), blocked NT-induced augmentation of AMPA EPSCs, whereas application of the NTS2 antagonist, levocabastine (30 μM), in the same fashion, failed to alter NT-induced facilitation of AMPA EPSCs. Up panel shows traces averaged from 10 EPSCs recorded at the time points indicated in the figure. B, Bath application of the selective NTS1 agonist, PD149163 (0.5 μM), significantly enhanced AMPA EPSC amplitudes. C, Bath application of NT failed to enhance AMPA EPSCs in slices cut from NTS1 KO mice, but still significantly increased AMPA EPSCs in slices cut from NTS2 KO mice. Figures are assembled in the same fashion.
3.2. NT increases presynaptic glutamate release
NT could increase AMPA EPSCs by facilitating presynaptic glutamate release or by up-regulating postsynaptic AMPA receptor function. We next differentiated between the pre- and postsynaptic effects of NT. First, we calculated the coefficient of variation (CV) of AMPA EPSCs recorded before and after the application of NT because alterations in presynaptic transmitter release are usually concomitant with a change of CV (Malinow and Tsien, 1990; McAllister and Stevens, 2000; Zucker and Regehr, 2002). NT significantly reduced the value of CV (Control: 0.212±0.016, NT: 0.135±0.011, n=9, P<0.001, Fig. 3A). Second, we compared the paired-pulse ratio (PPR) before and after the application of NT. NT significantly reduced PPR (Control: 1.24±0.03, NT: 1.10±0.04, n=14, P=0.001, Fig. 3B). These results indicate that NT increases AMPA EPSCs by facilitating presynaptic glutamate release.
Fig. 3.
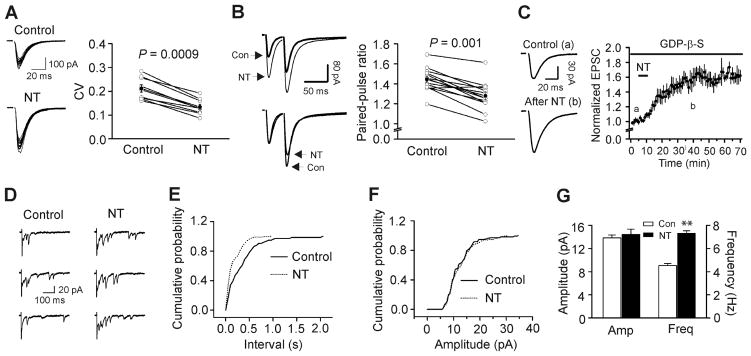
NT increases AMPA EPSCs by facilitating presynaptic glutamate release. A, NT reduced the coefficient of variation (CV=standard deviation/mean) of AMPA EPSCs. Standard deviation and mean were obtained by averaging 15 consecutive EPSCs. Left panel shows 15 consecutive EPSCs recorded before (upper) and after (lower) the application of NT. Right panel shows the calculated CVs from 9 cells (open circles) and their averages (solid circles). B, NT reduced paired-pulse ratio (PPR=P2/P1, P1 and P2 are the EPSCs evoked by two stimuli at an interval of 40 ms). Left upper, EPSCs averaged from ~20 current traces before (bold) and after (thin) the application of NT. Left lower, EPSCs recorded before and after the application of NT were scaled to the first EPSC. Note that the second EPSC after the application of NT is smaller than control. Right, PPRs recorded from 14 cells (open circles) and their averages (solid circles). C, Intracellular dialysis of GDP-β-S (4 mM) failed to block NT-mediated increases in AMPA EPSCs. D, Evoked AMPA EPSCs recorded from the same synapse in the presence of Sr2+ (6 mM) before and after application of NT. Note that AMPA EPSCs recorded as asynchronous events in the extracellular solution containing Sr2+ and application of NT increased the frequency of asynchronous events recorded in the presence of Sr2+. E, Cumulative frequency distribution of asynchronous EPSCs in the presence of Sr2+ before and after the application of NT. F, Cumulative amplitude distribution of asynchronous EPSCs in the presence of Sr2+ before and after the application of NT. G, Summarized asynchronous EPSC frequency and amplitude (n=6).
We next explored whether NT augments glutamate release via activation of presynaptic or postsynaptic NTS1 receptors at the PP-GC synapses because it is possible that NT interacts with postsynaptic NTS1 receptors to generate some retrograde messengers to enhance glutamate release. We differentiated the pre- and postsynaptic localization of NTS1 receptors by intracellular application of GDP-β-S, a G-protein inactivator, via the recording pipettes because application of GDP-β-S via the recording pipettes should block the postsynaptic but spare the presynaptic NTS1 receptors. We included GDP-β-S (4 mM) in the recording pipettes and waited for ~20 min after the formation of whole-cell configuration. Under this condition, application of NT still increased AMPA EPSC amplitudes significantly (146±8% of control, n=6, P=0.003 vs. baseline, P=0.81 vs. NT alone, Fig. 3C) suggesting that NT increases glutamate release by activating presynaptic NTS1 receptors.
3.3. NT enhances quantal content without altering quantal size
We next measured the effects of NT on the quantal properties of glutamate release at the PP-GC synapses by using the method of replacing extracellular Ca2+ with strontium (Sr2+) to induce asynchronous transmitter release (Goda and Stevens, 1994; Oliet et al., 1996). The amplitude of the asynchronous events when extracellular Ca2+ is replaced by Sr2+ is equal to the quantal size (q) (Goda and Stevens, 1994; Oliet et al., 1996). After forming whole-cell recordings, AMPA EPSCs evoked by stimulation of PP in the presence of extracellular Ca2+ were initially recorded. To avoid the confounding effects of spontaneous EPSCs on the asynchronous events in the presence of Sr2+, experiments were limited to those cells showing spontaneous events less than 0.5 Hz. The extracellular Ca2+ was then substituted with Sr2+ (6 mM) to induce asynchronous release from the same synapse. After recording sufficient events for quantal analysis in the control condition, we applied NT in the extracellular solution containing the same concentration of Sr2+ for 5 min and then washed in the same extracellular solution without NT for ~ 20 min to observe the maximal effect of NT. The results are shown in Figure 3D. When extracellular Ca2+ was replaced by Sr2+, asynchronous release occurred and NT significantly enhanced the frequency of asynchronous EPSCs (Control: 4.5±0.4 Hz, NT: 7.3±0.5 Hz, n=6, P<0.001, Fig. 3E, 3G) without altering the amplitude of asynchronous EPSCs (Control: 13.8±0.5 pA, NT: 14.4±0.9 pA, n=6, P=0.44, Fig. 3F, 3G), demonstrating that NT augments quantal content without altering quantal size.
3.4. NT increases the size of the readily releasable pool and release probability without changing the rate of recovery from vesicle depletion
Increases in presynaptic transmitter release can result from an increase in the size of the readily releasable pool (Nq, the product of the number of readily releasable quanta, N, and the quantal size, q) and/or an increase in release probability (Pr). We next used the method of high-frequency stimulation (Schneggenburger et al., 1999; Taschenberger et al., 2002) to evaluate NT-induced changes in Nq and Pr. This method is based on the assumption that high-frequency-stimulation-induced depression is primarily caused by the depletion of readily releasable quanta. The zero time intercept of a line fitted to a cumulative amplitude plot of EPSCs equals to the size of the readily releasable pool (Nq) (Schneggenburger et al., 1999; Taschenberger et al., 2002). Pr can be estimated from the first EPSC amplitude divided by Nq. To exclude any potential effects of high-frequency-stimulation-induced long-term plasticity because of the activations of NMDA and metabotropic glutamate receptors, we included, in the extracellular solution, dl-APV (100 μM) to block NMDA and MCPG (1 mM) to block metabotropic glutamate receptors. NT increased the EPSCs evoked by 20 stimuli at 40 Hz (Fig. 4A). The average data from 6 cells for the 20 stimuli are shown in Figure 4B. Figure 4C shows the cumulative amplitude histogram. NT increased Nq by 36±8% (n=6, P=0.008, Fig. 4D) and Pr by 17±5% (n=6, P=0.005, Fig. 4E). These data demonstrate that NT increases both the size of the readily releasable pool and release probability. Because NT did not change quantal size (q) (Fig. 3), NT-induced increases in the size of readily releasable pool (Nq) are likely due to an increase in the number of readily releasable quanta (N).
Fig. 4.
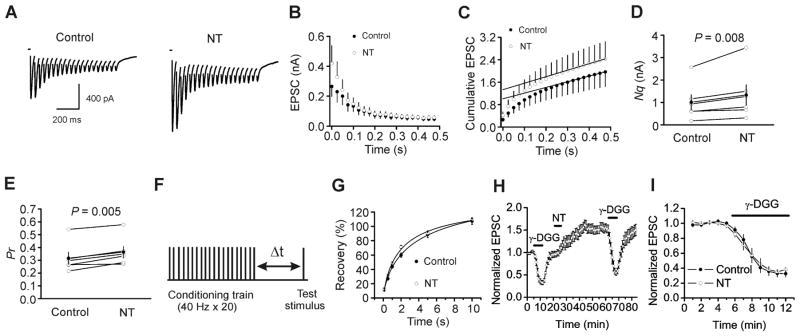
NT increases the size of readily releasable pool and release probability without changing the rate of recovery from vesicle depletion, but does not induce multivesicular release. A, EPSC trains averaged from 15 traces evoked by 20 stimuli at 40 Hz before (left) and after (right) the application of NT. Stimulation artifacts were blanked for clarity. B, EPSC amplitudes averaged from 6 cells in response to 20 stimuli at 40 Hz before and after the application of NT. The amplitude of EPSC evoked by each stimulus was measured by resetting the base line each time at a point within 0.5 ms before the beginning of each stimulation artifact. C, Cumulative amplitude histogram of EPSCs. For each cell, the last 6 EPSC amplitudes were fit with a linear regression line and extrapolated to time 0 to estimate the readily releasable pool size (Nq). D, NT increases Nq (n=6). E, NT increases the release probability (Pr, n=6). For each cell, Pr was calculated as the ratio of the first EPSC amplitude divided by its Nq obtained by linear fitting of the cumulative EPSC histogram. F, Experimental protocol. A conditioning train (20 stimuli at 40 Hz) was followed by a test stimulus at various intervals (Δt= 0.1–10 s). This protocol was repeated every 30 s. G, Time course of recovery from depletion before and during the application of NT expressed as percentage recovery = (Itest − Iss)/(I1st − Iss) × 100, where Itest is the EPSC evoked by the test pulse, Iss is the steady-state current left after the conditioning train (the average of the last 5EPSC evoked by the conditioning train), I1st is the EPSC evoked by the 1st stimulus of the conditioning train. Data before (solid circle) and after (empty circle) the application of NT from 6 cells were fit by a single exponential function. H, Pooled time course of normalized AMPA EPSCs in response to the application of γ-DGG (1 mM) before and after the application of NT. I, γ-DGG-induced percentage of inhibition on AMPA EPSCs from the same cells before and after the application of NT.
Increases in the size of the readily releasable pool can occur with or without a concomitant increase in the rate of vesicle replenishment. We next tested whether NT increases the rate of recovery from vesicle depletion. We employed a protocol comprising a train of stimulation (40 Hz, 20 stimuli) to deplete the readily releasable pool followed by a test pulse at various intervals (0.1 s, 0.5 s, 1 s, 2 s, 5 s, 10 s) to evaluate the replenishment of synaptic vesicles from depletion (Fig. 4F). The time course of recovery after the 40 Hz train could be fitted by a single exponential function with a time constant of 7.9±3.3 s before and 3.1±1.2 s after the application of NT (n=7, P=0.24, Fig. 4G) indicating that NT does not increase the rate of recovery from vesicle depletion.
Multivesicular release has been detected in hippocampal synapses (Christie and Jahr, 2006; Oertner et al., 2002; Smejkalova and Woolley, 2010; Tong and Jahr, 1994). We next tested whether NT enhances glutamate release by facilitating multivesicular release at the PP-GC synapses. If so, the glutamate transient concentration in the synaptic cleft after applying NT should be increased and application of γ-D-glutamylglycine (γ-DGG), a low-affinity AMPA receptor antagonist, should exert less inhibition as demonstrated previously (Christie and Jahr, 2006; Smejkalova and Woolley, 2010; Wadiche and Jahr, 2001). However, application of γ-DGG (1 mM) before (33±5% of control, n=7) and after (38±3% of control, n=7) the application of NT inhibited AMPA EPSCs to a comparable level (P=0.37, Fig. 4H, I), suggesting that NT does not increase multivesicular release.
3.5. NT-mediated enhancement of glutamate release is dependent on increases in presynaptic Ca2+ concentration
Because elevation of intracellular Ca2+ can result in increases in Pr and the readily releasable pool size (Zucker and Regehr, 2002), we tested whether NT-mediated augmentation of glutamate release at the PP-GC synapse is due to an elevation of intracellular Ca2+ concentration. Bath application of BAPTA-AM (200 μM) significantly reduced AMPA EPSCs (59±4% of control, n=10, P<0.001 vs. baseline, Fig. 5A) and blocked the enhancement of AMPA EPSCs induced by subsequent application of NT (102±10% of control, n=10, P=0.81 vs. baseline in the presence of BAPTA-AM alone, Fig. 5A) suggesting that NT-mediated enhancement of AMPA EPSCs is dependent on an increase in intracellular Ca2+. To ensure that the effect of BAPTA-AM was mediated presynaptically, we included BAPTA (10 mM) in the recording pipettes and waited for ~20 min after the formation of whole-cell recording configuration. Under these circumstances, application of NT still significantly increased AMPA EPSCs (155±7% of control, n=7, P<0.001 vs. baseline, P=0.77 vs. NT alone, Fig. 5B). These results together demonstrate that elevation of presynaptic, not postsynaptic, Ca2+ is responsible for NT-induced increases in glutamate release.
Fig. 5.

NT-mediated enhancement of glutamate release is related to the presynaptic Ca2+ influx via L-type Ca2+ channels and requires the function of calmodulin. A, Application of BAPTA-AM (200 μM) by itself decreased AMPA EPSCs and blocked NT-induced augmentation of AMPA EPSCs. B, Intracellular perfusion of BAPTA (10 mM) via the recording pipettes failed to alter significantly NT-mediated augmentation of AMPA EPSCs. C, Pretreatment of slices with and continuous bath application of thapsigargin (10 μM) failed to change NT-induced enhancement of AMPA EPSCs significantly. D, Pretreatment of slices with and continuous bath application of nimodipine (10 μM) blocked NT-induced increases in AMPA EPSCs. E, Pretreatment of slices with and continuous bath application of verapamil (10 μM) blocked NT-induced increases in AMPA EPSCs. F, Application of W7 (50 μM), the calmodulin inhibitor, in the same fashion blocked NT-induced increases in AMPA EPSCs.
Increases in the intracellular Ca2+ concentration could arise from release from intracellular Ca2+ stores and/or extracellular Ca2+ influx. We next tested the potential roles of intracellular Ca2+ release in the presynaptic terminals in NT-induced facilitation of glutamate release. Slices were pretreated with thapsigargin (10 μM, a sarcoplasmic reticulum Ca2+-ATPase inhibitor) and the same concentration of thapsigargin was continuously bath-applied. Under these circumstances, application of NT still significantly increased AMPA EPSC amplitude (152±15% of control, n=10, P=0.007 vs. baseline, P=0.93 vs. NT alone, Fig. 5C). These results suggest that elevation of Ca2+ in the presynaptic terminals in response to NT must be from the extracellular side.
3.6. NT-mediated enhancement of glutamate release requires L-type Ca2+ channels
NT has been shown to increase L-type Ca2+ channels and many effects of NT are dependent on L-type Ca2+ channels (Li et al., 2008; Mule and Serio, 1997). We therefore tested the effects of L-type Ca2+ channels on NT-induced increases in AMPA EPSCs. Slices were pretreated with L-type Ca2+ channel blocker, nimodipine (10 μM), and the same concentration of nimodipine was included in the extracellular solution. In the presence of nimodipine, application of NT did not significantly increase AMPA EPSCs (105±7% of control, n=8, P=0.54 vs. baseline, Fig. 5D). Likewise, application of verapamil (10 μM), another L-type Ca2+ channel blocker, blocked NT-induced increases in AMPA EPSCs (87±15% of control, n=8, P=0.43 vs. baseline, Fig. 5E). These data together demonstrate that Ca2+ influx via L-type Ca2+ channels is required for NT-induced increases in glutamate release.
3.7. Calmodulin and MLCK are required for NT-induced increases in glutamate release
Because calmodulin is an important target for intracellular Ca2+, we then tested whether calmodulin is required for NT-mediated facilitation of glutamate release. Slices were pretreated with calmodulin inhibitor, W7 (50 μM), and the same concentration of W7 was included in the extracellular solution. Under these circumstances, application of NT failed to increase AMPA EPSCs significantly (86±9% of control, n=7, P=0.18 vs. baseline, Fig. 5F), demonstrating that calmodulin is required for NT-induced enhancement of glutamate release.
Our results have demonstrated that NT increases the size of the readily releasable pool, suggesting that NT facilitates the delivery of synaptic vesicles from the reserve pool to the readily releasable pool. Because myosin has been reported to participate in the delivery of vesicles from the reserve pool to the readily releasable pool (Mochida, 1995; Mochida et al., 1994; Ryan, 1999) and the function of myosin is controlled by MLCK, we tested the roles of MLCK in NT-mediated facilitation of AMPA EPSCs. Treatment of slices with MLCK inhibitor, wortmannin (10 μM) which inhibits the activity of enzyme possibly by acting at or near the ATP binding site of the enzyme (Nakanishi et al., 1992), blocked NT-induced increases in AMPA EPSCs (100±5% of control, n=6, P=0.98 vs. baseline, Fig. 6A). Because wortmannin at the concentration used also inhibits phosphatidylinositol 3-kinase (PI3K), we used the selective PI3K inhibitor, LY294002 which competitively inhibits the ATP-binding site of PI3K (Vlahos et al., 1994), to test whether the effect of wortmannin is mediated by inhibition of PI3K instead. Slices were pretreated with LY294002 (10 μM) and the same concentration of LY294002 was continuously applied in the extracellular solution. Under these circumstances, application of NT still significantly enhanced AMPA EPSCs (158±11% of control, n=7, P=0.002, Fig. 6B), suggesting that the effect of wortmannin is not mediated by PI3K, but by MLCK.
Fig. 6.

NT-induced augmentation of AMPA EPSCs requires the function of MLCK. A, Pretreatment of slices with and continuous bath application of wortmannin (10 μM) which inhibits both MLCK and PI3K at this concentration, blocked NT-induced increases in AMPA EPSCs. B, Application of LY294002 (10 μM), a selective PI3K inhibitor, did not block NT-induced enhancement of AMPA EPSCs.
4. Discussion
Our results demonstrate that NT increases AMPA EPSCs via activating NTS1 receptors at the PP-GC synapse. NT-induced augmentation of AMPA EPSCs is mediated by increasing presynaptic glutamate release. Activation of NTS1 receptors increases the size of readily releasable pool and release probability without altering the recovery rate from vesicle depletion. NT-mediated augmentation of glutamate release does not involve multivesicular release or an alteration of quantal size. The effects of NT require an influx of Ca2+ in the presynaptic terminals via L-type Ca2+ channels and the functions of calmodulin and MLCK. Our results support a scenario in which activation of NTS1 results in increases in intracellular Ca2+ concentration via activation of L-type Ca2+ channels, leading to activation of MLCK. MLCK further phosphorylates and changes the conformation of myosin, a motor protein, to facilitate the delivery of vesicles from the reserve pool to the readily releasable pool to increase glutamate release. A working hypothesis of the mechanism based on the available evidence has been proposed (Fig. 7).
Fig. 7.

A working hypothesis of the mechanism based on the available evidence. Activation of presynaptic NTS1 receptors results in Ca2+ influx via L-type Ca2+ channels. Ca2+ then activates calmodulin leading to the activation of MLCK. Phosphorylation of myosin light chain changes the conformation of myosin facilitating the delivery of synaptic vesicles to the readily releasable pool.
Although we have demonstrated previously that activation of NTS1 receptors increases the excitability of layer II stellate neurons of the EC (Xiao et al., 2014), the exact actions of NT on the terminals of the stellate neurons, i.e. the PP, have not been determined. The following lines of anatomical evidence suggest that NT acts in both the EC and the dentate gyrus. First, the NT-containing cell bodies are observed in the EC (Atoji et al., 1995) which innervates the dentate gyrus. Second, a very large population of the molecular layer of the dentate gyrus where the PP-GC synapses form contains numerous NT-immunoreactive varicose axons (Sakamoto et al., 1986). Third, the dentate gyrus exhibits a high density of binding sites for NT (Cadet et al., 1993; Kohler et al., 1987; Moyse et al., 1987; Rowe et al., 2006). The anatomic expression of NT and NT receptors in the dentate gyrus region suggests a physiological role of NT in this region. In the present study, we used Cs+-gluconate-containing intracellular solution in the recording pipettes to block any effects of NT on postsynaptic K+ channels and probed the actions of NT on glutamatergic transmission at the PP-GC synapse. Our results demonstrate that NT enhances glutamate release at the PP-GC synapses via activation of NTS1 receptors. Because NT-mediated enhancement of AMPA EPSCs is still observed when GDP-β-S is included in the recording pipettes to inactivate postsynaptic NTS1 receptors, these results demonstrate that the involved NTS1 receptors are localized presynaptically, i.e. at the PP. Consistent with our results, NT has been shown to enhance glutamatergic transmission in the ventral tegmental area (Kempadoo et al., 2013), in rat spinal cord dorsal horn (Kadiri et al., 2011) and in globus pallidus (Chen et al., 2006). Moreover, endogenously released NT is also required for dopamine D1 receptor-mediated LTP at GABA synapses (Krawczyk et al., 2013).
Whereas it has been determined that NT facilitates glutamatergic transmission, the underlying cellular and molecular mechanisms have not been elucidated. Our results demonstrate that extracellular Ca2+ influx via L-type Ca2+ channels is required for NT-induced facilitation of glutamate release. The next question is how L-type Ca2+ channels are activated by NT. We consider two potential mechanisms whereby NT opens L-type Ca2+ channels. First, activation of NTS1 receptors directly opens L-type Ca2+ channels resulting in Ca2+ influx to facilitate glutamate release. Second, as we have demonstrated previously, activation of NTS1 receptors in layer II stellate neurons results in persistent depolarization (Xiao et al., 2014), which could further activate L-type Ca2+ channels leading to increases in Ca2+ influx. Consistent with the second mechanism, subthreshold depolarization of presynaptic terminals activates Ca2+ channels to elevate intracellular Ca2+ concentration resulting in increases in glutamate release (Awatramani et al., 2005). Because NT-induced enhancements of both the excitability of layer II stellate neurons and the glutamate release at the PP-GC synapses are persistent, it seems likely that the effect of NT on glutamate release is secondary to its action on the excitability of layer II stellate neurons. However, in the horizontal slices, it is almost impossible to keep the integrity of the soma of layer II stellate neurons and their axons (the PP) in complete. One explanation is that NT-induced increases in neuronal excitability also happen at the axons of the layer II stellate neurons, i.e. at the PP. Because it is difficult, if not impossible, to record directly from the PP, other methods would be sought to differentiate these two mechanisms later.
What are the targets for NT-induced elevation of intracellular Ca2+? We demonstrate that calmodulin and MLCK are the targets for NT-induced increases in intracellular Ca2+. Because our results also demonstrate that an increase in the readily releasable pool size is a mechanism whereby NT facilitates glutamate release at the PP-GC synapses, one reasonable hypothesis to reconcile these data is that calmodulin and MLCK enhance the delivery of synaptic vesicles from the reserve pool to the readily releasable pool by phosphorylation of myosin which is a motor protein. This hypothesis is supported by the following lines of evidence. First, NT has been shown to activate calmodulin in neostriatal neurons (Kasckow et al., 1991), suggesting that calmodulin is an intracellular signaling molecule in response to NT. Second, increases in the function of presynaptic calmodulin enhances the readily releasable pool size at the calyx of Held synapse (Lee et al., 2010; Wu et al., 2009) and application of the calmodulin antagonist, calmidazolium, blocks nicotine-induced increase in the size of the readily releasable pool at dopamine terminals (Turner, 2004). Third, myosin and MLCK have been demonstrated to be involved in the mobilization of synaptic vesicles at a variety of synapses (Mochida, 1995; Mochida et al., 1994) including the hippocampal synapses (Ryan, 1999).
Because the hippocampal synapses are capable of multivesicular release (Christie and Jahr, 2006; Oertner et al., 2002; Tong and Jahr, 1994) and application of estradiol (Smejkalova and Woolley, 2010) or LTP-induction protocols (Bender et al., 2009) facilitates multivesicular release, it is rational to hypothesize that NT facilitates glutamate release by increasing multivesicular release. If this is the case, the transient concentration of glutamate in the synaptic cleft after the application of NT would be higher. Higher transient concentration of glutamate would reduce the percentage of inhibition mediated by the low-affinity AMPA receptor antagonist, γ-DGG. However, there is no significant difference for the percentage of inhibition mediated by γ-DGG before and after the application of NT at the same synapses, suggesting that it is unlikely that NT facilitates glutamate release at the PP-GC synapse by enhancing multivesicular release.
In this study, we have found that brief (5 min) application of NT induces persistent enhancement of glutamate release at the PP-GC synapses, although it takes ~20 min to observe the maximal effect of NT. Whereas we currently do not have an explanation for the delayed action of NT, one possibility is that the time course of the NT may reflect the activation of intracellular signaling molecules and/or delivery of vesicles from reserve pool to the readily releasable pool. Furthermore, NT-induced persistent enhancement of glutamate release resemble LTP. Therefore, it is reasonable to speculate that NT-mediated augmentation of glutamate release may serve as a cellular and molecular mechanism to explain its roles in facilitating learning and memory. Because we have shown previously that microinjection of NT into layer II of the EC augments spatial memory (Xiao et al., 2014), it is not irrational to conjecture that activation of the NTS1 receptors in the soma or dendrites of layer II stellate neurons increases neuronal excitability which is then propagated to the terminal, i.e. the PP. At the PP, Ca2+ influx via L-type Ca2+ channels in response to NT-induced depolarization would activate calmodulin and MLCK to increase the readily releasable pool size resulting in increases in glutamate release. Facilitation of glutamatergic transmission in the hippocampal circuitry likely augments spatial learning and memory. Our results in this study, therefore, at least refine the previously proposed cellular and molecular mechanisms whereby NT facilitates learning and memory.
5. Conclusion
In conclusion, our results demonstrate that NT increases glutamate release at the PP-GC synapse via activation of presynaptic NTS1 receptors. NT-induced increases in glutamate release are mediated by enhancing the size of readily releasable pool and release probability. Presynaptic Ca2+ influx via L-type Ca2+ channels, calmodulin and MLCK are required for NT-induced augmentation of glutamate release. NT-mediated augmentation of glutamatergic transmission likely contributes to its facilitatory effects on learning and memory.
Highlights.
NT persistently increases glutamate release at PP-GC synapse in dentate gyrus
The effect of NT is mediated by presynaptic NTS1 receptors
NT increases the release probability and the number of readily releasable vesicles
Ca2+ influx via L-type Ca2+ channels is involved
Functions of calmodulin and myosin light chain kinase are required
Acknowledgments
This work was supported by National Institutes of Mental Health (MH082881 to S.L.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atoji Y, Watanabe H, Yamamoto Y, Suzuki Y. Distribution of neurotensin-containing neurons in the central nervous system of the dog. J Comp Neurol. 1995;353:67–88. doi: 10.1002/cne.903530108. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Azmi N, Norman C, Spicer CH, Bennett GW. Effects of a neurotensin analogue (PD149163) and antagonist (SR142948A) on the scopolamine-induced deficits in a novel object discrimination task. Behav Pharmacol. 2006;17:357–362. doi: 10.1097/01.fbp.0000224382.63744.20. [DOI] [PubMed] [Google Scholar]
- Bender VA, Pugh JR, Jahr CE. Presynaptically expressed long-term potentiation increases multivesicular release at parallel fiber synapses. J Neurosci. 2009;29:10974–10978. doi: 10.1523/JNEUROSCI.2123-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boules M, Li Z, Smith K, Fredrickson P, Richelson E. Diverse roles of neurotensin agonists in the central nervous system. Front Endocrinol (Lausanne) 2013;4:36. doi: 10.3389/fendo.2013.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceda R, Kinkead B, Nemeroff CB. Neurotensin: role in psychiatric and neurological diseases. Peptides. 2006;27:2385–2404. doi: 10.1016/j.peptides.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Kujirai K, Carlson E, Epstein CJ. Autoradiographic distribution of [3H]neurotensin receptors in the brains of superoxide dismutase transgenic mice. Synapse. 1993;14:24–33. doi: 10.1002/syn.890140105. [DOI] [PubMed] [Google Scholar]
- Chen L, Yung KK, Yung WH. Neurotensin selectively facilitates glutamatergic transmission in globus pallidus. Neuroscience. 2006;141:1871–1878. doi: 10.1016/j.neuroscience.2006.05.049. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Lei S. Serotonin increases GABA release in rat entorhinal cortex by inhibiting interneuron TASK-3 K+ channels. Mol Cell Neurosci. 2008;39:273–284. doi: 10.1016/j.mcn.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers RA, Zhang Y, Hellmich MR, Evers BM. Neurotensin-mediated activation of MAPK pathways and AP-1 binding in the human pancreatic cancer cell line, MIA PaCa-2. Biochem Biophys Res Commun. 2000;269:704–708. doi: 10.1006/bbrc.2000.2335. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans E, Maloteaux JM. Mechanisms of regulation of neurotensin receptors. Pharmacol Ther. 1998;79:89–104. doi: 10.1016/s0163-7258(98)00009-6. [DOI] [PubMed] [Google Scholar]
- Hwang JI, Kim DK, Kwon HB, Vaudry H, Seong JY. Phylogenetic history, pharmacological features, and signal transduction of neurotensin receptors in vertebrates. Ann N Y Acad Sci. 2009;1163:169–178. doi: 10.1111/j.1749-6632.2008.03636.x. [DOI] [PubMed] [Google Scholar]
- Kadiri N, Rodeau JL, Schlichter R, Hugel S. Neurotensin inhibits background K+ channels and facilitates glutamatergic transmission in rat spinal cord dorsal horn. Eur J Neurosci. 2011;34:1230–1240. doi: 10.1111/j.1460-9568.2011.07846.x. [DOI] [PubMed] [Google Scholar]
- Kasckow J, Cain ST, Nemeroff CB. Neurotensin effects on calcium/calmodulin-dependent protein phosphorylation in rat neostriatal slices. Brain Res. 1991;545:343–346. doi: 10.1016/0006-8993(91)91311-n. [DOI] [PubMed] [Google Scholar]
- Kempadoo KA, Tourino C, Cho SL, Magnani F, Leinninger GM, Stuber GD, Zhang F, Myers MG, Deisseroth K, de Lecea L, Bonci A. Hypothalamic neurotensin projections promote reward by enhancing glutamate transmission in the VTA. J Neurosci. 2013;33:7618–7626. doi: 10.1523/JNEUROSCI.2588-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C, Radesater AC, Chan-Palay V. Distribution of neurotensin receptors in the primate hippocampal region: a quantitative autoradiographic study in the monkey and the postmortem human brain. Neurosci Lett. 1987;76:145–150. doi: 10.1016/0304-3940(87)90706-3. [DOI] [PubMed] [Google Scholar]
- Krawczyk M, Mason X, Debacker J, Sharma R, Normandeau CP, Hawken ER, Di Prospero C, Chiang C, Martinez A, Jones AA, Doudnikoff E, Caille S, Bezard E, Georges F, Dumont EC. D1 Dopamine Receptor-Mediated LTP at GABA Synapses Encodes Motivation to Self-Administer Cocaine in Rats. J Neurosci. 2013;33:11960–11971. doi: 10.1523/JNEUROSCI.1784-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo K, Toth K, Kertes E, Peczely L, Ollmann T, Lenard L. Effects of neurotensin in amygdaloid spatial learning mechanisms. Behav Brain Res. 2010;210:280–283. doi: 10.1016/j.bbr.2010.02.038. [DOI] [PubMed] [Google Scholar]
- Lee JS, Ho WK, Lee SH. Post-tetanic increase in the fast-releasing synaptic vesicle pool at the expense of the slowly releasing pool. J Gen Physiol. 2010;136:259–272. doi: 10.1085/jgp.201010437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen C, He Q, Li H, Moyzis RK, Xue G, Dong Q. Neurotensin receptor 1 gene (NTSR1) polymorphism is associated with working memory. PLoS One. 2011;6:e17365. doi: 10.1371/journal.pone.0017365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Geiger JD, Lei S. Neurotensin enhances GABAergic activity in rat hippocampus CA1 region by modulating L-type calcium channels. J Neurophysiol. 2008;99:2134–2143. doi: 10.1152/jn.00890.2007. [DOI] [PubMed] [Google Scholar]
- Liu F, Yang P, Baez M, Ni B. Neurotensin negatively modulates Akt activity in neurotensin receptor-1-transfected AV12 cells. J Cell Biochem. 2004;92:603–611. doi: 10.1002/jcb.20098. [DOI] [PubMed] [Google Scholar]
- Maeno H, Yamada K, Santo-Yamada Y, Aoki K, Sun YJ, Sato E, Fukushima T, Ogura H, Araki T, Kamichi S, Kimura I, Yamano M, Maeno-Hikichi Y, Watase K, Aoki S, Kiyama H, Wada E, Wada K. Comparison of mice deficient in the high- or low-affinity neurotensin receptors, Ntsr1 or Ntsr2, reveals a novel function for Ntsr2 in thermal nociception. Brain Res. 2004;998:122–129. doi: 10.1016/j.brainres.2003.11.039. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Mazella J. Sortilin/neurotensin receptor-3: a new tool to investigate neurotensin signaling and cellular trafficking? Cell Signal. 2001;13:1–6. doi: 10.1016/s0898-6568(00)00130-3. [DOI] [PubMed] [Google Scholar]
- Mazella J, Botto JM, Guillemare E, Coppola T, Sarret P, Vincent JP. Structure, functional expression, and cerebral localization of the levocabastine-sensitive neurotensin/neuromedin N receptor from mouse brain. J Neurosci. 1996;16:5613–5620. doi: 10.1523/JNEUROSCI.16-18-05613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Stevens CF. Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci U S A. 2000;97:6173–6178. doi: 10.1073/pnas.100126497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry A, Vijayan E. Differential responses of hypothalamic cAMP and cGMP to substance P and neurotensin in ovariectomized rats. Brain Res Bull. 1987;18:169–173. doi: 10.1016/0361-9230(87)90187-0. [DOI] [PubMed] [Google Scholar]
- Mochida S. Role of myosin in neurotransmitter release: functional studies at synapses formed in culture. J Physiol Paris. 1995;89:83–94. doi: 10.1016/0928-4257(96)80555-9. [DOI] [PubMed] [Google Scholar]
- Mochida S, Kobayashi H, Matsuda Y, Yuda Y, Muramoto K, Nonomura Y. Myosin II is involved in transmitter release at synapses formed between rat sympathetic neurons in culture. Neuron. 1994;13:1131–1142. doi: 10.1016/0896-6273(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Moyse E, Rostene W, Vial M, Leonard K, Mazella J, Kitabgi P, Vincent JP, Beaudet A. Distribution of neurotensin binding sites in rat brain: a light microscopic radioautographic study using monoiodo [125I]Tyr3-neurotensin. Neuroscience. 1987;22:525–536. doi: 10.1016/0306-4522(87)90350-2. [DOI] [PubMed] [Google Scholar]
- Mule F, Serio R. Mode and mechanism of neurotensin action in rat proximal colon. Eur J Pharmacol. 1997;319:269–272. doi: 10.1016/s0014-2999(96)00943-0. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Kakita S, Takahashi I, Kawahara K, Tsukuda E, Sano T, Yamada K, Yoshida M, Kase H, Matsuda Y, et al. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem. 1992;267:2157–2163. [PubMed] [Google Scholar]
- Navarro V, Martin S, Sarret P, Nielsen MS, Petersen CM, Vincent J, Mazella J. Pharmacological properties of the mouse neurotensin receptor 3. Maintenance of cell surface receptor during internalization of neurotensin. FEBS Lett. 2001;495:100–105. doi: 10.1016/s0014-5793(01)02367-5. [DOI] [PubMed] [Google Scholar]
- Nouel D, Sarret P, Vincent JP, Mazella J, Beaudet A. Pharmacological, molecular and functional characterization of glial neurotensin receptors. Neuroscience. 1999;94:1189–1197. doi: 10.1016/s0306-4522(99)00354-1. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci. 2002;5:657–664. doi: 10.1038/nn867. [DOI] [PubMed] [Google Scholar]
- Ohinata K, Sonoda S, Inoue N, Yamauchi R, Wada K, Yoshikawa M. beta-Lactotensin, a neurotensin agonist peptide derived from bovine beta-lactoglobulin, enhances memory consolidation in mice. Peptides. 2007;28:1470–1474. doi: 10.1016/j.peptides.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Bidirectional control of quantal size by synaptic activity in the hippocampus. Science. 1996;271:1294–1297. doi: 10.1126/science.271.5253.1294. [DOI] [PubMed] [Google Scholar]
- Pelaprat D. Interactions between neurotensin receptors and G proteins. Peptides. 2006;27:2476–2487. doi: 10.1016/j.peptides.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Poinot-Chazel C, Portier M, Bouaboula M, Vita N, Pecceu F, Gully D, Monroe JG, Maffrand JP, Le Fur G, Casellas P. Activation of mitogen-activated protein kinase couples neurotensin receptor stimulation to induction of the primary response gene Krox-24. Biochem J. 1996;320 (Pt 1):145–151. doi: 10.1042/bj3200145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan G, Cilz NI, Kurada L, Hu B, Wang X, Lei S. Vasopressin facilitates GABAergic transmission in rat hippocampus via activation of V(1A) receptors. Neuropharmacology. 2012;63:1218–1226. doi: 10.1016/j.neuropharm.2012.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe WB, Kar S, Meaney MJ, Quirion R. Neurotensin receptor levels as a function of brain aging and cognitive performance in the Morris water maze task in the rat. Peptides. 2006;27:2415–2423. doi: 10.1016/j.peptides.2006.03.036. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–1323. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto N, Michel JP, Kiyama H, Tohyama M, Kopp N, Pearson J. Neurotensin immunoreactivity in the human cingulate gyrus, hippocampal subiculum and mammillary bodies. Its potential role in memory processing. Brain Res. 1986;375:351–356. doi: 10.1016/0006-8993(86)90756-0. [DOI] [PubMed] [Google Scholar]
- Sarret P, Gendron L, Kilian P, Nguyen HM, Gallo-Payet N, Payet MD, Beaudet A. Pharmacology and functional properties of NTS2 neurotensin receptors in cerebellar granule cells. J Biol Chem. 2002;277:36233–36243. doi: 10.1074/jbc.M202586200. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Zacco AE, Maslanski JA, Norris TE, McLane MW, Moore WC, Rogers NE, Ignarro LJ. The cloned neurotensin receptor mediates cyclic GMP formation when coexpressed with nitric oxide synthase cDNA. Mol Pharmacol. 1994;46:115–121. [PubMed] [Google Scholar]
- Smejkalova T, Woolley CS. Estradiol acutely potentiates hippocampal excitatory synaptic transmission through a presynaptic mechanism. J Neurosci. 2010;30:16137–16148. doi: 10.1523/JNEUROSCI.4161-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Gelais F, Jomphe C, Trudeau LE. The role of neurotensin in central nervous system pathophysiology: what is the evidence? J Psychiatry Neurosci. 2006;31:229–245. [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leao RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Tirado-Santiago G, Lazaro-Munoz G, Rodriguez-Gonzalez V, Maldonado-Vlaar CS. Microinfusions of neurotensin antagonist SR 48692 within the nucleus accumbens core impair spatial learning in rats. Behav Neurosci. 2006;120:1093–1102. doi: 10.1037/0735-7044.120.5.1093. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Turner TJ. Nicotine enhancement of dopamine release by a calcium-dependent increase in the size of the readily releasable pool of synaptic vesicles. J Neurosci. 2004;24:11328–11336. doi: 10.1523/JNEUROSCI.1559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler-McMahon BM, Boules M, Richelson E. Neurotensin: peptide for the next millennium. Regul Pept. 2000;93:125–136. doi: 10.1016/s0167-0115(00)00183-x. [DOI] [PubMed] [Google Scholar]
- Vincent JP, Mazella J, Kitabgi P. Neurotensin and neurotensin receptors. Trends Pharmacol Sci. 1999;20:302–309. doi: 10.1016/s0165-6147(99)01357-7. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Wang S, Chen X, Kurada L, Huang Z, Lei S. Activation of group II metabotropic glutamate receptors inhibits glutamatergic transmission in the rat entorhinal cortex via reduction of glutamate release probability. Cereb Cortex. 2012;22:584–594. doi: 10.1093/cercor/bhr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kurada L, Cilz NI, Chen X, Xiao Z, Dong H, Lei S. Adenosinergic depression of glutamatergic transmission in the entorhinal cortex of juvenile rats via reduction of glutamate release probability and the number of releasable vesicles. PLoS One. 2013;8:e62185. doi: 10.1371/journal.pone.0062185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang AP, Kurada L, Matsui T, Lei S. Cholecystokinin facilitates neuronal excitability in the entorhinal cortex via activation of TRPC-like channels. J Neurophysiol. 2011;106:1515–1524. doi: 10.1152/jn.00025.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MA, Yamada M, Cusack B, Veverka K, Bolden-Watson C, Richelson E. The rat neurotensin receptor expressed in Chinese hamster ovary cells mediates the release of inositol phosphates. J Neurochem. 1992;59:1967–1970. doi: 10.1111/j.1471-4159.1992.tb11035.x. [DOI] [PubMed] [Google Scholar]
- Wu XS, McNeil BD, Xu J, Fan J, Xue L, Melicoff E, Adachi R, Bai L, Wu LG. Ca(2+) and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat Neurosci. 2009;12:1003–1010. doi: 10.1038/nn.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Cilz NI, Kurada L, Hu B, Yang C, Wada E, Combs CK, Porter JE, Lesage F, Lei S. Activation of neurotensin receptor 1 facilitates neuronal excitability and spatial learning and memory in the entorhinal cortex: beneficial actions in an Alzheimer’s disease model. J Neurosci. 2014;34:7027–7042. doi: 10.1523/JNEUROSCI.0408-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Deng PY, Yang C, Lei S. Modulation of GABAergic transmission by muscarinic receptors in the entorhinal cortex of juvenile rats. J Neurophysiol. 2009;102:659–669. doi: 10.1152/jn.00226.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Watson MA, Richelson E. Neurotensin stimulates cyclic AMP formation in CHO-rNTR-10 cells expressing the cloned rat neurotensin receptor. Eur J Pharmacol. 1993;244:99–101. doi: 10.1016/0922-4106(93)90064-g. [DOI] [PubMed] [Google Scholar]
- Yamauchi R, Wada E, Kamichi S, Yamada D, Maeno H, Delawary M, Nakazawa T, Yamamoto T, Wada K. Neurotensin type 2 receptor is involved in fear memory in mice. J Neurochem. 2007;102:1669–1676. doi: 10.1111/j.1471-4159.2007.04805.x. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]