

Abstract
Animal models of disease have been used extensively by the research community for the past several decades to better understand the pathogenesis of different diseases as well as assess the efficacy and toxicity of different therapeutic agents. Retrospective analyses of numerous preclinical intervention studies using mouse models of acute and chronic inflammatory diseases reveal a generalized failure to translate promising interventions or therapeutics into clinically-effective treatments in patients. Although several possible reasons have been suggested to account for this generalized failure to translate therapeutic efficacy from the laboratory bench to the patient’s bedside, it is becoming increasingly apparent that the mouse immune system may not adequately recapitulate the immuno-pathological mechanisms observed in human diseases. Indeed, it is well-known that >80 major differences exist between mouse and human immunology; all of which contribute to significant differences in immune system development, activation and responses to challenges in innate and adaptive immunity. This inconvenient reality has prompted investigators to attempt to humanize the mouse immune system in order to address important, human-specific questions that are impossible to study in patients. The successful long-term engraftment of human hemato-lymphoid cells in mice would provide investigators with a relatively inexpensive, small animal model to study clinically-relevant mechanisms as well as facilitate the evaluation of human-specific therapies in vivo. The discovery that targeted mutation of the IL-2 receptor common gamma chain in lymphopenic mice allows for the long-term engraftment of functional human immune cells has advanced greatly our ability to humanize the mouse immune system. The objective of this review is to present a brief overview of the recent advances that have been made in the development and use of humanized mice with special emphasis on autoimmune and chronic inflammatory diseases. In addition, we discuss current challenges and possible solutions for utilizing these unique mouse models to define the human-specific immuno-pathological mechanisms responsible for the induction and perpetuation of chronic gut inflammation.
Keywords: hematopoietic stem cells, fetal thymus, fetal liver, interleukin-2 common gamma receptor, NK cells, NOD mice, SCID mice, human immune system, rheumatoid arthritis, graft vs. host disease, diabetes, allograft rejection
INTRODUCTION
Animal models of disease have been used extensively by the research community over the past several decades as surrogates to better understand the pathogenetic mechanisms responsible for the induction of different diseases, to assess toxicity, determine pharmacokinetics of different compounds and to evaluate the efficacy of novel therapeutic agents. Surprisingly, retrospective meta-analyses of preclinical intervention studies using animal models of a variety of different acute and chronic inflammatory diseases such as stroke, sepsis, diabetes, multiple sclerosis, and rheumatoid arthritis report that only a small fraction of promising preclinical interventions and novel therapeutics translate to clinical efficacy (1–7). A cursory search of PubMed using the keywords mouse and colitis identifies >5,800 studies that have been published using mouse models of the inflammatory bowel diseases (IBD; Crohn’s disease, ulcerative colitis). Of these, hundreds of studies report significant anti-inflammatory effects of numerous small molecules, biologics, genetic alterations or immune manipulations in these models of IBD. Yet very few of the potential “targets” or therapeutic interventions identified in this voluminous literature have been taken to the next level and evaluated in clinical studies. In fact, of the more than 50 novel small molecules, biologics and cell-based therapies that have been reported to be effective in preclinical animal studies and have been or are currently being evaluated in several hundred phase I–III clinical studies, only monoclonal antibodies directed against TNF (i.e. inflixamab, adlimumab, certolizumab, golimumab) or α4(β7) integrins (i.e. natalizumab, vedolizumab) have been shown to be effective in clinical studies and approved for treatment of patients with IBD (Reviewed in (8); http://wwwclinicaltrialsgov). The reasons for the disconnect between preclinical studies and therapeutic efficacy have not been clearly delineated; however several possible factors are thought to be involved including: a) the use of animal models that do not adequately mimic the chronic immunopathology of human IBD, b) the use of inbred strains of mice as surrogates for heterogeneous human populations, c) differences in intestinal microbiota d) flawed experimental design and/or data analyses and e) publication bias (1–7;9). In addition to these shortcomings in the design and evaluation of preclinical studies, a particularly troubling situation has emerged over the past few years that has garnered a great deal of attention by funding agencies and the publishing community: the inability of academic and industry investigators to reproduce published studies demonstrating therapeutic efficacy of novel small molecules and biologics in animal models of disease (2;10–15).
One potential strategy for improving the bench-to-bedside transition for promising therapeutics is to identify and utilize the most immunologically relevant mouse models of IBD and pharmacologic strategies that most closely mimic the clinical situation (1). However, even with more rigorous standardization of preclinical studies, we are faced with the reality that mice are not humans and thus the immuno-pathogenetic mechanisms observed in mouse models of chronic inflammation may not necessarily recapitulate those for human disease. It is well-known that the structure and function of the mouse immune system is, in many instances, significantly different from humans (16;17). For example, mice possess relatively few lymph nodes that are grouped together in small numbers of “chains”. Humans on the other hand, possess larger numbers of lymph nodes that are organized into complex chains that drain relatively small areas of tissue when compared to mouse (16). In addition to peripheral lymphoid structures, the structure and function of mouse bronchus-, nasal- and gut-associated lymphoid tissues may differ dramatically from those of the human depending upon its anatomic location (16). Given the structural differences in the immune systems between species, it is not surprising to learn that certain functions of the immune system are also quite different in mice when compared to humans. It is well-appreciated that >80 major differences exist between mouse and human immunology that are involved in immune system development, activation and responses to immune challenges (17). Major differences between the two species include hematopoietic activity of the spleen, circulating levels of lymphoid and myeloid leukocytes, innate immune mechanisms [e.g. Toll Like Receptors (TLRs), NOD Like Receptors (NLRs), defensins], T cell signaling pathways, B cell function, IgA production and isotype, intestinal intraepithelial and lamina propria cell composition, gut-associated dendritic cell and Natural Killer T (NKT) cell subsets and inducible nitric oxide synthase (9;17–21). Indeed, these differences may account, in part, for the lack of consensus as to whether mouse models of inflammation can be used as surrogates for human inflammatory diseases. Although one recent study suggests that genomic responses in mouse models of inflammation do not recapitulate those observed in human diseases (22), another equally compelling study concluded that gene expression patterns in mouse models closely mirror human inflammatory responses (23).
This inconvenient reality, coupled to the fact that animal models have been underwhelming in their ability to predict clinical trial outcomes in a variety of human diseases, reinforces the need to develop small animal models that more closely recapitulate human immunity. The development of mice with a fully functional human immune system via the long-term engraftment of the human hemato-lymphoid cells (e.g. hematopoietic stem cells, lymphocytes) would provide investigators a preclinical model to more accurately define disease pathogenesis as well as facilitate the evaluation of human-specific therapies in vivo. This approach has been successfully utilized for mechanistic and intervention studies in models of human-specific infectious diseases (e.g. HIV)(9;24–26). In addition, great strides have been made in using humanized mice to study the pathogenesis and human-specific therapies in different diseases such as diabetes, arthritis, graft vs. host disease, transplant rejection and cancer (25–31). A major breakthrough in the evolution of the humanization of the mouse immune system came with the discovery that targeted mutation of the IL-2 receptor common gamma chain (IL-2γ) in NOD/scid mice (termed NOD/scid-IL2rγ−/− or NSG mice) greatly enhanced the long-term engraftment of human hemato-lymphoid cells in these severely immuno-deficient mice (32–35). The objective of this review is to present a brief overview of the recent advances that have been made in the development and use of humanized mice with special emphasis on their use in autoimmune and inflammatory diseases. In addition, we discuss the challenges and possible solutions for utilizing these unique mouse models to define the human-specific immuno-pathological mechanisms responsible for the induction and perpetuation of chronic gut inflammation.
The Use of Immuno-Deficient Mice as Recipients for Human Hemato-lymphoid Cells: A Look Back
Adoptive transfer of xenogeneic (human) hemato-lymphoid cells (e.g. hematopoietic stem cells or peripheral blood mononuclear cells) into healthy wild type mice results in rapid rejection of these cells and, depending upon the type of cells injected, may result in lethal xenogeneic graft vs. host disease. In order to limit these damaging immune responses, immunological tolerance of recipient mice for donor human cells as well as tolerance of human cells for mouse tissue antigens is required for successful engraftment and survival of human progenitor and/or immune cells (9). Thus, the ideal hosts for creating this “bidirectional tolerance” are mice devoid of their adaptive and innate immune systems and major histocompatibility (MHC) complexes I and II. Although these mice have not yet been generated, substantial progress has been made in creating severely immuno-deficient mice that support the long term engraftment of human progenitor and immune cells (9;25;26;30). These new generation immuno-deficient mice are being used as platforms to study the immunological mechanisms associated with hematopoiesis, infectious diseases, cancer, autoimmune diseases, gene therapy and regenerative medicine (9;25;26;36;37). In addition, these humanized mice are providing preclinical models to evaluate the therapeutic efficacy of new drug and vaccine therapies (9;25;26). Below, we provide a brief history of development of immuno-deficient mice and highlight those discoveries that proved to be instrumental for enhancing the long-term engraftment of human hemato-lymphoid cells.
Nude Mice
The engraftment of human cells or tissue into immuno-deficient mice began more than 50 years ago following the discovery that T cell-deficient mice failed to reject normal and malignant human tissue (38). These hairless “nude” (nu/nu) mice were originally observed in a colony of albino mice operated by Dr. N.R. Grist in 1962 at the Virus Laboratory in Ruchill Hospital in Glasgow, Scotland. Dr. Grist subsequently sent the mice to the Institute of Animal Genetics in Edinburgh where Dr. S.P. Flanagan provided a detailed description of the hairless phenotype as well as described several abnormalities associated with these mice including problems with fertility and neonatal viability, liver disease and susceptibility to systemic toxoplasmosis (38). In 1968, Pantelouris reported that nu/nu mice lacked a thymus and thus were devoid of functional T cells (39) (Table 1). A subsequent series of investigations revealed that the nu/nu phenotype arose from a mutation of the Foxn1 gene previously known as the whn gene (winged-helix nude or Hfh11nu)(40;41). This pleotropic transcription factor is critical for both thymus development and the expression of different hair keratin genes. These T cell-deficient mice have proven to be one of the most popular mouse models used for engrafting normal or malignant human tissue. Unfortunately, nu/nu mice are of limited use for humanization of the mouse immune system as they do not support engraftment of human hemato-lymphoid cells (42). As correctly surmised by Ganick et. al., the failure of human immune cells to survive and proliferate in nu/nu mice may be due to the presence of functional murine NK cells (42)(see below).
Table 1.
Immuno-deficient Mice Used for Human Cell and Tissue Engraftment
| Strain | Phenotype | Limitations |
|---|---|---|
| Nude (nu/nu) | Lack T cells |
|
| SCID (Severe Combined Immuno-deficient) | Lack T & B cells |
|
| RAG-1−/− and RAG-2−/− (Recombination Activating Gene-1 or -2 Deficient) | Lack T & B cells |
|
| NOD (Nonobese Diabetic) |
|
|
| NOD/SCID |
Enhanced human cell engraftment |
|
Severe Combined Immuno-deficient and Recombination-Activating Gene-1 or -2-Deficient Mice
The severe combined immunodeficiency (scid) mutation was originally described by Bosma et. al. in C.B.-17 mice (43). It was ultimately revealed that the scid gene encodes for protein kinase, DNA activated catalytic polypeptide (Prkdc)(44;45). Mutation of the Prkdc gene prevents the expression of rearranged antigen receptors thereby preventing the development of mature T and B cells (46;47;47–51) (Table 1). Because scid mice lacked both cell-mediated and humoral immunity, they became the strain-of-choice for engrafting different populations of human hemato-lymphoid cells to study hematopoiesis, infectious disease, gene therapy protocols and tumor cell trafficking (reviewed in (52)). Several variations of the human-scid (hu-scid) chimeric mouse model were developed that used different routes of administration of the human cells (or tissue) into irradiated or un-irradiated scid mice (52). Although exciting new data were generated using the different hu-scid models, the numbers of human cells that engrafted and proliferated in these mice were very low (0.5–5% of total scid bone marrow cells)(52). Although young scid mice were devoid of both T and B cells, investigators soon found that T and B cell receptors undergo age-dependent rearrangements resulting the in formation of functional T and B cells, a phenomenon called “leakiness”(53–55). In addition, these immuno-deficient mice developed life-shortening thymic lymphomas (47;53;56). Furthermore, Bosma et. al. observed an increase in the development of thymic tumors in mice that became leaky as measured by the appearance of serum immunoglobulin (Ig+)(53). They observed that the incidence of thymic lymphomas increased from 8% in 3–5 month old (Ig−)scid mice to approximately 32% in age- matched (Ig+)scid mice (53). Furthermore, they found that almost 60% of the scid(Ig+) mice developed these malignant lymphomas at 5–9 months of age (53).
The inability of scid mice to support robust engraftment of xenogeneic immune cells was not specific for the scid strain or to their age-dependent propensity to generate functional T and B cells as human cell engraftment in recombination-activating gene-2-deficient (RAG-2−/−) mice was also very low or absent (52;57). Both the RAG-1 and RAG-2 genes are responsible for the activation of the V(D)J recombination reaction such that deletion of either gene produces mice devoid of T and B lymphocytes (58;59) (Table 1). Unlike scid mice, RAG-1−/− and RAG-2−/− mice (collectively referred to as RAG−/− mice) are not “leaky”, do not develop thymic lymphomas and are less sensitive to radiation (58;59). The reasons for the disappointingly low engraftment of human cells in scid or RAG−/− mice were found to be due to: a) lack of cross-reactivity between human progenitor cells and essential murine growth factors and b) the presence of robust innate immune systems in the lymphopenic recipients. It was clearly demonstrated that human hematopoietic stem cells were incapable of interacting with critical mouse cytokines and growth factors that are required for progenitor cell survival and proliferation (52). Surprisingly, neither exogenous administration nor transgenic expression of human hematopoietic growth factors (e.g. IL-3, GM-CSF, SCF) increased substantially the engraftment of human cells in scid recipients (52;60). Another confounding limitation that became apparent during this time was that both scid and RAG−/− mice maintained normal (or increased) numbers of myeloid cells (neutrophils, monocytes, macrophages) and NK cells as well as normal or increased hemolytic complement; any one of which were capable of destroying xenogeneic progenitor cells (52;55;60).
Nonobese Diabetic/scid Mice
Recognition of the critical role that innate immune cells (particularly NK cells) played in restricting the engraftment of human immune cells in scid and RAG−/− mice prompted investigators to develop new stocks of immuno-deficient mice that would allow for greater repopulation xenogeneic immune cells. A major turning point in these efforts came in 1994 when Shultz et. al. reported that back crossing nonobese diabetic (NOD) mice onto the scid background produced offspring (NOD/scid mice) that were devoid of both T and B cells, produced little or no serum immunoglobulin, had low numbers of functional innate immune cells (e.g. NK and myeloid cells) and lacked the hemolytic complement component C5(55) (Table 1). Unlike their scid relatives, <10% of the NOD/scid mice developed functional T and B cells at 200 days of age (55). The rationale for generating NOD/scid mice was based upon previous studies that were investigating the immuno-pathological mechanisms responsible for the spontaneous development of insulin-dependent diabetes mellitus (IDDM) in NOD mice (61). These earlier studies demonstrated that while NOD mice possessed autoreactive CD4+ and CD8+ T cells, these animals displayed defective innate immune function including reduced numbers and function of NK and myeloid cells, defective dendritic cell and macrophage function and absence of the hemolytic complement C5(52;62–64) (Table 1). In addition, it was found that NOD mouse-derived macrophages expressed the sigma regulatory protein alpha (SIRPα) receptor that was able to bind to CD47 expressed on human immune cells (65;66). This receptor-ligand interaction induces an inhibitory or “do not eat me” signal within murine macrophages thereby preventing these phagocytes from engulfing and destroying human progenitor or immune cells (9;65;66). Thus, the progeny of the NOD-scid cross were not only lymphopenic but they also possessed phagocytic tolerance as well as a defective innate immune system making them more likely to support long term engraftment of xenogeneic immune cells (55;67) (Table 1).
The generation of the NOD/scid stock initiated one of the most active periods of investigations in the humanization of the mouse immune system. Indeed, for more than 10 years, the NOD/scid mouse was considered the “gold standard” for long-term repopulation of human hemato-lymphoid cells (25;26;55;68). Investigators found that the engraftment of human peripheral blood mononuclear cells (PBMCs) or hematopoietic stem cells (HSCs) was 5–10-fold greater in NOD/scid mice when compared to scid mice (25;52;55;69). During the ensuing decade of investigations using the NOD/scid stock, it soon became clear that despite its numerous advantages over scid or RAG−/− recipients, the NOD/scid platform possessed its own set of shortcomings that limited its overall utility for humanization studies. For example, it was found that NOD/scid mice had a relatively short life span (8 months) due to the development of thymic lymphomas in approximately 70% of these mice by 40 weeks of age (52;55;70). In addition, while significantly reduced, residual NK and myeloid cell activities limited robust engraftment of human immune cells in these mice (55). These shortcomings made it clear that additional molecular and/or genetic alterations of this promising mouse strain would have to be made in order to enhance further the engraftment of human hemato-lymphoid cells.
Immuno-deficient Mice Devoid of Functional IL-2 Receptor Common γ Chain
A fundamental breakthrough that greatly enhanced the long-term engraftment of even greater numbers of human hemato-lymphoid cells came in 2002 with the development of NOD/scid mice that lacked a functional IL-2 receptor common gamma chain (IL-2rγ−/−)(33). As early as 1995, investigators knew that IL-2rγ was a critical component of the high-affinity receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21(71;72) (Figure 1). Because these cytokines are required for T, B and NK cell development, targeted mutation of IL-2rγ resulted in the loss of receptor-ligand signaling thereby creating mice devoid of T, B and NK cells (73–75). Over the next few years, several laboratories developed immunodeficient mice devoid of IL-2rγ that differed with respect to strain background and the type of IL-2rγ mutation (26;28;68) (Table 2). The nomenclature for describing and categorizing the different IL-2rγ−/− strains may appear a bit confusing for those readers who are not mouse geneticists. Therefore, we have found it helpful to group the most popular IL-2rγ−/− strains into three major categories depending upon whether the IL-2rγ mutation has been bred onto the NOD/scid, NOD/RAG−/− backgrounds (Table 2). For example, the major NOD/scid-IL-2rγ−/− strains include the NOD.Cg-PrkdcscidIl2rgtm1Wjl (NSG) mouse and the NOD.cg-PrkdcscidIl2rgtm1Sug (NOG) mouse. The IL-2rγ mutation in NSG mice is a complete null mutation and thus is not expressed whereas NOG mice express a truncated/inactive form of the γ chain that can bind cytokines but does not promote cell signaling. Similarly, the NOD/RAG1−/−IL-2rγ−/− mouse (NOD.Cg-Rag1tm1MomIl2rgtm1Wjl; or NRG) contains a complete null mutation of IL-2rγ. The third major group of IL-2rγ−/− mice currently being used for humanization studies consists of BALB/c/Rag1−/− IL-2rγ−/− and BALB/c/Rag2−/− IL-2rγ−/− mice (both are called BRG). These mice express a truncated/inactive form of IL-2rγ similar to that expressed in NOG mice. We refer readers to several recent reviews that describe the generation and unique characteristics of these different (26;28;68;69).
Figure 1. The common ƒÁ chain plays a critical role in interleukin signaling and the development of immune cells.
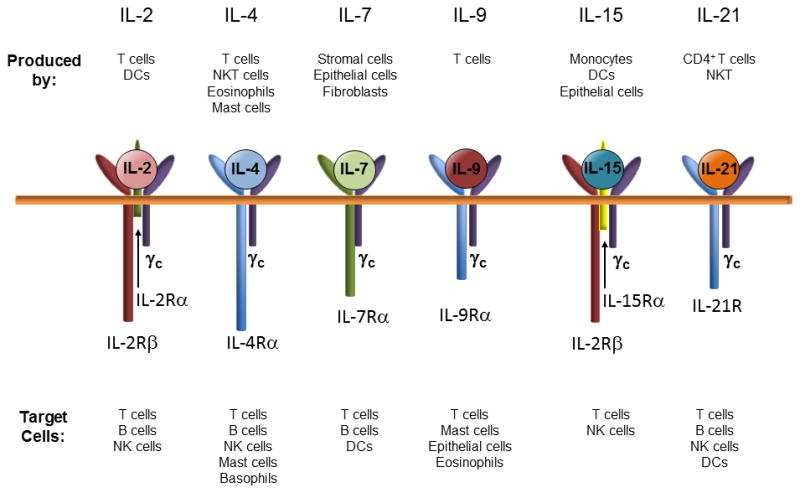
The common γ chain is associated with receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21. These cytokines are produced by different immune, stromal cells and epithelial cells. Receptor/ligand signaling is critically important for the generation of different populations of immune cells (Modified from reference 75 with permission).
Table 2.
Immuno-Deficient Mice Lacking Functional IL-2rγ
| Group | Strain | Name | Characteristics |
|---|---|---|---|
| NOD/scid-IL-2rγ−/− | NOD.Cg-PrkdcscidIl2rgtm1Wjl | NSG | The Il2rγ mutation is a complete null such that IL-2rγ is not expressed. |
| NOD.cg-PrkdcscidIl2rgtm1Sug | NOG | The Il2rγ mutation encodes a truncated form of the γ chain such that it lacks the intracytoplasmic domain; it can bind cytokines but will not promote cell signaling | |
| NOD/RAG-1−/−IL-2rγ−/− | NOD.Cg-Rag1tm1MomIl2rgtm1Wjl | NRG | The Il2rγ mutation is a complete null such that IL-2rγ is not expressed. |
| BALB/c/RAG−/−IL-2rγ−/− | C.Cg-Rag1tm1MomIl2rgtm1Wjl | BRG | This strain is generated on a BALB/c background that has the RAG-1−/− mutation. The IL-2rγ chain lacks the intracytoplasmic domain thereby preventing intracellular signaling. |
| C.Cg-Rag2tm1FwaIl2rgtm1Sug | BRG | This strain is generated on a BALB/c background that has the RAG-2−/− mutation. The IL-2rγ chain lacks the intracytoplasmic domain thereby preventing intracellular signaling. |
The development of these immuno-deficient IL-2rγ−/− strains was a defining moment in the quest to humanize the mouse immune system as each of these stocks were shown to engraft human hematopoietic stem cells (HSCs) or peripheral blood mononuclear cells (PBMCs) to a much greater extent than had been observed for any of the previous immuno-deficient strains (26;28;68;69). In addition, these new-generation IL-2rγ−/− mice do not develop thymic lymphomas and live a normal lifespan of 2 years. These novel platform strains have been particularly useful for studies involved in human immunity, hematopoiesis, infectious disease, autoimmune disease, regenerative medicine and cancer (9;26;69).
Humanized Mouse Models
There are currently three major humanized mouse models that utilize the immuno-deficient IL-2rγ−/− strains described above (Table 2). As with any animal model, each of these models has advantages and limitations. These humanized mouse models have been shown to possess different capabilities of engrafting human hemato-lymphoid cells. For example, investigators have determined that NSG and NRG mice engraft human HSCs and promote T cell development to a greater extent than do BRG mice (76) whereas NSG mice support engraftment of larger numbers of human HSCs than do NOG mice (77). Therefore, the choice of a specific humanized model will depend upon the specific questions being posed by the investigator. Theoretically, it should be possible to model most if not all of the human autoimmune and chronic inflammatory diseases using one or more of the immuno-deficient IL-2rγ−/− strains. This section describes the generation, uses and limitations of the 3 major humanized mouse models
Engraftment of Human Peripheral Blood Mononuclear Cells into Lymphopenic IL-2rγ−/− Mice
In 1988, Mosier et. al. demonstrated that intra-peritoneal (i.p.) injection of human peripheral blood leukocytes (PBLs) into scid mice (termed Hu-PBL-scid mice) resulted in the stable reconstitution of a functional human immune system (78). Although all populations of circulating leukocytes were injected in the Hu-PBL-scid model, the large majority of stably engrafted cells were activated T cells with only very small numbers of human B cells, myeloid cells or other immune cells present in the scid recipients. As discussed in the preceding section, the total numbers of human T cells that engraft and proliferate in scid mice are actually quite low due to the age-related development of mouse T and B cells and the presence of an active innate immune system that together destroy xenogeneic cells (52;55;60). In addition, scid mice developed life-shortening thymic lymphomas thereby limiting the usefulness of this model (43;53;56). We would like to caution the reader that the use of the term Hu-PBL-scid has been used and continues to be used to describe any humanized mouse model where human PBMCs are injected into lymphopenic recipients such as a scid, NOD/scid or the newer immuno-deficient IL-2rγ−/− strains. In order to avoid any confusion, we have chosen to focus primarily on the use of lymphopenic IL-2rγ−/− strains as recipients for human PBMCs in this and the following sections (Table 3). Indeed, the generation of these immuno-deficient IL-2rγ−/− strains greatly advanced the field as investigators observed much greater levels of engraftment of T cells than had been documented in other lymphopenic recipients. In addition, lymphopenic IL-2rγ−/− strains possessed a much greater lifespan due to the lack of thymic lymphoma development (26;68;69). Intravenous injection of human PBMCs into irradiated (conditioned) or naive IL-2rγ−/− strains produces the most robust engraftment of human T cells than in any previous immuno-deficient recipient (79). The Hu-PBMC-IL-2rγ−/− mouse model is currently used to study human immunity, autoimmunity, infectious diseases, regenerative medicine, allograft rejection and cancer (9;26;28;68;80) (Table 3). In addition, this model has been used to assess novel cell-based therapies (36). Although this model is well-suited for investigating T cell-mediated immune responses of patients with immunologic disorders as well as autoimmune and chronic inflammatory diseases, the “window of opportunity” for performing these types of studies is limited by the development of lethal xenogeneic graft-versus-host disease (GVHD) within 3–4 weeks following T cell transfer (81). King and coworkers have demonstrated that xenogeneic GVHD results from the ability of human T cells to recognize and mount aggressive immune responses towards murine MHC class I and II (81). These investigators have reported a significant delay in xenogenic GVHD when human PBMCs are engrafted into NSG mice devoid of MHC class I and II (81). Despite this limitation, the Hu-PBL-IL-2rγ−/− model has been used successfully to explore immuno-pathological mechanisms involved in different autoimmune and infectious diseases.
Table 3.
Models of Human Hemato-Lymphoid Cell Engraftment into Lymphopenic IL-2rγ−/− Mice
| Model Name | Description | Advantages | Applications | Limitations |
|---|---|---|---|---|
| Hu-PBMC-IL-2rγ−/− (Human Peripheral Blood Mononuclear Cells engrafted into lymphopenic IL-2rγ−/− mice) | Lymphopenic IL-2rγ−/− mice are injected with human peripheral mononuclear cells |
|
|
|
| Hu-HSC-IL-2rγ−/− (Human Hematopoietic Stem Cells engrafted into lymphopenic IL-2rγ−/− mice) | Lymphopenic IL-2rγ−/− mice are injected with CD34+ HSCs derived from fetal liver, cord blood, bone marrow, or from peripheral blood following G-CSF-mediated mobilization |
|
|
|
| BLT-IL-2rγ−/− (Bone marrow, Liver, Thymus engrafted into lymphopenic IL-2rγ−/− mice) | Lymphopenic IL-2rγ−/− mice are implanted with small pieces of human fetal liver and autologous thymus under the renal capsule; the mice are then injected with human CD34+ HSCs purified from the same fetal liver sample. |
|
|
|
Engraftment of Human Hematopoietic Stem Cells into Lymphopenic IL-2rγ−/− Mice
Following the early successes of engrafting PBMCs into scid recipients, investigators initiated studies to ascertain the ability of scid mice to support the engraftment and differentiation of human CD34+ hematopoietic stem cells (HSCs) derived from human fetal tissue or bone marrow (82;83). Lapidot and coworkers reported that adoptive transfer of human CD34+ HSCs into scid recipients resulted in the generation of small numbers of immature lymphocytes and myeloid cells (83)(Table 3). This model, called the human scid repopulating cell scid or Hu-SRC-scid model, has been used and continues to be used for exploring different immunological aspects of human hematopoiesis, infectious diseases, and adaptive immunity. However, the utility of this model was limited by disappointingly low levels of HSC engraftment and lack of formation of functional T and B lymphocytes, myeloid cells, and dendritic cells. The development of immuno-deficient IL-2rγ−/− mice provided a much needed catalyst for developing a more robust humanized immune system in mice (32–35). A series of studies demonstrated that HSCs derived from fetal liver, umbilical cord blood (UC), bone marrow, and G-CSF mobilized peripheral blood could be used to engraft different lymphopenic IL-2rγ−/− mice resulting in dramatic increases in engraftment and differentiation of HSCs into different immune cell subsets (76;84;85). Brehm and coworkers recently compared the engraftment efficiency of CD34+ HSCs obtained from cord blood in three different immuno-deficient IL-2rγ−/− mouse strains at different ages (76). They found that engraftment was more robust in irradiated newborns compared to irradiated adults and that T and B cell numbers were significantly greater in NSG and NRG mice compared to BRG recipients. Other investigators have found that intrahepatic injection of HSCs also provides an effective mode of delivery of human HSCs for humanizing the immune system (86;87). Indeed, Misharin et al reported that NSG mice engrafted via intrahepatic injection of human CD34+ HSCs spontaneously produced human immunoglobulins as well as generated specific IgG following immunization (87). In addition to T and B cells, adoptive transfer of human HSCs into IL-2rγ−/− recipients promotes the formation of small but significant numbers of myeloid cells and DCs (32–34;88–90).
A major advantage of Hu-HSC-IL-2rγ−/− models is the absence of xenogeneic GVHD (83). Because human T cells undergo positive and negative selection within the mouse thymus, these lymphocytes are restricted to mouse MHC and thus, do not react to highly antigenic murine proteins. However, this advantage is also its primary drawback i.e. human T cells are not human leukocyte antigen (HLA) restricted. This means that T cells will not recognize nor react to antigen presented by human antigen presenting cells that develop within tissue (91). In addition, HSC engraftment in immuno-deficient IL-2rγ−/− mice have poor T cell development most likely due to inefficient human thymopoeisis in the mouse thymus (89). Furthermore, very low numbers of circulating platelets, erythrocytes and polymorphonuclear neutrophils (PMNs) are produced in following transfer of human HSCs (26;80;91;91). Despite these shortcomings, the Hu-HSC-IL-2rγ−/− models have provided invaluable information on many aspects of human immuno-biology.
Engraftment of Human Hematopoietic Stem Cells into Lymphopenic IL-2rγ−/− Mice Implanted with Human Fetal Thymic and Liver Tissues
To create a more robust humanized environment that would allow for more complete differentiation of engrafted human HSCs, the Bone marrow-Liver-Thymus (BLT) humanized mouse model was developed (Table 3). In this model, animals are first conditioned using sublethal irradiation and then are implanted with small fragments (~1 mm3) of autologous human fetal liver and thymus under the kidney capsules (9;26;92;93). Recipients of the human tissues are then injected (i.v.) with 1–5 ×105 autologous human HSCs obtained from the fetal human liver implanted into the recipients (94). The BLT model, originally developed in NOD/scid mice, convincingly showed long-term engraftment of multi-lineage human cells including T and B cells, DCs, monocytes, macrophages, erythrocytes and platelets (9;69;92;93;95). In addition, these studies demonstrated that human T cells were HLA restricted and were capable, along with human B cells, of mounting adaptive immune responses in vivo (9;69;92;93;95). Furthermore, BLT mice developed secondary lymphoid tissue (e.g. mesenteric lymph nodes) and human mucosal immune systems thereby making this the model of choice for HIV investigators (9;69;92;93;95). Following the development of immuno-deficient IL-2rγ−/− mice, investigators compared the relative engraftment efficiencies of human HSCs in the NOD/scid-BLT vs. NSG-BLT model with varying results. Although some studies reported similar levels of reconstitution of human HSCs in blood, spleen, liver and lung in the two BLT models (77;96;97), other studies reported superior human immune cell engraftment in NSG-BLT vs. NOD/scid-BLT mice (32;33;98;99). One important difference between the two BLT models reported by Denton and coworkers was the relative lack of intraepithelial and lamina propria T cells within the intestines of NSG-BLT mice when compared to NOD/scid BLT mice (97). In a more recent study, Nochi et. al. demonstrated that cryptopatches and gut associated lymphoid tissue (GALT) failed to develop in NSG-BLT mice whereas intestinal T cells and cryptopatches were readily observed in NOD/scid-BLT mice (100). Furthermore, these investigators demonstrated that cryptopach development was required for the generation of a functional GALT. Taken together, these two studies highlight the importance of the IL-2rγ chain in IL-7/IL-7 receptor signaling for normal GALT development (100).
Despite model variability, the NOD/scid-BLT and NSG-BLT models are considered two of the best humanized mouse models for studies involved in human hematopoiesis, infectious diseases, acute and chronic diseases and therapeutic/vaccine development. Despite the fact that BLT mice have a “blended” immune system composed of murine myeloid cells co-existing with a human adaptive immune system, these humanized animals are capable of mounting both direct and indirect delayed type hypersensitivity (DTH) responses to two different antigenic challenges (101). Because DTH responses are considered the most stringent test for a fully functional and well-integrated immune system, this model is suitable for studies evaluating T cell- dependent immune responses. As with the other humanized mouse models, the BLT model has a few significant drawbacks including the requirement for small animal surgical skills, the use of human fetal tissue, generation of relatively low levels of mature/functional human granulocytes, myeloid cells, B cells and a delayed xenogeneic GVHD (94;102–105). It is not clear why GVHD develops by 4–5 months post-reconstitution however, a loss of tolerance of human immune cells to murine MHC antigens is considered a distinct possibility. A recent study demonstrates that deletion of either MHC class-I or -II fails to delay the development of GVHD in the NSG-BLT model (94). Interestingly, Lavender and coworkers have demonstrated that deletion of CD47 in C57Bl/6-RAG-2−/−IL-2rγ−/− mice renders these triple knock out (TKO; CD47−/−RAG-2−/−IL-2rγ−/−) animals resistant to GVHD in the BLT model (106;107). Based upon previous work by Wang et. al.(108), the authors hypothesize that during development, phagocytes (i.e. macrophages, DCs) within TKO mice become tolerized to xenogeneic HSCs (106). Because the expression of SIRPα on macrophages derived from CD47−/− mice is very similar, if not identical to that observed on WT macrophages, tolerance observed in TKO-BLT mice is unlikely to be caused by SIRPα-CD47 signaling (106;108). The major advantages of using this C57Bl/6-based, TKO-BLT model vs. the NOD/scid-BLT or NSG-BLT model are: a) C57Bl/6-TKO mice maintain an intact complement system, b) C57Bl/6-TKO mice are more radiation resistant allowing for more efficient conditioning prior to human cell engraftment and c) genetic manipulations (e.g. gene inactivation; introduction of transgenes) are more easily accomplished due to the ready availability of a variety of different mutant mice on the C57Bl/6 background (106).
Current Strategies for Improving Engraftment of Human Hemato-lymphoid Cells in Immuno-deficient Mice
The advances that have been made in humanizing the immune system of immunodeficient mice over the past 20 years has been truly remarkable. Nevertheless, a number of limitations remain that prevent the development of a fully functional human immune system. For example, virtually all humanized mouse models possess only small numbers of human granulocytes (e.g. PMNs, eosinophils), myeloid cells (e.g. monocytes, macrophages), erythrocytes and platelets as well as reduced numbers of fully functional T and B cells (9;28;80). Many of these deficiencies are most likely due to the lack of cross reactivity of certain murine cytokines and/or growth factors with human progenitor cell receptors that are known to be important for hematopoiesis and immune system development (9). However, poor development of peripheral lymph nodes, GALT and mucosal immune systems undoubtedly contribute to an underdeveloped immune system (28;80;97;100). The following section discusses some of the new approaches that are currently being used to improve engraftment and differentiation of human HSCs in immuno-deficient mice.
Strategies for enhancing the human innate immune system
The development, numbers and function of human PMNs, monocytes, macrophages and NK cells are generally quite low in the currently used models of humanized mice. For example, human myeloid cells have been found to be less than 5–10% of what is found in humans[reviewed in (9)]. Although some studies report that these myeloid cells are reasonably functional (90;109), other studies have described these myeloid cells as phenotypically immature and functionally defective (110). Furthermore, human NK cell maturation, function and homeostasis are also defective in humanized mice owing in part, to NK cell susceptibility to phagocytosis by murine macrophages (98) (discussed below) and lack of trans-presentation hIL15-hIL15Rα(111). Many of the cytokines and growth factors required for innate immune system development and function are produced by non-hematopoietic cells and thus are of murine origin. A recent review by Rongvaux et. al. presents an extensive list of these cytokines and factors and highlights the significant differences in sequence homologies between the mouse and human proteins (9). The lack of effective receptor-ligand interactions between human progenitor cells and mouse-derived cytokines and growth factors would be expected to greatly diminish hemato-lymphoid cell development in immuno-deficient mice (9).
In order to improve innate immune cell development and function, several genetic and technological approaches are currently being employed to promote and/or enhance expression of crucial human cytokines and growth factors that are known to be important in innate immune system development and function including: a) use of cDNA constructs expressing human genes that can be controlled by ubiquitous or tissue-specific promoters; b) use of bacteria artificial chromosomes (BACs) that have regulatory elements for gene expression; and c) use of knock-in technology to replace mouse genes with specific human genes. The cDNA construct approach can lead to transgene expression at non-physiological levels or without temporal and tissue specific regulation. BACs often include regulatory elements for control of gene expression facilitating gene expression at physiologic levels and at the appropriate time and location. It should be noted that both the cDNA and BAC approaches result in expression of both the mouse and human gene(s) of interest because the homologous mouse gene is left intact. However, knock-in technology replaces the mouse gene with the equivalent human gene resulting in human gene expression without expression of the mouse protein. Indeed, knock-in technology has been used to increase the numbers of myeloid cells in humanized mice expressing human interleukin 3 (IL3) and granulocyte-macrophage colony-stimulating factor (GM-CSF)(112), thrombopoietin (113) or macrophage colony-stimulating factor (M-CSF)(114). In a more recent study, Rongvaux et. al.(115) report the generation of mice expressing human genes for M-CSF, human IL-3, human GM-CSF and human thrombopoietin that were knocked-in to their corresponding mouse loci in immunodeficient RAG-2−/− IL-2rγ −/− mice (termed MITRG mice). They also generated MITRG mice that also express human SIRPα encoded in a BAC (termed MISTRG). Engraftment of human fetal liver-derived CD34+ HSCs into the MITRG, MISTRG, RAG-2−/− IL-2rγ −/− and NSG mice resulted in significantly greater myeloid cells in the blood and BM and nearly 10-fold increases in myeloid cells in the lung, liver and colon of MITRG and MISTRG mice compared to engrafted RAG-2−/− IL-2rγ −/− and NSG mice (115). Human peripheral blood myeloid cell populations in of MITRG and MISTRG mice were composed predominantly of monocytes indicating that the mouse environment remained insufficient for the development and/or survival of human PMNs. Human NK cells were also detected in tissues at numbers approximately 10-fold higher in MISTRG engrafted mice compared to NSG engrafted mice. These exciting results were tempered by the poor development and survival of human RBCs in MITRG and MISTRG mice resulting in progressive anemia that developed due to poor mouse erythropoesis after irradiation preconditioning. Both the MITRG and MISTRG mice are currently commercially available.
Hydrodynamic delivery (via i.v. injection) of plasmids encoding different human cytokines has also been used in humanized mouse models to transiently increase expression of important cytokines and factors known to be important for hemato-lymphoid system development and function. Using this delivery system, Chen et. al. demonstrated that transient expression of human IL-15 and Flt-3/Flk-2 ligand in NSG mice increased the numbers of human NK cells for more than 30 days following HSC engraftment (116). These investigators also demonstrated that transient expression of GM-CSF and IL-4, macrophage colony stimulating factor (M-CSF) or erythropoietin and IL-3 significantly enhanced the generation of DCs, monocytes/macrophages or erythrocytes, respectively in Hu-HSC-NSG mice (116). In more recent studies, Li et. al. reported that hydrodynamic delivery of a plasmid encoding for human M-CSF enhanced the generation of mature and functional monocytes and tissue-associated macrophages in Hu-HSC-NSG mice (109). The use of transgenic technology is another approach that is currently being used to enhance expression of human cytokines and growth factors in immuno-deficient mice. For example, Billerbeck and coworkers have reported that transgenic expression of human stem cell factor (SCF), GM-CSF and IL-3 in NSG mice greatly enhanced the generation of myeloid cells following engraftment with CD34+ HSCs (117). Surprisingly, these investigators also observed an increase in the numbers of functional CD4+Foxp3+ regulatory T cells (Tregs) in peripheral tissues of these transgenic mice when compared to humanized NSG animals. Another group of investigators have shown accelerated differentiation of human granulocytes as well as generation of myeloid cells in NSG mice expressing the human transgene encoding SCF (118).
Strategies for enhancing the human adaptive immune system
The currently used humanized mouse models generate, to varying degrees, all lineages of human immune cells. The Hu-HSC models have been found to possess relatively weak primary and secondary immune responses including lack of immunoglobulin (Ig) class switching whereas NOD/scid-BLT and NSG-BLT models demonstrate more robust adaptive immune responses that include the generation of both IgM and IgG (9;26;80;92;93;95). As pointed out above, factors that limit the development of a well-integrated and fully functional adaptive immune system include defective cytokine and growth factor-mediated signaling in human progenitor cells, underdeveloped peripheral and mucosal lymphoid tissues and susceptibility of human lymphocytes to phagocytosis by mouse macrophages (9;26). A few studies have demonstrated that humanized mice develop weak antigen-specific T cell responses (26;119). Human T cells isolated from the spleens of NSG-BLT mice reveal functional deficiencies characterized by loss of expression of certain co-stimulatory molecules such as CD27(120). This specific deficiency could be partially reversed in vitro by addition of human IL-2 and IL-7. However, in general, development, maintenance and function of human T cells in current humanized mice are suboptimal (9). A few studies have shown, with varying degrees of success, that the development and maintenance of human T cells may be enhanced using delivery of human recombinant IL-7 or IL-15(34;121;122). Another factor that is critical for T cell development and function is the interaction between T cell receptors (TCR) and MHC class I and II. Several groups of investigators have recently reported the generation of transgenic IL-2rγ−/− mice (e.g. NSG, NRG, NOG) expressing human MHC I or II molecules termed human leukocyte antigen (HLA)-I or II, respectively (9;28;98;123–129). Transgenic expression of HLA-I or -II increased the numbers and function of human T cells as well as enhanced antigen-specific immune responses following viral infection or immunization. The future development of transgenic IL-2rγ−/− mice expressing both HLA-I and II but devoid of both murine MHC class I and II will allow human T cells to develop in a HLA- restricted manner (28).
In contrast to the relative paucity of functional T cells in Hu-HSC-IL-2rγ−/− mouse models, human B cell numbers are actually quite reasonable. However, a number of different investigators have reported that B cell development is incomplete and antigen specific B cell responses are significantly reduced in this model compared to human responses (96;120;130–132). Investigators have observed a skewing of B cell development towards a more immature phenotype with steady state levels of human IgG at ~1% of that observed in humans (96;131;132). Although some investigators have shown that immunization or infection of Hu-HSC or BLT mice may increase production of human IgM and IgG, the results have been variable (132). For example, IgG formation following immunization of humanized mice with tetanus toxoid or viral antigens has not been consistently observed whereas immunization of BLT mice with 2,4-dinitrophyl-hapten-keyhole limpet hemocyanin produced antigen-specific generation of IgG1 and IgG2 (95;132). Because B cells isolated from Hu-HSC-IL-2rγ−/− mice exhibit a highly diverse immunoglobulin repertoire that is very similar to that found in human B cells, it has been hypothesized that humanized mice have the “genetic potential” to mount antibody responses similar to humans (132). Studies to enhance human B cell maturation in humanized mice are currently being explored by different laboratories.
The lack of fully mature B cells in humanized mice can be explained in part by the apparent inability of murine B lymphocyte stimulating factor (BLyS or BAFF) to interact with human progenitor cells. Administration of human recombinant BLyS to NOD RAG-2−/− Prf1−/− mice engrafted with human PBMCs increased both human B and T cell engraftment (133). Treatment with human BLyS for 14 days resulted in 40 fold greater IgM and a 10 fold greater IgG production than controls and mounted 10-fold higher IgM and IgG de novo antibody response to pneumococcal thymus-independent antigens. Knock-in of human BLyS into immuno-deficient mice should be beneficial to enhance humoral and cell mediated immune responses in humanized mice in Hu-PBMC-IL-2rγ−/−, Hu-HSC-IL-2rγ−/− and BLT mice. Hydrodynamic injection of plasmids encoding for human GM-CSF and IL-4 has been reported to increase production of antigen-specific IgG following immunization in Hu-HSC mice (134). Another reason for reduced humoral responses in Hu-HSC-IL-2rγ−/− mice may be due to defective T cell help since human T cells are selected within the mouse thymus based upon murine MHC-II rather than HLA-2 as described above. In support of the importance of HLA-II restricted T cells in humoral responses is a recent study reporting that engraftment of human HSCs into immuno-deficient IL-2rγ−/− mice expressing the human HLA-2 (HLA-DR4) transgene results in the generation of IgM, IgG (all four subclasses), IgA and IgE following immunization (123).
Another important determinant for the development of a fully functional adaptive immune system is the presence of peripheral and mucosal lymphoid tissue. While immuno-deficient mice devoid of functional IL2rγ allow for greater engraftment of human hemato-lymphoid cells, there are few if any peripheral lymph node (PLN) or gut-associated lymphoid tissue (GALT) development in these animals due to impaired IL-7/IL-7R signaling (9;28;97;100). Absence of this important signaling pathway prevents the generation of lymphoid tissue inducer (LTi) cells that are required for PLN and GALT development. Transgenic overexpression of thymic stromal lymphopoietin (TSLP), a protein that has functional overlapping activity with IL-7, increased LTi numbers and restored PLN development in IL-7−/− and RAG-2−/−IL-2rγ−/− mice (135). Thus, treatment with or overexpression of TSLP may be used to restore PLN and GALT development in humanized NSG mice. Although the effect of this approach on PLN and GALT development in immuno-deficient IL2rγ−/− mice reconstituted with human hemato-lymphoid cells has not been reported, hTSLP transgenic NSG mice have been developed and reported in abstract form (136).
An under-investigated aspect of humanized mouse models is the role that intestinal microbiota play in immune system development. Chung and coworkers elegantly demonstrated that the immune system in germ-free mice fully develops in the presence of mouse but not rat or human microbiota (137). Germ-free mice colonized with human microbiota had markedly reduced CD4+ and CD8+ T cells in the intestine along with fewer proliferating T cells, dendritic cells, and low antimicrobial peptide expression compared to those colonized with mouse microbiota. Colonization of the mice with rat microbiota also failed to fully expand the numbers of intestinal T cells. However, mouse-segmented filamentous bacteria partially restored T cell numbers in the human microbiota colonized mice. Thus, human-specific microbiota is likely required for healthy human immune system development. Whether microbial-host interaction is required between the microbial cells and immune cells or microbial cells and host non-hematopoietic cells or both will need to be determined and humanized mice should be an excellent system to define these interactions.
Eradication of residual murine innate immune cells
Residual mouse innate immune function in immuno-deficient IL2Rγ−/− mice continues to limit robust hemato-lymphoid cell engraftment. Although these mice have a complete absence of NK cells due to the deletion of the IL2rγ chain that is necessary for IL15R signaling, it is clear that their absence is not sufficient for full human immune cell engraftment.(9;80). Most human cells (especially lymphocytes and erythrocytes) are particularly susceptible to phagocytosis and destruction by murine macrophages and possibly other phagocytic leukocytes (e.g. PMNs). A major component of the innate immune system that prevents phagocytic destruction of its hemato-lymphoid cells is the interaction between the CD47 ligand and SIRPα rector. As briefly described previously, the SIRPα receptor is highly expressed on macrophages, dendritic cells and PMNs whereas its ligand CD47 is expressed on virtually all other cell types. Interaction of CD47-expressing cells with phagocyte-associated SIRPα initiates anti-phagocytosis signaling thereby preventing the inadvertent auto-destruction of nonphagocytic cells (9;65;66). Because murine SIRPα does not recognize human CD47, murine phagocytes readily engulf and destroy human hemato-lymphoid cells. The NOD strain contains a SIRPα polymorphism making it functionally similar to human protein that partially accounts for the improved engraftment in the NOD/scid and NSG strains (65;66). Genetically-modified strains have been developed with transgenic overexpression of human SIRPα (98) or that express murine CD47 in the human xenograft (138) that have enhanced engraftment and immune system development. As discussed previously, Lavender and coworkers have demonstrated that CD47−/−RAG-2−/−IL-2rγ−/−-BLT mice are resistant to GVHD (106;107).
A number of other innovative approaches have been used in an attempt to reduce or eliminate macrophage-mediated destruction of engrafted human cells. Clodronate liposomes have been used to kill macrophages in vivo in NOD/scid or NSG mice receiving human CD34+ fetal liver cells resulting in enhanced survival of human red cells (139). However, clodronate also kills human macrophages which will limit it’s use in studies of immune function with its long-term use limited by toxicity. Brehm, Shultz and colleagues are evaluating CD11b or CD11c promoter-driven diphtheria toxin receptor expression as a means to selectively eliminate murine myeloid or dendritic cell populations in NSG mice (28;80). Murine macrophages and PMNs can also be depleted with antibodies such as RB6–8C5 (specific for macrophages and PMNs) or 1A8 (specific for PMNs alone)(140;141).
The ability to effectively study innate immune responses in autoimmunity or infectious diseases is complicated by the presence of murine Toll-like receptor (TLR) expression as well as reactive oxygen production. In order to address this, NSG mice devoid of TLR4, MYD88 cytosolic adapter protein, and type 1 interferon (IFN) receptors are currently being generated (80); however, this approach may have unintended consequences due to disruption of physiologic homeostatic and inflammatory roles for TLR expression in a variety of host cells including intestinal epithelial cells and myofibroblast (142;143). Additionally, NSG mice deficient for neutrophil cytosolic factor 1 (Ncf1), a gene that controls macrophage and PMN superoxide production (141;144), are currently being generated to reduce the cytotoxic activity of murine phagocytic leukocytes (80).
Use of Humanized Mice to Study Autoimmune and Inflammatory Diseases
The National Institutes of Health (NIH) estimates that ~24 million Americans suffer from one of the major autoimmune diseases such as Type 1 diabetes, lupus, rheumatoid arthritis, multiple sclerosis or the inflammatory bowel diseases. In contrast, the American Autoimmune Related Diseases Association (AARDA) reports that the actual number may be closer to 50 million individuals because the NIH only tracks and reports only those 24 autoimmune diseases with well-documented etiology studies (http://www.aarda.org/autoimmune-information/autoimmune-statistics/). In fact, the AARDA states that there are greater than 100 different autoimmune diseases that contribute to direct health care costs of more than $100 billion per year. Thus, there is an urgent need to develop new and more potent therapeutic agents to treat these lifelong diseases. The development of the new generation humanized mouse models has produced a great deal of excitement within the autoimmunity research community as in theory, it should be possible to model many of the major autoimmune and inflammatory diseases. The following section describes some of the more recent studies evaluating the pathogenesis and new drug therapies in humanized mouse models of autoimmune and chronic inflammatory diseases.
Graft-Versus-Host and Host-Versus-Graft Disease
Allogeneic bone marrow transplantation (BMT) may, in some instances, cure certain types of blood cancers and genetic disorders. Despite the long-term success of BMT, the development of graft-versus-host disease (GVHD) remains a major cause of morbidity and mortality in these recipients (68;145;146). Although GVHD is known to result from donor innate and adaptive immune cells responding to recipient antigens (ie MHC complexes), donor T cells are thought to be the most important disease-producing effector cells. Two types of GVHD have been described in humans and mice including acute GVHD (aGVHD) and chronic GVHD (cGVHD) (68;145;146). Development of aGVHD in mouse models is characterized by systemic inflammatory tissue injury of the intestine, liver, lungs and skin whereas cGVHD involves oral and salivary tissue, intestine, joints, liver, lungs and connective tissue (68;145;146). Several of the newer immuno-deficient IL-2rγ−/− mice have been used to examine the immuno-pathogeneis of GVHD and evaluate new drug therapies to treat this systemic inflammatory disorders. In general, the Hu-PBMC-IL-2rγ−/− mouse models appear to model a Th1/Th17-driven aGVHD (81;103;147–149). One concern with the use of conventional Hu-PBMC-IL-2rγ−/− model is that it is, in actuality, a model of xenogeneic not allogenic GVHD. Covassin et. al. recently reported the generation of a novel humanized mouse model of allogeneic GVHD (150). Using NSG mice devoid of murine MHC class II but expressing the human transgene HLA DRB1-0401(called NSG-Ab° DR4 mice), these investigators found that injection of human DR4-positive CD4+ T cells failed to induce GVHD in NSG-Ab° DR4 mice whereas injection of CD4+ T cells obtained from human DR4-negative donors induced allogeneic GVHD (150). Although the majority of Hu-PBMC-IL-2rγ−/− mice do not develop auto-antibody-mediated cGVHD, a few studies have reported cGVHD-like immunopathology (with fibrosis) in the BLT-IL2rγ−/− model (68;94;102–105;151). These models have been particularly useful in testing a variety of different human-specific therapies such as anti-TNF antibodies, thymus-derived or induced regulatory T cells (Tregs) or mesenchymal stem cells (81;152–155). A more recent study demonstrates that administration of an antibody directed against HLA-A*02:01 to NSG mice injected with PBMCs expressing this human antigen, enhanced survival of these recipients (156). These exciting new data may provide a new approach to treating GVHD in HLA mismatched BMT.
Transplantation of allogeneic organs or tissues also induces potent immune responses in which the host immune system reacts vigorously to the engrafted tissue creating host versus graft disease, more commonly referred to as allograft rejection. (68;157). Despite decades of immunosuppressive therapy, allograft rejection remains a major hurdle for the long-term survival of the transplanted tissue. The use of humanized mice to study the immunopathology of xenograft or allograft rejection has been driven by the realization that the immune responses to human allografts in these animals differs substantially to those observed in the same species (mouse into mouse) allograft rejection models[reviewed in (68)]. However, the inability to transplant entire human organs into the mouse has limited allograft rejection studies to using human blood vessels, skin or islet cells. It should be noted that although a number of important studies have been performed using Balb/c-scid beige (SCID/beige) recipients, relatively few studies have utilized the newer immuno-deficient IL-2rγ−/− platforms such as the NSG, NOG, NRG or BRG mice (Table 2). When human vascular allograft rejection has been studied in a Hu-PBMC-IL-2rγ−/− model, investigators have found that engraftment of human arterial vessels and human PBMCs were significantly better than what occurs in the well-characterized Hu-PMC-SCID/beige model (158–160). The use of Hu-PBMC-IL-2rγ−/− models has allowed investigators to evaluate new, human-specific cell-based therapies in treating vascular allograft rejection (159;160). In addition to vascular allograft rejection, investigators have utilized the newer humanized mouse models to explore the immuno-pathogenesis of skin allograft rejection. Transplantation of human skin in mice is technically easy and once engrafted, the allograft exhibits viable epidermis and dermis as well as a functioning vasculature. In addition, skin is a highly immunogenic tissue that elicits vigorous immune responses by the recipient (157;161). Racki et. al. have reported an important study in which they utilized NSG mice to examine human skin allograft rejection in absence or presence of human PBMCs (162). Surprisingly, they found that transplantation of human skin into NSG recipients elicited extensive perivascular infiltration of large numbers of mouse Gr1+ granulocytes that ultimately lead to the allograft injury and rejection (162). Treatment of NSG-engrafted mice with an antibody specific for Gr1 remarkably reduced the leukocyte infiltration as well as promoted wound healing and graft remodeling. Using this modification to allow allograft survival, the authors were able to demonstrate human PBMC-mediated allograft rejection following human PBMC administration (162). These studies have allowed investigators to evaluate the therapeutic efficacy of human specific therapies such as the use of human Tregs (163;164). The new and exciting work being performed on allogeneic islet cell transplantation is discussed in the following section.
Type 1 Diabetes
Type 1 diabetes (T1D) is a multifactorial autoimmune disease that arises from a complex interaction among genetics, the immune system and environment. As with other autoimmune and chronic inflammatory diseases, the incidence and prevalence of T1D has increased dramatically over the past 50–60 years especially in “modernized” societies (165). These data coupled with those demonstrating that concordance rates of <50% in genetically identical twins suggest that environmental factors (e.g. intestinal microbiota) may play an important role in the pathogenesis of T1D in genetically-susceptible individuals (165;166). Numerous animal and human studies suggest that immune-mediated destruction of pancreatic β cells involves both CD4+ and CD8+ T cells (165;166). A variety of different rat and mouse models of T1D have been used over the past several decades to investigate the immuno-pathogenesis and treatment of this autoimmune disease. Because a T1D-like disease was found to spontaneously develop in NOD mice without the need for addition of toxic chemicals or infectious agents, it became and continues to be the most popular animal model for T1D preclinical studies (19;167;168). Despite the wealth of information that has been generated related to the immuno-pathogenesis and treatment of T1D using this mouse model, no therapy has been shown to prevent or cure patients with T1D. The differences between mouse and human immune systems as well as differences in islet cell composition and function may be major contributors to this lack of bench to bedside transition (19;168). The development of humanized mouse models based upon the different immuno-deficient IL2rγ−/− stocks provides investigators with immunologically more relevant animal models for investigating immuno-pathogenesis and treatment of T1D. King and coworkers developed a chemically-induced model of T1D in NSG mice in which mice are rendered hyperglycemic via injection (i.p) of the β cell toxin streptozotocin (STZ). These investigators found that transplant of human islet cells into STZ-treated NSG mice reduced the hyperglycemia created in these mice (79;169). Injection of human allogeneic PBMCs into NSG engrafted with islet cells abrogated the protective effect resulting in allograft rejection and return to hyperglycemia. Unfortunately, the use of STZ to induce β cell injury is problematic given that the response to STZ can be quite variable and mouse islets/β cells are known recover from the chemical injury (79;157;169). In an interesting variation of this model, Zhao et. al. demonstrates that adoptive transfer of irradiated splenic mononuclear cells (SMCs) derived from NOD mice with active diabetes into NSG mice prior to engraftment of human T cells induces a mild insulitis that acts to trigger severe insulitis and β cell destruction following engraftment of human PBMCs derived from T1D patients (170). These investigators show that human T cells migrate to pancreatic islets via the interaction between chemokine stromal cell-derived factor 1 and chemokine receptor CXCR-4. Severe insulitis and β cell destruction correlated with the infiltration of large numbers of human CD4+ and CD8+ T cells into the pancreatic islets (170). Engraftment of irradiated, NOD-derived PBMCs or human T1D PBMCs alone did not induce diabetes in the NSG recipients. Taken together, these data suggest that this approach may provide a more immunologically-relevant mouse model to study the role of T cells in the pathogenesis of T1D. However, it must be remembered that the Hu-PBMC-IL-2rγ−/− models utilized to investigate the immunological mechanisms responsible for induction of T1D as well as islet allograft rejection restrict investigator to studying only the of role of human T cells in these processes.
A major step forward in generating mouse models to study islet allograft rejection in mice with a fully humanized immune system has been described by Brehm and colleagues (171). This model is based upon the discovery that mice possessing a spontaneous point mutation in the insulin 2 gene spontaneously develop diabetes by 7 weeks of age (172;173). Investigators determined that disease was due to insulin 2 protein misfolding resulting in induction of the unfolded protein response followed ultimately by β cell apoptosis (174;175). The diabetes/hyperglycemia that develops in these mice (called Ins2Akita mice) are not associated with obesity or insulitis. Backcrossing Ins2Akita mice with NRG mice produced immuno-deficient offspring that develop spontaneous hyperglycemia by 3–6 weeks of age (19;157;171). Investigators demonstrated that normoglycemia could be achieved by transplantation of human or mouse islet cells (171). They also found that within one month following engraftment of allogenic human HSCs into normoglycemic, human islet cell-containing NRG-Ins2Akita mice, the majority (60%) of mice became hyperglycemic following rejection of allogeneic islet cells (171). Thus, this modification of the Hu-HSC-IL-2rγ−/− model may offer new insights into the treatment and immuno-pathogenesis of allograft rejection. It should be noted that these data contrast with those reported by Jacobson and coworkers who found that human islet cells were not rejected by BRG mice engrafted with allogeneic HSCs (176). The reasons for these differences are not readily apparent at the current time. With refinement of these current humanized models and development of new models that more accurately mimic the rejection processes, investigators will be in a position to evaluate novel, human-specific therapies to reverse or even prevent allograft rejection. New cell-based therapies that include administration of ex vivo-expanded Tregs or bone-marrow-derived mesenchymal stem cells are showing great promise in prolonging islet allograft survival in diabetic mice (177–179).
As discussed above, T1D is a polygenic disease that appears to be mediated by both CD4+ and CD8+ T cells (165;166). Recent genome-wide association studies have confirmed earlier reports demonstrating that the greatest genetic susceptibility to T1D is localized to the class II region of the HLA complex on chromosome 6(165;166). Indeed, >90% of all pediatric patients with T1D cases possess HLA-DR3,DQB1*0201 (called DR3-DQ2) or HLA-DR4,DQB1*0302 (called DR4-DQ8)(165;166). There is good evidence to suggest that priming and activation of CD4+ T cells is critical to providing help to CD8+ cytotoxic T cells that ultimately destroy islet β cells (reviewed in (165;166)). Activation of CD4+ T cells occurs via the interaction between the CD4+ T cell receptor (TCR) and autoantigens presented by the HLA-DR or HLA-DQ complexes residing on antigen presenting cells (APCs) in the draining lymph nodes and/or within the pancreatic islets. Examples of known T1D-related autoantigens include preproinsulin (PPI), insulinoma-associated antigen-2, glutamic acid decarboxylase, zinc transporter and islet-specific glucose-6-phosphatase catalytic subunit (IGRP) to name just a few (165;180).
The generation of transgenic IL-2rγ−/− mice expressing HLA class I or II has provided investigators the tools to evaluate the roles of different T cell subsets in autoantigen-driven insulitis and diabetes in vivo. Recent work by Viehman-Milan et. al. shows that engraftment of HLA-matched human PBMCs (DRB1*0401) that were pulsed (ex vivo) with autoantigen-derived peptides (PPI or IGRP) into NSG-DR tg mice induced the infiltration of T cells into mouse islets (180). Although T cell infiltration was observed following transfer of healthy, autoantigen-pulsed PBMCs, insulitis was found to be more severe following engraftment of autoantigen-pulsed PBMCs obtained from diabetic donors. Furthermore, the authors observed decreased insulin staining and β cell injury in NSG-DR tg mice engrafted with diabetic vs. healthy PBMCs. Although overt diabetes was not observed in this model, this is a very good mouse model for investigating the mechanisms responsible for the selective trafficking of autoreactive T cells to pancreatic islets as well as the mechanisms by which they damage the tissue. Other investigators have used a similar approach to ascertain the role that CD8+ T cells play in the induction of insulitis in the humanized IL-2rγ−/− mouse models. Whitfield-Larry et. al. demonstrated that transfer of HLA-A2–matched PBMCs from patients with T1D but not from healthy donors into NSG-HLA-A2 transgenic mice induced selective and robust islet inflammation (181). The inflammatory infiltrate was composed of both CD4+ and CD8+ T cells. Importantly, the authors showed that invading CD8+ T-cells were autoantigen-specific that could be stimulated to produce large amounts of interferon-γ following ex vivo stimulation with different T1D-associated autoantigens (181). Similar to the model described above, these mice did not develop overt diabetes. Nevertheless, this model represents another important step forward that will be useful for defining the cellular and immunological mechanisms responsible for islet-specific lymphocyte trafficking and HLA-restricted antigen responses. In a third study, Unger and coworkers reported that injection of a IGRP-specific CD8+ T-cell clone generated from diabetic donor blood into NSG-HLA-A2 tg mice induced severe insulitis and islet cell damage (182). These data clearly showed that autoreactive CD8+ T-cell clones are capable of trafficking specifically to pancreatic islets where they mediated injury to HLA-expressing beta-cells. The fact that none of the three NSG-HLA transgenic mouse models described above develop hyperglycemia/overt diabetes suggest that engraftment with both CD4+ and CD8+ T cells may be required for full expression of disease. Transgenic expression of both HLA class I and II molecules may help to promote development of diabetes provided that disease can develop within 4–5 weeks i.e. before the onset of GVHD. Furthermore, the use of HLA tg versions of the Hu-HSC-NSG or BLT models may provide the necessary innate and adaptive immune cells required for full expression of disease in the absence of GVHD.
Multiple Sclerosis (MS)
MS is an autoimmune disease characterized by chronic inflammation and demyelination of the central nervous system (183). Although the precise etiology of MS remains to be defined, there is accumulating evidence that the inflammation and consequent neuronal demyelination occurs from aberrant T cell responses to myelin-derived antigens, such as proteolipid protein (PLP) and myelin basic protein (MBP)(183–185). The strongest genetic factor associated with the development of MS is expression of certain HLA class II molecules such as the HLA-DRB1*1501 allele which confers a ~3 fold increase in risk (186). A large number of studies, using different mouse models of MS have been used over the past 30 years that have greatly advanced our understanding of the immuno-pathological mechanisms responsible for CNS inflammation and injury (183). The most popular mouse models have included induced models based upon autoimmune encephalomyelitis (EAE) studies, spontaneous models of MS and more recently, transgenic mice expressing different human HLA alleles (183). Although these models have been instrumental in revealing important cellular and molecular pathways responsible for induction of CNS inflammation, they did not recapitulate human MS. An obvious reason for the differences is that CNS inflammation in mice is mediated by “mouse-specific” effector cell mechanisms. More recently, investigators have utilized the Hu-PBL-NSG model to produce a subclinical autoimmune model of encephalomyelitis (187). For this model, investigators co-transferred myelin oligodendrocyte glycoprotein (MOG)-pulsed, human DCs together with healthy human PBMCs obtained from the same donor as DCs into NSG mice. Control mice were injected with both human PBMCs and DCs that were not pulsed with antigen or pulsed with irrelevant peptides. Following the initial injections, the humanized mice received a “booster” injection (s.c.) of immature/nonpulsed DCs followed 12 hrs later with a subcutaneous injection of MOG (or irrelevant peptide) suspended in Freund’s complete adjuvant (FCA) to induce DC maturation. The authors found that at 24 days following engraftment of MOG-pulsed PBMCs, significant CNS inflammation could be observed. The CD45+ inflammatory infiltrate was composed primarily of human CD4+ and CD8+ T cells. Although human T cells obtained from spleens of humanized NSG mice were shown to mount robust MOG-specific immune responses, the classic signs of EAE (e.g. paralysis and paresis) were not observed in this novel model (187). Nevertheless, this new model of myelin-induced CNS inflammation will be a valuable tool to assess new drug therapies targeted at T cell trafficking and inflammatory cytokine production in a humanized system in vivo.
Autoimmune and Inflammatory Rheumatic Diseases
Autoimmune and inflammatory rheumatic diseases represents the largest subset of autoimmune diseases affecting over 45 million individuals in the US (188–190). These chronic inflammatory diseases are characterized by painful swelling of the joints, muscle and/or tissue. The most prevalent of the rheumatic diseases include rheumatoid arthritis (juvenile and adult), systemic lupus erythematosus, Sjogren’s syndrome, the spondylarthritides (e.g. ankylosing spondylitis, reactive arthritis, psoriatic arthritis, enteropathic arthritis) and systemic sclerosis (190). Although a number of different mouse models have been developed to act as surrogates for some of the major rheumatic diseases, the use of humanized mice to study human-specific immune mechanisms and treatment modalities in these disease models have only recently been attempted. Below we describe the use of immuno-deficient IL-2rγ−/− stocks to generate mouse models of some the major rheumatic diseases.
Rheumatoid Arthritis
A variety of mouse models have been used to study various aspects of induced or spontaneous arthritis; however none of these models completely recapitulate the immuno-pathogenesis of human rheumatoid arthritis (RA)(191). Similar to diabetes, the susceptibility to RA is associated with certain HLA class II alleles. Previous studies have demonstrated that immunization of transgenic mice expressing the human HLA-DR4 or -DR1 allele with type II collagen (CII) induces chronic joint inflammation. As with other mouse models of autoimmunity, interpretation of data generated using these tg models is limited by the fact that joint inflammation is driven entirely by the mouse immune system (192;193). Recent studies have attempted to utilize the new-generation, humanized mouse models to examine arthritis pathogenesis and treatment. Based upon intriguing clinical observations demonstrating that Epstein-Barr virus (EBV) infection is associated with the development of RA in humans (194;195), Kuwana et. al. have developed an interesting model of erosive arthritis based upon the Hu-HSC-NOG mouse model (196). These investigators observed that 65% of the NOG mice that were engrafted with human HSCs and then infected with EBV exhibited extensive arthritis in knee and ankle joints beginning at 26 days post-infection. No joint inflammation was observed in the absence of EBV infection. Histopathological analysis revealed remarkable synovial proliferation as well as extensive bone destruction and edema all of which appear very similar to those observed in human RA (196). In addition, they found that the inflamed joint and synovial tissue contained large numbers of human CD4+ and CD8+ T cells, B cells and macrophages. These interesting results demonstrate that EBV infection is capable of inducing human immune cell-mediated erosive arthritis that is quite similar to human RA. In another study, Misharin and coworkers report the development of a humanized mouse model of acute arthritis (87). These investigators used a similar humanized mouse model in which NSG mice were engrafted with human CD34+ HSCs and then subjected to intra-articular injection of FCA 16 weeks following HSC engraftment to induce acute arthritis. At 7 days post FCA injection, mice developed histological evidence of arthritis that was characterized by erythema, edema, pannus formation and infiltration of human lymphocytes as well as human and mouse PMNs and macrophages. Furthermore, these investigators demonstrated that FCA-induced arthritis could be attenuated by administration of the TNF inhibitor Etanercept (87).
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a multi-organ, autoimmune disease that affects primarily the joints, kidneys and skin but may also involve the lungs and central nervous system. This rheumatic disease is characterized by production various autoantibodies and deposition of immune complexes into the different tissues. Although investigators believe that one or more of the autoantibodies generated during the development of disease is/are important in disease pathogenesis, the precise etiology of SLE has yet to be defined. As with RA and other autoimmune diseases, induced and spontaneous mouse models of SLE have greatly advanced our understanding of those immune cells and mediators that promote disease; however, development of disease in many of the mice is strain-specific and do not recapitulate certain immuno-pathological aspects of human SLE (197;198). In an attempt to model SLE in mice with a partially humanized immune system, Andrade et. al. have utilized the Hu-PBMC-BRG model to create mice that develop most or all of the pathological hallmarks observed in human SLE (197). These investigators found that human PBMCs obtained from either SLE or healthy donors (HD) engrafted very well in NRG recipients at 3–4 weeks post transfer. In addition, the circulating numbers of CD4+ T cells was remarkably reduced in SLE- vs. HD-engrafted mice whereas CD8+ levels were substantially greater in SLE vs. HD engrafted animals (197). The authors found no significant differences in human IgG and anti-dsDNA antibody levels in SLE vs. HD-engrafted mice. However, if mice were injected with PBMCs obtained from a SLE donor that was expressing high levels of anti-cardiolipin antibodies (aCL), all engrafted mice expressed 2–3 times more aCL than did the other SLE or HD engrafted mice. At 4–8 weeks post engraftment, SLE-NRG mice were found to develop proteinuria and human IgG immune complex deposition within the kidney glomeruli. Microthrombi as well as CD8+ T cell and B cell infiltration were observed in NRG mice engrafted with PBMCs obtained from SLE donors that expressed high titers of aCL. Taken together, this novel model appears to recapitulate some of the pathological features observed in human SLE.
Sjogren’s Syndrome
Sjogren’s syndrome (SS) is one of the most prevalent autoimmune diseases that affect ~3% of the population. Individuals who suffer from SS experience systemic inflammation however disease is usually localized to salivary and lacriminal glands (199). Histopathological analyses reveal the infiltration of large numbers of CD4+ T cells and the upregulation of different pro-inflammatory cytokines (199). Because the pathology observed in these mouse models of SS do not reproduce some of the key alterations described in human disease, investigators are just beginning to explore the use of humanized mice to study the immuno-pathological mechanisms responsible for induction of this prevalent disease. In the first and only study reporting the use of humanized mice in SS investigations, Andrade and coworkers showed that adoptive transfer PBMCs obtained from SS donors into NSG mice resulted in increased serum levels of a variety of human inflammatory cytokines including IFN-γ, IL-17, Il-6 and TNFα when compared to NSG engrafted with healthy donor (HD) PBMCs (200). In addition, these investigators observed the infiltration of significantly larger numbers of human CD4+ T cells into the salivary and lacriminal glands of the SS- vs HD-engrafted NSG mice. Although significantly larger numbers of human CD8+ T cells and B cells were observed in both glands in SS- vs. HD-engrafted animals, the absolute numbers of these lymphocytes were much smaller than those observed for CD4+ T cells (200). This inflammatory infiltrate correlated well with loss of target organ function as saliva production was significantly decreased by 36% at 4 weeks post transfer of SS-derived PBMCs. This exciting new model will likely provide investigators the unique opportunities to not only explore T cell-dependent mechanisms of tissue injury but will also provide an important platform to assess new therapeutic agents to treat this rheumatic disease.
Inflammatory Bowel Diseases
Crohn’s disease (CD) and ulcerative colitis (UC) are two of the best-characterized inflammatory bowel diseases. They are chronic and unrelenting inflammatory disorders of the small bowel and/or colon that affect approximately 1.5 million people in the US with a calculated annual cost for both medical expenses and work loss of almost $4 billion dollars (201). Although the etiologies of CD and UC have yet to be fully elucidated, there is growing experimental and clinical evidence to suggest that chronic gut inflammation results from a dysregulated immune response to components of the normal gut flora in genetically-susceptible individuals. This paradigm is based upon both animal and human studies demonstrating that: a) intestinal bacteria are required for the development of chronic gut inflammation in genetically-susceptible mice; b) increased numbers of epithelial-associated bacteria as well as enhanced bacterial translocation occur during chronic gut inflammation; c) certain subsets of patients with IBD express polymorphisms in genes that are involved in intracellular processing and killing of bacteria and d) IBD is associated with major alterations in the luminal composition of the microbiota, a situation termed dysbiosis. Whether dysbiosis is a cause or consequence of IBD has yet to be determined. Neither CD nor UC are considered classical autoimmune diseases since no convincing data have been reported demonstrating that these diseases develop from auto-aggressive immune responses to intestinal tissue self-antigens. If however, we consider the trillions of intestinal microbes as a “virtual organ” that is present for our entire lifetime, we may wish to redefine our microbiota as “self”(202;203). Using this definition of self, it is quite reasonable to consider IBD as just another type of autoimmune disease. Few autoimmune or chronic inflammatory diseases have garnered more attention from their respective research communities in such a short period of time as has IBD. In just the last 20 years, thousands of studies have been published using mouse models of IBD in order to define disease pathogenesis and potential treatment modalities. However, the success of translating the hundreds of promising preclinical studies into effective new therapies to treat IBD has been unimpressive. The reasons for the lack of bench-to-bedside transition have already been described in previous sections and are beyond the scope of this overview. We do however, refer the reader to recent reviews that examine this subject (1;8).
In view of the large number of preclinical studies that have been and continue to be performed, it is rather surprisingly to find that very few studies have attempted to model IBD in humanized mice. Nolte et. al. recently described a model of chemically-induced IBD using the Hu-PBMCs-NSG mouse model (204). These investigators engrafted NSG mice with human PBMCs obtained from healthy donors (HD) or from individuals suffering from UC or atopic dermatitis (AD). At 7 days post transfer, mice were sensitized, via dermal application, to the vehicle (ethanol) or the hapten oxazalone (OXA). The mice were then challenged 24 hrs later via intrarectal administration of either 50% ethanol or OXA in 50% ethanol (204;205). Histopathological analyses of the different groups revealed that mice challenged with ethanol or OXA developed the same degree of colonic inflammation that consisted of edema, fibrosis, crypt dropout and infiltration of T cells into the lamina propria (204). No significant differences were observed in the histopathology scores among NSG mice engrafted with HD, UC or CD PBMCs and challenged with OXA. Although this study presents interesting data related to “hapten”-induced colitis in humanized mice, it illustrates a major problem associated with mouse models of chemically-induced colitis: the use of erosive/toxic chemicals such as ethanol creates severe inflammation that may mask more subtle alterations induced in humanized mice. Another limitation with the use of this type of chemical model is that lacks participation by the human innate system that will likely be important to faithfully model human IBD.
Using a similar approach as described for the OXA model, Goettel et. al. recently reported in abstract form only the use of transgenic NSG mice that expressed human HLADR1 but lacked the murine MHC class II (called NSGAb0DR1 mice) as a platform for inducing acute colitis (206). These investigators engrafted the NSGAb0DR1 mice with HLA-matched CD4+ T cells and then sensitized (via dermal administration) the mice with the hapten, trinitrobenzene sulfonic acid (TNBS). One week later, these mice received an intra-rectal challenge with TNBS in ethanol resulting in clinical and histologic evidence of intestinal inflammation that was not observed in the NSGAb0DR1 mice receiving TNBS in ethanol but no human T cells (206). Although promising, the TNBS model suffers from many of the same limitations as does the do other chemically-induced models of gut inflammation that use erosive/toxic substances to “break” the mucosal barrier (1). A second preliminary study reported by this same group (abstract form only) showed that engraftment of NSGAb0DR1 mice with HLA-matched CD34+ HSCs from a patient with immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome induced systemic and lethal autoimmunity similar to that observed in humans with IPEX and mice that lack functional Foxp3 transcription factor (207). It will be of great interest to see how this type of humanized mouse model may be used in the future to model human IBD in a well-controlled and statistically-powered study.
A novel mouse model of allergen-induced intestinal inflammation has recently been developed by Weigmann et. al.(208). These investigators engrafted PBMCs from donors with clinical allergy to grass, birch pollen or hazelnut together with their respective allergen (ip) into NSG mice. At 21 days post engraftment, mice were challenged orally or rectally with their specific allergen. Mice engrafted with allergic PBMCs developed colonic inflammation that consisted primarily of human lymphocytes and neutrophils in both the oral or rectal challenged groups but not following saline challenge. Mice engrafted with non-allergic (healthy) PBMCs did not develop colitis when challenged with the allergens (208). Furthermore, the colitis could be significantly attenuated by administration of anti-human IgE antibody but not with control human IgG. Although it was not clear whether the PMNs infiltrating the colon were of human and/or murine origin, this study clearly demonstrates that human leukocyte trafficking to the intestine is largely intact and sufficient to develop colonic inflammation. Whether trafficking is sufficient to allow the full spectrum of IBD pathology remains to be determined.
Looking Forward
The development of humanized mouse models of IBD offers investigators the ability to study the pathophysiology of CD or UC in a manner not possible in humans. Pre-clinical therapies can be tested, and the environmental influences such as diet and the gut microbiome can be tested and defined. However, a number of issues and fundamental questions remain regarding our ability to generate these humanized mouse models. It may be a bit naïve to believe that chronic gut inflammation can be generated by simply engrafting HSCs obtained from patients with CD or UC into any of the immuno-deficient IL-2rγ−/− mice. Because genetically-identical twins express relatively low concordance rates for CD (~30–35%) or UC (~15–20%), environmental factors (e.g. intestinal bacteria, diet, housing) most likely will play important roles in model development (209–214). Although mouse and human flora are in general, fairly similar in composition, significant differences do exist (9). Indeed, the enormous impact that mouse vs. human microbiota has on murine intestinal immune system development (137) suggests that the antigenic stimuli may require colonization with human flora, disease specific flora or autologous flora specific to the PBMC or HSC donors. If donor specific flora is required for induction of disease, antibiotic treatment followed by human fecal transfer may be sufficient and alleviate the need for a gnotobiotic facility (215). Transfer of autologous PBMCs into immuno-deficient mice engrafted with HSCs from IBD patients may also prove useful in providing reactive T-cells with immunologic memory to induce disease. Further refinements could include treatment with indomethacin or piroxicam to initiate and synchronize the onset of disease, an approach that has been used in the IL10−/− model of chronic colitis (216;217). Another concern relates to the extent of engraftment of CD34+-derived endothelial cells in mice (9). Although the incomplete engraftment of human endothelial cells may influence trafficking of human leukocytes to lymphoid and nonlymphoid tissue, current evidence suggests that leukocyte infiltration and intestinal inflammation do occur in the xenogeneic GVHD that develops in the Hu-PBMC-IL-2rγ−/− and BLT-IL-2rγ−/− mouse models (104;155). In summary, the outlook for the generation of even better humanized mouse models to study autoimmune and chronic inflammatory diseases is very bright. It will be very interesting to following the evolution of this exciting aspect of animal model development over the next few years.
Acknowledgments
Some work reported in this review is supported by grants from the DOD (W81XWH-11-1-0666; MBG) and the NIH (R01-DK091269; MBG)
Reference List
- 1.Koboziev I, Karlsson F, Zhang S, Grisham MB. Pharmacological intervention studies using mouse models of the inflammatory bowel diseases: translating preclinical data into new drug therapies. Inflamm Bowel Dis. 2011;17:1229–1245. doi: 10.1002/ibd.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeVoss J, Diehl L. Murine models of inflammatory bowel disease (IBD): challenges of modeling human disease. Toxicol Pathol. 2014;42:99–110. doi: 10.1177/0192623313509729. [DOI] [PubMed] [Google Scholar]
- 3.Hackam DG, Redelmeier DA. Translation of research evidence from animals to humans. JAMA. 2006;296:1731–1732. doi: 10.1001/jama.296.14.1731. [DOI] [PubMed] [Google Scholar]
- 4.Hackam DG. Translating animal research into clinical benefit. BMJ. 2007;334:163–164. doi: 10.1136/bmj.39104.362951.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sena ES, van der Worp HB, Bath PM, Howells DW, Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. 2010;8:e1000344. doi: 10.1371/journal.pbio.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sena ES, Currie GL, McCann SK, Macleod MR, Howells DW. Systematic reviews and meta-analysis of preclinical studies: why perform them and how to appraise them critically. J Cereb Blood Flow Metab. 2014;34:737–742. doi: 10.1038/jcbfm.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, Macleod MR. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7:e1000245. doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valatas V, Vakas M, Kolios G. The value of experimental models of colitis in predicting efficacy of biological therapies for inflammatory bowel diseases. Am J Physiol Gastrointest Liver Physiol. 2013;305:G763–G785. doi: 10.1152/ajpgi.00004.2013. [DOI] [PubMed] [Google Scholar]
- 9.Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, Manz MG. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. 2013;31:635–674. doi: 10.1146/annurev-immunol-032712-095921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Receptive to replication. Nat Biotech. 2013;31:943. doi: 10.1038/nbt.2748. [DOI] [PubMed] [Google Scholar]
- 11.Arrowsmith J. Trial watch: Phase II failures: 2008–2010. Nat Rev Drug Discov. 2011;10:328–329. doi: 10.1038/nrd3439. [DOI] [PubMed] [Google Scholar]
- 12.Arrowsmith J. Trial watch: phase III and submission failures: 2007–2010. Nat Rev Drug Discov. 2011;10:87. doi: 10.1038/nrd3375. [DOI] [PubMed] [Google Scholar]
- 13.Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature. 2012;483:531–533. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- 14.Couzin-Frankel J. When mice mislead. Science. 2013;342:922–3. 925. doi: 10.1126/science.342.6161.922. [DOI] [PubMed] [Google Scholar]
- 15.Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 16.Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49–71. doi: 10.1016/s0300-483x(03)00043-x. [DOI] [PubMed] [Google Scholar]
- 17.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 18.Ariffin JK, Sweet MJ. Differences in the repertoire, regulation and function of Toll-like Receptors and inflammasome-forming Nod-like Receptors between human and mouse. Curr Opin Microbiol. 2013;16:303–310. doi: 10.1016/j.mib.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Brehm MA, Powers AC, Shultz LD, Greiner DL. Advancing animal models of human type 1 diabetes by engraftment of functional human tissues in immunodeficient mice. Cold Spring Harb Perspect Med. 2012;2:a007757. doi: 10.1101/cshperspect.a007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbons DL, Spencer J. Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol. 2011;4:148–157. doi: 10.1038/mi.2010.85. [DOI] [PubMed] [Google Scholar]
- 21.Mann ER, Landy JD, Bernardo D, Peake ST, Hart AL, Al-Hassi HO, Knight SC. Intestinal dendritic cells: their role in intestinal inflammation, manipulation by the gut microbiota and differences between mice and men. Immunol Lett. 2013;150:30–40. doi: 10.1016/j.imlet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1401965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brehm MA, Jouvet N, Greiner DL, Shultz LD. Humanized mice for the study of infectious diseases. Curr Opin Immunol. 2013 doi: 10.1016/j.coi.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 26.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brehm MA, Shultz LD, Greiner DL. Humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes. 2010;17:120–125. doi: 10.1097/MED.0b013e328337282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brehm MA, Wiles MV, Greiner DL, Shultz LD. Generation of improved humanized mouse models for human infectious diseases. J Immunol Methods. 2014 doi: 10.1016/j.jim.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearson T, Greiner DL, Shultz LD. Creation of “humanized” mice to study human immunity. Curr Protoc Immunol. 2008;Chapter 15(Unit) doi: 10.1002/0471142735.im1521s81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shultz LD, Brehm MA, Bavari S, Greiner DL. Humanized mice as a preclinical tool for infectious disease and biomedical research. Ann N Y Acad Sci. 2011;1245:50–54. doi: 10.1111/j.1749-6632.2011.06310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, Facciponte J, Jin M, Shen Q, Lin Q. Humanized NOD-SCID IL2rg−/− mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer Lett. 2014;344:13–19. doi: 10.1016/j.canlet.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–1573. doi: 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 34.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 35.Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, Manz MG. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304:104–107. doi: 10.1126/science.1093933. [DOI] [PubMed] [Google Scholar]
- 36.Spranger S, Frankenberger B, Schendel DJ. NOD/scid IL-2Rg(null) mice: a preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J Transl Med. 2012;10:30. doi: 10.1186/1479-5876-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vudattu NK, Waldron-Lynch F, Truman LA, Deng S, Preston-Hurlburt P, Torres R, Raycroft MT, Mamula MJ, Herold KC. Humanized mice as a model for aberrant responses in human T cell immunotherapy. J Immunol. 2014;193:587–596. doi: 10.4049/jimmunol.1302455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flanagan SP. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res. 1966;8:295–309. doi: 10.1017/s0016672300010168. [DOI] [PubMed] [Google Scholar]
- 39.Pantelouris EM. Absence of thymus in a mouse mutant. Nature. 1968;217:370–371. doi: 10.1038/217370a0. [DOI] [PubMed] [Google Scholar]
- 40.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- 41.Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature. 1994;372:103–107. doi: 10.1038/372103a0. [DOI] [PubMed] [Google Scholar]
- 42.Ganick DJ, Sarnwick RD, Shahidi NT, Manning DD. Inability of intravenously injected monocellular suspensions of human bone marrow to establish in the nude mouse. Int Arch Allergy Appl Immunol. 1980;62:330–333. doi: 10.1159/000232530. [DOI] [PubMed] [Google Scholar]
- 43.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 44.Jeggo PA, Jackson SP, Taccioli GE. Identification of the catalytic subunit of DNA dependent protein kinase as the product of the mouse scid gene. Curr Top Microbiol Immunol. 1996;217:79–89. doi: 10.1007/978-3-642-50140-1_6. [DOI] [PubMed] [Google Scholar]
- 45.Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, Oettinger MA, Brown JM. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 46.Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci U S A. 1996;93:10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bosma MJ, Carroll AM. The SCID mouse mutant: definition, characterization, and potential uses. Annu Rev Immunol. 1991;9:323–350. doi: 10.1146/annurev.iy.09.040191.001543. [DOI] [PubMed] [Google Scholar]
- 48.Finnie NJ, Gottlieb TM, Blunt T, Jeggo PA, Jackson SP. DNA-dependent protein kinase defects are linked to deficiencies in DNA repair and V(D)J recombination. Philos Trans R Soc Lond B Biol Sci. 1996;351:173–179. doi: 10.1098/rstb.1996.0014. [DOI] [PubMed] [Google Scholar]
- 49.Taccioli GE, Amatucci AG, Beamish HJ, Gell D, Xiang XH, Torres Arzayus MI, Priestley A, Jackson SP, Marshak RA, Jeggo PA, et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity. 1998;9:355–366. doi: 10.1016/s1074-7613(00)80618-4. [DOI] [PubMed] [Google Scholar]
- 50.Lieber MR, Hesse JE, Lewis S, Bosma GC, Rosenberg N, Mizuuchi K, Bosma MJ, Gellert M. The defect in murine severe combined immune deficiency: joining of signal sequences but not coding segments in V(D)J recombination. Cell. 1988;55:7–16. doi: 10.1016/0092-8674(88)90004-9. [DOI] [PubMed] [Google Scholar]
- 51.Malynn BA, Blackwell TK, Fulop GM, Rathbun GA, Furley AJ, Ferrier P, Heinke LB, Phillips RA, Yancopoulos GD, Alt FW. The scid defect affects the final step of the immunoglobulin VDJ recombinase mechanism. Cell. 1988;54:453–460. doi: 10.1016/0092-8674(88)90066-9. [DOI] [PubMed] [Google Scholar]
- 52.Greiner DL, Hesselton RA, Shultz LD. SCID mouse models of human stem cell engraftment. Stem Cells. 1998;16:166–177. doi: 10.1002/stem.160166. [DOI] [PubMed] [Google Scholar]
- 53.Bosma GC, Fried M, Custer RP, Carroll A, Gibson DM, Bosma MJ. Evidence of functional lymphocytes in some (leaky) scid mice. J Exp Med. 1988;167:1016–1033. doi: 10.1084/jem.167.3.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nonoyama S, Smith FO, Bernstein ID, Ochs HD. Strain-dependent leakiness of mice with severe combined immune deficiency. J Immunol. 1993;150:3817–3824. [PubMed] [Google Scholar]
- 55.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154:180–191. [PubMed] [Google Scholar]
- 56.Custer RP, Bosma GC, Bosma MJ. Severe combined immunodeficiency (SCID) in the mouse. Pathology, reconstitution, neoplasms. Am J Pathol. 1985;120:464–477. [PMC free article] [PubMed] [Google Scholar]
- 57.Martin A, Valentine M, Unger P, Yeung SW, Shultz LD, Davies TF. Engraftment of human lymphocytes and thyroid tissue into scid and rag2-deficient mice: absent progression of lymphocytic infiltration. J Clin Endocrinol Metab. 1994;79:716–723. doi: 10.1210/jcem.79.3.8077352. [DOI] [PubMed] [Google Scholar]
- 58.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 59.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 60.Bock TA, Orlic D, Dunbar CE, Broxmeyer HE, Bodine DM. Improved engraftment of human hematopoietic cells in severe combined immunodeficient (SCID) mice carrying human cytokine transgenes. J Exp Med. 1995;182:2037–2043. doi: 10.1084/jem.182.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 62.Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 63.Kataoka S, Satoh J, Fujiya H, Toyota T, Suzuki R, Itoh K, Kumagai K. Immunologic aspects of the nonobese diabetic (NOD) mouse. Abnormalities of cellular immunity. Diabetes. 1983;32:247–253. doi: 10.2337/diab.32.3.247. [DOI] [PubMed] [Google Scholar]
- 64.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- 65.Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, Dick JE, Danska JS. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- 66.Takizawa H, Manz MG. Macrophage tolerance: CD47-SIRP-alpha-mediated signals matter. Nat Immunol. 2007;8:1287–1289. doi: 10.1038/ni1207-1287. [DOI] [PubMed] [Google Scholar]
- 67.Shultz LD, Lang PA, Christianson SW, Gott B, Lyons B, Umeda S, Leiter E, Hesselton R, Wagar EJ, Leif JH, et al. NOD/LtSz-Rag1null mice: an immunodeficient and radioresistant model for engraftment of human hematolymphoid cells, HIV infection, and adoptive transfer of NOD mouse diabetogenic T cells. J Immunol. 2000;164:2496–2507. doi: 10.4049/jimmunol.164.5.2496. [DOI] [PubMed] [Google Scholar]
- 68.Hogenes M, Huibers M, Kroone C, de WR. Humanized mouse models in transplantation research. Transplant Rev (Orlando) 2014;28:103–110. doi: 10.1016/j.trre.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 69.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 70.Prochazka M, Gaskins HR, Shultz LD, Leiter EH. The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci U S A. 1992;89:3290–3294. doi: 10.1073/pnas.89.8.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci U S A. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Ohbo K, Nakamura M, Takeshita T. The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol. 1996;14:179–205. doi: 10.1146/annurev.immunol.14.1.179. [DOI] [PubMed] [Google Scholar]
- 73.Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 74.Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001;1:200–208. doi: 10.1038/35105066. [DOI] [PubMed] [Google Scholar]
- 75.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brehm MA, Cuthbert A, Yang C, Miller DM, Diiorio P, Laning J, Burzenski L, Gott B, Foreman O, Kavirayani A, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin Immunol. 2010;135:84–98. doi: 10.1016/j.clim.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116:193–200. doi: 10.1182/blood-2010-02-271841. [DOI] [PubMed] [Google Scholar]
- 78.Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335:256–259. doi: 10.1038/335256a0. [DOI] [PubMed] [Google Scholar]
- 79.King M, Pearson T, Shultz LD, Leif J, Bottino R, Trucco M, Atkinson MA, Wasserfall C, Herold KC, Woodland RT, et al. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin Immunol. 2008;126:303–314. doi: 10.1016/j.clim.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 80.Brehm MA, Shultz LD, Luban J, Greiner DL. Overcoming current limitations in humanized mouse research. J Infect Dis. 2013;208(Suppl 2):S125–S130. doi: 10.1093/infdis/jit319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, Laning J, Fodor W, Foreman O, Burzenski L, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157:104–118. doi: 10.1111/j.1365-2249.2009.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241:1632–1639. doi: 10.1126/science.241.4873.1632. [DOI] [PubMed] [Google Scholar]
- 83.Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science. 1992;255:1137–1141. doi: 10.1126/science.1372131. [DOI] [PubMed] [Google Scholar]
- 84.Lepus CM, Gibson TF, Gerber SA, Kawikova I, Szczepanik M, Hossain J, Ablamunits V, Kirkiles-Smith N, Herold KC, Donis RO, et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/gammac−/−, Balb/c-Rag1−/−gammac−/−, and C.B-17-scid/bg immunodeficient mice. 2009:790–802. doi: 10.1016/j.humimm.2009.06.005. 2009/06/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsumura T, Kametani Y, Ando K, Hirano Y, Katano I, Ito R, Shiina M, Tsukamoto H, Saito Y, Tokuda Y, et al. Functional CD5+ B cells develop predominantly in the spleen of NOD/SCID/gammac(null) (NOG) mice transplanted either with human umbilical cord blood, bone marrow, or mobilized peripheral blood CD34+ cells. Exp Hematol. 2003;31:789–797. doi: 10.1016/s0301-472x(03)00193-0. [DOI] [PubMed] [Google Scholar]
- 86.Choi B, Chun E, Kim M, Kim SY, Kim ST, Yoon K, Lee KY, Kim SJ. Human T cell development in the liver of humanized NOD/SCID/IL-2Rgamma(null)(NSG) mice generated by intrahepatic injection of CD34(+) human (h) cord blood (CB) cells. Clin Immunol. 2011;139:321–335. doi: 10.1016/j.clim.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 87.Misharin AV, Haines GK, III, Rose S, Gierut AK, Hotchkiss RS, Perlman H. Development of a new humanized mouse model to study acute inflammatory arthritis. J Transl Med. 2012;10:190. doi: 10.1186/1479-5876-10-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shultz LD, Pearson T, King M, Giassi L, Carney L, Gott B, Lyons B, Rossini AA, Greiner DL. Humanized NOD/LtSz-scid IL2 receptor common gamma chain knockout mice in diabetes research. Ann N Y Acad Sci. 2007;1103:77–89. doi: 10.1196/annals.1394.002. [DOI] [PubMed] [Google Scholar]
- 89.Yahata T, Ando K, Nakamura Y, Ueyama Y, Shimamura K, Tamaoki N, Kato S, Hotta T. Functional human T lymphocyte development from cord blood CD34+ cells in nonobese diabetic/Shi-scid, IL-2 receptor gamma null mice. J Immunol. 2002;169:204–209. doi: 10.4049/jimmunol.169.1.204. [DOI] [PubMed] [Google Scholar]
- 90.Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, Shultz LD, Kiyono H, Ishikawa F. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rgammaKO mice. Journal of immunology (Baltimore, Md : 1950) 2012:6145–6155. doi: 10.4049/jimmunol.1103660. 2012/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, Ito R, Ito M, Minegishi M, Minegishi N, et al. The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice) Int Immunol. 2009;21:843–858. doi: 10.1093/intimm/dxp050. [DOI] [PubMed] [Google Scholar]
- 92.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108:487–492. doi: 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- 93.Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, Wege AK, Haase AT, Garcia JV. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 2006;12:1316–1322. doi: 10.1038/nm1431. [DOI] [PubMed] [Google Scholar]
- 94.Covassin L, Jangalwe S, Jouvet N, Laning J, Burzenski L, Shultz LD, Brehm MA. Human immune system development and survival of NOD-scid IL2rgamma (NSG) mice engrafted with human thymus and autologous hematopoietic stem cells. Clin Exp Immunol. 2013 doi: 10.1111/cei.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tonomura N, Habiro K, Shimizu A, Sykes M, Yang YG. Antigen-specific human T-cell responses and T cell-dependent production of human antibodies in a humanized mouse model. Blood. 2008;111:4293–4296. doi: 10.1182/blood-2007-11-121319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, Shin HS, Brooks SF, Knight HL, Eichbaum Q, et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. 2009;83:7305–7321. doi: 10.1128/JVI.02207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Denton PW, Nochi T, Lim A, Krisko JF, Martinez-Torres F, Choudhary SK, Wahl A, Olesen R, Zou W, Di Santo JP, et al. IL-2 receptor gamma-chain molecule is critical for intestinal T-cell reconstitution in humanized mice. Mucosal Immunol. 2012;5:555–566. doi: 10.1038/mi.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, Philbrick W, Eynon EE, Manz MG, Flavell RA. Transgenic expression of human signal regulatory protein alpha in Rag2−/−γc−/− mice improves engraftment of human hematopoietic cells in humanized mice. 2011;108:13218–13223. doi: 10.1073/pnas.1109769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stoddart CA, Maidji E, Galkina SA, Kosikova G, Rivera JM, Moreno ME, Sloan B, Joshi P, Long BR. Superior human leukocyte reconstitution and susceptibility to vaginal HIV transmission in humanized NOD-scid IL-2Rgamma(−/−) (NSG) BLT mice. Virology. 2011;417:154–160. doi: 10.1016/j.virol.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nochi T, Denton PW, Wahl A, Garcia JV. Cryptopatches are essential for the development of human GALT. Cell reports. 2013;3:1874–1884. doi: 10.1016/j.celrep.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rajesh D, Zhou Y, Jankowska-Gan E, Roenneburg DA, Dart ML, Torrealba J, Burlingham WJ. Th1 and Th17 immunocompetence in humanized NOD/SCID/IL2rgammanull mice. Hum Immunol. 2010;71:551–559. doi: 10.1016/j.humimm.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Villaudy J, Schotte R, Legrand N, Spits H. Critical assessment of human antibody generation in humanized mouse models. J Immunol Methods. 2014;410:18–27. doi: 10.1016/j.jim.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 103.Ali N, Flutter B, Sanchez RR, Sharif-Paghaleh E, Barber LD, Lombardi G, Nestle FO. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS One. 2012;7:e44219. doi: 10.1371/journal.pone.0044219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Greenblatt MB, Vrbanac V, Tivey T, Tsang K, Tager AM, Aliprantis AO. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. 2012:e44664. doi: 10.1371/journal.pone.0044664. 2012/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lockridge JL, Zhou Y, Becker YA, Ma S, Kenney SC, Hematti P, Capitini CM, Burlingham WJ, Gendron-Fitzpatrick A, Gumperz JE. Mice engrafted with human fetal thymic tissue and hematopoietic stem cells develop pathology resembling chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2013;19:1310–1322. doi: 10.1016/j.bbmt.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lavender KJ, Pang WW, Messer RJ, Duley AK, Race B, Phillips K, Scott D, Peterson KE, Chan CK, Dittmer U, et al. BLT-humanized C57BL/6 Rag2−/−gammac−/−CD47−/− mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood. 2013;122:4013–4020. doi: 10.1182/blood-2013-06-506949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lavender KJ, Messer RJ, Race B, Hasenkrug KJ. Production of bone marrow, liver, thymus (BLT) humanized mice on the C57BL/6 Rag2(−/−)gammac(−/−)CD47(−/−) background. J Immunol Methods. 2014;407:127–134. doi: 10.1016/j.jim.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang H, Madariaga ML, Wang S, Van RN, Oldenborg PA, Yang YG. Lack of CD47 on nonhematopoietic cells induces split macrophage tolerance to CD47null cells. Proc Natl Acad Sci U S A. 2007;104:13744–13749. doi: 10.1073/pnas.0702881104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li Y, Chen Q, Zheng D, Yin L, Chionh YH, Wong LH, Tan SQ, Tan TC, Chan JK, Alonso S, et al. Induction of functional human macrophages from bone marrow promonocytes by M-CSF in humanized mice. J Immunol. 2013;191:3192–3199. doi: 10.4049/jimmunol.1300742. [DOI] [PubMed] [Google Scholar]
- 110.Gille C, Orlikowsky TW, Spring B, Hartwig UF, Wilhelm A, et al. Monocytes derived from humanized neonatal NOD/SCID/IL2Rγnull mice are phenotypically immature and exhibit functional impairments. 2012;73:346–54. doi: 10.1016/j.humimm.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 111.Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, Corcuff E, Mortier E, Jacques Y, Spits H, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. The Journal of experimental medicine. 2009;206:25–34. doi: 10.1084/jem.20082013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, Auerbach W, Eynon EE, Stevens S, Manz MG, et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2390–2395. doi: 10.1073/pnas.1019682108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rongvaux A, Willinger T, Takizawa H, Rathinam C, Auerbach W, Murphy AJ, Valenzuela DM, Yancopoulos GD, Eynon EE, Stevens S, et al. Human thrombopoietin knockin mice efficiently support human hematopoiesis in vivo. Proc Natl Acad Sci U S A. 2011;108:2378–2383. doi: 10.1073/pnas.1019524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rongvaux A, Eynon EE, Manz MG, Flavell RA. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood. 2011;118:3119–3128. doi: 10.1182/blood-2010-12-326926. [DOI] [PubMed] [Google Scholar]
- 115.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc Natl Acad Sci U S A. 2009;106:21783–21788. doi: 10.1073/pnas.0912274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgamma(null) humanized mice. Blood. 2011;117:3076–3086. doi: 10.1182/blood-2010-08-301507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Takagi S, Saito Y, Hijikata A, Tanaka S, Watanabe T, Hasegawa T, Mochizuki S, Kunisawa J, Kiyono H, Koseki H, et al. Membrane-bound human SCF/KL promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood. 2012;119:2768–2777. doi: 10.1182/blood-2011-05-353201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marodon G, Desjardins D, Mercey L, Baillou C, Parent P, Manuel M, Caux C, Bellier B, Pasqual N, Klatzmann D. High diversity of the immune repertoire in humanized NOD. SCID.gamma c−/− mice. Eur J Immunol. 2009;39:2136–2145. doi: 10.1002/eji.200939480. [DOI] [PubMed] [Google Scholar]
- 120.Biswas S, Chang H, Sarkis PT, Fikrig E, Zhu Q, Marasco WA. Humoral immune responses in humanized BLT mice immunized with West Nile virus and HIV-1 envelope proteins are largely mediated via human CD5+ B cells. Immunology. 2011;134:419–433. doi: 10.1111/j.1365-2567.2011.03501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Unsinger J, McDonough JS, Shultz LD, Ferguson TA, Hotchkiss RS. Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized” mice. 2009:219–227. doi: 10.1189/jlb.1008615. 2009/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huntington ND, Alves NL, Legrand N, Lim A, Strick-Marchand H, Mention JJ, Plet A, Weijer K, Jacques Y, Becker PD, et al. IL-15 transpresentation promotes both human T-cell reconstitution and T-cell-dependent antibody responses in vivo. Proc Natl Acad Sci U S A. 2011;108:6217–6222. doi: 10.1073/pnas.1019167108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Danner R, Chaudhari SN, Rosenberger J, Surls J, Richie TL, Brumeanu TD, Casares S. Expression of HLA class II molecules in humanized NOD. Rag1KO.IL2RgcKO mice is critical for development and function of human T and B cells. PLoS One. 2011;6:e19826. doi: 10.1371/journal.pone.0019826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jaiswal S, Pearson T, Friberg H, Shultz LD, Greiner DL, Rothman AL, Mathew A. Dengue virus infection and virus-specific HLA-A2 restricted immune responses in humanized NOD-scid IL2rgammanull mice. PLoS One. 2009;4:e7251. doi: 10.1371/journal.pone.0007251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jaiswal S, Pazoles P, Woda M, Shultz LD, Greiner DL, Brehm MA, Mathew A. Enhanced humoral and HLA-A2-restricted dengue virus-specific T-cell responses in humanized BLT NSG mice. 2012:334–343. doi: 10.1111/j.1365-2567.2012.03585.x. 2012/03/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M, Doi T, Sone A, Suzuki N, Fujiwara H, et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci U S A. 2010;107:13022–13027. doi: 10.1073/pnas.1000475107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Strowig T, Gurer C, Ploss A, Liu YF, Arrey F, Sashihara J, Koo G, Rice CM, Young JW, Chadburn A, et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med. 2009;206:1423–1434. doi: 10.1084/jem.20081720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Suzuki M, Takahashi T, Katano I, Ito R, Ito M, Harigae H, Ishii N, Sugamura K. Induction of human humoral immune responses in a novel HLA-DR-expressing transgenic NOD/Shi-scid/gammacnull mouse. Int Immunol. 2012;24:243–252. doi: 10.1093/intimm/dxs045. [DOI] [PubMed] [Google Scholar]
- 129.Serra-Hassoun M, Bourgine M, Boniotto M, Berges J, Langa F, Michel ML, Freitas AA, Garcia S. Human Hematopoietic Reconstitution and HLA-Restricted Responses in Nonpermissive Alymphoid Mice. J Immunol. 2014 doi: 10.4049/jimmunol.1400412. [DOI] [PubMed] [Google Scholar]
- 130.Gorantla S, Sneller H, Walters L, Sharp JG, Pirruccello SJ, West JT, Wood C, Dewhurst S, Gendelman HE, Poluektova L. Human immunodeficiency virus type 1 pathobiology studied in humanized BALB/c-Rag2−/−gammac−/− mice. J Virol. 2007;81:2700–2712. doi: 10.1128/JVI.02010-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vuyyuru R, Patton J, Manser T. Human immune system mice: current potential and limitations for translational research on human antibody responses. Immunol Res. 2011;51:257–266. doi: 10.1007/s12026-011-8243-9. [DOI] [PubMed] [Google Scholar]
- 132.Seung E, Tager AM. Humoral immunity in humanized mice: a work in progress. J Infect Dis. 2013;208(Suppl 2):S155–S159. doi: 10.1093/infdis/jit448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schmidt MR, Appel MC, Giassi LJ, Greiner DL, Shultz LD, Woodland RT. Human BLyS facilitates engraftment of human PBL derived B cells in immunodeficient mice. PloS one. 2008;3:e3192. doi: 10.1371/journal.pone.0003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen Q, He F, Kwang J, Chan JK, Chen J. GM-CSF and IL-4 stimulate antibody responses in humanized mice by promoting T, B, and dendritic cell maturation. J Immunol. 2012;189:5223–5229. doi: 10.4049/jimmunol.1201789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chappaz S, Finke D. The IL-7 signaling pathway regulates lymph node development independent of peripheral lymphocytes. Journal of immunology. 2010;184:3562–3569. doi: 10.4049/jimmunol.0901647. [DOI] [PubMed] [Google Scholar]
- 136.Payne KJ, Su RJ, Francis OL, Martinez SR, Bennett T, Arogyaswamy K, Morris CL, Dovat S. A human-mouse xenogaft model to study the role of TSLP in CRLF2d-ALL. 2012;72(supplement) [Google Scholar]
- 137.Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–1593. doi: 10.1016/j.cell.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, de Geus SJ, Pouw SM, Böhne M, Voordouw A, et al. Functional CD47/signal regulatory protein alpha (SIRPα) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. 2011;108:13224–13229. doi: 10.1073/pnas.1101398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hu Z, Van Rooijen N, Yang YG. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood. 2011;118:5938–5946. doi: 10.1182/blood-2010-11-321414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. Journal of leukocyte biology. 2008:64–70. doi: 10.1189/jlb.0407247. 2007/09/22. [DOI] [PubMed] [Google Scholar]
- 141.Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, Annamali M, Piganelli JD, Moldawer LL, Mathews CE. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes. 2011:2144–2151. doi: 10.2337/db10-1222. 2011/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. 2013:812–827. doi: 10.1016/j.cell.2013.04.020. 2013/05/15. [DOI] [PubMed] [Google Scholar]
- 143.Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proceedings of the National Academy of Sciences of the United States of America. 2011:4607–4614. doi: 10.1073/pnas.1000092107. 2010/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283:16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Holtan SG, Pasquini M, Weisdorf DJ. Acute graft-versus-host disease: a bench-to-bedside update. Blood. 2014;124:363–373. doi: 10.1182/blood-2014-01-514786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Markey KA, MacDonald KP, Hill GR. The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood. 2014;124:354–362. doi: 10.1182/blood-2014-02-514745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Abraham S, Choi JG, Ye C, Manjunath N, Shankar P. IL-10 exacerbates xenogeneic GVHD by inducing massive human T cell expansion. Clin Immunol. 2015;156:58–64. doi: 10.1016/j.clim.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ito R, Katano I, Kawai K, Hirata H, Ogura T, Kamisako T, Eto T, Ito M. Highly sensitive model for xenogenic GVHD using severe immunodeficient NOG mice. Transplantation. 2009;87:1654–1658. doi: 10.1097/TP.0b013e3181a5cb07. [DOI] [PubMed] [Google Scholar]
- 149.van Rijn RS, Simonetti ER, Hagenbeek A, Hogenes MC, de Weger RA, Canninga-van Dijk MR, Weijer K, Spits H, Storm G, van Bloois L, et al. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2−/− gammac−/− double-mutant mice. 2003:2522–2531. doi: 10.1182/blood-2002-10-3241. 2003/06/07. [DOI] [PubMed] [Google Scholar]
- 150.Covassin L, Laning J, Abdi R, Langevin DL, Phillips NE, Shultz LD, Brehm MA. Human peripheral blood CD4 T cell-engrafted non-obese diabetic-scid IL2rgamma(null) H2-Ab1 (tm1Gru) Tg (human leucocyte antigen D-related 4) mice: a mouse model of human allogeneic graft-versus-host disease. Clin Exp Immunol. 2011;166:269–280. doi: 10.1111/j.1365-2249.2011.04462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hogenes MC, van DS, van KJ, Monteiro FR, ter HN, van Dijk MR, Martens AC, de Weger RA. Histological assessment of the sclerotic graft-versus-host response in the humanized RAG2−/−gammac−/− mouse model. Biol Blood Marrow Transplant. 2012;18:1023–1035. doi: 10.1016/j.bbmt.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 152.Hannon M, Lechanteur C, Lucas S, Somja J, Seidel L, Belle L, Bruck F, Baudoux E, Giet O, Chantillon AM, et al. Infusion of clinical-grade enriched regulatory T cells delays experimental xenogeneic graft-versus-host disease. Transfusion. 2013 doi: 10.1111/trf.12279. [DOI] [PubMed] [Google Scholar]
- 153.Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, June CH, Miller JS, Wagner JE, Blazar BR. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplant. 2011;11:1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tobin LM, Healy ME, English K, Mahon BP. Human mesenchymal stem cells suppress donor CD4(+) T cell proliferation and reduce pathology in a humanized mouse model of acute graft-versus-host disease. Clin Exp Immunol. 2013;172:333–348. doi: 10.1111/cei.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zheng J, Liu Y, Liu Y, Liu M, Xiang Z, Lam KT, Lewis DB, Lau YL, Tu W. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Science translational medicine. 2013;5:168ra9. doi: 10.1126/scitranslmed.3004943. [DOI] [PubMed] [Google Scholar]
- 156.Nakauchi Y, Yamazaki S, Napier SC, Usui JI, Ota Y, Takahashi S, Watanabe N, Nakauchi H. Effective treatment against severe Graft-versus-Host Disease with allele-specific anti-HLA monoclonal antibody in a humanized-mouse model. Exp Hematol. 2014 doi: 10.1016/j.exphem.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 157.Brehm MA, Shultz LD. Human allograft rejection in humanized mice: a historical perspective. Cell Mol Immunol. 2012;9:225–231. doi: 10.1038/cmi.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Abele-Ohl S, Leis M, Mahmoudian S, Weyand M, Stamminger T, Ensminger SM. Rag2−/− gamma-chain−/− mice as hosts for human vessel transplantation and allogeneic human leukocyte reconstitution. Transpl Immunol. 2010;23:59–64. doi: 10.1016/j.trim.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 159.Feng G, Nadig SN, Backdahl L, Beck S, Francis RS, Schiopu A, Whatcott A, Wood KJ, Bushell A. Functional regulatory T cells produced by inhibiting cyclic nucleotide phosphodiesterase type 3 prevent allograft rejection. Sci Transl Med. 2011;3:83ra40. doi: 10.1126/scitranslmed.3002099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, Schiopu A, Taggart DP, Wood KJ. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med. 2010;16:809–813. doi: 10.1038/nm.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee WP, Yaremchuk MJ, Pan YC, Randolph MA, Tan CM, Weiland AJ. Relative antigenicity of components of a vascularized limb allograft. Plast Reconstr Surg. 1991;87:401–411. doi: 10.1097/00006534-199103000-00001. [DOI] [PubMed] [Google Scholar]
- 162.Racki WJ, Covassin L, Brehm M, Pino S, Ignotz R, Dunn R, Laning J, Graves SK, Rossini AA, Shultz LD, et al. NOD-scid IL2rgamma(null) mouse model of human skin transplantation and allograft rejection. Transplantation. 2010;89:527–536. doi: 10.1097/TP.0b013e3181c90242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Issa F, Hester J, Goto R, Nadig SN, Goodacre TE, Wood K. Ex vivo-expanded human regulatory T cells prevent the rejection of skin allografts in a humanized mouse model. Transplantation. 2010;90:1321–1327. doi: 10.1097/TP.0b013e3181ff8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. 2011;3:83ra42. doi: 10.1126/scitranslmed.3002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Todd JA. Etiology of type 1 diabetes. 2010:457–467. doi: 10.1016/j.immuni.2010.04.001. 2010/04/24. [DOI] [PubMed] [Google Scholar]
- 166.Morran MP, Vonberg A, Khadra A, Pietropaolo M. Immunogenetics of type 1 diabetes mellitus. Mol Aspects Med. 2015 doi: 10.1016/j.mam.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 168.Reed JC, Herold KC. Thinking bedside at the bench: the NOD mouse model of T1DM. Nat Rev Endocrinol. 2015 doi: 10.1038/nrendo.2014.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.King M, Pearson T, Rossini AA, Shultz LD, Greiner DL. Humanized mice for the study of type 1 diabetes and beta cell function. 2008:46–53. doi: 10.1196/annals.1447.009. 2009/01/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhao Y, Guo C, Hwang D, Lin B, Dingeldein M, Mihailescu D, Sam S, Sidhwani S, Zhang Y, Jain S, et al. Selective destruction of mouse islet beta cells by human T lymphocytes in a newly-established humanized type 1 diabetic model. Biochem Biophys Res Commun. 2010;399:629–636. doi: 10.1016/j.bbrc.2010.07.128. [DOI] [PubMed] [Google Scholar]
- 171.Brehm MA, Bortell R, Diiorio P, Leif J, Laning J, Cuthbert A, Yang C, Herlihy M, Burzenski L, Gott B, et al. Human immune system development and rejection of human islet allografts in spontaneously diabetic NOD-Rag1null IL2rgammanull Ins2Akita mice. Diabetes. 2010;59:2265–2270. doi: 10.2337/db10-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mathews CE, Langley SH, Leiter EH. New mouse model to study islet transplantation in insulin-dependent diabetes mellitus. Transplantation. 2002;73:1333–1336. doi: 10.1097/00007890-200204270-00024. [DOI] [PubMed] [Google Scholar]
- 173.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. 1997:887–894. doi: 10.2337/diab.46.5.887. 1997/05/01. [DOI] [PubMed] [Google Scholar]
- 174.Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52:409–416. doi: 10.2337/diabetes.52.2.409. [DOI] [PubMed] [Google Scholar]
- 175.Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest. 2002;109:443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jacobson S, Heuts F, Juarez J, Hultcrantz M, Korsgren O, Svensson M, Rottenberg M, Flodstrom-Tullberg M. Alloreactivity but failure to reject human islet transplants by humanized Balb/c/Rag2gc mice. Scand J Immunol. 2010;71:83–90. doi: 10.1111/j.1365-3083.2009.02356.x. [DOI] [PubMed] [Google Scholar]
- 177.Wu DC, Hester J, Nadig SN, Zhang W, Trzonkowski P, Gray D, Hughes S, Johnson P, Wood KJ. Ex vivo expanded human regulatory T cells can prolong survival of a human islet allograft in a humanized mouse model. Transplantation. 2013;96:707–716. doi: 10.1097/TP.0b013e31829fa271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu H, Wen D, Mahato RI. Third-party Mesenchymal Stem Cells Improved Human Islet Transplantation in a Humanized Diabetic Mouse Model. Mol Ther. 2013 doi: 10.1038/mt.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yi S, Ji M, Wu J, Ma X, Phillips P, Hawthorne WJ, O’Connell PJ. Adoptive transfer with in vitro expanded human regulatory T cells protects against porcine islet xenograft rejection via interleukin-10 in humanized mice. Diabetes. 2012;61:1180–1191. doi: 10.2337/db11-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Viehmann Milam AA, Maher SE, Gibson JA, Lebastchi J, Wen L, Ruddle NH, Herold KC, Bothwell AL. A humanized mouse model of autoimmune insulitis. Diabetes. 2014;63:1712–1724. doi: 10.2337/db13-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Whitfield-Larry F, Young EF, Talmage G, Fudge E, Azam A, Patel S, Largay J, Byrd W, Buse J, Calikoglu AS, et al. HLA-A2-matched peripheral blood mononuclear cells from type 1 diabetic patients, but not nondiabetic donors, transfer insulitis to NOD-scid/gammac(null)/HLA-A2 transgenic mice concurrent with the expansion of islet-specific CD8+ T cells. Diabetes. 2011:1726–1733. doi: 10.2337/db10-1287. 2011/04/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Unger WW, Pearson T, Abreu JR, Laban S, van der Slik AR, der Kracht SM, Kester MG, Serreze DV, Shultz LD, Griffioen M, et al. Islet-specific CTL cloned from a type 1 diabetes patient cause beta-cell destruction after engraftment into HLA-A2 transgenic NOD/scid/IL2RG null mice. PloS one. 2012;7:e49213. doi: 10.1371/journal.pone.0049213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, Hohlfeld R, Wekerle H. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun. 2014;54:33–50. doi: 10.1016/j.jaut.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 184.Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. 1990:183–187. doi: 10.1038/346183a0. 1990/07/12. [DOI] [PubMed] [Google Scholar]
- 185.Sospedra M, Martin R. Immunology of multiple sclerosis. 2005:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. 2005/03/18. [DOI] [PubMed] [Google Scholar]
- 186.Lill CM. Recent advances and future challenges in the genetics of multiple sclerosis. 2014:130. doi: 10.3389/fneur.2014.00130. 2014/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zayoud M, El Malki K, Frauenknecht K, Trinschek B, Kloos L, Karram K, Wanke F, Georgescu J, Hartwig UF, Sommer C, et al. Subclinical CNS inflammation as response to a myelin antigen in humanized mice. 2013:1037–1047. doi: 10.1007/s11481-013-9466-4. 2013/05/04. [DOI] [PubMed] [Google Scholar]
- 188.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 189.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shapira Y, Agmon-Levin N, Shoenfeld Y. Geoepidemiology of autoimmune rheumatic diseases. Nat Rev Rheumatol. 2010;6:468–476. doi: 10.1038/nrrheum.2010.86. [DOI] [PubMed] [Google Scholar]
- 191.Kobezda T, Ghassemi-Nejad S, Mikecz K, Glant TT, Szekanecz Z. Of mice and men: how animal models advance our understanding of T-cell function in RA. Nat Rev Rheumatol. 2014;10:160–170. doi: 10.1038/nrrheum.2013.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Andersson EC, Hansen BE, Jacobsen H, Madsen LS, Andersen CB, Engberg J, Rothbard JB, McDevitt GS, Malmstrom V, Holmdahl R, et al. Definition of MHC and T cell receptor contacts in the HLA-DR4restricted immunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. 1998:7574–7579. doi: 10.1073/pnas.95.13.7574. 1998/06/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gumanovskaya M, Zaller DM, Woods A, Altmann DM, Stuart JM, Kang AH. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. 1997:1113–1122. doi: 10.1084/jem.185.6.1113. 1997/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 195.Toussirot E, Roudier J. Pathophysiological links between rheumatoid arthritis and the Epstein-Barr virus: an update. Joint Bone Spine. 2007;74:418–426. doi: 10.1016/j.jbspin.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 196.Kuwana Y, Takei M, Yajima M, Imadome K, Inomata H, Shiozaki M, Ikumi N, Nozaki T, Shiraiwa H, Kitamura N, et al. Epstein-Barr virus induces erosive arthritis in humanized mice. PloS one. 2011;6:e26630. doi: 10.1371/journal.pone.0026630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Andrade D, Redecha PB, Vukelic M, Qing X, Perino G, Salmon JE, Koo GC. Engraftment of peripheral blood mononuclear cells from systemic lupus erythematosus and antiphospholipid syndrome patient donors into BALB-RAG-2−/− IL-2Rgamma−/− mice: a promising model for studying human disease. Arthritis Rheum. 2011;63:2764–2773. doi: 10.1002/art.30424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Crampton SP, Morawski PA, Bolland S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis Model Mech. 2014;7:1033–1046. doi: 10.1242/dmm.016451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Singh N, Cohen PL. The T cell in Sjogren’s syndrome: force majeure, not spectateur. J Autoimmun. 2012;39:229–233. doi: 10.1016/j.jaut.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Young NA, Wu LC, Bruss M, Kaffenberger BH, Hampton J, Bolon B, Jarjour WN. A chimeric human-mouse model of Sjogren’s syndrome. Clin Immunol. 2015;156:1–8. doi: 10.1016/j.clim.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Longobardi T, Jacobs P, Bernstein CN. Work losses related to inflammatory bowel disease in the United States: results from the National Health Interview Survey. Am J Gastroenterol. 2003;98:1064–1072. doi: 10.1111/j.1572-0241.2003.07285.x. [DOI] [PubMed] [Google Scholar]
- 202.Koboziev I, Reinoso WC, Furr KL, Grisham MB. Role of the enteric microbiota in intestinal homeostasis and inflammation. Free Radic Biol Med. 2014;68:122–133. doi: 10.1016/j.freeradbiomed.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mathis D, Benoist C. Microbiota and autoimmune disease: the hosted self. Cell Host Microbe. 2011;10:297–301. doi: 10.1016/j.chom.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 204.Nolte T, Zadeh-Khorasani M, Safarov O, Rueff F, Gülberg V, Herbach N, Wollenberg A, Mueller T, Siebeck M, Wolf E, et al. Oxazolone and ethanol induce colitis in non-obese diabetic-severe combined immunodeficiency interleukin-2Rγnull mice engrafted with human peripheral blood mononuclear cells. 2013;172:349–362. doi: 10.1111/cei.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nolte T, Zadeh-Khorasani M, Safarov O, Rueff F, Varga R, Herbach N, Wanke R, Wollenberg A, Mueller T, Gropp R, et al. Induction of oxazolone-mediated features of atopic dermatitis in NOD-scid IL2Rgamma(null) mice engrafted with human peripheral blood mononuclear cells. Dis Model Mech. 2013;6:125–134. doi: 10.1242/dmm.009167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Goettel JA, Shouval DS, Lexmond W, Muise AM, Fiebiger E, Snapper SB. 134 Development of Novel Humanized Murine Models to Assess Mucosal Homeostasis: Human anti-CD3 Antibody or TNBS Administration Leads to Small and Large Bowel Inflammation Respectively in Immunodeficient Mice Transferred With Human T Cells. Inflamm Bowel Dis. 2013;144:S–32. [Google Scholar]
- 207.Goettel JA, Biswas S, Lexmond WS, Sun J, Ouahed J, McCann K, Shouval DS, Milford E, Fiebiger E, Muise A, et al. Human Hematopoietic Stem Cells With a Defined Immunodeficiency and Enteropathy Transfer Clinical Phenotype to a Novel Humanized Mouse Strain. Inflamm Bowel Dis. 2014;146:S–81. [Google Scholar]
- 208.Weigmann B, Schughart N, Wiebe C, Sudowe S, Lehr HA, Jonuleit H, Vogel L, Becker C, Neurath MF, Grabbe S, et al. Allergen-induced IgE-dependent gut inflammation in a human PBMC-engrafted murine model of allergy. The Journal of allergy and clinical immunology. 2012;129:1126–1135. doi: 10.1016/j.jaci.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 209.Jess T, Riis L, Jespersgaard C, Hougs L, Andersen PS, Orholm MK, Binder V, Munkholm P. Disease concordance, zygosity, and NOD2/CARD15 status: follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. The American journal of gastroenterology. 2005;100:2486–2492. doi: 10.1111/j.1572-0241.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 210.Bernstein CN, Shanahan F. Disorders of a modern lifestyle: reconciling the epidemiology of inflammatory bowel diseases. Gut. 2008;57:1185–1191. doi: 10.1136/gut.2007.122143. [DOI] [PubMed] [Google Scholar]
- 211.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–1794. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 212.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 214.Ventham NT, Kennedy NA, Nimmo ER, Satsangi J. Beyond Gene Discovery in Inflammatory Bowel Disease: The Emerging Role of Epigenetics. Gastroenterology. 2013 doi: 10.1053/j.gastro.2013.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hintze KJ, Cox JE, Rompato G, Benninghoff AD, Ward RE, Broadbent J, Lefevre M. Broad scope method for creating humanized animal models for animal health and disease research through antibiotic treatment and human fecal transfer. 2014;5:183–191. doi: 10.4161/gmic.28403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Berg DJ, Zhang J, Weinstock JV, Ismail HF, Earle KA, Alila H, Pamukcu R, Moore S, Lynch RG. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 217.Hale LP, Gottfried MR, Swidsinski A. Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and mucosal exposure to intestinal bacteria. Inflammatory bowel diseases. 2005;11:1060–1069. doi: 10.1097/01.mib.0000187582.90423.bc. [DOI] [PubMed] [Google Scholar]