

Abstract
Purpose of review
The glomerulus is a unique structure required for filtration of blood, while retaining plasma proteins based on size and charge selectivity. Distinct cell types form the structural unit that creates the filtration barrier. Structurally, fenestrated endothelial cells line the capillary loops and lie in close contact with mesangial cells. Podocytes are connected by specialized intercellular junctions known as slit diaphragms and are separated from the endothelial compartment by the glomerular basement membrane. In order for this highly specialized structure to function, cross-communication between these cells must occur.
Recent findings
While classical studies have established key roles for vascular endothelial- and platelet derived-growth factors in glomerular cross-communication, novel paracrine signaling pathways within the glomerulus have recently been identified. In addition, unique cellular pathways of established signaling cascades have been identified that are important for maintaining glomerular barrier function in health and disease.
Summary
Here, we will review our current understanding of the processes of cross-communication between the unique cellular constituents forming the glomerular filtration unit. We will highlight recent findings of cellular crosstalk via signaling pathways that regulate glomerular barrier function in pathophysiological conditions.
Keywords: Vascular Endothelial Growth Factor, Angiopoietin, C-X-C chemokine ligand 12, Activated protein C, Endothelin-1
Introduction
Failure to adequately maintain glomerular barrier function result in loss of the size/charge selectivity of filtration, resulting in excretion of critical blood components into the urine. Loss of barrier function is characterized by proteinuria and observed in a large number of diseases. A number of cellular and structural defects lead to alterations in the glomerular filter, some of these result from impairment of the molecular signaling processes within and between the cells forming the glomerulus.
Development of the mature glomerulus consists of a series of tightly coordinated and well-defined stages (reviewed in detail by [1]). The cells that form the mature glomerulus include the podocytes that wrap around the glomerular capillaries, interdigitate, and connect by slit diaphragms that bridge the filtration slits (Figure 1). The intraglomerular mesangial cells remain situated in close contact with the glomerular endothelium that populate the side of the glomerular basement membrane opposite to the podocytes. The specialized endothelial cells lining the glomerular capillary loops are flattened and fenestrated, allowing high flux required by filtration [1] (Figure 1). Advances in the development of targeted genomic deletion strategies in select cell types of the glomerulus, most notably the podocytes, have greatly increased our understanding of the communication within the glomerulus and identified the importance of specific signaling molecules that allow the formation and maintenance of the glomerular filtration barrier, in health and disease. This review aims to highlight recent discoveries of signaling systems that mediate intraglomerular crosstalk. [2]
Figure 1.
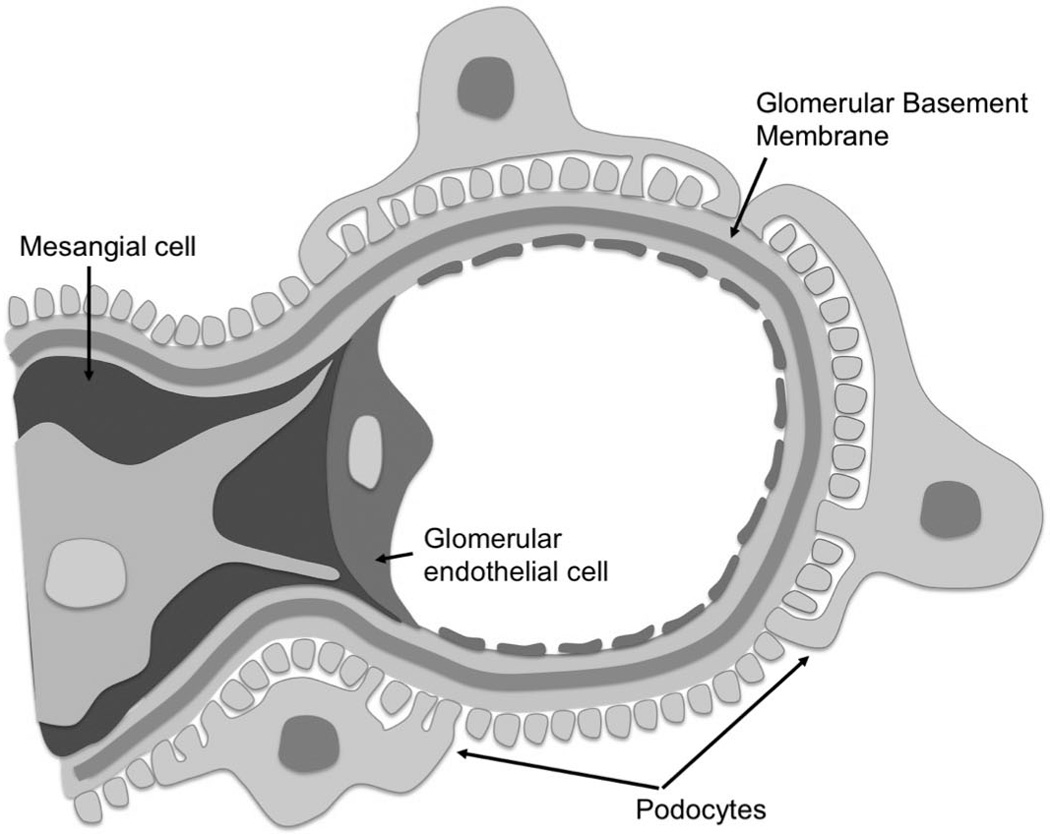
Composition of the glomerulus. Parts of this figure is adapted from [2].
Vascular Endothelial Growth Factor A (VEGFA) signaling
Vegfa is of key importance for angiogenesis in multiple organs systems, and remains important for the formation and maintenance of select microvascular beds within the kidney [3–5]. Vegfa expression by the podocytes has been shown to be a critical regulator of glomerular development and function and precise regulation of the amount of Vegfa produced by podocytes is required for adequate barrier function [3,6,7]. The complex dosage sensitivity of Vegfa on glomerular health is further demonstrated in mouse models of diabetic nephropathy. In patients with diabetic renal sequelae, glomerular VEGFA increases. In mice, inducible deletion of Vegfa from podocytes results in endothelial injury and an accelerated progression of the glomerular damage in diabetes [8]. Conversely, overexpression of Vegfa in podocytes in diabetic mice worsens diabetic glomerulopathy [9]. Thus, achieving optimal levels of Vegfa in the podocytes in diabetic nephropathy may be a prerequisite for minimizing progression of renal disease.
Given the important function and dosage sensitivity of Vegfa in the glomerulus, it is perhaps not surprising that mutations in genes that affect glomerular health also promote changes in glomerular Vegfa expression. However, in these settings, it remains to be determined whether the changes in Vegfa abundance occur as a cause or a consequence of glomerular disease. Recent findings have described that deletion of TGF-β-activated kinase 1 from podocytes results in early onset proteinuria, due to delayed glomerulogenesis and abnormal capillary formation. Importantly, these mice show an increased expression of Vegfa by podocytes [10]. Expression profiling of glomeruli from mice following podocyte-specific deletion of the transcription factor, Tcf21/Pod1 showed a reduction in angiogenic factors including that of Vegfa and associated receptors, suggesting this might be a regulatory factor for maintaining podocyte-endothelial vascular pathways [11].
VEGFA produced in podocytes signals to the endothelium via VEGF receptors. Here, the VEGF receptor 2 (VEGFR2/Kdr/FLK-1) expressed by endothelial cells appears most important as global deletion of Vegfr2 results in vascular defects, with glomerular thrombotic microangiopathy [12]. Signaling by VEGFA to the endothelial compartment via VEGFR1 (FLT1) as well as neuropilin-1, that can act as a coreceptor, appears less important, as kinase-deficient VEGFR1 mice do not develop glomerular disease [13]. Intriguingly, podocytes produce the soluble form of Vegfr1, named soluble Flt1 (sFlt1) [13]. Deletion of Vegfr1 from podocytes leads to massive proteinuria, ultimately progressing to renal failure. Unexpectedly, the apparent phenotype resulted from effacement of podocyte foot processes, suggesting a primary defect in podocyte function. Importantly, the phenotype of the mice lacking Vegfr1 in podocytes could be rescued by breeding to a mouse carrying a truncated Vegfr1 allele lacking the kinase domain. As sFlt1 is transcribed from the Vegfr1 gene by alternative splicing, these observations demonstrate an important role for the soluble form of the receptor in glomerular health. It was found that sFlt1 secreted by the podocytes acts via an autocrine pathway, binding to glycosphingolipids in lipid rafts, initiating an intracellular signaling cascade, that facilitates actin reorganization and cell adhesion [13]. In experimental diabetes, inducible overexpression of sFlt1 in podocytes of mice has also been found to reduce albuminuria and ameliorate diabetic glomerulopathy [14]. Collectively, the Vegfa/Vegfr system plays a number of critical roles in intraglomerular communication, ultimately determining glomerular barrier function in health and disease (Figure 2).
Figure 2.

Summary of signaling pathways between the different cellular components of the glomerulus.
The recent observations have confirmed and expanded important roles for the Vegfa signaling system within the glomerulus, both for cross-communication with endothelial cells and via autocrine pathways. Few recent examples have demonstrated reverse signaling from the endothelial compartment to the podocytes. Vasohibin-1 has recently been described as an endothelial gene that increases its expression in response to Vegfa. Vasohibin-1 affects endothelial cells in an autocrine manner, but may also have effects on podocytes and mesangial cells. Heterozygous deletion of Vasohibin-1 in mice has been shown to exacerbate the effects of experimental diabetes by augmenting albuminuria and promoting podocyte injury [15], suggesting that the protein may counteract Vegfa signaling, thereby ameliorating the effects of diabetic glomerulopathy.
Tie2/Angiopoietin signaling
Similar to the Vegf-Vegfr paracrine system, Angiopoietin-1 (Angpt1) is expressed by podocytes and mesangial cells and its cognate tyrosine kinase receptor, Tie2/Tek is expressed by the glomerular endothelial cells. Both the Angpt1 and Angpt2 bind the Tie2 receptor and have classically been viewed as having opposing effects on microvascular development, with Angpt1 stabilizing the vasculature, and Angpt2 antagonizing these effects by binding the Tie2 receptor in a competitive manner. Recent data have however suggested that Angpt1 and Angpt2 may cooperate to regulate Tie2 in lymphatic endothelium of the eye [16]. Global deletion of Angpt1 results in early embryonic lethality [17], as does induced global deletion of Angpt1 before embryonic day (E) 12.5 [18]. In mice with induced deletion of Angpt1 at E10.5, defects were observed in the glomerulus, with dilated capillary loops and a disrupted structure of the subendothelial glomerular basement membrane [18]. Reductions in mesangial cells were noted, while podocytes seemed intact. Later induction of global Angpt1 deletion produced no overt phenotype in Angpt1-deficient mice. However, when these mice developed diabetes, the loss of Angpt1 led to profound changes in glomerular barrier function. As compared to diabetic wildtype mice, increased proteinuria was present in diabetic Angpt1 deficient mice, accompanied by mesangial matrix expansion and glomerulosclerosis. Importantly, using various Cre recombinase driver lines, it was found that only deletion of Angpt1 in both the podocytic and mesangial compartment could recapitulate the effects seen in global Angpt1 deficient mice after a diabetic challenge [18]. A reduction in intraglomerular Angpt1 expression has been noted in diabetes, suggesting that altered growth factor expression could influence glomerular changes accompanying the disease [19]. Repletion of the glomerular Angpt1, using selective podocyte-specific overexpression of Angpt1 in diabetic mice, retarded the development of albuminuria as well as glomerular endothelial cell proliferation [19]. Thus, Angpt1 plays an important role in glomerular health and signaling of the Tie2/Angiopoietin pathway appears to be indispensable for the maintenance of the filtration barrier during renal development and in pathophysiological conditions [18,19]. Conversely, overexpression of Angpt2 in podocytes promotes apoptosis of glomerular endothelial cells and albuminuria, suggesting that within the glomerulus, Angpt2 may act in a competitive manner with Angpt1 [20] (Figure 2).
Recent studies have also shown important roles for angiopoietin-like-4 (Angptl4). The family of angiopoietin-like proteins share structural similarity with the angiopoietins, but do not signal via Tie2 receptors. Angptl4 is secreted as a glycoprotein from the cell. Podocyte expression of Angptl4 was substantially increased in rat models of puromycin-induced nephrosis [21]. Targeted overexpression of Angptl4 in podocytes of rats lead to marked albuminuria, foot process effacement, and loss of ionic charges within the glomerular basement membrane. Importantly, when Angptl4 was expressed transgenically in rat adipose tissue, circulating levels of Angptl4 were markedly increased, but no change in albuminuria was noted. It was subsequently discovered that the Angptl4 secreted by podocytes in these forms of disease, lacked normal sialylation (modification of the amount of sialic acids carried by glycoproteins). Increasing sialylation of Angptl4 decreased proteinuria in transgenic rats overexpressing Angptl4 in podocytes [21]. Thus, the expression and secretion of hyposialylated Angptl4 by podocytes is detrimental for glomerular ultrafiltration in puromycin-induced nephrosis. In a subsequent study the authors discovered that proteinuria-induced increases in circulating normosialylated Angptl4 was protective in nephrotic models by binding to αvβ5 integrins on the glomerular endothelium, presumably protecting the endothelial cells from oxidative injury [22]. However, as a consequence of high circulating levels of Angptl4 in nephrotic syndrome, free fatty acid uptake was also reduced in target organs and produced hypertriglyceridemia. Accordingly, normosialylated Angptl4 appears to contribute to glomerular health by communication with the endothelium, while the hyposialylated form accelerates oxidative injury in these disease states [22] (Figure 2).
CXCL12/CXCR4/CXCR7 signaling
The stromal cell-derived factor 1/ C-X-C chemokine ligand 12 (CXCL12) is a chemokine identified to play an important role in glomerular cross-communication. Cxcl12 secretion has been noted in podocytes, acting on glomerular endothelial cells expressing the C-X-C chemokine receptor type 4 (CXCR4) [23]. Consistent with a role in cross-communication, mice lacking either Cxcl12 or Cxcr4 had similar renal phenotypes with altered blood vessel formation. Both transgenic strains were found to have ballooning of glomerular capillaries and altered pattering of the renal vasculature [23]. The role of Cxcl12 was evaluated using a selective Cxcl12 inhibitor in a mouse model of type 2 diabetes [24]. Inhibition of Cxcl12 significantly retarded the development of diabetic glomerulopathy. In particular, blockade of Cxcl12 signaling reduced diffuse glomerulosclerosis and prevented albuminuria in the diabetic model [24]. Subsequent studies have identified an additional factor in the signaling cascade, namely CXCR7. Deletion of Cxcr7 in mice recapitulates the phenotype of the Cxcr4 deficient mice, with ballooning of glomerular capillaries. In contrast to Cxcr4, the Cxcr7 receptor is expressed by podocytes. Cxcr7 gene deletion also reduced the expression of Cxcr4, potentially explaining the observed phenotype. The authors speculate that Cxcr7 may function as a scavenger, thereby influencing Cxcl12 communication with Cxcr4 on endothelial cells [25]. The Cxcl12 signaling pathway may also play a role in potentiating Shiga toxin-induced renal injury. In fact, Shiga toxin increased the secretion of Cxcl12 and induced albuminuria likely by altering endothelial permeability, while inhibition of the Cxcl12 /Cxcr4 interaction led to improved survival and lower creatinine levels [26] (Figure 2).
Interleukin-6 signaling
Interleukin-6 secreted by podocytes has been shown important for recruitment of leukocytes in vitro [27]. Here it was discovered that the presence of podocytes, which release Interleukin-6, reduced TNF-α induced neutrophil recruitment to the glomerular endothelial cells. This immunosuppressive effect of Interleukin-6 appeared to alter the presentation of chemokines on the glomerular endothelial cells [27]. Whether this cross-communication also occurs in the intact animal remains to be determined.
Activated protein C/protease-activated receptor signaling
Protein C is activated by components of the thrombomodulin-thrombin system, which reside on endothelial cells, including glomerular capillaries. Activated Protein C (aPC) has been shown to play an important role in the setting of diabetic glomerulopathy. Mice carrying a point mutation in thrombomodulin, which has a severely reduced ability to generate aPC shows an exaggerated glomerular response to diabetic induction, with increased apoptosis of endothelial cells and podocytes [28]. However, when these mice were backcrossed with mice carrying a gain of function activating mutation that allow conversion of protein C in the absence of thrombomodulin, the accelerated phenotype was attenuated [28], strongly implicating a role for aPC signaling in the maintenance of glomerular barrier function in diabetes. The protective effects of aPC seem to depend on several receptor systems, where the protease-activated receptors convey the signal in podocytes. Genetic ablation of the protease-activated receptor-3 in the lipopolysaccharide–induced podocyte injury model, reduced the protective effects of aPC on podocytes [29] (Figure 2). These protective effects of aPC on podocytes may result from epigenetic changes that reduce oxidative stress [30].
Transforming growth factor-β receptor 1 (Tgfbr1) /Endothelin-1 (ET-1) signaling
The ET-1 system has recently been implicated in podocyte to endothelial cross-communication. Here it was found that inducible overexpression of the Tgfbr1 gene in podocytes, produced a phenotype with albuminuria and glomerulosclerosis in mice [31]. Altered glomerular endothelial cell architecture appeared before evidence of foot process effacement and depletion of podocytes. Importantly, mitochondrial dysfunction and oxidative stress were noted in endothelial cells from transgenic mice overexpressing podocyte Tgfbr1. Expression profiling revealed that Tgfbr1 overexpression in podocytes increased ET-1 precursor abundance in podocytes and ET-1 receptor A (ETA) in adjacent endothelial cells [31]. Additional proof that this signaling pathway is biologically important was obtained using an ETA antagonist in the setting of increased Tgfbr1 signaling in podocytes. Here the antagonist prevented both mitochondrial dysfunction in the endothelium, but also ameliorated the podocyte defects. Thus, endothelial ETA activation and resulting mitochondrial dysfunction appeared to be a prerequisite for podocyte apoptosis after stimulation of the Tgfbr1 pathway in podocytes [31] (Figure 2). Aside from the effect of ET-1 on the endothelium, ET-1 receptors on the podocyte appear important for maintaining glomerular integrity in diabetes. As such, deletion of ETA and ETB receptors in the podocyte protected the mice from diabetic glomerulopathy [32].
Endothelial generated nitric oxide signaling
In the setting of experimental diabetes, genetic deletion of endothelial nitric oxide synthase (eNos) leads to increased albuminuria that cannot be attenuated by Vegfa receptor blockade [33]. This must be viewed in comparison to wildtype mice, where blockade of the Vegfa receptor decreased diabetic glomerulopathy, which is surprising since Vegfa has classically been described to signal via eNos. Interestingly, inducible overexpression of podocytic Vegfa in eNos deficient mice results in marked proteinuria and glomerulosclerosis, further confirming nitric oxide independent effects of Vegfa [34]. Thus, loss of eNos may aggravate the diabetic condition independent of Vegfa. In fact, lack of eNos results in early onset albuminuria and changes in podocyte architecture in diabetic mice, not observed in wildtype diabetics (Figure 2). Furthermore, these sequelae can be blocked by inhibiting the renin-angiotensin-aldosterone system (RAAS) [33]. As eNos is situated on endothelial cells, some form of cross-communication must occur with the podocytes and eNos deficiency may thus trigger harmful responses in a RAAS-dependent manner.
Cross-communication between intraglomerular endothelial and mesangial cells
Platelet-derived growth factor B (Pdgfb) signaling
Classical studies have delineated a key role for the Pdgfb in communication between the glomerular endothelium and nearby mesangial cells. As such, endothelial cells express Pdgfb, while its receptor (Pdgfrb) resides on pericytes, including the specialized mesangial cells [35]. Genetic ablation of Pdgfb in mice disrupts the formation of capillaries, due to failure to attract pericyte progenitors, resulting in early embryonic lethality [36]. In these animals, absence of mesangial cells leads to the formation of a single ballooned glomerular capillary loop [35]. Targeted deletion of Pdgfrb generates a similar glomerular phenotype, indicating true endothelial- mesangial communication [37]. Selective deletion of endothelial derived Pdgfb produces a milder phenotype, where most animals reached adulthood. However, they did still develop glomerular abnormalities [38] (Figure 2).
Integrin αvβ8 and Tgf-β signaling
Signal transduction in the mesangial-endothelial direction must also occur. The close proximity of the mesangial and endothelial cells likely allows for efficient signaling, without the presence of massive amounts of ligand. Currently, few mesangial-secreted factors have been identified that partake in glomerular endothelial cross-communication in vivo. As noted above, deletion of Angpt1 from both podocytes and mesangial cells were necessary to obtain similar dramatic alterations in barrier function after experimental diabetes as observed in mice with a global deletion of Angpt1 [18]. Another example of mesangial to endothelial communication involves Integrin αvβ8. The integrin is expressed by mesangial cells, where it sequesters Tgf-β, thereby reducing Tgf-β signaling. Deletion of integrin αvβ8 results in early mortality, albuminuria and glomerulopathy in mice. These effects were ascribed to increased local bioavalibility of Tgf-β, which promoted apoptosis by acting on glomerular endothelial cells [39]. Regulation of Integrin αvβ8 by the mesangial cells may therefore amend Tgf-β signaling to the endothelial cells (Figure 2).
Cross-communication between podocytes and mesangial cells
Till now, direct in vivo evidence for communication between podocytes and mesangial cells is scarce. However, some signaling is likely to occur as specific injury of the podocytes clearly affects mesangial cell morphology and function. Yet, it cannot be excluded that disrupted mesangial function develops due to altered endothelial signaling after podocyte injury, as endothelial Pdgfb remains essential for mesangial cell function. Several signaling pathways have been suggested to be involved in this type of signaling, including ET-1, CCR7 and its ligand SLC/CCL21, platelet-derived growth factor, connective tissue growth factor, hepatocyte growth factor and Tgf-β [40] (Figure 2).
Conclusion
The increased application of cell-specific transgenic models and better methods to identify signaling pathways and regulators have greatly advanced our understanding of glomerular cross-communication in vivo. However, novel ligands allowing paracrine signaling, especially those released from intraglomerular mesangial and endothelial cells, likely remain to be discovered. The identification of these mediators as well as a better understanding of already established signaling pathways, should provide new targets for the prevention and treatment of glomerular diseases.
Key points.
-
-
Glomerular cross-communication is essential for adequate development and maintenance of the glomerular filtration barrier.
-
-
Pathological alterations in intraglomerular signaling may facilitate or aggravate glomerulopathy.
-
-
Understanding these intraglomerular signaling pathways may allow identification of novel pharmacological targets to help alleviate glomerular disease progression.
Acknowledgements
Financial support and sponsorship
The laboratory of H. Dimke is supported by the Novo Nordisk Foundation, the Carlsberg Foundation, and the A.P. Møller Foundation for the Advancement of Medical Science. Y. Maezawa is supported by Grants-in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan. S.E. Quaggin holds the Charles Mayo Chair of Medicine at the Feinberg School of Medicine and a Finnish Distinguished Professorship at the Oulu Biocenter. The laboratory of S.E. Quaggin is funded by NIH, NHLBI grant HL1241200, CIHR grants M0P62931 and M0P77756, E-Rare Joint Translational Call (JTC 2011) for European Research Projects on Rare Diseases and TF grant 016002.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
*of special interest
**of outstanding interest
- 1.Vaughan MR, Quaggin SE. How do mesangial and endothelial cells form the glomerular tuft? J Am Soc Nephrol. 2008;19:24–33. doi: 10.1681/ASN.2007040471. [DOI] [PubMed] [Google Scholar]
- 2.Maezawa Y, Cina D, Quaggin SE. Glomerular Cell Biology. In: Alpern R, Caplan M, editors. Seldin and Giebisch's The Kidney: Physiology & Pathophysiology. Vol. 1. Moe O: Elsevier; 2012. pp. 721–756. [Google Scholar]
- 3.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 5.Dimke H, Sparks MA, Thomson BR, et al. Tubulovascular Cross-Talk by Vascular Endothelial Growth Factor A Maintains Peritubular Microvasculature in Kidney. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veron D, Villegas G, Aggarwal PK, et al. Acute podocyte vascular endothelial growth factor (VEGF-A) knockdown disrupts alphaVbeta3 integrin signaling in the glomerulus. PLoS One. 2012;7:e40589. doi: 10.1371/journal.pone.0040589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sivaskandarajah GA, Jeansson M, Maezawa Y, et al. Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 2012;61:2958–2966. doi: 10.2337/db11-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veron D, Bertuccio CA, Marlier A, et al. Podocyte vascular endothelial growth factor (Vegf(1)(6)(4)) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia. 2011;54:1227–1241. doi: 10.1007/s00125-010-2034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim SI, Lee SY, Wang Z, et al. TGF-beta-activated kinase 1 is crucial in podocyte differentiation and glomerular capillary formation. J Am Soc Nephrol. 2014;25:1966–1978. doi: 10.1681/ASN.2013030252. *The paper explores the role of TGF-β-activated kinase 1 (TAK1) in podocytes. The work also implicates a role for glomerular Vegfa communication in the phenotype as deletion of TAK1 from podocytes results in glomerulosclerosis and proteinuria, which may in part result from increased Vegfa expression by the podocyte and altered cappilary architecture.
- 11.Maezawa Y, Onay T, Scott RP, et al. Loss of the Podocyte-Expressed Transcription Factor Tcf21/Pod1 Results in Podocyte Differentiation Defects and FSGS. J Am Soc Nephrol. 2014;25:2459–2470. doi: 10.1681/ASN.2013121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sison K, Eremina V, Baelde H, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21:1691–1701. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin J, Sison K, Li C, et al. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell. 2012;151:384–399. doi: 10.1016/j.cell.2012.08.037. [DOI] [PubMed] [Google Scholar]
- 14.Ku CH, White KE, Dei Cas A, et al. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes. 2008;57:2824–2833. doi: 10.2337/db08-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinamoto N, Maeshima Y, Yamasaki H, et al. Exacerbation of diabetic renal alterations in mice lacking vasohibin-1. PLoS One. 2014;9:e107934. doi: 10.1371/journal.pone.0107934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomson BR, Heinen S, Jeansson M, et al. A lymphatic defect causes ocular hypertension and glaucoma in mice. J Clin Invest. 2014;124:4320–4324. doi: 10.1172/JCI77162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suri C, Jones PF, Patan S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 18.Jeansson M, Gawlik A, Anderson G, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121:2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dessapt-Baradez C, Woolf AS, White KE, et al. Targeted glomerular angiopoietin-1 therapy for early diabetic kidney disease. J Am Soc Nephrol. 2014;25:33–42. doi: 10.1681/ASN.2012121218. * The manuscript illustrates a critical role of angiopoietin-1 dependent cross-communication in maintaining glomerular barrier function in experimental diabetes. Replenishing podocytic angiopoietin-1 amiliorates glomerular alterations in diabetic kidney disease.
- 20.Davis B, Dei Cas A, Long DA, et al. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. doi: 10.1681/ASN.2006101093. [DOI] [PubMed] [Google Scholar]
- 21.Clement LC, Avila-Casado C, Mace C, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. 2011;17:117–122. doi: 10.1038/nm.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clement LC, Mace C, Avila-Casado C, et al. Circulating angiopoietin-like 4 links proteinuria with hypertriglyceridemia in nephrotic syndrome. Nat Med. 2014;20:37–46. doi: 10.1038/nm.3396. * This paper demonstrates how Angptl4 can reduce proteinuria in nephrotic syndrome by acting on integrin αvβ5 on the glomerular endothelial cells. However, elevated levels of Angptl4 facilitates hypertriglyceridemia by inhibiting free fatty acid uptake in other organs and explain the link between hyperlipidemia and proteinuria in this disease.
- 23.Takabatake Y, Sugiyama T, Kohara H, et al. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol. 2009;20:1714–1723. doi: 10.1681/ASN.2008060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sayyed SG, Hagele H, Kulkarni OP, et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia. 2009;52:2445–2454. doi: 10.1007/s00125-009-1493-6. [DOI] [PubMed] [Google Scholar]
- 25.Haege S, Einer C, Thiele S, et al. CXC chemokine receptor 7 (CXCR7) regulates CXCR4 protein expression and capillary tuft development in mouse kidney. PLoS One. 2012;7:e42814. doi: 10.1371/journal.pone.0042814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petruzziello-Pellegrini TN, Yuen DA, Page AV, et al. The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J Clin Invest. 2012;122:759–776. doi: 10.1172/JCI57313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuravi SJ, McGettrick HM, Satchell SC, et al. Podocytes regulate neutrophil recruitment by glomerular endothelial cells via IL-6-mediated crosstalk. J Immunol. 2014;193:234–243. doi: 10.4049/jimmunol.1300229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 29.Madhusudhan T, Wang H, Straub BK, et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood. 2012;119:874–883. doi: 10.1182/blood-2011-07-365973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bock F, Shahzad K, Wang H, et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A. 2013;110:648–653. doi: 10.1073/pnas.1218667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daehn I, Casalena G, Zhang T, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest. 2014;124:1608–1621. doi: 10.1172/JCI71195. ** This paper illustrates the need for cross-communication between the podocytes and endothelial cells within the glomerulus. Activation of the Tgfb1 receptor in podocytes promotes crosstalk via the ET-1 pathway, which leads to mitochondrial oxidative stress in the ETA receptor expressing endothelial cells and subsequent podocyte apoptosis. Importantly, by blocking ETA or administering mitochondria-targeted antioxidants prevents podocyte injury, confirming an important functional relationship between the two cell types.
- 32.Lenoir O, Milon M, Virsolvy A, et al. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J Am Soc Nephrol. 2014;25:1050–1062. doi: 10.1681/ASN.2013020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuen DA, Stead BE, Zhang Y, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol. 2012;23:1810–1823. doi: 10.1681/ASN.2011121170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veron D, Aggarwal PK, Velazquez H, et al. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J Am Soc Nephrol. 2014;25:1814–1824. doi: 10.1681/ASN.2013070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindahl P, Hellstrom M, Kalen M, et al. Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in kidney glomeruli. Development. 1998;125:3313–3322. doi: 10.1242/dev.125.17.3313. [DOI] [PubMed] [Google Scholar]
- 36.Lindahl P, Johansson BR, Leveen P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 37.Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 38.Bjarnegard M, Enge M, Norlin J, et al. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–1857. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- 39.Khan S, Lakhe-Reddy S, McCarty JH, et al. Mesangial cell integrin alphavbeta8 provides glomerular endothelial cell cytoprotection by sequestering TGF-beta and regulating PECAM-1. Am J Pathol. 2011;178:609–620. doi: 10.1016/j.ajpath.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlondorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179–1187. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]