

Abstract
Purpose of review
Foam cells in human glomeruli can be encountered in various renal diseases including focal segmental glomerulosclerosis and diabetic nephropathy. Although foam cells are a key participant in atherosclerosis, surprisingly little is known about their pathogenicity in the kidney. We review our understanding (or lack thereof) of foam cells in the kidney as well as insights gained in studies of foam cells and macrophages involved in atherosclerosis, to suggest areas of investigation that will allow better characterization of the role of these cells in renal disease.
Recent findings
There is a general dearth of animal models of disease with renal foam cell accumulation, limiting progress in our understanding of the pathobiology of these cells. Recent genetic modifications of hyperlipidemic mice have resulted in some new disease models with renal foam cell accumulation. Recent studies have challenged older paradigms by findings that indicate many tissue macrophages are derived from cells permanently residing in the tissue from birth rather than circulating monocytes.
Summary
Renal foam cells remain an enigma. Extrapolating from studies of atherosclerosis suggests that therapeutics targeting mitochondrial ROS production or modulating cholesterol and lipoprotein uptake or egress from these cells may prove beneficial for kidney diseases in which foam cells are present.
Keywords: kidney, glomerulus, macrophage, foam cell, atherosclerosis
INTRODUCTION
While it is established that monocyte/macrophage derived foam cells have a central role in the development of atherosclerosis, the pathophysiological significance of these cells in the kidney has been a puzzle for many years. Renal pathologists commonly encounter these cells in human renal biopsies in diverse disease settings. These include diabetic nephropathy, and the tip and cellular variant forms of focal and segmental glomerulosclerosis (FSGS), in which glomerular capillaries may appear occluded by an infiltrate of these cells (Fig.1) [1,2]. Dissolution of the normally compact mesangial matrix (“mesangiolysis”) frequently is present in conjunction with glomerular foam cell accumulation, but whether these processes are mechanistically linked remains unknown. Rare glomerular diseases in humans characterized by foam cell accumulation include immune-mediated acquired lecithincholesterol acyltransferase deficiency [3] and a glomerulopathy associated with a homozygous mutation of apolipoprotein E (apolipoprotein E2) [4]. Foam cells commonly populate the interstitium in nephrotic states and in Alport syndrome, and at least one study has suggested that these cells contribute to progressive tubulointerstitial injury and are associated with a higher prevalence of concurrent FSGS regardless of primary disease etiology [5]. However, the evidence linking renal interstitial foam cells to chronic kidney disease (CKD) in human or experimental disease is scanty. Indeed, it is our impression that many observers regard these cells as bystanders in renal disease states which are unlikely to have clinical significance. While the central questions of whether or how these cells are pathogenic in the kidney cannot be answered definitively at present, new insights concerning monocyte and macrophage biology may provide guidance in our thinking about how these cells may participate in kidney disease.
Figure 1.
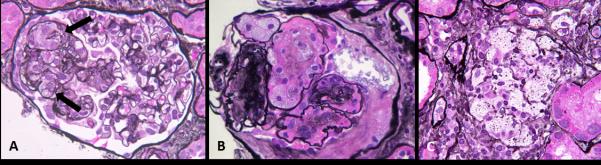
Foam cell accumulation in human glomerular disease. A, The “tip variant of focal segmental glomerulosclerosis” often is characterized by accumulation of intracapillary foam cells (arrow). B, Intracapillary foam cell accumulation may also be present in diabetic nephropathy, where they are usually associated with mesangiolysis and distension of capillaries adjacent mesangiolytic foci. C, Aggregates of foam cells are often encountered in the interstitium in Alport syndrome as shown here, and may also be present in nephrotic states resulting from multiple etiologies.
Although lipid-laden foam cells may be derived from a variety of cell types, including smooth muscle cells, mesangial cells, epithelial cells, endothelial cells and resident phagocytic cells such as Kupffer cells in the liver, it is the lipid-laden monocyte/macrophage derived foam cell that remains the best studied, with well-established pathogenic properties [6,7■■,8■,9■■,10]. These cells are central to the development and progression of atherosclerotic plaques [11■■], and it is these cells that are the subject of this review. This class of foam cells arises in response to metabolic conditions that promote intracellular uptake of lipoproteins and cholesterol and/or concurrent dysfunction of homeostatic mechanisms that normally allow egress of cholesterol and lipids from cells.
FOAM CELLS ARE PATHOGENIC IN ATHEROSCLEROSIS
Generation of foam cells derived from monocytes/macrophages is the result of three broad processes: excessive influx of cholesterol and lipid into the cell; metabolic modification of cholesterol, primarily by esterification, and impaired egress of cholesterol back into the circulation (a process known as reverse cholesterol transport, or RCT). The details of these processes have been well reviewed [7■■,12]. In the setting of atherosclerosis, the first steps in foam cell formation are the uptake of local accumulations in artery walls of oxidized low-density lipoproteins (oxLDL) and acetylated low-density lipoproteins (AcLDL) by monocytes/macrophages. Binding to matrix proteins such as proteoglycans is a major means that causes these lipoproteins to lodge in the arterial intima where atherosclerosis is initiated. Uptake by monocytes/macrophages occurs primarily through several scavenger receptors, principally CD36 and scavenger receptor A (SR-A), although other processes including macropinocytosis and phagocytosis contribute [13]. There is no data currently available that addresses the issue of whether local accumulation of foam cells in the kidney is similarly initiated. The nature of scavenger receptor interactions with lipoproteins is not straightforward. For instance, the disorder lipoprotein glomerulopathy is characterized by abnormal accumulations of ApoE containing “lipoprotein thrombi” within glomerular capillaries, but typically without prominent foam cell formation [14]. Lipid accumulation per se (so-called “fatty kidney”) is not sufficient for renal macrophage foam cell accumulation [9■■]. Foam cell formation in arteries is regulated in part by cytokines and mediators of inflammation including interleukin-33, interferon-γ and transforming growth factor-β (TGF-β), contributing to the concept that this process and atherosclerosis can be considered, at least in part, as an inflammatory injury [12,13]. As reviewed by Randolph and Moore, there is evidence that macrophages and foam cells in atherosclerosis may be both the result of engagement of both innate and adaptive immune pathways, and in turn these cells likely contribute to amplification of these pathways by such functions as antigen processing and presentation and providing damage associated molecular pattern signals (DAMPs) [15,16■■].
Foam cell accumulation in the intima of large arteries marks the initiation of atherosclerosis in both human disease and in such well-established animal models as fat-fed non-human primates and rabbits, and in genetically mutated mice with hyperlipidemia such as apolipoprotein E (ApoE−/−) and low density lipoprotein receptor (LDLR−/−) deficient mice [17]. Necrobiosis of foam cells leads to instability and rupture of atherosclerotic plaques. Maneuvers that reduce or eliminate macrophage accumulation in experimental atherosclerosis also reduce or prevent the formation of atherosclerotic plaques, as exemplified by ApoE−/− and LDLR−/− mice in which the two main components of the principle chemokine homing system for macrophages (monocyte chemoattractant protein-1, MCP-1 and/or its receptor CCR2) have also been genetically deleted [18,19]. Finally, the phenotypic characterization of foam cells in atherosclerotic plaques has revealed several mechanisms of their pathogenicity, including production and secretion of reactive oxygen species, production and secretion of oxidized lipoproteins, activation and secretion of metalloproteinases, and secretion of pro-inflammatory cytokines and growth factors that promote sclerosis [10,11■■,16■■,20-24].
THE NEED FOR ANIMAL MODELS OF FOAM CELLS IN THE KIDNEY
The genesis of this review began as a reconsideration of a once-influential hypothesis paper of Diamond and Karnovsky that proposed that FSGS was analogous to atherosclerosis [25]. Their conclusion was based on identified similarities of mesangial cells in cases of FSGS to the vascular smooth muscle-like cells that are one of the principal cell types found in atherosclerotic plaques, and the ubiquitous presence of monocyte-derived foam cells, the other major cell type found in atherosclerotic plaques [25]. More recently, a substantial body of epidemiologic evidence clearly demonstrates strong links between chronic kidney disease and atherosclerotic cardiovascular disease (ASCVD), and it has been shown that the increased cardiovascular mortality in patients with diabetes is attributable to the nephropathy that occurs in the subset of patients with this complication [26-28]. Indeed, the tight link between ASCVD and CKD extends beyond diabetes, as it now well established that ASCVD is the main cause of death in CKD patients [29]. The need to identify and understand common pathogenic mechanisms that may underlie atherosclerosis and chronic kidney disease (CKD) remains unmet. We must ask: Is it correct that what is true in atherosclerosis can be reliably extrapolated to the kidney? Are foam cells important contributors to chronic and sclerosing renal injury? Are foam cells in aortas and large arteries biologically identical to those found in the kidney, and are those found in the glomerulus identical to those found in the renal interstitium?
The answers to these critical questions are unknown and rarely addressed in the renal literature. This likely is the result of a lack of good model systems in which these concepts can be tested. Unlike the case with atherosclerosis, there are no robust animal models of foam cell accumulation in the kidney. We have published our experience with three models of hyperlipidemic renal injury: the ApoE−/− mouse [30], the LDLR−/− mice [31] and the fat fed B6.ROP Os/+ mouse with reduced nephron number [32]. We have shown that these mice, when maintained for extended periods of time on a “Western” high fat diet (>32 weeks), spontaneously develop progressive glomerular foam cell accumulation, mesangiolysis and tubular injury (Fig. 2), but neither we nor others have studied these models for lengthier periods of time to determine whether this additional step will lead to the development of CKD. While we have established temporal sequences of foam cell accumulation and mesangiolysis and sclerosis in our mouse models, neither we nor others have tested whether these associations are indeed causal.
Figure 2.
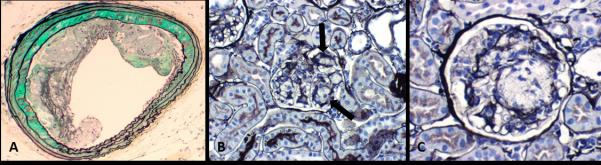
Foam cell accumulation in the mice model. A, Foam cells are normally encountered in apolipoprotein E (ApoE) null mice as part of the atherosclerosis (in this example, involving aorta) that occurs in these mice. B, ApoE null mice show irregular accumulations of foam cells in glomeruli (arrow). C, At higher magnification, the foam cell accumulation are associated with segmental mesangiolysis and microaneurysmal dilatation of adjacent capillary loops due in part to the loss of capillary anchoring into the mesangium.
There are important deficiencies of these models that need to be overcome in order to facilitate the testing of concepts of renal pathogenicity of foam cells. The development of foam cell lesions has not been sufficiently widespread in glomeruli in the models cited above to allow robust experimental studies of foam cell pathogenicity to the kidneys. The structural changes that are present have not led to progressive CKD, and so without further modification these mice cannot serve as good model systems for the combined CKD and ASCVD that is so lethal to patients with kidney disease. In efforts to overcome this deficiency, animal models of ASCVD such as ApoE−/− and LDLR−/− mice have been subjected to further modifications such as partial nephrectomy [33] or administration of a renal toxin such as adriamycin, or introduction of specific gene mutations in order to induce CKD in the setting of ASCVD that is already present in order to model the situation in humans with CKD [34■,35]. Recent examples of such modifications include the study of Tavori et al. [35] which overexpressed variants of apoE specifically in macrophages in both ApoE−/− and combined ApoE−/−/ LDLR−/− mice. This study found macrophage specific expression of a variant of ApoE (ApoESendai) linked to the development of lipoprotein glomerulopathy in humans is protective against atherosclerosis but causes lesions of lipoprotein glomerulopathy to develop in combined ApoE−/−/ LDLR−/− mice. Interestingly, the glomerular lesions were reported to include foci of mesangiolysis, but the presence of foam cells was not described. The study of Kennedy et al. [34■] focused on ApoE−/− mice with the additional deletion of the CD 36 scavenger receptor. Fat fed ApoE−/−/ CD36−/− mice had less macrophage accumulation, less glomerular foam cell accumulation and less interstitial fibrosis and improved creatinine clearance as compared to fat fed ApoE−/− controls. However, as noted above, CKD has not been shown to reliably develop in the ApoE−/− mouse model, and the degree of interstitial fibrosis was not substantial in any of the study groups. The concurrence of diminished foam cell prevalence with improvement in measured renal function offers support for the proposition that these cells are indeed pathogenic in the kidney, and so this model offers potential for further studies to validate this injury process.
Studies by Levi and colleagues have shown that altered lipid metabolism in general, and increased expression of sterol regulatory element binding proteins 1 and 2 (SREBP-1 and 2) contribute to diabetic kidney disease [36]. SREBP-1 and 2 are transcription factors that promote lipid and cholesterol accumulation in the kidney; mice that overexpress SREBP-1 develop a sclerosing glomerulopathy and proteinuria. Studies involving these proteins, particularly the nuclear hormone receptor farnesoid X receptor (FXR) which regulates SREBP expression, indicate manipulating this pathway by overexpressing FXR activity can have a beneficial effect in an experimental model of diabetic nephropathy [36]. Particularly noteworthy is the accumulation of foam cells, accompanied by mesangiolysis, in FXR deficient mice made diabetic by streptozotocin administration [36]. We are unaware of other mouse models of diabetic nephropathy that have such a prominent accumulation of foam cells. Curiously, this finding occurred in a mouse model (streptozotocin treated C57BL/6 mice) known to be resistant to developing structural changes of diabetic nephropathy [37]. This is one of the few mouse models in which foam cell formation and mesangiolysis can be induced, and is deserving of further studies that might identify mechanisms that link these two processes. The availability of specific antagonists to FXR may be especially useful in pursuing such studies.
GLOMERULAR FOAM CELLS AND FSGS
In FSGS, glomerular capillaries are segmentally occluded by relatively acellular material including matrix proteins and hyalin. The tip lesion and cellular variants of FSGS in particular may manifest expansile lesions stuffed with foam cells [1,2]. These variants of FSGS often demonstrate a confluence of swollen and hypertrophied podocytes in conjunction with the intracapillary foam cell accumulation. To date, there has been little evidence to link glomerular foam cell accumulation to podocyte injury per se, but a recent abstract presented at the 2014 annual meeting of American Society of Nephrology by the group of Nagata used a dual mouse model of hyperlipidemia (LDLR−/− mice) crossed with the NEP25 model of podocyte depletion induced by administration of the toxin LMB. An exciting finding was the preferential accumulation of foam cells to those glomerular capillaries exhibiting significant podocyte loss [38]. The authors postulated a sequence of injury whereby initial podocyte injury leads to disordered lipid metabolism and to endothelial and mesangial injury, in turn leading to induction of cytokines that then leads to recruitment of macrophages and foam cell formation at the site of injury [38]. This would imply that foam cells are relative latecomers to the injury process, but establishing the chronology of podocyte injury and foam cell accumulation in FSGS will require further testing. Indeed, it might be possible to use such a model to test an alternate scenario whereby foam cell formation occurs early in the injury process, and then test whether local uptake of oxLDL or AcLDL by macrophages initiates the process of foam cell formation as it does in atherosclerosis. These studies are particularly exciting in that they offer a plausible model for studies of lesions such as tip and cellular variants of human FSGS in which both foam cell accumulation and podocyte injury are important features.
FOAM CELLS: TRANSIENT VISITORS OR INDIGENOUS POPULATION?
It has been commonly thought that mature tissue macrophages originate from bone marrow derived hematopoietic cells, traveling through the circulation as monocytes and settling in tissues as part of either injury or reparative processes. Such cells may reside in the kidney for short periods of time, leaving by a return to the circulation or by cell death, or they may extend their stay but with little capacity for self-renewal. Very recent studies, reviewed by Sieweke and Allen [39], challenge this paradigm. In aggregate, these studies indicate that populations of tissue-based macrophages may originate from embryonic precursors that are tissue specific and hence may not be of bone marrow origin. Furthermore, mature tissue-based macrophages have a capacity to self-renew by local proliferation [40■]. This suggests that macrophage-derived foam cells in the kidney may not necessarily come from the circulation, and hence strategies to block monocyte/macrophage recruitment to sites of injury, such as blocking local production of chemokines or blockade of chemokine receptors may have little or no effect on the accumulation of such cells in disease states. It also raises the possibility that a fixed or locally self-renewing population of cells may acquire and/or abandon a foam cell phenotype in response to local stimuli.
FOAM CELLS AS THERAPEUTIC TARGETS
The pathogenicity of foam cells in atherosclerosis has led to exploration of multiple therapeutic strategies to modulate their pathogenic activities. Such strategies could prove useful should such cells be shown to have effects in the kidney that promote disease. One major approach includes enhancement of egress of cholesterol from these cells. Cholesterol egress can occur by several routes, mediated by membrane transport proteins, including adenosine, triphosphate binding cassette transporter A1 (ABCA1), adenosine triphosphate transporter G1 (ABCG1), and scavenger receptor class B member-1 (SR B-1) [6,12,15] . The liver X receptor (LXR) is a transcription factor that promotes reverse cholesterol transport out of the cell through these pathways [15,24]. When these pathways are disrupted (e.g., deletion of ABCA1 and ABCG1 in macrophages), these cells show increased lipid accumulation and increased atherosclerotic lesion severity [41]. Several therapeutic agents have been shown to be atheroprotective in model systems acting directly or indirectly on these pathways. These include taurine, the AMPK activator AICAR, activators of heme-oxygenase-1, and peroxisome proliferator-activated receptors α and γ agonists such as glitazones [12,15,24].
Mitochondrial oxidative stress has been increasingly identified as a key early pathogenic event in glomerular diseases (diabetic nephropathy and FSGS) that can have foam cell accumulation [42-44]. While reactive oxygen species (ROS) have been identified in lesional monocytes/macrophage of atherosclerosis [45] for some time, exciting recent studies by Wang et al. [46■■], have shown that enhanced expression of catalase, a scavenger of ROS, when restricted to mitochondria of monocytes/macrophages, reduces oxidative stress and results in marked reduction of foam cell accumulation and other features of atherosclerosis in LDLR−/− mice. Reduction of ROS is an attractive therapeutic strategy for amelioration of potential deleterious effects of foam cells in the kidney, as such strategies are already being tested for their potential benefit in some forms of kidney disease, such as acute kidney injury and diabetic nephropathy. Following the example in the LDLR−/− mouse, interventions to reduce mitochondrial oxidative stress could be used to both establish pathogenicity of renal foam cells and potentially provide therapeutic benefit if a robust model of foam cell accumulation in kidney disease could be developed for testing such strategies.
CONCLUSION
The goal of this review was to assess whether renal foam cells are pathogenic in the kidney, or not. Sadly, the information currently available does not allow a meaningful answer to this question. A critical issue has been the limited availability of animal models that robustly produce foam cell accumulations in kidney disease settings analogous to human diseases. The animal models of FSGS commonly employed (renal ablation; administration of glomerular toxins such as adriamycin or puromycin aminonucleoside, and genetic deletions of podocyte or basement membrane proteins) do not possess this feature. Indeed, good animal models of the tip variant of FSGS await development, although the mouse model of podocyte ablation in a hyperlipidemic setting may prove to be such a model. While a useful animal model of Alport syndrome, the COL4 null mouse has been developed, descriptions of this model to date have not identified a component of interstitial foam cell accumulation. There are opportunities for model development that may enable an answer to the question of pathogenicity. If models of foam cell accumulation in glomeruli such as the ApoE−/− mouse and the LDLR−/− mouse could be made more robust, the pathogenicity of the foam cells could then be studied by such currently available techniques as macrophage ablation or modulation of specific monocyte/macrophage function strategies akin to those employed in studies of atherosclerosis.
KEY POINTS.
Foam cells are encountered in common glomerular diseases such as focal and segmental glomerulosclerosis and diabetic nephropathy, but their pathogenic significance is unknown.
Foam cells are also commonly encountered in the renal interstitium in Alport syndrome and in nephrotic states.
Progress in understanding foam cell pathogenicity is hampered by the paucity of robust animal models of human renal diseases with foam cells.
It is not established whether foam cells arise from resident cells in tissue or from monocytes that have emigrated from the circulation.
Acknowledgments
None
Financial support and sponsorship
This work was supported by a grant from the National Institute of Health (DK83391) and supported in the past by a grant from the Genzyme GRIP program.
Footnotes
Conflicts of interest
None.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlightened as:
• of special interest
•• of outstanding interest
- 1.Stokes MB, Valeri AM, Markowitz GS, D'Agati VD. Cellular focal segmental glomerulosclerosis: clinical and pathologic features. Kidney Int. 2006;70:1783–1792. doi: 10.1038/sj.ki.5001903. [DOI] [PubMed] [Google Scholar]
- 2.D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi S, Hiromura K, Tsukida M, et al. Nephrotic syndrome caused by immune-mediated acquired LCAT deficiency. J Am Soc Nephrol. 2014;24:1305–1312. doi: 10.1681/ASN.2012090913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawanishi K, Sawada A, Ochi A, et al. Glomerulopathy with homozygous apolipoprotein e2: a report of three cases and review of the literature. Case Rep Nephrol Urol. 2013;3:128–135. doi: 10.1159/000356849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Y, Chen Y, Chen D, et al. Presence of foam cells in kidney interstitium is associated with progression of renal injury in patients with glomerular diseases. Nephron Clin Pract. 2009;113:c155–c161. doi: 10.1159/000232596. [DOI] [PubMed] [Google Scholar]
- 6.Yu XH, Fu YC, Zhang DW, et al. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245–252. doi: 10.1016/j.cca.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 7••.Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93:1317–1542. doi: 10.1152/physrev.00004.2012. [An almost unimaginably comprehensive review on the pathophysiology of atherosclerosis, including a current review of the mechanism of foam cell formation.] [DOI] [PubMed] [Google Scholar]
- 8•.Chaabane C, Coen M, Bochaton-Piallat ML. Smooth muscle cell phenotypic switch: implications for foam cell formation. Curr Opin Lipidol. 2014;25:374–379. doi: 10.1097/MOL.0000000000000113. [A reminder that not all foam cells are of macrophage origin!.] [DOI] [PubMed] [Google Scholar]
- 9••.de Vries AP, Ruggenenti P, Ruan XZ, et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014;2:417–426. doi: 10.1016/S2213-8587(14)70065-8. [An important review of pathology by which lipid may have deleterious effect on the kidney and with an overall focus on obesity related renal injury.] [DOI] [PubMed] [Google Scholar]
- 10.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- 11••.Zeller I, Srivastava S. Macrophage functions in atherosclerosis. Circ Res. 2014;115:e83–e85. doi: 10.1161/CIRCRESAHA.114.305641. [A succinct review of the pathogenicity of macrophages in atherosclerosis, with a focus on the development of foam cells.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaren JE, Michael DR, Ashlin TG, Ramji DP. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res. 2011;50:331–347. doi: 10.1016/j.plipres.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Michael DR, Ashlin TG, Davies CS, et al. Differential regulation of macropinocytosis in macrophages by cytokines: implications for foam cell formation and atherosclerosis. Cytokine. 2013;64:357–361. doi: 10.1016/j.cyto.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito T, Matsunaga A. Lipoprotein glomerulopathy may provide a key to unlock the puzzles of renal lipidosis. Kidney Int. 2014;85:243–245. doi: 10.1038/ki.2013.404. [DOI] [PubMed] [Google Scholar]
- 15.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res. 2014;114:1757–1771. doi: 10.1161/CIRCRESAHA.114.301174. [A comprehensive review of macrophage biology in the setting of atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 18.Rollins BJ. Chemokines and atherosclerosis: what Adam Smith has to say about vascular disease. J Clin Invest. 2001;108:1269–1271. doi: 10.1172/JCI14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 20.Abrass CK. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol. 2004;24:46–53. doi: 10.1159/000075925. [DOI] [PubMed] [Google Scholar]
- 21.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 23.Rader DJ, Pure E. Lipoproteins, macrophage function, and atherosclerosis: beyond the foam cell? Cell Metab. 2005;1:223–230. doi: 10.1016/j.cmet.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Uitz E, Bahadori B, McCarty MF, Moghadasian MH. Practical strategies for modulating foam cell formation and behavior. World J Clin Cases. 2014;2:497–506. doi: 10.12998/wjcc.v2.i10.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diamond JR, Karnovsky MJ. Focal and segmental glomerulosclerosis: analogies to atherosclerosis. Kidney Int. 1988;33:917–924. doi: 10.1038/ki.1988.87. [DOI] [PubMed] [Google Scholar]
- 26.Afkarian M, Sachs MC, Kestenbaum B, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol. 2013;24:302–308. doi: 10.1681/ASN.2012070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groop PH, Thomas MC, Moran JL, et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes. 2009;58:1651–1658. doi: 10.2337/db08-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orchard TJ, Secrest AM, Miller RG, Costacou T. In the absence of renal disease, 20 year mortality risk in type 1 diabetes is comparable to that of the general population: a report from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetologia. 2010;53:2312–2319. doi: 10.1007/s00125-010-1860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 30.Wen M, Segerer S, Dantas M, et al. Renal injury in apolipoprotein E-deficient mice. Lab Invest. 2002;82:999–1006. doi: 10.1097/01.lab.0000022222.03120.d4. [DOI] [PubMed] [Google Scholar]
- 31.Spencer MW, Muhlfeld AS, Segerer S, et al. Hyperglycemia and hyperlipidemia act synergistically to induce renal disease in LDL receptor-deficient BALB mice. Am J Nephrol. 2004;24:20–31. doi: 10.1159/000075362. [DOI] [PubMed] [Google Scholar]
- 32.Muhlfeld AS, Spencer MW, Hudkins KL, et al. Hyperlipidemia aggravates renal disease in B6.ROP Os/+ mice. Kidney Int. 2004;66:1393–1402. doi: 10.1111/j.1523-1755.2004.00854.x. [DOI] [PubMed] [Google Scholar]
- 33.Zuo Y, Yancey P, Castro I, et al. Renal dysfunction potentiates foam cell formation by repressing ABCA1. Arterioscler Thromb Vasc Biol. 2009;29:1277–1282. doi: 10.1161/ATVBAHA.109.188995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.Kennedy DJ, Chen Y, Huang W, et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension. 2013;61:216–224. doi: 10.1161/HYPERTENSIONAHA.112.198770. [A unique finding of this study linked a reduction in glomerular foam cell accumulation to improvements in renal function.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tavori H, Fan D, Giunzioni I, et al. Macrophage-derived apoESendai suppresses atherosclerosis while causing lipoprotein glomerulopathy in hyperlipidemic mice. J Lipid Res. 2014;55:2073–2081. doi: 10.1194/jlr.M049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang XX, Jiang T, Shen Y, et al. Diabetic nephropathy is accelerated by farnesoid X receptor deficiency and inhibited by farnesoid X receptor activation in a type 1 diabetes model. Diabetes. 2010;59:2916–2927. doi: 10.2337/db10-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brosius FC, 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hara S, Kobayashi N, Manabe S, et al. Podocyte injury promotes glomerular lipid peroxidation and foam cell infiltration under hypercholesterolemia.. Presented at the American Society of Nephrology Annual Meeting; Philadelphia, PA.. November 2014. [Google Scholar]
- 39.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342:1242974. doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- 40•.Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. [An important study that challenges a paradigm that circulating macrophages are the main source of lesional macrophages in atherosclerosis, and instead indicates accumulated macrophages are the result of local proliferation and thereby offers new possibilities for therapeutics.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westerterp M, Murphy AJ, Wang M, et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sedeek M, Nasrallah R, Touyz RM, Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol. 2013;24:1512–1518. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holterman CE, Thibodeau JF, Kennedy CR. NADPH oxidase 5 and renal disease. Curr Opin Nephrol Hypertens. 2015;24:81–87. doi: 10.1097/MNH.0000000000000081. [DOI] [PubMed] [Google Scholar]
- 44.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–2340. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madamanchi NR, Zhou RH, Vendrov AE, et al. Does oxidative DNA damage cause atherosclerosis and metabolic syndrome?: new insights into which came first: the chicken or the egg. Circ Res. 2010;107:940–942. doi: 10.1161/CIRCRESAHA.110.230904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ Res. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [This study demonstrates a role for macrophage mitochondrial oxidative stress in atherosclerosis progression in mice. Reduction of oxidative stress by macrophage specific expression of mitochondrial catalase diminished foam cell content in atherosclerosis-prone mice.] [DOI] [PMC free article] [PubMed] [Google Scholar]