

Summary
Pseudomonas aeruginosa causes chronic lung infections in the airways of cystic fibrosis (CF) patients. Psl is an extracellular polysaccharide expressed by non-mucoid P. aeruginosa strains, which are believed to be initial colonizers. We hypothesized that Psl protects P. aeruginosa from host defences within the CF lung prior to their conversion to the mucoid phenotype. We discovered that serum opsonization significantly increased the production of reactive oxygen species (ROS) by neutrophils exposed to a psl-deficient mutant, compared with wild-type (WT) and Psl overexpressing strains (Psl++). Psl-deficient P. aeruginosa were internalized and killed by neutrophils and macrophages more efficiently than WT and Psl++ variants. Deposition of complement components C3, C5 and C7 was significantly higher on psl-deficient strains compared with WT and Psl++ bacteria. In an in vivo pulmonary competition assay, there was a 4.5-fold fitness advantage for WT over psl-deficient P. aeruginosa. Together, these data show that Psl inhibits efficient opsonization, resulting in reduced neutrophil ROS production, and decreased killing by phagocytes. This provides a survival advantage in vivo. Since phagocytes are critical in early recognition and control of infection, therapies aimed at Psl could improve the quality of life for patients colonized with P. aeruginosa.
Introduction
Pseudomonas aeruginosa is a versatile opportunistic pathogen that can cause chronic pulmonary infection in cystic fibrosis (CF) patients and is also a significant cause of disease in immune-compromised patients (Lyczak et al., 2002). The ability of P. aeruginosa to form biofilms that are recalcitrant to treatment is a major cause of mortality and morbidity in these patients. Neutrophils are the first phagocytic cells mobilized to clear pathogenic bacteria during lung infection, yet pulmonary infection in CF is complicated by the robust recruitment, activation and damage caused by these cells (Walker et al., 2005). During colonization and infection of the CF airway, P. aeruginosa undergoes phenotypic conversion from a non-mucoid to mucoid morphology. This phenotype is due to the overproduction of alginate, a capsular polysaccharide that confers a selective advantage for P. aeruginosa. Alginate produced by mucoid P. aeruginosa scavenges bactericidal reactive oxygen species (ROS), and interferes with complement activation, chemotaxis, and neutrophil and macrophage phagocytic killing (Learn et al., 1987; Pedersen et al., 1990; Mai et al., 1993; Leid et al., 2005). However, in most CF patients, mucoid conversion occurs months or years after initial colonization (Govan and Deretic, 1996). There remain significant gaps in our understanding of how P. aeruginosa survives the inflammatory-rich environment of the CF lung prior to converting to the alginate-producing phenotype.
Psl is a recently discovered exopolysaccharide (EPS) of non-mucoid P. aeruginosa and the genes encoding this EPS are highly conserved among P. aeruginosa isolates (Wolfgang et al., 2003). Psl is composed of repeating pentasaccharide units of d-mannose, d-glucose and l-rhamnose (Byrd et al., 2009). Psl is critical for biofilm formation and believed to be expressed in P. aeruginosa isolates that are the first to colonize CF patients (Jackson et al., 2004; Ma et al., 2006). This implies that during initial infection, biofilm formation and Psl synthesis likely precede the switch to mucoidy. Polysaccharides in several pathogenic bacteria can promote phagocyte evasion by modulating deposition of complement or antibody on the microbe’s surface (Clay et al., 2008; Goebel et al., 2008). However, there have been no studies investigating if P. aeruginosa Psl modulates interactions with cells of the innate immune system.
We focused our study on neutrophils and complement since these innate immune effectors are critical in host defence against P. aeruginosa (Jesaitis et al., 2003; Mueller-Ortiz et al., 2004). Neutrophils are recruited early in infection and directly kill invading bacteria using both oxidative and non-oxidative mechanisms. The complement system is a highly regulated and multifunctional pathway that promotes recognition of pathogens by innate immune cells and is also capable of direct bacterial killing upon insertion of the terminal membrane attack complex (MAC). Opsonization by complement is a central mechanism for bacterial clearance and is crucial for maintaining mucosal immunity against infection (Gross et al., 1978). Once bacteria are taken up by neutrophils, the subsequent oxidative burst leads to the production of toxic ROS (Kanegasaki, 1986; Nauseef, 2007). The processes of degranulation, phagocytosis and oxidative killing furnish the neutrophil with a varied arsenal of antimicrobial effectors that are critical for an effective innate immune response.
A central hypothesis in the present study is that surface-bound Psl impairs phagocytosis by reducing complement-mediated opsonization. To test this, we utilized a collection of isogenic P. aeruginosa strains expressing variable amounts of surface Psl polysaccharide. Compared with WT bacteria, P. aeruginosa lacking Psl demonstrated increased complement-mediated opsonization. Lack of Psl expression led to enhanced uptake, oxidative burst response, and reduced intracellular bacterial survival in phagocytic cells. The presence of Psl provided P. aeruginosa a fitness advantage over psl mutants in an acute murine pulmonary model of infection. Enzymatic degradation of surface Psl also resulted in increased complement deposition, suggesting that pharmacological agents aimed at reducing Psl levels may enhance recognition and clearance of P. aeruginosa by innate immune effectors.
Results
Both serum opsonins and Psl polysaccharide affect the oxidative burst response generated by human neutrophils
To determine if Psl affects the oxidative burst response generated by human neutrophils, we used three isogenic P. aeruginosa strains previously developed (Ma et al., 2006) including WFPA801, an inducible Psl overexpressing variant, the psl-deficient strain WFPA800, and WT strain PAO1. Psl expression by these strains was verified with an immunoblot of surface extracted polysaccharide (Fig. 1A). To measure the oxidative burst response generated by human neutrophils towards these P. aeruginosa strains, we incubated fresh serum-opsonized bacteria with human neutrophils in the presence of luminol and monitored the oxidative burst over time (Fig. 1B). Luminol is known to react with both extra- and intracellular superoxide anions generated by neutrophils (Briheim et al., 1984). Kinetic analyses of neutrophils exposed to serum-opsonized psl mutants showed a more rapid and robust oxidative burst response compared with neutrophils incubated with equivalent numbers of WT or Psl overexpressing strains (Fig. 1B). The highest oxidative burst response was observed at 25 min. Therefore, this time point was chosen for comparing the response of neutrophils exposed to bacteria under serum-opsonized (NHS), unopsonized and heat-inactivated serum (HIS) opsonized conditions (Fig. 1C). The oxidative burst response of neutrophils exposed to serum-opsonized WT, psl mutant and a Psl overexpressing P. aeruginosa strain was significantly higher compared with non-opsonized strains (Fig. 1C). Furthermore, there was a significant reduction but not elimination of the oxidative burst response generated by neutrophils incubated with P. aeruginosa opsonized with HIS, which is devoid of complement activity (Fig. 1C). These studies were also performed utilizing the fluorescence probe dichlorohydro-fluorescein (DCF) to detect ROS and similar results were observed (data not shown). This suggests that serum opsonins (both complement and immunoglobulin) and the Psl status significantly affect the activity of neutrophils, which are critical in the innate immune responses towards P. aeruginosa. The remainder of this study focused only on the contribution of complement as the opsonin.
Fig. 1.
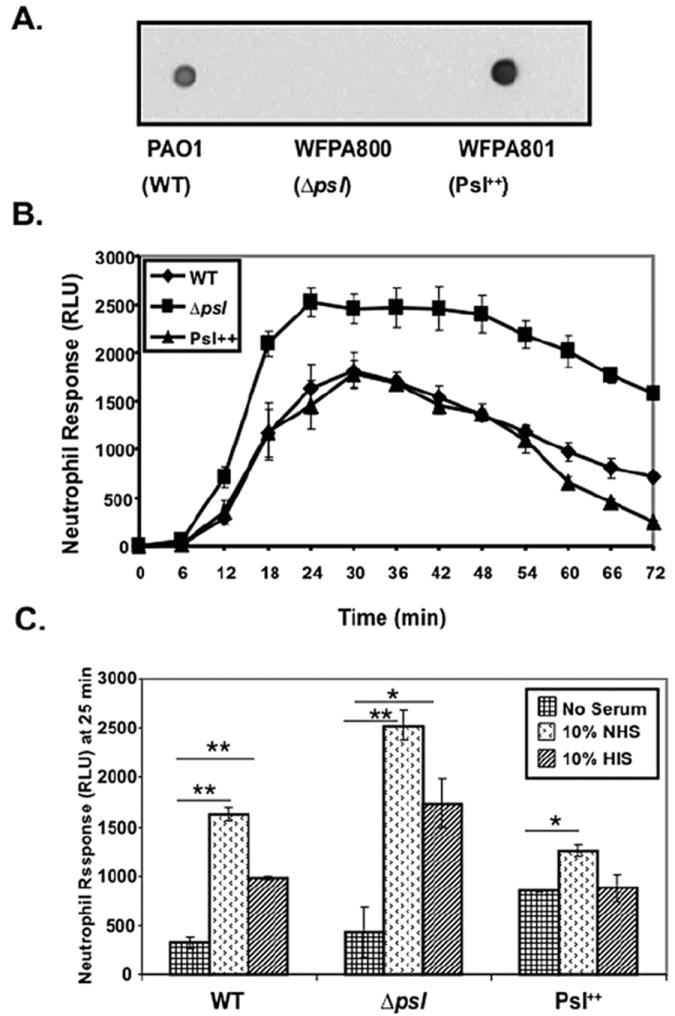
Serum opsonization of a Psl-deficient mutant of P. aeruginosa increases the oxidative burst by human neutrophils. A. Immunoblot showing expression of Psl from EPS extracts derived from strains PAO1, WFPA800 and WFPA801 (Byrd et al., 2009). These extracts were blotted on a nitrocellulose membrane, probed with anti-Psl antiserum and detected by chemiluminescence using a HRP-conjugated secondary antibody. B. Kinetic analysis of the chemiluminescence response (measurement of oxidative burst) of neutrophils generated against pre-opsonized Psl variant strains of P. aeruginosa. Each experiment was performed in quadruplet wells (n = 3 with 10% fresh normal human serum opsonization). RLU, relative luminescence unit. C. Oxidative burst response of neutrophils induced by Psl variant strains of P. aeruginosa under fresh serum opsonized (NHS), heat-inactivated serum (HIS)-opsonized and non-opsonized conditions. Bacteria were incubated with serum or no serum, washed and then incubated with neutrophils (moi 50:1) at 37°C for 25 min in presence of luminol and the chemiluminescence response of neutrophils was measured. Each experiment was performed in quadruplet wells (n = 5 for 10% NHS opsonization, HIS opsonization and non-opsonization). Values are mean ± SEM for five experiments. A statistical analysis was carried out using an unpaired two-tailed student’s t-test to compare the chemiluminescence response of neutrophils induced by HIS, opsonized and unopsonized bacteria. The asterisks indicate P-values (*P ≤ 0.05; **P ≤ 0.005). RLU, relative luminescence unit.
We previously showed that some Psl is released from cells during growth of P. aeruginosa (Byrd et al., 2009). One potential explanation for the results in Fig. 1 is that released Psl may be directly inhibiting the oxidative burst of neutrophils. This is the case for EPS produced by Burkholderia cenocepacia (Bylund et al., 2006). However, there was no significant difference in the oxidative burst response of neutrophils stimulated with phorbol myristate acetate (PMA) either in the presence or absence of purified Psl (Fig. S1). This suggests that Psl is not sufficient to inhibit neutrophil activation.
The presence of Psl reduces opsonophagocytosis of P. aeruginosa by human innate immune cells
As outlined in Fig. 1, the presence of Psl limits the oxidative burst response by neutrophils. We reasoned that these differential burst responses might be due to differences in the phagocytosis of P. aeruginosa strains. To evaluate this, serum opsonized P. aeruginosa WT and psl-deficient strains were incubated with human neutrophils and THP-1 derived macrophages. THP-1 macrophages have been frequently used to study bacterial internalization (Longhi et al., 2004; Ferrer et al., 2010). Unattached bacteria were removed by washing extensively and bacterial association and internalization assessed by differential immunofluorescence staining (see Experimental procedures). The amount of psl-defective bacteria taken up by THP-1 macrophages was 4.5-fold higher compared with that of the WT strain (Fig. 2A). In human neutrophils, there was an approximately threefold higher number of psl-deficient P. aeruginosa internalized when compared with WT bacteria (Fig. 2B). Thus, the increased phagocytosis of the psl-deficient strain correlates well with the increased oxidative burst observed when neutrophils are exposed to the same mutant strain.
Fig. 2.
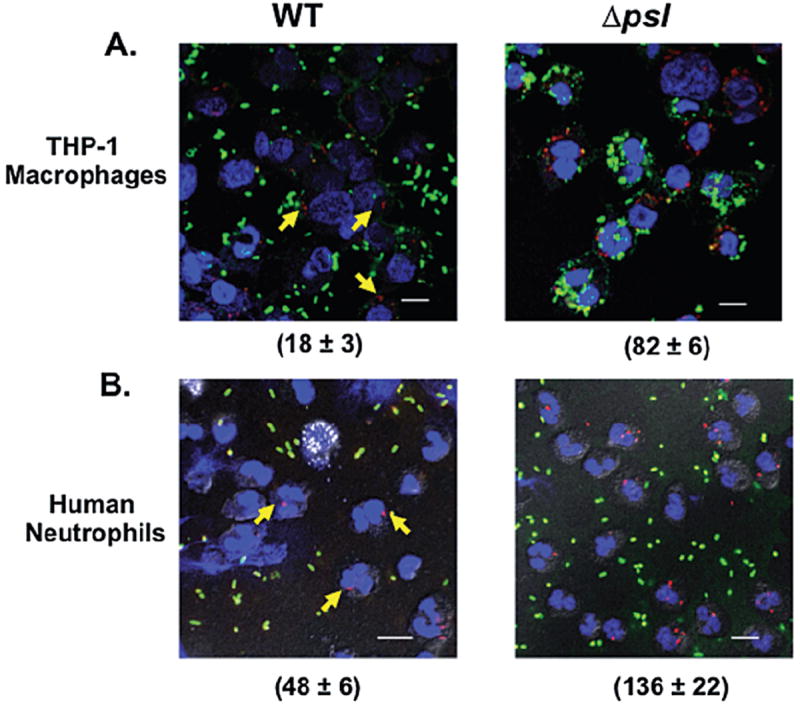
The presence of Psl reduces the uptake of P. aeruginosa by human phagocytic cells. Immunofluorescence images of (A) THP-1 cells and (B) human neutrophils, infected with pre-opsonized WT or a psl-deficient strain of P. aeruginosa. Bacteria were incubated with neutrophils at an moi of 5:1 and processed as described in Experimental procedures. Attached bacteria exposed to the antibodies before permeabilization stained green. Those exposed to the antibodies only after permeabilization stained red (internalized bacteria). Blue (DAPI) fluorescence was used to stain the nuclei of THP-1 cells and neutrophils (60× oil objective). Arrows indicate the internalized bacteria in the PAO1 samples. Scale bar is equal to 10 μm. The number (mean ± SD) of internalized bacteria is indicated in parentheses and is representative of 100 infected cells (neutrophils and macrophages) examined from triplicate coverslips.
The presence of Psl inhibits deposition of complement component C3, C5 and C7 on the P. aeruginosa surface
The activation of complement can cause direct lysis of bacteria or increase opsonophagocytosis (Pangburn and Muller-Eberhard, 1980; van Lookeren Campagne et al., 2007; Clay et al., 2008). Data in Fig. 1C show that opsonization of P. aeruginosa with heat-inactivated human serum reduced the oxidative burst response relative to fresh human serum, indicating a significant contribution of complement in the recognition of P. aeruginosa bacteria by neutrophils. We subsequently evaluated the effect of Psl on deposition of complement component C3, which promotes opsonization and is central to all three complement cascade pathways (Pangburn and Muller-Eberhard, 1980). P. aeruginosa strains were opsonized and C3 deposition was evaluated by flow cytometry. There was a significant increase in C3 deposition on psl-deficient P. aeruginosa compared with WT and Psl overexpressing strains (Fig. 3A). Following a 5 min opsonization period, C3 complement deposition on psl-deficient P. aeruginosa was increased 2.1 ± 0.34- and 4.1 ± 0.28-fold, compared with WT and Psl overexpressing bacteria respectively. C3 deposition on psl-deficient P. aeruginosa was also significantly higher compared with WT and Psl overexpressing P. aeruginosa after 20 min of opsonization (Fig. 3A).
Fig. 3.
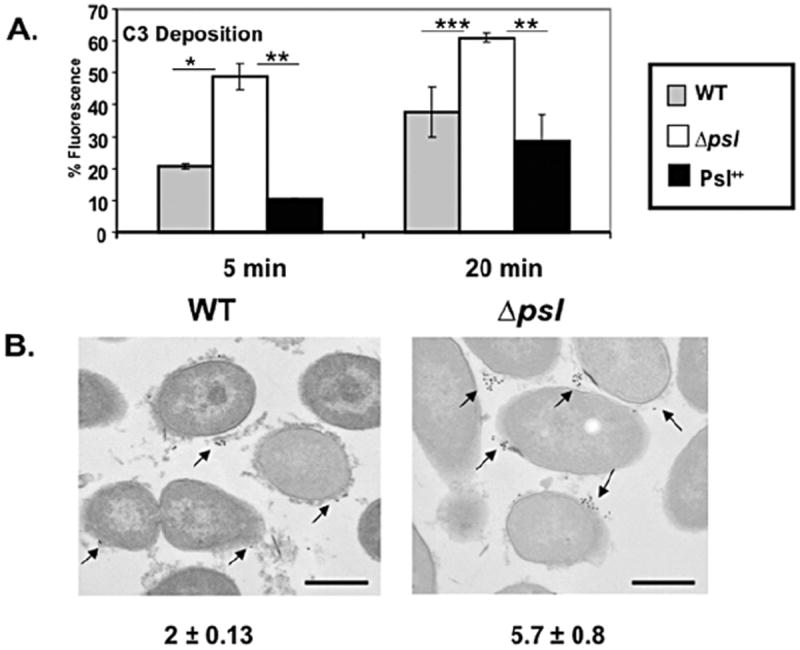
Increased C3 complement component deposition on psl-deficient P. aeruginosa. A total of 2 × 108 mid-log phase bacteria were opsonized with 20% fresh human serum at 37°C for 5 or 20 min, followed by the addition of 10 mM EDTA to stop the reaction. Bacteria were fixed and stained with anti-human C3 antibody (A) counter stained with fluorescent antibody and analysed by flow cytometry (B) counter stained with 15-nm colloidal gold rabbit anti-goat antibody and analysed by transmission electron microscopy (TEM), as outlined in Experimental procedures. Bar represents 500 nm. Arrows indicate the C3 complement deposition on the bacterial surface. The number in parentheses (mean ± SD) indicated deposition of C3 complement component on individual bacteria (averaged from 100 bacterial cells evaluated). Statistical analysis was carried out using the unpaired two-tailed Student’s t-test to compare the deposition of C3 complement component on the surface of the psl-deficient strain with WT and the Psl++ strains of P. aeruginosa. Mean ± SEM are given (n = 3). The asterisks indicate P-values (*P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.0005).
We confirmed enhanced C3 deposition on serum opsonized psl-deficient P. aeruginosa compared with WT strains using immunogold transmission electron microscopy (TEM). These data also revealed C3 deposition on the psl-deficient P. aeruginosa surface was elevated threefold compared with C3 binding to WT bacteria (Fig. 3B).
While one role of complement is to facilitate opsonophagocytosis, complement components also can directly kill bacteria through generation of the membrane attack complex (MAC), which requires complement components C5-C9. To examine the effect of Psl on complement components associated with the terminal lytic pathway, C5 and C7 deposition was evaluated. When opsonized for 20 min, C5 and C7 deposition was significantly higher on psl-deficient P. aeruginosa compared with WT and Psl over-expressing strains (Fig. 4A and B respectively). Increased C5 deposition occurred as early as 5 min, similar to the results with C3 and thus highlighting the rapid kinetics of complement activation on the psl-deficient strain. C7 deposition was somewhat delayed (Fig. 4B). Since complement deposition occurs sequentially, the observed increase in C5 and C7 binding on Psl-deficient P. aeruginosa (Fig. 4A and B) is likely due to the fact that C3 deposition was elevated on these bacteria.
Fig. 4.
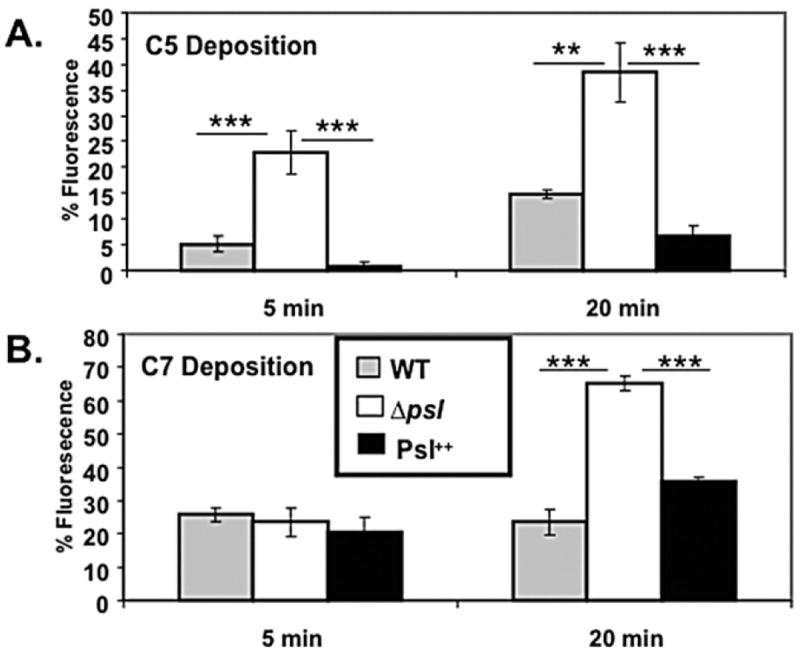
Psl limits C5 and C7 complement component deposition on P. aeruginosa. A total of 2 × 108 mid-log phase bacteria were opsonized with 20% fresh human serum at 37°C for 5 and 20 min, followed by the addition of 10 mM EDTA to stop the reaction. Bacteria were fixed and stained with (A) anti-human C5 antibody and (B) anti-human C7 antibody as outlined in Experimental procedures. Mean ± SEM are given (n = 3). Statistical analysis was carried out using the unpaired two-tailed Student’s t-test to compare the deposition of C3 complement component on the surface of the psl-deficient strain with WT and the Psl++ strains of P. aeruginosa. The asterisks indicate P-values (**P ≤ 0.005; ***P ≤ 0.0005).
We next examined the binding of C9 to P. aeruginosa strains. Although C5b, C6, C7, C8 and C9 are necessary for forming the MAC complex, the multimeric form of C9 is involved in the actual disruption of the phospholipid membrane (Inoue and Yonemasu, 1968; Goldman et al., 1969; Schreiber et al., 1979; Joiner et al., 1985). In contrast to C3, C5 and C7 deposition, we found little difference in the amount of C9 bound to P. aeruginosa WT, psl-deficient, or Psl overexpressing strains (Fig. 5A), suggesting that differences observed in upstream complement components do not translate into increased C9 deposition on psl-deficient cells.
Fig. 5.
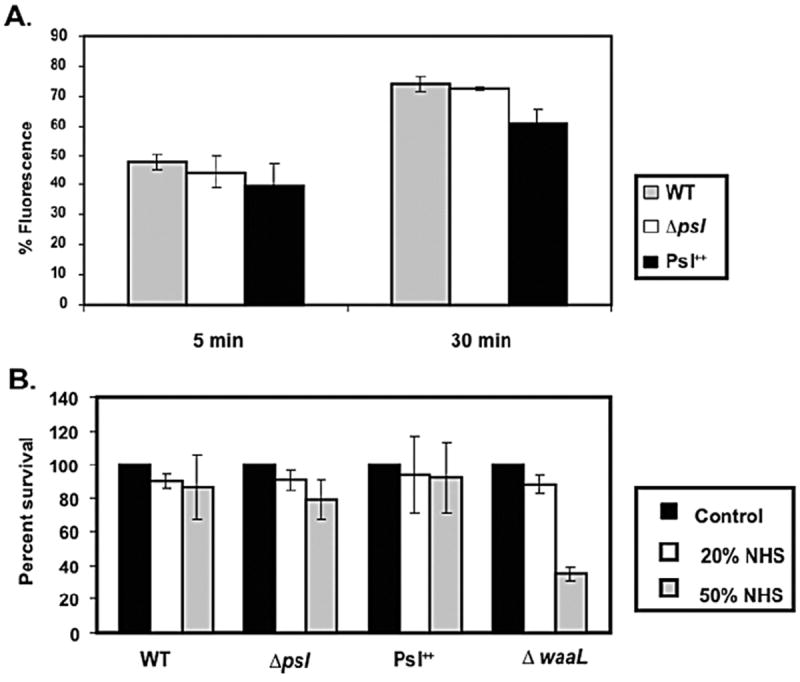
Psl polysaccharide has no effect on C9 deposition and complement-mediated killing.
A. C9 complement component deposition was measured on WT, psl-deficient and Psl overexpressing strains of P. aeruginosa. Results are expressed as percentage of fluorescence positive cells as in Fig. 4, which is indicative of complement deposition on the bacterial surface. The experiment shown is representative of three independent experiments performed in triplicate. Data are mean ± SEM.
B. Sensitivity of WT, psl-deficient and Psl overexpressing strains of P. aeruginosa to complement-mediated killing in the presence of 0, 20 and 50 percent fresh normal human serum (NHS). Bacterial survival is expressed as the percentage of bacteria recovered from the initial inoculation amount (incubated with buffer only without serum), which is set at 100%. Data represent mean ± SEM from a representative experiment (n = 3).
C9 assembly on the surface of bacteria is essential for MAC formation and serum-mediated killing (Joiner et al., 1985). To test whether the presence of Psl affected serum-mediated killing, a bacterial survival assay was performed in the presence of fresh human serum. We also included an LPS O-antigen-deficient P. aeruginosa strain (ΔwaaL) as a control since an intact O-antigen is necessary for serum resistance (Hancock et al., 1983). Addition of normal human serum at two concentrations (20% and 50%) failed to kill WT or any of the Psl variant strains (Fig. 5B). As expected, P. aeruginosa ΔwaaL was more serum sensitive than the parental strain. These results indicate little role for Psl in promoting serum resistance.
Disruption of Psl by addition of exogenous cellulase increases C3 complement binding on the bacterial surface
Since psl mutants are more efficiently opsonized and taken up by phagocytes than are WT P. aeruginosa, agents aimed at removal of Psl may promote clearance of these bacteria during infection. As a proof of principle we utilized cellulase, which disrupts β1–3 and β1–4 linkages in Psl (Ma et al., 2009), and examined the effect on C3 binding. Bacteria were left untreated or treated with cellulase, extensively washed, opsonized and C3 binding examined (see Experimental procedures). When opsonized for 5 min, cellulase-treated WT bacteria exhibited a 10-fold increase in C3 binding on the surface compared with untreated cells. WT P. aeruginosa treated with phosphate-buffered saline (PBS) or PBS + cellulase showed 6.31 ± 2.58% and 66.78 ± 0.66% C3 complement deposition respectively. This effect was reduced but still significant with longer opsonization times, indicating that either cellulase did not remove all Psl or that C3 binding at later time points becomes saturated. Importantly, treatment of the psl-deficient strain with cellulase did not further increase C3 binding (Δpsl with PBS or PBS + cellulose showed 58.49 ± 12% and 69.57 ± 2.7% C3 deposition respectively). This suggests that Psl is the primary cell surface molecule targeted by cellulase.
Psl promotes the intracellular survival of P. aeruginosa
To study the role of Psl in protection of bacteria from neutrophil killing, we incubated normal serum opsonized WT and psl-deficient strains of P. aeruginosa with human neutrophils and performed a gentamicin survival assay (see Experimental procedures). Compared with the WT and Psl overexpressing strains, there was an eightfold reduction in intracellular survival of psl mutants (Fig. 6A). We reasoned that this reduced survival of psl mutants in neutrophils was likely due to their increased opsonophagocytosis, and more intense oxidative burst compared with that generated in response to WT bacteria. To address any potential additional role for Psl in protection from intracellular killing by neutrophils, we normalized the bacterial uptake in neutrophils by adjusting the multiplicity of infection (moi) of WT and psl-deficient P. aeruginosa and repeated the intracellular killing assay as above. Under these conditions, equivalent numbers of WT and psl mutant bacteria were taken up by neutrophils (Fig. 6B). Our result revealed that WT P. aeruginosa bacteria had a sixfold increase in survival compared with the psl-deficient strain (Fig. 6A). These data indicate an additional role for Psl in providing protection of bacteria from intracellular neutrophil-mediated killing.
Fig. 6.
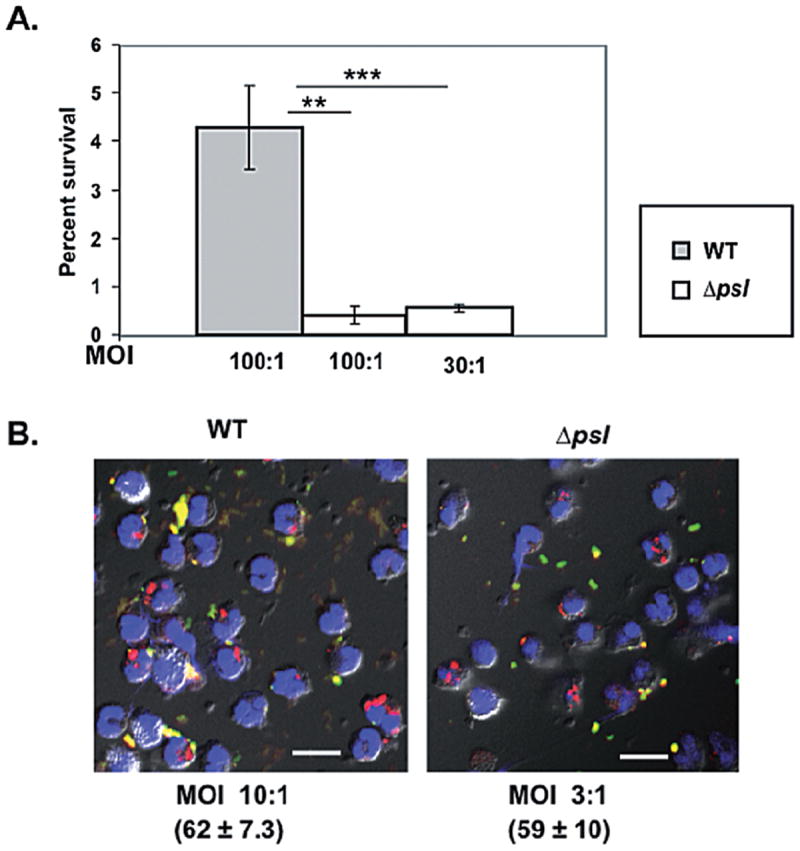
Psl enhances intracellular survival of P. aeruginosa in human neutrophils.
A. Opsonized WT PAO1 and psl-deficient P. aeruginosa were incubated with 2 × 105 human neutrophils at the indicated moi for 30 min. Extracellular and attached bacteria were killed by treatment with gentamicin, then neutrophils were lysed with 0.1% Triton X-100 and lysates plated to enumerate viable bacteria. Results are expressed as the percentage of bacteria that survived relative to total cell-associated bacteria and are representative of three independent experiments performed in triplicate. Bars represent mean ± SEM. Statistical analysis was carried out using the unpaired two-tailed Student’st-test. The asterisks indicate P-values (**P ≤ 0.005; ***P ≤ 0.0005).
B. Immunofluorescence images (60× oil objective) showing equal uptake of bacteria (stained red) by neutrophils when the moi of WT and psl-deficient bacteria were adjusted. The moi and number of internalized bacteria (mean ± SD) per 100 neutrophils are indicated. Colours indicate: green, attached bacteria; red, internalized bacteria; blue, neutrophil nucleus. Scale bar represent 10 μm.
The presence of Psl provides a fitness advantage for P. aeruginosa during an acute murine pulmonary infection
Since Psl appears to limit ospsonophagocytosis and promotes intracellular survival in neutrophils, we designed a study to determine if WT P. aeruginosa had an in vivo fitness advantage compared with a psl-deficient strain. Mice were co-infected intranasally with a 1:1 mixture of isogenic WT and psl-deficient strains of P. aeruginosa. These bacteria were distinguished based on differential antibiotic susceptibility (see Experimental procedures). At 4 and 12 h post infection mice were sacrificed and the bacterial burden was enumerated from the lung homogenate. The mean competitive index (CI) value was 1.2 at 4 h post infection, indicating similar delivery of both WT and psl mutants. However, this declined to 0.3 at 12 h post infection, indicating that psl-deficient bacteria had a significantly reduced fitness advantage in the lungs of mice when co-infected with WT strains. An in vitro growth competition experiment with co-culturing PAO1 Smr and psl-deficient bacteria did not show a growth advantage for either strain with a CI value of 1.0. These results support the conclusion that Psl contributes to protection of P. aeruginosa from innate immune cells.
Discussion
Psl polysaccharide plays a crucial role in initial attachment and biofilm formation by non-mucoid P. aeruginosa strains (Friedman and Kolter, 2004; Jackson et al., 2004; Matsukawa and Greenberg, 2004). Non-mucoid, Psl-expressing bacteria are believed to be the first to colonize the CF airway and are challenged with a harsh inflammatory- and neutrophil-rich lung environment. There have been no studies addressing the potential role of EPS expressed by non-mucoid P. aeruginosa in protecting bacteria from the phagocytic activity of neutrophils or other effectors of innate immunity. In this study we show that (i) serum opsonins (both complement and immunoglobulin) and the Psl status are critical components in dictating the host innate immune response towards non-mucoid P. aeruginosa; (ii) the presence of Psl reduces opsonophagocytosis of P. aeruginosa by human innate immune cells; (iii) Psl inhibits deposition of complement component C3, C5 and C7 on the P. aeruginosa surface; (iv) disruption of Psl by addition of exogenous cellulase increases C3 complement binding on the bacterial surface; (v) Psl promotes the intracellular survival of P. aeruginosa; and (vi) Psl-proficient bacteria have a competitive advantage over Psl-deficient strains in vivo.
Phagocytosis by innate immune cells is a multi-step process, which involves microbe recognition and ingestion by neutrophils and subsequent degradation by generating ROS and other digestive enzymes. The sequential production of ROS due to bacterial and neutrophil interaction is collectively known as an oxidative burst (Lorenzen et al., 2000; Roos et al., 2003; Nauseef, 2007). Bacterial polysaccharides produced by B. cenocepacia and Staphylococcus aureus also prevent phagocytosis and killing by immune cells (Karakawa et al., 1988; Thakker et al., 1998; Kampen et al., 2005; Saldias et al., 2009). Our data clearly demonstrate that Psl reduces bacterial uptake and the oxidative burst response generated by neutrophils when challenged with opsonized P. aeruginosa. We also observed an enhanced oxidative burst response to non-opsonized Psl overexpressing strains compared with that of non-opsonized WT P. aeruginosa (Fig. 1C). This might be due to an interaction of Psl with non-opsonic receptors on neutrophils. Mannose receptors of monocyte-derived macrophages facilitate phagocytosis of non-opsonized P. aeruginosa (Speert et al., 1988), and Psl is mannose-rich (Byrd et al., 2009). Heale et al. also suggested independent involvement of CR3 or CD14 in non-opsonic phagocytosis of P. aeruginosa strains (Heale et al., 2001). Importantly, purified Psl exhibited no effect on PMA induced neutrophil burst responses in vitro, indicating that Psl does not likely directly inhibit neutrophil function.
Gondwe et al. showed the importance of opsonins for induction of the oxidative burst response of neutrophils and monocytes by invasive non-typhoidal Salmonella (Gondwe et al., 2010). Similarly, P. aeruginosa opsonization clearly affects the response of neutrophils (Fig. 1C). The results in Figs 3 and 4 also provide an explanation for the data in Fig. 1, since elevated opsonization of the psl mutants is expected to enhance opsonophagocytosis with a resultant increase in the oxidative burst response. Opsonization of bacteria requires deposition of activated C3b on the surface. C3b forms stable thioester bonds, leading to the covalent attachment of C3b with free hydroxyl or amine groups present on the surface of bacteria (Abdul Ajees et al., 2006). We currently favour the hypothesis that Psl functions by masking the accessibility of a ligand(s) present on the bacterial surface for interaction with C3. The identity of the P. aeruginosa surface ligand(s) to which C3 binds is not known.
Our observation that WT and psl-deficient P. aeruginosa show differential C3 deposition when incubated with human serum is similar to the findings of Clay et al. (2008). Their data provided evidence that the LPS O-antigen regulates complement C3 deposition on the surface of Francisella tularensis and is a major determinant of complement activation. A role of O-antigen in preventing C3 deposition on Bordetella parapertussis has also been established (Goebel et al., 2008). Given the importance of C3 in host defence (Clay et al., 2008; Barker et al., 2009), the inhibitory role of Psl for C3 deposition on the bacterial surface may provide additional protection of the pathogen in the environment of the CF lung and could help promote a chronic infection.
In contrast to other polysaccharides, i.e. Escherichia coli K2 capsule and the O-antigen of F. tularensis, which contribute to serum resistance (Clay et al., 2008; Buckles et al., 2009), Psl EPS does not appear to protect P. aeruginosa against complement-mediated killing. This could be due to several reasons. Bacterial killing by complement is associated with partial or complete dissolution of the outer membrane, and the molecular form of the C5-9 complex is crucial for optimal killing (Feingold et al., 1968a,b). Joiner et al. confirmed that multimeric C9 is necessary for E. coli J5 killing and that sufficient polymerization of C9 rendered disruption of the multilamellar detergent resistant bacterial cell wall (Joiner et al., 1985). An average of at least three C9 molecules is required for one C5-8 complex to kill susceptible cells (Joiner et al., 1985; Bloch et al., 1987). Trypanosoma cruzi amastigotes escape complement mediated killing because C9, while bound efficiently, is not inserted deep enough in the parasite lipid bilayer and fails to form pores in the membrane (Iida et al., 1989). Therefore, the correct stoichiometric ratio of MAC complement components may be suboptimal for lysis of both WT and Psl-defective P. aeruginosa. Alternatively, one or more molecules on the bacterial surface might be inhibiting the polymerization of C9 resulting in protection from serum killing. Multiple factors likely contribute to preventing the effect of MAC-mediated killing of P. aeruginosa. While our results do not support a role for Psl in protection against complement-mediated killing, it is intriguing that there were significant differences in deposition of C3, C5 and C7 complement components on the surface of WT versus psl mutants. The observation that there were no apparent differences in C9 binding among the strains could be explained by the fact that multiple copies of C9 are normally found in the MAC (Kolb et al., 1972; Kondos et al., 2010), and the sensitivity of our binding assays may not be sufficient to resolve such differences.
Since our data suggested a protective role for Psl during interactions with neutrophils in vitro, we performed an in vivo acute murine pulmonary infection experiment. These results also revealed that Psl provides a fitness advantage to P. aeruginosa in vivo (Fig. 7). To our knowledge, this is a first study that addresses the impact of Psl during interaction with cells of the innate immune system in vivo and in vitro.
Fig. 7.
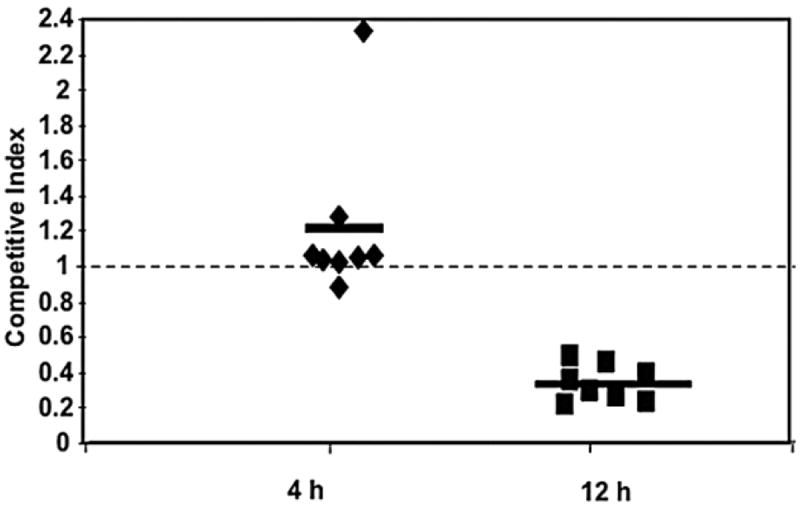
The presence of Psl provides a fitness advantage for P. aeruginosa during an acute murine pulmonary infection. Six-week-old female C57BL/6 mice were co-infected at a 1:1 ratio (total of 108 bacteria) with psl mutant and WT streptomycin resistant P. aeruginosa strains. Bacteria were harvested from the lungs at 4 and 12 h post infection. Viable cell counts and the competitive index (CI) of the two strains were determined as outlined in the Experimental procedures. Dashed line represents a CI of 1.0 (no advantage for WT or psl mutants).
In conclusion, our results demonstrate that deletion of Psl leads to increased complement deposition on the bacterial surface and increased opsonophagocytosis, oxidative burst, and bacterial killing by neutrophils. Complement components play an important role in the recognition and clearance of bacteria by facilitating phagocytosis by innate immune cells. Using a murine model of pneumonia, Stacey et al. showed that C3 deficient mice had more robust P. aeruginosa colonization compared with wild-type mice (Mueller-Ortiz et al., 2004). The reduction of CI at 12 h post infection in our in vivo model could suggest that Psl is important for adaptation to host conditions that exist later in infection. This may be one mechanism by which non-mucoid P. aeruginosa persists during lung infection in CF patients even in the presence of a robust neutrophil response (Fig. 7). The psl operon appears to be highly conserved in clinical and environmental isolates of P. aeruginosa (Wolfgang et al., 2003; Jackson et al., 2004). Given the importance of Psl in colonization, biofilm formation and avoidance of host defence mechanisms, Psl may represent a therapeutic target for prevention of persistent P. aeruginosa infection in CF patients.
Experimental procedures
Strains and growth media
Wild-type P. aeruginosa (PAO1), Psl-deficient P. aeruginosa WFPA800 (Δpsl), the arabinose-inducible Psl overexpressing strain WFPA801 (Ma et al., 2006), a streptomycin resistant variant of PAO1 (Jackson et al., 2004), and the O-antigen deficient strain of P. aeruginosa (ΔwaaL) (Abeyrathne et al., 2005), were grown in Luria–Bertani (LB) without NaCl (LBNS). Arabinose was added to WFPA801 for induction of Psl (2% w/v final concentration) and also included in the WT (PAO1) and Psl-deficient (WFPA800) culture as a control. No antibiotic selection was used for routine growth of these strains.
Neutrophil isolation and oxidative burst studies
Human neutrophils and serum were obtained from healthy adult donors using an approved IRB protocol (2009H0314) at The Ohio State University. Heparinized blood was placed on a Ficoll Hypaque density cushion (Amersham Biosciences), centrifuged at 400 g and the resultant RBC-rich pellet was resuspended 1:1 in normal saline. To obtain a leucocyte-rich supernatant, RBCs were allowed to sediment in the presence of 1.5% dextran at 4°C for 30 min. The supernatant was gently removed and centrifuged at 550 g for 10 min at 4°C. Pelleted RBCs were subjected to hypotonic lysis with 10 ml of distilled water and the isotonicity was restored by the addition of an equal amount of 1.8% NaCl. Neutrophils were resuspended in Hank’s buffered salt solution (HBSS), counted on a haemocytometer, and kept on ice for use.
The luminol chemiluminescence assay was used to detect intracellular and extracellular ROS generated by neutrophils upon interaction with P. aeruginosa (Engels et al., 1985). WT P. aeruginosa, Δpsl and Psl-overexpressing bacterial suspensions were opsonized with 10% fresh human serum at 37°C for 30 min, washed and compared for generation of the ROS response by neutrophils. PMA (10 ng) was used as a positive control for generation of the ROS by neutrophils. Neutrophils (2 × 105) were seeded on a 96-well microtitre plate and a bacterial suspension of mid-log phase culture was added (moi of 50 bacteria to 1 neutrophil) to the wells in the presence of 50 μM of luminol. The microtitre plate was centrifuged at 100 g for 2 min at 12°C to synchronize the infection. The relative amount of ROS generated by neutrophils was detected at regular intervals over 72 min by measuring the luminescence. Relative light units (RLU) were plotted as a function of time to evaluate chemiluminescence (CL) rate (Mohapatra et al., 2010). The DCF assay for detecting the respiratory burst was performed as outlined elsewhere (Crowther et al., 2004). Psl was purified according to Byrd et al. (2009) and 500 μg or 1 mg was used to study the oxidative burst response by PMA-activated neutrophils.
Complement component deposition and bacterial killing
Methods by Balagopal et al. (2006) were used to harvest fresh complement sufficient serum. Mid-log phase grown bacteria were opsonized with 20% fresh human serum at 37°C for 5 and 20 min, and reactions stopped by the addition of 10 mM EDTA and incubation on ice. Bacteria were washed, fixed and stained using anti-human C3 (Quidel), C5, C7 or C9 polyclonal antibodies (Complement Tech.) at a 1:1000 dilution. Following incubation, samples were washed and detected using Alexa Fluor 647-conjugated secondary antibody (Invitrogen) at a 1:500 dilution. After final washes, the pelleted bacteria were analysed by flow cytometry (FACS Calibur; BD Biosciences) for quantification of complement deposition on the bacterial surface. Non-opsonized bacteria were used in each experiment to set the gate for analysis. Data from 30 000-gated events were collected and the percentage of fluorescence (percentage positive cells) was calculated for each sample. For the bacterial killing assays, mid-log phase WT, Δpsl, Psl-overexpressing and ΔwaaL strains of P. aeruginosa were opsonized with PBS or 20% and 50% pooled normal human serum (Complement Tech.) for 30 min at 37°C with slow agitation. The tubes were placed on ice for 5 min to stop the reaction, bacteria were washed extensively, and 10-fold serial dilutions were plated to determine surviving colony-forming units (cfu).
Electron microscopy
Mid-log phase grown bacteria were opsonized with 20% pooled human serum (Complement Tech.) at 37°C for 20 min. Reactions were stopped by adding 10 mM EDTA, bacteria were washed, fixed, blocked and stained with anti-human C3 antibody (1:500 dilution) for 1 h. Following incubation, samples were washed and counter stained with 15 nm colloidal gold labelled rabbit anti-goat antibody (Abcam) at a dilution of 1:500 for 1 h. Labelled samples were washed, fixed and embedded in Eponate 12 resin, followed by sectioned on a Leica EMU 6 ultramicrotome. Sections were collected onto grids and viewed by transmission electron microscopy (FEI Tecnai G2 Spirit) operating at 80 kV. Images in Fig. 3B are representative of cells in two independent experiments. For quantification of C3 binding, 100 random bacterial cells were evaluated in two independent experiments.
Bacterial uptake by neutrophils
Neutrophils (2 × 105 per well) were seeded on poly-l-lysine coated coverslips and mid-log phase P. aeruginosa, opsonized with 20% human serum, were added at an moi (bacteria : neutrophil) of 10:1, 5:1 and 3:1 onto the neutrophil monolayers in the appropriate experiment. The mixture was incubated for 30 min at 37°C, 5% CO2. Cells were washed extensively with HBSS to remove non-adherent bacteria, fixed and stained with anti-Pseudomonas antibody (diluted to 1:1000) before and after cell membrane permeabilization with methanol (Balagopal et al., 2006). The anti-Pseudomonas antibody was generated against whole surface antigens of strain PAO1. Visualization of bacteria–phagocyte association by confocal microscopy (Olympus FV 1000) using 60× oil objective allowed us to differentiate attached P. aeruginosa from internalized bacteria. Attached bacteria exposed to the antibodies before permeabilization stain green due to counter staining with Alexa Fluor 488 (diluted to 1:500), while internalized bacteria exposed to the antibodies after permeabilization stain red due to Alexa Fluor 647 as a secondary stain (diluted to 1:500). For quantification, 100 neutrophils were chosen at random and the cell-associated versus internalized bacteria counted and averaged by two readers blinded to the treatment conditions. Bacterial uptake was also studied in THP-1-derived macrophages by growing THP-1 cells in RPMI supplemented with 10% FBS. Cultured THP-1 cells were stimulated with 100 nM PMA for 36 h prior to the addition of bacterial suspension. The resultant THP-1 derived macrophages were incubated with bacteria for 90 min and bacterial attachment and internalization were analysed as above.
P. aeruginosa-neutrophil killing assay
Neutrophils (2 × 105) were seeded onto poly-l-lysine coated 24-well plates in HBSS, and mid-log phase cultures of bacteria were added at an moi of 100:1. To normalize the uptake of bacteria by neutrophil, this experiment was also performed with reduced moi of psl-deficient P. aeruginosa (30:1). After a 30 min incubation, neutrophils were washed vigorously and treated with 150 μg ml−1 gentamicin for 30 min at 37°C in 5% CO2 to kill extracellular and attached bacteria. Samples were washed and lysed with 0.1% Triton X-100 for 2 min immediately prior to plating for cfu. The percentage of killed bacteria was calculated relative to the total number of cell-associated bacteria. To estimate the total number of associated bacteria, wells were not treated with gentamicin.
Cellulase treatment
Mid-log phase bacteria were incubated with 10 mg ml−1 cellulase (1.38 unit per mg, Sigma) in PBS for 20 h at 37°C. After extensive washing with PBS bacteria were opsonized with 20% normal human pooled serum and C3 deposition on the bacterial surface was evaluated at both 5 min and 30 min time points as above.
In vivo mixed infection competition
All experimental animals were handled in accordance with OSU institutional guidelines (IACUC approval number 2009A0177). P. aeruginosa strains grown to OD600 = 0.5 at 37°C, harvested by centrifugation, washed with PBS, and diluted to an estimated concentration of 1 × 108 bacteria in 30 μl. Groups of 5 female C57BL/6 mice were mildly sedated by isoflurane inhalation and infected intranasally with ~ 1 × 108 total bacteria of a 1:1 mixture of streptomycin resistant P. aeruginosa PAO1 and its isogenic psl-deficient strain. A parallel control group received the same inoculum of PBS. At 4 and 12 h post infection, five mice from each group were euthanized and lung homogenates plated in the presence and absence of streptomycin for differential enumeration of bacterial strains. The CI was calculated by dividing the ratio of the amount of psl-deficient bacteria recovered by the amount of PAO1 Smr recovered (CI values < 1 indicate a competitive advantage of the WT strain). An in vitro growth competition experiment with co-culturing PAO1 Smr and WFPA800 did not show a growth advantage for either strain or a CI value of 1.0.
Supplementary Material
Fig. S1. Purified Psl has no effect on the oxidative burst response generated by human neutrophils. The oxidative burst response of PMA induced neutrophils when incubated with Psl (0.5 or 1.0 mg) prepared from P. aeruginosa strain WFPA801. Each experiment was performed in quadruplet wells (n = 3). RLU, relative luminescence unit.
Acknowledgments
This work was supported by Public Health Service grants AI061396 and HL058334 (D.J.W.), a WFUHS intramural grant to L.C.M., CFF fellowship MISHRA11F0 (M.M.), and NRSA fellowship AI07870002 (M.S.B.). We would like to acknowledge Richard Montione from the Campus Microscopy and Imaging Facility of the Ohio State University for excellent technical help with TEM. We thank Preston Hill for the preparation of Psl, and Dominique Limoli, Christopher Jones and Kurtis Host for providing assistance with the animal experiments.
Footnotes
Supporting information
Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abdul Ajees A, Gunasekaran K, Volanakis JE, Narayana SV, Kotwal GJ, Murthy HM. The structure of complement C3b provides insights into complement activation and regulation. Nature. 2006;444:221–225. doi: 10.1038/nature05258. [DOI] [PubMed] [Google Scholar]
- Abeyrathne PD, Daniels C, Poon KK, Matewish MJ, Lam JS. Functional characterization of WaaL, a ligase associated with linking O-antigen polysaccharide to the core of Pseudomonas aeruginosa lipopolysaccharide. J Bacteriol. 2005;187:3002–3012. doi: 10.1128/JB.187.9.3002-3012.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopal A, MacFarlane AS, Mohapatra N, Soni S, Gunn JS, Schlesinger LS. Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect Immun. 2006;74:5114–5125. doi: 10.1128/IAI.00795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JH, McCaffrey RL, Baman NK, Allen LA, Weiss JP, Nauseef WM. The role of complement opsonization in interactions between F. tularensis subsp. novicida and human neutrophils. Microbes Infect. 2009;11:762–769. doi: 10.1016/j.micinf.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch EF, Schmetz MA, Foulds J, Hammer CH, Frank MM, Joiner KA. Multimeric C9 within C5b-9 is required for inner membrane damage to Escherichia coli J5 during complement killing. J Immunol. 1987;138:842–848. [PubMed] [Google Scholar]
- Briheim G, Stendahl O, Dahlgren C. Intra- and extracellular events in luminol-dependent chemiluminescence of polymorphonuclear leukocytes. Infect Immun. 1984;45:1–5. doi: 10.1128/iai.45.1.1-5.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckles EL, Wang X, Lane MC, Lockatell CV, Johnson DE, Rasko DA, et al. Role of the K2 capsule in Escherichia coli urinary tract infection and serum resistance. J Infect Dis. 2009;199:1689–1697. doi: 10.1086/598524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund J, Burgess LA, Cescutti P, Ernst RK, Speert DP. Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J Biol Chem. 2006;281:2526–2532. doi: 10.1074/jbc.M510692200. [DOI] [PubMed] [Google Scholar]
- Byrd MS, Sadovskaya I, Vinogradov E, Lu H, Sprinkle AB, Richardson SH, et al. Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol Microbiol. 2009;73:622–638. doi: 10.1111/j.1365-2958.2009.06795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay CD, Soni S, Gunn JS, Schlesinger LS. Evasion of complement-mediated lysis and complement C3 deposition are regulated by Francisellatularensis lipopolysaccharide O antigen. J Immunol. 2008;181:5568–5578. doi: 10.4049/jimmunol.181.8.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther JE, Kutala VK, Kuppusamy P, Ferguson JS, Beharka AA, Zweier JL, et al. Pulmonary surfactant protein a inhibits macrophage reactive oxygen intermediate production in response to stimuli by reducing NADPH oxidase activity. J Immunol. 2004;172:6866–6874. doi: 10.4049/jimmunol.172.11.6866. [DOI] [PubMed] [Google Scholar]
- Engels W, Endert J, Kamps MA, van Boven CP. Role of lipopolysaccharide in opsonization and phagocytosis of Pseudomonas aeruginosa. Infect Immun. 1985;49:182–189. doi: 10.1128/iai.49.1.182-189.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold DS, Goldman JN, Kuritz HM. Locus of the action of serum and the role of lysozyme in the serum bactericidal reaction. J Bacteriol. 1968a;96:2118–2126. doi: 10.1128/jb.96.6.2118-2126.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold DS, Goldman JN, Kuritz HM. Locus of the lethal event in the serum bactericidal reaction. J Bacteriol. 1968b;96:2127–2131. doi: 10.1128/jb.96.6.2127-2131.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer NL, Gomez AB, Neyrolles O, Gicquel B, Martin C. Interactions of attenuated Mycobacterium tuberculosis phoP mutant with human macrophages. PLoS ONE. 2010;5:e12978. doi: 10.1371/journal.pone.0012978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol. 2004;186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel EM, Wolfe DN, Elder K, Stibitz S, Harvill ET. O antigen protects Bordetella parapertussis from complement. Infect Immun. 2008;76:1774–1780. doi: 10.1128/IAI.01629-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JN, Ruddy S, Austen KF, Feingold DS. The serum bactericidal reaction. 3. Antibody and complement requirements for killing a rough Escherichia coli. J Immunol. 1969;102:1379–1387. [PubMed] [Google Scholar]
- Gondwe EN, Molyneux ME, Goodall M, Graham SM, Mastroeni P, Drayson MT, MacLennan CA. Importance of antibody and complement for oxidative burst and killing of invasive nontyphoidal Salmonella by blood cells in Africans. Proc Natl Acad Sci USA. 2010;107:3070–3075. doi: 10.1073/pnas.0910497107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GN, Rehm SR, Pierce AK. The effect of complement depletion on lung clearance of bacteria. J Clin Invest. 1978;62:373–378. doi: 10.1172/JCI109138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock RE, Mutharia LM, Chan L, Darveau RP, Speert DP, Pier GB. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: a class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect Immun. 1983;42:170–177. doi: 10.1128/iai.42.1.170-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale JP, Pollard AJ, Stokes RW, Simpson D, Tsang A, Massing B, Speert DP. Two distinct receptors mediate nonopsonic phagocytosis of different strains of Pseudomonas aeruginosa. J Infect Dis. 2001;183:1214–1220. doi: 10.1086/319685. [DOI] [PubMed] [Google Scholar]
- Iida K, Whitlow MB, Nussenzweig V. Amastigotes of Trypanosoma cruzi escape destruction by the terminal complement components. J Exp Med. 1989;169:881–891. doi: 10.1084/jem.169.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Yonemasu K. Studies on the third component (C’3) of guinea pig complement. II. Decay and generation of SAC’ la,4,2a,3. Biken J. 1968;11:181–191. [PubMed] [Google Scholar]
- Jackson KD, Starkey M, Kremer S, Parsek MR, Wozniak DJ. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J Bacteriol. 2004;186:4466–4475. doi: 10.1128/JB.186.14.4466-4475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, et al. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. 2003;171:4329–4339. doi: 10.4049/jimmunol.171.8.4329. [DOI] [PubMed] [Google Scholar]
- Joiner KA, Schmetz MA, Sanders ME, Murray TG, Hammer CH, Dourmashkin R, Frank MM. Multimeric complement component C9 is necessary for killing of Escherichia coli J5 by terminal attack complex C5b-9. Proc Natl Acad Sci USA. 1985;82:4808–4812. doi: 10.1073/pnas.82.14.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampen AH, Tollersrud T, Lund A. Staphylococcus aureus capsular polysaccharide types 5 and 8 reduce killing by bovine neutrophils in vitro. Infect Immun. 2005;73:1578–1583. doi: 10.1128/IAI.73.3.1578-1583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanegasaki S. Studies on phagocyte response to bacteria: conditions for efficient interaction and mechanism of oxygen radical production. Tokai J Exp Clin Med. 1986;11(Suppl):97–102. [PubMed] [Google Scholar]
- Karakawa WW, Sutton A, Schneerson R, Karpas A, Vann WF. Capsular antibodies induce type-specific phagocytosis of capsulated Staphylococcus aureus by human polymorphonuclear leukocytes. Infect Immun. 1988;56:1090–1095. doi: 10.1128/iai.56.5.1090-1095.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb WP, Haxby JA, Arroyave CM, Muller-Eberhard HJ. Molecular analysis of the membrane attack mechanism of complement. J Exp Med. 1972;135:549–566. doi: 10.1084/jem.135.3.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondos SC, Hatfaludi T, Voskoboinik I, Trapani JA, Law RH, Whisstock JC, Dunstone MA. The structure and function of mammalian membrane-attack complex/perforin-like proteins. Tissue Antigens. 2010;76:341–351. doi: 10.1111/j.1399-0039.2010.01566.x. [DOI] [PubMed] [Google Scholar]
- Learn DB, Brestel EP, Seetharama S. Hypochlorite scavenging by Pseudomonas aeruginosa alginate. Infect Immun. 1987;55:1813–1818. doi: 10.1128/iai.55.8.1813-1818.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol. 2005;175:7512–7518. doi: 10.4049/jimmunol.175.11.7512. [DOI] [PubMed] [Google Scholar]
- Longhi C, Conte MP, Penta M, Cossu A, Antonini G, Superti F, Seganti L. Lactoferricin influences early events of Listeria monocytogenes infection in THP-1 human macrophages. J Med Microbiol. 2004;53:87–91. doi: 10.1099/jmm.0.05367-0. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Wiesmann C, Brown EJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol. 2007;9:2095–2102. doi: 10.1111/j.1462-5822.2007.00981.x. [DOI] [PubMed] [Google Scholar]
- Lorenzen DR, Gunther D, Pandit J, Rudel T, Brandt E, Meyer TF. Neisseria gonorrhoeae porin modifies the oxidative burst of human professional phagocytes. Infect Immun. 2000;68:6215–6222. doi: 10.1128/iai.68.11.6215-6222.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Jackson KD, Landry RM, Parsek MR, Wozniak DJ. Analysis of Pseudomonas aeruginosa conditional psl variants reveals roles for the psl polysaccharide in adhesion and maintaining biofilm structure postattachment. J Bacteriol. 2006;188:8213–8221. doi: 10.1128/JB.01202-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Conover M, Lu H, Parsek MR, Bayles K, Wozniak DJ. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009;5:e1000354. doi: 10.1371/journal.ppat.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai GT, Seow WK, Pier GB, McCormack JG, Thong YH. Suppression of lymphocyte and neutrophil functions by Pseudomonas aeruginosa mucoid exopolysaccharide (alginate): reversal by physicochemical, alginase, and specific monoclonal antibody treatments. Infect Immun. 1993;61:559–564. doi: 10.1128/iai.61.2.559-564.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa M, Greenberg EP. Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J Bacteriol. 2004;186:4449–4456. doi: 10.1128/JB.186.14.4449-4456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra NP, Soni S, Rajaram MV, Dang PM, Reilly TJ, El-Benna J, et al. Francisella acid phosphatases inactivate the NADPH oxidase in human phagocytes. J Immunol. 2010;184:5141–5150. doi: 10.4049/jimmunol.0903413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Ortiz SL, Drouin SM, Wetsel RA. The alternative activation pathway and complement component C3 are critical for a protective immune response against Pseudomonas aeruginosa in a murine model of pneumonia. Infect Immun. 2004;72:2899–2906. doi: 10.1128/IAI.72.5.2899-2906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- Pangburn MK, Muller-Eberhard HJ. Relation of putative thioester bond in C3 to activation of the alternative pathway and the binding of C3b to biological targets of complement. J Exp Med. 1980;152:1102–1114. doi: 10.1084/jem.152.4.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SS, Kharazmi A, Espersen F, Hoiby N. Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infect Immun. 1990;58:3363–3368. doi: 10.1128/iai.58.10.3363-3368.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos D, van Bruggen R, Meischl C. Oxidative killing of microbes by neutrophils. Microbes Infect. 2003;5:1307–1315. doi: 10.1016/j.micinf.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Saldias MS, Ortega X, Valvano MA. Burkholderia cenocepacia O antigen lipopolysaccharide prevents phagocytosis by macrophages and adhesion to epithelial cells. J Med Microbiol. 2009;58:1542–1548. doi: 10.1099/jmm.0.013235-0. [DOI] [PubMed] [Google Scholar]
- Schreiber RD, Morrison DC, Podack ER, Muller-Eberhard HJ. Bactericidal activity of the alternative complement pathway generated from 11 isolated plasma proteins. J Exp Med. 1979;149:870–882. doi: 10.1084/jem.149.4.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speert DP, Wright SD, Silverstein SC, Mah B. Functional characterization of macrophage receptors for in vitro phagocytosis of unopsonized Pseudomonas aeruginosa. J Clin Invest. 1988;82:872–879. doi: 10.1172/JCI113692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker M, Park JS, Carey V, Lee JC. Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect Immun. 1998;66:5183–5189. doi: 10.1128/iai.66.11.5183-5189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, et al. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun. 2005;73:3693–3701. doi: 10.1128/IAI.73.6.3693-3701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, et al. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2003;100:8484–8489. doi: 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Purified Psl has no effect on the oxidative burst response generated by human neutrophils. The oxidative burst response of PMA induced neutrophils when incubated with Psl (0.5 or 1.0 mg) prepared from P. aeruginosa strain WFPA801. Each experiment was performed in quadruplet wells (n = 3). RLU, relative luminescence unit.