

Abstract
Aims
Reactive oxygen species (ROS)-mediated formation of mixed disulfides between critical cysteine residues in proteins and glutathione, a process referred to as protein S-glutathionylation, can lead to loss of enzymatic activity and protein degradation. Since mitochondria are a major source of ROS and a number of their proteins are susceptible to protein-S-glutathionylation, we examined if overexpression of mitochondrial thioltranferase glutaredoxin 2a (Grx2a) in macrophages of dyslipidemic atherosclerosis-prone mice would prevent mitochondrial dysfunction and protect against atherosclerotic lesion formation.
Methods and results
We generated transgenic Grx2aMacLDLR−/− mice, which overexpress Grx2a as an EGFP fusion protein under the control of the macrophage-specific CD68 promoter. Transgenic mice and wild type siblings were fed a high fat diet for 14 weeks at which time we assessed mitochondrial bioenergetic function in peritoneal macrophages and atherosclerotic lesion formation. Flow cytometry and Western blot analysis demonstrated transgene expression in blood monocytes and peritoneal macrophages isolated from Grx2aMacLDLR−/− mice, and fluorescence confocal microscopy studies confirmed that Grx2a expression was restricted to the mitochondria of monocytic cells. Live-cell bioenergetic measurements revealed impaired mitochondrial ATP turnover in macrophages isolated from Grx2aMacLDLR−/− mice compared to macrophages isolated from non-transgenic mice. However, despite impaired mitochondrial function in macrophages of Grx2aMacLDLR−/− mice, we observed no significant difference in the severity of atherosclerosis between wildtype and Grx2aMacLDLR−/− mice.
Conclusion
Our findings suggest that increasing Grx2a activity in macrophage mitochondria disrupts mitochondrial respiration and ATP production, but without affecting the proatherogenic potential of macrophages. Our data suggest that macrophages are resistant against moderate mitochondrial dysfunction and rely on alternative pathways for ATP synthesis to support the energetic requirements.
Keywords: glutaredoxin 2, reactive oxygen species, mitochondria, thiols, macrophage, atherosclerosis
1. Introduction
The recruitment of circulating blood monocytes to sites of local inflammation, followed by their differentiation into mature macrophages is a rate limiting step in multiple physiological processes including wound repair, inflammation, pathogen clearance, and replacement of tissue-derived resident macrophages1–3. Macrophages, particularly M2 polarized macrophages involved in inflammation resolution, rely on mitochondria for the energetically efficient production of ATP via oxidative phosphorylation (OXPHOS)4. However, these cells can survive and function in hypoxic inflamed tissues and meet their energy demands during hypoxic stress via anaerobic glycolysis5. In the process of OXPHOS, electrons are transferred through complexes I-IV, generating the proton gradient that drives ATP synthesis. During this process, 0.2% −2.0% of the electrons leak from the respiratory chain, primarily from complex I and complex III, resulting in a constant low level of reactive oxygen species (ROS) formation6. These ROS are detoxified by mitochondrial antioxidant enzymes, including superoxide dismutase, peroxiredoxin(s) and glutathione peroxidase. However, there is evidence that during conditions of metabolic stress, ROS production increases dramatically, resulting in oxidative stress, mitochondrial dysfunction and damage7–9. During oxidative stress, ROS promote the oxidation of glutathione (GSH) to glutathione disulfides (GSSG), either directly or through the reduction of peroxides by glutathione peroxidases, which use GSH as an electron donor10,11. Under physiological conditions, GSSG generated within the mitochondria is reduced by mitochondrial glutathione reductase (GR) and is converted back to GSH. However, under conditions of increased ROS production, GR is oxidatively inactivated, resulting in the accumulation of GSSG12,13. The lack of a GSSG efflux system within the mitochondria makes this cellular compartment highly susceptible to dramatic decreases in the GSH/GSSG ratio, particularly under conditions of high ROS production and thus prone to oxidative thiol modifications of redox-sensitive proteins through a thiol-disulfide exchange process referred to as protein S-glutathionylation. This reversible formation of mixed disulfides between protein thiols and GSH had originally been proposed to be a defense mechanism against irreversible thiol oxidation, protecting mitochondria against oxidative damage. However, more recent evidence now suggests that protein-S-glutathionylation plays a critical role in enzyme regulation and redox-sensitive signaling pathways.
Protein-S-glutathionylation has now emerged as a major reversible posttranslational cysteine modification that can become perturbed under conditions of oxidative stress, leading to changes in the activity of proteins involved in transcription, DNA synthesis, protein turnover, apoptosis, signal transduction, and mitochondrial function14–17. The reduction of S-glutathionylated protein thiols under physiological conditions is relatively slow and requires enzymatic catalysis10. Thioredoxins (Trx) and protein-disulfide isomerases can catalyze protein deglutathionylation, but glutaredoxins (Grxs) have been shown to be far more effective and highly specific for GSH-containing mixed disulfides18. Two Grx isoforms are present in mammalian cells, Grx1 and Grx2. Grx1 is localized in the cytosol and mitochondrial intermembrane space and is a well-characterized, specific and efficient catalyst of the reduction of protein-GSH mixed disulfides19,20. The more recently identified Grx2 has three splice variants, Grx2a, Grx2b and Grx2c in humans and Grx2a, Grx2c and Grx2d in mice21,22. Grx2a contains a mitochondrial localization sequence and is expressed ubiquitously in all tissues of both mice and humans. Grx2b and Grx2c in humans are localized in both the cytosol and nucleus and have been detected primarily in testes and cancer cell lines23. Mice, however, possess a Grx2c splice variant that is ubiquitously expressed in most tissues and a testes specific variant, Grx2d, which is not enzymatically active.
Grx2a is the best studied Grx2 splice variant in mammals and shares many characteristic features of Grx1, including a high specificity for glutathione-containing mixed disulfide and a double displacement kinetic mechanism24. Under oxidative stress conditions, (high ROS flux, low GSH, high GSSG levels), however, Grx1 can become inactivated due to the oxidation of non-catalytic cysteine residues, whereas Grx2a is highly stable and resistant to oxidative inactivation25. Inactivation of mitochondrial GR under these conditions is likely to further exacerbate thiol oxidative stress and promote S-glutathionylation of protein thiols. These observations suggest that Grx2a may play an important role in redox homeostasis of mitochondrial protein thiols, particularly under pathological conditions associated with increased oxidative stress such as atherosclerosis and other chronic inflammatory diseases.
The glutathione-dependent antioxidant system plays a critical role in protecting monocytes and macrophages against dysfunction and cell injury, thereby limiting macrophage accumulation at sites of vascular inflammation and preventing atherosclerotic lesion formation in mice. We reported that increased expression of Grx1 protects monocytes from priming, dysregulation and hyper-sensitization to chemokines induced by metabolic stress. We also showed that increased expression of either cytosolic or mitochondrial GR in macrophages reduces atherogenic lesion size in mice26–28. However, the role of mitochondrial Grx2a in macrophages and atherogenesis has not been studied.
To determine the role of monocytic mitochondrial Grx2a in atherosclerosis, we generated a novel transgenic mouse model (CD68-Grx2aTg mice), in which human mitochondrial Grx2a is expressed as an EGFP fusion protein under the control of the macrophage-specific CD68 promoter. To test our hypothesis that increased expression of Grx2a protects monocyte mitochondria from oxidative damage induced by metabolic stress, prevents monocyte priming and thus reduces atherosclerotic lesion formation, we crossed our transgenic mice (CD68-Grx2aTg mice) into the atherosclerosis-prone LDLR−/− mice (Grx2aMacLDLR−/−) and fed them a high fat diet for 14 weeks to induce atherosclerotic lesion formation. To our surprise, we found that increased expression of Grx2a was not atheroprotective and resulted in abnormal mitochondrial respiratory profiles and impaired mitochondrial function. Mitochondrial dysfunction in monocytes and macrophages, however, did not increase atherosclerotic lesion formation in these mice, suggesting that the partial loss of mitochondrial function is not sufficient to promote proatherogenic activities in monocytes and macrophages.
2. Materials and Methods
2.1. Animals
C57BL/6 mice overexpressing a fusion protein of mitochondrial Grx2a fused with EGFP under the control of the macrophage-specific CD68 promoter were generated in collaboration with the Transgenic Core Facility at UT Southwestern Medical Center (Fig. 1). Briefly, human Grx2a cDNA was excised by restriction enzyme and ligated to the EGFP sequence (Clontech), generating a fusion protein construct containing Grx2a and EGFP in the C-terminal. EGFP was then used as a marker for transgene expression. The Grx2a-EGFP sequence was then ligated into a vector containing the macrophage-specific CD68 promoter29, which carried the transgene on one allele, were crossed with atherosclerosis-prone LDLR−/− null mice to generate transgenic mice (Grx2aMacLDLR−/−) that overexpress Grx2a in monocytic cells and are prone to the development of atherosclerosis. Ten to thirteen week-old transgenic Grx2aMacLDLR−/− and wild type (Wt) littermates were fed a high fat diet (HFD; fat: 21% wt/wt and cholesterol: 0.15% wt/wt; AIN-76A, BioServe) for 14 weeks, at which time we assessed mitochondrial bioenergetic function in peritoneal macrophages, monocyte subset distribution and atherosclerotic lesion formation.
Fig. 1. Schematic diagram of the macrophage-specific CD68 promoter Grx2a-EGFP construct.
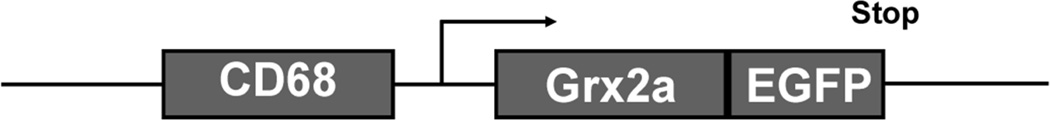
The CD68 promoter restricts expression of the construct to monocytes and macrophages. Grx2a is expressed as EGFP fusion protein to allow for visualization of transgene expression in live cells and tissues.
2.2. Blood analysis
For monocyte subset analysis, blood was obtained by venous tail bleed prior to sacrifice. Plasma cholesterol and triglyceride levels were determined using enzymatic assay kits (Wako Chemicals) using blood obtained by cardiac puncture. For differential blood cell counts, cardiac blood was collected and blood cell counts were obtained on an Abaxis VetScan HM2 Complete Blood Count Analyzer.
2.3. Western blot
Transgene expression in CD68-Grx2aTg mice was evaluated by Western blot analysis. Briefly, peritoneal macrophages were isolated and lysed in MES-buffered saline with 1% triton X-100(MBST) and supplemented with protease inhibitor cocktail (Roche). Cell lysate proteins were separated by SDS-PAGE (12%) and both endogenous and transgenic Grx2a were detected using a rabbit-anti-human Grx2a antibody (a kind gift from Dr. John Mieyal, Case Western Reserve University, Cleveland) followed by anti-rabbit-HRP antibodies. Chemiluminescence was imaged and analyzed on a Kodak Imaging Station 4000MM (Carestream Health).
2.4. Peritoneal macrophage isolation and purification
Resident peritoneal cells were harvested by lavage of the peritoneal cavity with 10 mL of ice-cold complete culture medium (RPMI 1640; Gibco BRL and Cellgro), final glucose concentration: 5 mM, supplemented with 2 mM/L L-alanyl-l-glutamine (GLUTAMAX-1; Gibco BRL), 1% v/v nonessential amino acids (Gibco BRL), 1 mM sodium pyruvate (Gibco BRL), penicillin G/streptomycin (100 U/L and 100 µg/ml, respectively; Gibco BRL), 10 mM HEPES (Fluka) and with 2% fetal bovine serum (FBS)26. To obtain a cell population highly enriched in macrophages, peritoneal macrophages were purified by negative selection using antibody-coated magnetic beads (Dynabeads® mouse pan B (B220) and Dynabeads® mouse pan T (Thy 1.2)). This procedure routinely increased the macrophage content of the isolate from approximately 40% CD68-positive cells to greater than 95% CD68-positive cells. Purified peritoneal macrophages were then plated and maintained in culture media with 10% FBS.
2.5. Mitochondrial bioenergetic measurements and extracellular flux (XF) analysis in peritoneal macrophages
Bioenergetic measurements were conducted using the XF analyzer (Seahorse Bioscience, MA). Purified peritoneal macrophages were seeded in Seahorse Bioscience XF24 cell culture plates at a density of 250,000 cells per well. Preliminary cell seeding experiments demonstrated that a seeding density of 250,000 purified peritoneal macrophages allowed the formation of a single macrophage monolayer. In addition, the coefficient of variance (CV) between wells was below the 20% recommended by the manufacturer, and cells exhibited a baseline oxygen consumption rate (OCR) of 280 ± 14.8 pMoles/min. Prior to mitochondrial bioenergetic measurements, culture media was replaced with XF assay media (unbuffered RPMI) and macrophages were allowed a 1 h equilibration period at 37°C in an ambient CO2 incubator not supplemented with additional CO2. OCR was monitored and real-time metabolic changes in cellular respiration were recorded in intact cells. To generate a mitochondrial respiratory profile, mitochondrial inhibitors including oligomycin (1.45 µM), FCCP (1.45 µM), and rotenone (10 µM) were sequentially added at 40 minute intervals, and mitochondrial responses were monitored every 10 minutes following a cycle of 4 minute measurement/4 minute mixing/2 minute delay. The following four respiratory parameters were utilized to analyze bioenergetic function according to the manufacturer’s instructions: basal respiration, ATP turnover, proton leak, and maximal respiratory capacity.
2.6. Analysis of atherosclerosis
Grx2aMacLDLR−/− and Wt littermates (10–13 weeks of age) were fed a high fat diet (HFD fat 21% wt/wt and cholesterol 0.15% wt/wt; AIN-76A, BioServe) for 14 weeks. Two vascular beds were used for the analysis of atherosclerosis as we described previously27. Briefly, mice were euthanized and hearts and aortas were perfused with PBS through the left ventricle. Aortas were dissected from the proximal ascending aorta to the bifurcation of the iliac artery and fixed with 4% paraformaldehyde in PBS. Hearts were embedded in OCT and frozen on dry ice. For en face analysis, the adventitial fat was removed and aortas were opened longitudinally and digitally photographed at a fixed magnification. Total aortic area and lesion areas were calculated using ImagePro Plus 6.0 (Media Cybernetics) and expressed as percent lesion area. As a second measure of atherosclerosis, lesions of the aortic root were analyzed. Serial sections were cut through an 800 µm segment of the aortic root. For each mouse, 8 sections (10 µm) separated by 80 µm were examined. Each section was stained with oil red O (ORO), counterstained with hematoxylin (Vector Labs), and digitized. Lesion area was measured using ImagePro Plus 6.0 (Media Cybernetics) and expressed as millimeters squared. Macrophages were detected with anti-CD68 antibody (Serotech) and macrophage content in lesions was determined as the average CD68-positive area per total lesion area30.
2.7. Flow cytometry
To verify EGFP expression in whole blood monocytes of transgenic mice, red blood cells were lysed and Tg EGFP expression was analyzed by flow cytometry. For monocyte subset analysis, whole blood (40 µL) was washed with FACS buffer (PBS with 2% FBS and 0.05% NaN3) and incubated in CD16/CD32 mAb for 15 minutes at 4°C to block nonspecific binding. Cells were incubated with the surface APC-conjugated Gr-1 (Ly6-C and Ly6-G) at 4°C for 15 minutes. Red blood cells were lysed using BD FACS lysing solution (1:10). Cells were washed, and then fixed and permeabilized with BD Fixation and Permeabilization solution. All subsequent washes contained saponin (PBS with 1% BSA, 0.1% saponin, and 0.1% NaN3) to maintain cell permeabilization. Cells were then stained with PE-conjugated CD68. Appropriate isotype controls were used to ensure specificity of antibodies used. The cells were analyzed using a BD FACSCalibur™ equipped for multi-color flow cytometry. Each measurement contained a defined number of 4 ×105 cells. Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
2.8. Confocal microscopy of EGFP and Cox-IV co-localization
To verify the exclusively mitochondrial localization of the Grx2a-EGFP transgene, isolated peritoneal macrophages were stained with a rabbit polyclonal antibody to cytochrome c oxidase subunit 4 (COX-IV; 1:500 Abcam #16056) and Cy3 donkey anti-rabbit IgG (1:500). Confocal images were taken on an Olympus FV-1000 (60×).
2.9. Viability Assays
Resident peritoneal macrophages were harvested from wild-type C57BL/6 and transgenic Grx2aMac mice, purified and cultured for 24 h as described in section 2.4. Macrophages were treated for 9 h with either 20 µg/mL or 40 µg/mL 7-ketocholesterol (7KC). Cells were then washed with PBS and lysed on ice in RIPA lysis buffer [50 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate] supplemented with protease inhibitors (Roche). To determine the extent of apoptosis induced by 7KC, lysates were assessed for caspase-3 activity using the fluorescent PARP substrate Ac-DECD-AFC (Enzo Life Sciences). The substrate was prepared as a 20 mM stock solution in DMSO according to manufacturer’s instructions. Ten µl of stock substrate was added to 30 µg of cell lysate diluted in reaction buffer (20mM HEPES, pH 7.5, 10% glycerol, 4mM DTT). Reactions were incubated for 2 h at 37° C in black-walled 96-well clear bottom plates, and fluorescence was measured using a plate reader (Spectra MAX Gemini EM). Wells containing only reaction buffer plus substrate or lysate plus reaction buffer were used to normalize the values. Additionally, a reaction containing recombinant human caspase-3 (Sigma) was used as a control to test for substrate cleavage under these experimental conditions.
2.10. Mitochondrial Superoxide Assay
Mitochondrial superoxide production was assessed using the MitoSOX™ Red reagent (Life Technology) in purified peritoneal macrophages isolated from WT and Grx2a transgenic mice. Macrophages were plated in a 96-well cell culture plates at a cell density of 200,000 cells per well, washed and incubated with the MitoSOX reagent (5 µM working solution in 1× HBSS generated from 5 mM stock solution in DMSO) for 10 min according to manufacturer’s instructions. Mitochondrial superoxide production was quantified using a fluorescence plate reader (SpectraMAX; λex = 510 nm; λem = 580 nm).
2.11. Statistical analysis
Data were analyzed using ANOVA (Sigma Stat), unless state otherwise. Data were tested for use of parametric or nonparametric post hoc analysis, and multiple comparisons were performed by using the least significant difference method. All data are presented as mean ± SE. Results were considered statistically significant at the P < 0.05 level.
3. Results
3.1. Transgenic Grx2a-EGFP expression is restricted to monocytic cells and localizes to mitochondria
To examine the role of the mitochondrial Grx2a on atherosclerosis, we generated a transgenic mouse model that overexpresses mitochondrially-targeted Grx2a as an EGFP fusion protein under the control of the monocyte/macrophage-specific CD68 promoter (Fig. 1). Flow cytometry analysis showed that in blood, the transgene was expressed in 5.5% of total leukocytes and that all EGFP+ cells were CD115+, i.e. blood monocytes (Fig. 2). To examine the expression of the transgene within the macrophages populations, purified peritoneal macrophages (CD68+) were gated based on the expression of CD115 and F4/80. EGFP expression was detected in more than 73.9% of CD115+F4/80+ cells (Fig. 3A), demonstrating a high percentage of transgene expression within the macrophage population. The presence of the intact Grx2a-EGFP fusion protein was confirmed by Western blot analysis of protein lysates generated from purified peritoneal macrophages isolated from wildtype and Grx2a-EGFP transgenic mice (Fig. 3B)31.
Fig. 2. Blood EGFP transgene expression was restricted to monocytes.

Flow cytometry analysis of peripheral blood monocytes from our CD68-Grx2a-EGFP transgenic (Tg) and wildtype (Wt) mice. In our CD68-Grx2a-EGFP transgenic mice, EGFP was expressed in 5.5% of total leukocytes. Within the EGFP (+) gate there was a shift in CD115+ expression thereby showing the blood transgene expression was restricted to CD115+ cells, i.e. blood monocytes. Within the EGFP (−) gate cells were CD115+ negative.
Fig. 3. Transgenic Grx2a-EGFP fusion protein is expressed in peritoneal macrophages.

(A) Flow cytometric analysis of peritoneal macrophages from wildtype (Wt) and transgenic CD68-Grx2aTg mice (Tg). Macrophages were identified based on F4/80 and CD115, and transgenic cells based on their expression of EGFP. Note 73.9% ± 3.0 of CD115+F4/80+ cells from transgenic mice were EGFP positive (n = 3). (B) Western Blot analysis using an anti-Grx2a antibody of purified peritoneal macrophages isolated from wildtype (Wt) and transgenic CD68-Grx2aTg mice (Tg). The 45 kDa band corresponding to the Grx2a-EGFP fusion protein is denoted with (*).
To confirm that the transgene was targeted to mitochondria, we stained adherent peritoneal macrophages from wildtype and Grx2a-EGFP transgenic mice with antibodies directed against cytochrome c oxidase subunit 4 (Cox-lV), a mitochondrial marker. Confocal microscopy showed punctate EGFP expression in transgenic macrophages, and we observed the EGFP signal co-localize with Cox-IV (Fig. 4).
Fig. 4. Transgenic Grx2a-EGFP fusion protein localizes to mitochondria.

Confocal microscopy images of peritoneal macrophages isolated from wildtype (Wt) and transgenic CD68-Grx2aTg (Tg) mice and labeled with the mitochondrial marker Cox-IV (red) and the nuclear dye DAPI (blue). EGFP expression is shown in green. Image overlay shows co-localization of EGFP with the mitochondrial marker Cox-IV.
Grx2a-EGFP transgenic mice showed no overt phenotype. Compared to their respective wildtype littermates, neither female nor male Tg mice showed any significant differences in bodyweight or total plasma cholesterol and triglyceride levels (Table 1). We also found no significant differences in the red blood cell, platelet or leukocyte counts or the lymphocyte and monocyte levels within the leukocyte population (Table 2).
Table 1.
Body weights and total plasma cholesterol and triglyceride levels.
| Parameter | Wt | Grx2aMacLDLR−/− |
|---|---|---|
| Females | n=10 | n=9 |
| Weight, g | 30.8 ± 1.4 | 28.1 ± 0.8 |
| Plasma total cholesterol, mg/dl | 586 ± 130 | 670 ± 98 |
| Triglycerides, mg/dl | 96 ± 20 | 106 ± 14 |
| Males | n=5 | n=11 |
| Weight, g | 44.3 ± 1.4 | 41.1 ± 1.4 |
| Plasma total cholesterol, mg/dl | 1244 ± 205 | 938 ± 183 |
| Triglycerides, mg/dl | 195 ± 44 | 238 ± 25 |
After 14 weeks of HFD-feeding of LDLR−/− (Wt) and transgenic Grx2aMacLDLR−/− mice (Grx2aMacLDLR−/−) was completed, mice were weighed and plasma was obtained after cardiac punctures. Total plasma cholesterol and triglycerides were determined as described under Material and Methods. Results are expressed as mean ± SE.
Table 2.
Blood cell counts.
| Parameter | Wt | Grx2aMacLDLR−/− |
|---|---|---|
| Red Blood Cells (10^12/l) | 9.3 ± 0.3 | 9.2 ± 0.2 |
| White Blood Cells (10^9/l) | 5.4 ± 0.7 | 4.1 ± 0.4 |
| Lymphocytes (%) | 90 ± 2 | 96 ± 1 |
| Monocytes (%) | 3.0 ± 0.7 | 2.2 ± 0.9 |
| Platelets (10^9/l) | 707 ± 69 | 565 ± 50 |
After HFD-feeding was complete and LDLR−/− (Wt) and transgenic Grx2aMacLDLR−/− mice (Grx2aMacLDLR−/−) were sacrificed, blood was collected by cardiac puncture. Differential blood cell counts were obtained on an Abaxis VetScan HM2 Complete Blood Count Analyzer as described under Material and Methods. Results are expressed as mean ± SE (n=5 per group).
3.2. Increased expression of mitochondrial Grx2a decreases ATP turnover and mitochondrial respiratory activity in macrophages from dyslipidemic mice
Mitochondria are a source of ROS formation, which when unchecked promotes S-glutathionylation, resulting in decreased mitochondrial protein function, decreased ATP-linked mitochondrial oxygen consumption and reduced cellular bioenergetic reserve capacity32. We therefore predicted that in dyslipidemic LDL-R−/− mice, increasing the expression of Grx2a, the enzyme that catalyzes the de-glutathionylation of proteins in mitochondria, would protect metabolically stressed mitochondria against loss of protein function and improve mitochondrial respiratory efficiency. To this end, LDL-R−/− and Grx2aMacLDLR−/− mice were fed a HFD for 14 weeks after which peritoneal macrophages were isolated and analyzed for their mitochondrial respiratory activity. To our surprise, macrophages from Grx2aMacLDLR−/− mice showed an ATP turnover rate that was 15% lower than the rate measured in macrophages from non-transgenic littermates (Fig. 5), indicating that mitochondria in metabolically stressed macrophages overexpressing Grx2a were significantly less efficient in generating ATP than mitochondria in non-transgenic macrophages. In agreement with this finding, we also observed a lower maximal respiratory capacity in transgenic peritoneal macrophages, although this difference did not quite reach statistical significance (P=0.057).
Fig. 5. ATP turnover is reduced in peritoneal macrophages isolated from mice fed a HFD for 14 weeks with increased expression of Grx2a.

Respiratory measurements of purified peritoneal macrophages from mice were conducted as described in Material and Methods. The following four respiratory parameters were utilized to analyze bioenergetic function and calculated as follows: Basal respiration (OCRbaseline – OCRrotenone), ATP turnover(OCRoligomycin − OCRbaseline), Proton leak (OCRrotenone − OCRoligomycin), and Maximal respiratory capacity (OCRFCCP − OCRrotenone). Wt (open bars; n = 5) versus Grx2aMacLDLR−/− (closed bars, n = 6).
The proton leak is a measure of the endogenous leak of protons across the inner mitochondrial membrane which limits the proton motive force and could account for reduced ATP turnover rate. The proton leak was not altered by increased Grx2a expression, suggesting reduced ATP production was the result of a defect in mitochondrial electron transfer rather than increased uncoupling. Surprisingly, we observed no differences in MitoSox-sensitive mitochondrial ROS production between peritoneal macrophages isolated from wildtype and Grx2aMacLDLR−/− mice (data not shown). Nevertheless, our data show that increased expression of Grx2a impairs mitochondrial function in macrophages.
3.3. Increased expression of mitochondrial Grx2a increases sensitivity to oxysterol-induced apoptosis
To determine whether the reduced ATP turnover rate actually impaired biological functions in macrophages overexpressing Grx2a, we assessed whether macrophages from Grx2a-EGFP transgenic mice were more susceptible to oxysterol induced apoptosis. Indeed, macrophages from Grx2a-EGFP transgenic were significantly more sensitive to 7-KC-induced caspase-3 activation (Fig. 6). These data suggest that mitochondrial dysfunction induced by increased Grx2a expression results in increased sensitivity to oxysterol-induced apoptosis, possibly due to limited cellular ATP levels.
Fig. 6. Increased expression of Grx2a sensitizes peritoneal macrophages to 7-ketocholesterol-induced apoptosis.

Measurements of 7-ketocholesterol-induced caspase-3 activity in purified peritoneal macrophages isolated from C57BL/6 (open bars, n=3) and Grx2aMac mice (closed bars, n=3) were conducted using the fluorescent PARP substrate Ac-DECD-AFC as described in Material and Methods. Values were normalized to the mean relative fluorescence intensity obtained with lysates from untreated C57BL/6 macrophages (0 µg/ml), and expressed as fold change. Results are mean ± SD (n=3). *: P < 0.05 versus wildtype macrophages; **: P < 0.05 versus vehicle-treated wildtype macrophages (0 µg/ml).
3.4. Increased expression of mitochondrial Grx2a does not prevent monocytosis induced by dyslipidemia
HFD-feeding induces monocytosis in atherosclerosis prone mice and elevated blood monocyte counts are a well-established independent risk factor in human vascular disease33–35. We therefore examined blood monocyte counts and monocyte subset distribution in Grx2aMacLDLR−/− mice and LDLR−/− littermates, both before and after feeding a HFD for 14 weeks. Circulating blood leukocytes were analyzed by flow cytometry and cell populations were distinguished based on their expression of the monocytic lineage marker CD68 and the myeloid differentiation marker Gr-1 expressed on neutrophils and monocytes. Leukocyte subsets were defined as CD68hi (monocytes) and CD68lo (neutrophils), and monocytes were further divided into three subsets based on their expression level of Gr-1: CD68hiGr-1hi, CD68hiGr-1int and CD68hiGr-1lo. After 14 weeks of high-fat diet feeding, female and male mice in both the LDL-R−/− and Grx2aMacLDLR−/− groups demonstrated monocytosis as evidenced by a 1.9-fold increase in the total monocyte population (CD68hi, Table 3) of females and a 2.6-fold increase in male mice. In all four groups, the increase in monocytes appeared to be primarily driven by an increase in patrolling monocytes (CD68hi/Gr-1lo) rather than inflammatory monocytes (CD68hi/Gr-1hi), distinguishing the monocyte response in HFD-fed LDL-R−/− mice from that reported in apoE−/− mice36. Overall, increased expression of Grx2a did not appear to affect monocytosis induced by metabolic stress, i.e. HFD-feeding, nor monocyte subset distributions. However, we noted a small but statistically significant difference in the percentage of patrolling monocytes between male LDL-R−/− and Grx2aMacLDLR−/− mice after HFD feeding. The decrease in patrolling monocytes in Grx2aMacLDLR−/− mice was small, but it is possible that with increasing gene dosage this effect may become larger and biologically significant.
Table 3.
Effect of HFD-feeding on blood monocyte subset distributions in LDLR−/− and transgenic Grx2aMacLDLR−/− mice.
| Monocyte Subset | Baseline [%] |
14 Weeks HFD [%] |
|---|---|---|
| Females | ||
| Total Monocytes (CD68hi) | ||
| Wt | 6.13 ± 0.55 | 11.44 ± 1.11 * |
| Tg | 4.87 ± 0.29 | 11.56 ± 1.50 * |
| Inflammatory Monocytes (CD68hi Gr-1hi) | ||
| Wt | 2.59 ± 0.47 | 2.91 ± 0.32 |
| Tg | 1.87 ± 0.17 | 2.66 ± 0.38 |
| Patrolling Monocytes (CD68hi Gr-1lo) | ||
| Wt | 2.13 ± 0.16 | 5.66 ± 0.58 * |
| Tg | 1.89 ± 0.16 | 5.71 ± 0.87 * |
| Males | ||
| Total Monocytes (CD68hi) | ||
| Wt | 5.61 ± 0.48 | 14.52 ± 0.90 * |
| Tg | 5.91 ± 0.49 | 11.16 ± 1.06 * |
| Inflammatory Monocytes (CD68hi Gr-1hi) | ||
| Wt | 1.76 ± 0.14 | 3.56 ± 0.19 |
| Tg | 2.60 ± 0.39 | 3.06 ± 0.40 |
| Patrolling Monocytes (CD68hi Gr-1lo) | ||
| Wt | 2.56 ± 0.24 | 7.52 ± 0.58 *‡ |
| Tg | 1.95 ± 0.15 | 5.39 ± 0.46 * |
Blood was obtained from the tail vein of LDLR−/− (Wt) and transgenic Grx2aMacLDLR−/− mice (Tg) both prior to switching the mice to a HFD diet and after 14 weeks on a HFD. Red blood cells were lysed, and leukocyte staining and FACS analysis was performed as described under Material and Methods. Results for total monocytes(CD68hi), inflammatory monocytes (CD68hi/Gr-1lo) and patrolling monocytes (CD68hi/Gr-1lo) are expressed as percent of total leukocyte counts.
: P<0.05, baseline vs. 14 weeks HFD;
: P<0.05, Wt (n= 10) vs. Tg (n= 13).
3.5. Increased expression of mitochondrial Grx2a in macrophages does not affect atherosclerosis
Monocyte recruitment to sites of vascular injury is a rate-limiting step in the development and progression of atherosclerotic lesions. Transmigration of monocytes through the endothelium requires the activation of a large number of energy-dependent processes. Impaired mitochondrial ATP production in monocytes and macrophages may therefore impact the development and progression of atherosclerosis. En face analysis of the aortic root did not demonstrate any statistically significant difference in lesion size between Grx2aMacLDLR−/− and non-transgenic littermates (Wt), neither in female nor male mice (Table 4). We also observed no significant differences in lesion formation in the aortic roots between Grx2aMacLDLR−/− and non-transgenic LDL-R−/− littermates (Fig. 7). Macrophage content within the lesions was also not different between Grx2aMacLDLR−/− and non-transgenic littermates, females; 0.40 +/− 0.04 mm2 versus 0.36 +/− 0.03 mm2; P=0.45 males; 0.24 +/− 0.04 mm2 versus 0.41 +/− 0.06 mm2; P=0.07.
Table 4.
En face analysis of isolated aortic arches.
| En face | Wt (%) |
Grx2aMacLDLR−/− (%) |
|---|---|---|
| n=10 | n=10 | |
| Females | 14.7 ± 2.2 | 16.1 ± 1.9 |
| n=9 | n=11 | |
| Males | 8.6 ± 2.7 | 14.8 ± 2.6 |
After 14 weeks of HFD-feeding of LDLR−/− (Wt) and transgenic Grx2aMacLDLR−/− mice (Grx2aMacLDLR−/−) was completed, aortas were opened longitudinally and lesion areas were calculated as described under Material and Methods. Results expressed as mean ±SE.
Fig. 7. Increased expression of Grx2a in monocytes and macrophages did not affect HFD-induced atherosclerotic lesion formation.

(A) Representative image of Oil red O (ORO)-stained sections of the aortic root from female and male LDLR−/− (wildtype; Wt) and transgenic Grx2aMacLDLR−/− mice (Tg) fed a HFD for 14 weeks. (B) Quantification of atherosclerosis in the aortic root wildtype and transgenic Grx2aMacLDLR−/− mice. There was no statistically significant difference in lesion size between LDLR−/− mice (open bars) and Grx2aMacLDLR−/− transgenic mice (closed bars). Females: n=9 per group; Males: n=12 per group.
4. Discussion
The microenvironment in atherosclerotic plaques is pro-inflammatory and oxidative and can increase monocyte recruitment and impact macrophage differentiation, polarization, metabolism and survival. Under such oxidative conditions, ROS-mediated protein thiol modifications in monocytes and macrophages, namely S-glutathionylation, has been reported to regulate protein activity and gene expression levels37–39. Protein S-glutathionylation is largely associated with increased cellular oxidative stress, however, more recently it has been recognized as an important redox signaling mechanism involved in the regulation of protein signaling cascades, regulating apoptosis, transcription, and metabolism and other cellular functions40–44. Glutaredoxins (Grx1 and Grx2) are largely responsible for catalyzing protein S-(de)glutathionylation reactions and altering the relative expression levels of these thiol transferases can have complex consequences on cellular processes. Overexpression of Grx2a, the mitochondrial form of Grx2, in human hepatocellular liver carcinoma cell line (HepG2) has profound protective effects with regard to cell viability, ROS production, and GSH/GSSG maintenance45. Doxorubicin-induced mitochondrial protein S-glutathionylation and cardiac injury in mice can also be prevented by overexpression of Grx2a46. Alternatively, cultured mouse lens epithelial cells isolated from Grx2 knockout animals display increased sensitivity to oxidative stress induced by hydrogen peroxide treatment47. Grx2 knockout cells develop mitochondrial dysfunction characterized by reduced ATP pools and enhanced complex 1 inactivation under both resting and oxidative stress conditions. Taken together, these observations support the notion that Grx2a plays a pivotal role in maintaining mitochondrial function under conditions of oxidative stress, suggesting the enzyme may also play important roles in the protection against chronic inflammatory diseases such as atherosclerosis.
We previously demonstrated that the cytosolic glutathione-dependent antioxidant system plays a critical role in the recruitment of macrophages into the vascular wall and that this recruitment was associated with the formation of atherosclerotic plaques28. We also showed that overexpression in bone marrow-derived cells of glutathione reductase (GR), a homodimeric flavoprotein that maintains cellular thiol redox state by catalyzing the reduction of glutathione disulfide to glutathione, reduced atherosclerotic lesion formation by 32%27. Mitochondrial GR was found to be equally protective as the cytosolic enzyme, demonstrating that it is critical for macrophages to maintain their thiol redox balance in both compartments to prevent cell dysfunction or injury. In addition, we found that the increased expression of the cytosolic glutaredoxin (Grx1) completely blocked monocyte priming and dysfunction induced by either oxidative or metabolic stress48. These results demonstrate that the detrimental effects of oxidative and metabolic stress on monocyte migration and macrophage functions contributing to atherosclerotic lesion formation are mediated at least in part by changes in protein-S-glutathionylation. However, the role of Grx2a in maintaining monocyte and macrophage function under oxidative and metabolic stress, and the development of atherosclerosis was unknown. To determine the role of macrophage Grx2a, we therefore generated transgenic mice that overexpress Grx2a in monocytic cells (Grx2aMacLDLR−/− mice) and are prone to the development of metabolic stress and atherosclerosis in response to high fat diet (HFD) feeding.
Surprisingly, we observed no beneficial effect of increased Grx2a expression in macrophages on atherosclerotic lesion formation in animals after 14 weeks of HFD (Fig. 7). This is in stark contrast to the beneficial effects we observed previously when overexpressing other members of the glutathione antioxidant system including GR and Grx1. Instead, we found that Grx2a overexpression in macrophages actually impairs mitochondrial respiration and ATP production (Fig. 5). However, the 15% reduction in mitochondrial ATP turnover we observed is likely to have only minor consequences for macrophages, particularly if one considers that macrophages, particularly M1-activated macrophages, rely heavily on glycolysis49. This may explain why in our transgenic mice key functionalities of macrophages related to atherogenesis did not appear to be impaired by Grx2a overexpression. Our results suggest that contrary to our initial hypothesis, increased expression of Grx2a not only does not protect mitochondria during metabolic stress, but that by regenerating critical free thiols and maintaining them in a reduced state may actually disrupt mitochondrial functions. This conclusion is supported by our findings that Grx2a overexpression also sensitizes peritoneal macrophages to 7-ketocholestrol-induced apoptosis (Fig. 6).
Harper and colleagues were among the first to identify protein S-glutathionylation and Grx2 activity as regulators of mitochondrial function. Reversible protein S-glutationylation was found to act as a control switch for uncoupling protein 2 (UCP2) and uncoupling protein 3 (UCP3) activity in skeletal muscle, maintaining mitochondrial respiratory function under fluctuating redox conditions50. Furthermore, Grx2a appears to be required for UCP3 S-glutathionylation, which results in its inactivation and a decrease in the proton leak. These observations demonstrate the importance of Grx2-mediated protein S-glutathionylation in regulating mitochondrial OXPHOS42. More recently, Grx2 was reported to also play a central role in regulating mitochondrial OXPHOS in cardiac muscle. Cardiomyocytes isolated from animals deficient in Grx2 demonstrated decreased ATP production along with increased (complex I, IV) and decreased (complex III, V) levels of mitochondrial complex proteins. Further analysis revealed that complex I levels could be restored by exogenous Grx1, indicating that deglutathionylation reactions mediated by Grx2 regulate mitochondrial complex levels14. Collectively, these observations, in conjunction with those reported in this study, suggest that Grx2a in cells such as monocytes and macrophages appears to play the role of a rheostat rather than a switch in regulating mitochondrial OXPHOS, and both its loss and/or over-expression may disrupt the subtle redox balance required for optimal mitochondrial function, and lead to mitochondrial dysfunction. Although previous reports have causally linked Grx1 and Grx2 activity to cardiovascular disease (enhanced cardioprotection in Grx1 and Grx2 transgenic mice), these studies have since been retracted, warranting further clarification of the exact role of these enzymes in maintaining protein thiol redox homeostasis in specific cell types (i.e monocytes and macrophages) during atherogenesis51,52.
Recent efforts to identify potential substrates of mammalian Grx2 not only led to the identification of numerous proteins with redox function but also, to an even larger number, of proteins with other functions in metabolism, nuclear transport, protein turnover, and transcription/translation53,54. We now found that overexpression of Grx2a in macrophages appears to perturb rather than protect the mitochondrial protein S-glutathionylation/redox state, leading to the disruption of key metabolic processes. The beneficial effects of overexpressing antioxidant “protective” enzymes such as GR, Grx1 and Grx2a may thus be context specific and their roles in regulating cellular function via (de)-glutathionylation events are likely multifaceted. In summary, our data suggest that tight control of protein S-glutathionylation is essential for mitochondrial function and that both oxidative and reductive perturbations of the S-glutathionylation state of mitochondrial proteins appear to be detrimental to macrophage mitochondrial function, albeit with only limited impact on their atherogenic activity in vivo.
Highlights.
Transgenic mice carrying a transgene encoding mitochondrial glutaredoxin 2a (Grx2a) as an EGFP fusion protein under the control of the macrophage-specific CD68 promoter show Grx2a expression that is restricted to mitochondria of monocytes and macrophages.
Overexpression of mitochondrial Grx2a disrupts mitochondrial bioenergetics in macrophages.
Overexpression of mitochondrial Grx2a impairs mitochondrial ATP turnover in macrophages and sensitizes macrophages to oxysterol-induced apoptosis.
Despite promoting mitochondrial dysfunction in macrophages, overexpression of mitochondrial Grx2a in macrophages had little or no effect on atherogenesis in high-fat diet fed LDLR-null mice.
ACKNOWLEDGEMENTS
We are very grateful to Dr. John Mieyal for providing us with the rabbit-anti-hGrx2a antibody. This study was funded by grants from the National Institutes of Health (to R. Asmis HL70963 and HL115858) and a Grant-in-Aid from the American Heart Association (to R. Asmis 0855011F).
FUNDING SOURCES
This work was supported by grants to R.A. from the NIH (HL-70963) and the AHA (0855011F) and training grant to D.A.Z. from NIH T32 (HL-007446)
ABBREVIATIONS
- CBC
Complete Blood Cell Counts
- EGFP
Enhanced Green Fluorescent Protein
- ORO
Oil Red O
- OXPHOS
Oxidative Phosphorylation
- GR
Glutathione Reductase
- GSH
Glutathione
- GSSG
Glutathione Disulfide
- Grx
Glutaredoxin
- Grx2a
Glutaredoxin 2a
- ROS
Reactive Oxygen Species
- Trx
Thioredoxins
- Wt
Wild Type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
There are no conflicts to disclose.
REFERENCES
- 1.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 2.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–995. [Google Scholar]
- 4.Tavakoli S, Zamora D, Ullevig S, Asmis R. Bioenergetic profiles diverge during macrophage polarization: Implications for the interpretation of 18F-FDG PET imaging of atherosclerosis. J Nucl Med. 2013;54(9):1661–1667. doi: 10.2967/jnumed.112.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakamoto T, Seiki M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J Biol Chem. 2010;285(39):29951–29964. doi: 10.1074/jbc.M110.132704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staniek K, Nohl H. H(2)O(2) detection from intact mitochondria as a measure for one-electron reduction of dioxygen requires a non-invasive assay system. Biochim Biophys Acta. 1999;1413(2):70–80. doi: 10.1016/s0005-2728(99)00083-3. [DOI] [PubMed] [Google Scholar]
- 7.Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118(2):789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandes RP. Triggering mitochondrial radical release: A new function for NADPH oxidases. Hypertension. 2005;45(5):847–848. doi: 10.1161/01.HYP.0000165019.32059.b2. [DOI] [PubMed] [Google Scholar]
- 10.Hurd TR, Costa NJ, Dahm CC, et al. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7(7–8):999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 11.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal. 2000;2(4):811–820. doi: 10.1089/ars.2000.2.4-811. [DOI] [PubMed] [Google Scholar]
- 12.Untucht-Grau R, Schirmer RH, Schirmer I, Krauth-Siegel RL. Glutathione reductase from human erythrocytes: Amino-acid sequence of the structurally known FAD-binding domain. Eur J Biochem. 1981;120(2):407–419. doi: 10.1111/j.1432-1033.1981.tb05718.x. [DOI] [PubMed] [Google Scholar]
- 13.Savvides SN, Scheiwein M, Bohme CC, et al. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J Biol Chem. 2002;277(4):2779–2784. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 14.Mailloux RJ, Xuan JY, McBride S, et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J Biol Chem. 2014;289(21):14812–14828. doi: 10.1074/jbc.M114.550574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demasi M, Netto LE, Silva GM, et al. Redox regulation of the proteasome via S-glutathionylation. Redox Biol. 2013;2:44–51. doi: 10.1016/j.redox.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: Role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7(3–4):348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 17.Go YM, Jones DP. Redox control systems in the nucleus: Mechanisms and functions. Antioxid Redox Signal. 2010;13(4):489–509. doi: 10.1089/ars.2009.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung CH, Thomas JA. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch Biochem Biophys. 1996;335(1):61–72. doi: 10.1006/abbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 19.Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32(13):3368–3376. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 20.Pai HV, Starke DW, Lesnefsky EJ, Hoppel CL, Mieyal JJ. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 2007;9(11):2027–2033. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- 21.Gladyshev VN, Liu A, Novoselov SV, et al. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J Biol Chem. 2001;276(32):30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 22.Hudemann C, Lonn ME, Godoy JR, et al. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid Redox Signal. 2009;11(1):1–14. doi: 10.1089/ars.2008.2068. [DOI] [PubMed] [Google Scholar]
- 23.Lonn ME, Hudemann C, Berndt C, et al. Expression pattern of human glutaredoxin 2 isoforms: Identification and characterization of two testis/cancer cell-specific isoforms. Antioxid Redox Signal. 2008;10(3):547–557. doi: 10.1089/ars.2007.1821. [DOI] [PubMed] [Google Scholar]
- 24.Gallogly MM, Starke DW, Leonberg AK, Ospina SM, Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: Implications for intracellular roles. Biochemistry. 2008;47(42):11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandes AP, Holmgren A. Glutaredoxins: Glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6(1):63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Qiao M, Mieyal JJ, Asmis LM, Asmis R. Molecular mechanism of glutathione-mediated protection from oxidized low-density lipoprotein-induced cell injury in human macrophages: Role of glutathione reductase and glutaredoxin. Free Radic Biol Med. 2006;41(5):775–785. doi: 10.1016/j.freeradbiomed.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 27.Qiao M, Kisgati M, Cholewa JM, et al. Increased expression of glutathione reductase in macrophages decreases atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27(6):1375–1382. doi: 10.1161/ATVBAHA.107.142109. [DOI] [PubMed] [Google Scholar]
- 28.Qiao M, Zhao Q, Lee CF, et al. Thiol oxidative stress induced by metabolic disorders amplifies macrophage chemotactic responses and accelerates atherogenesis and kidney injury in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2009;29(11):1779–1786. doi: 10.1161/ATVBAHA.109.191759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greaves DR, Quinn CM, Seldin MF, Gordon S. Functional comparison of the murine macrosialin and human CD68 promoters in macrophage and nonmacrophage cell lines. Genomics. 1998;54(1):165–168. doi: 10.1006/geno.1998.5546. [DOI] [PubMed] [Google Scholar]
- 30.Huby T, Doucet C, Dachet C, et al. Knockdown expression and hepatic deficiency reveal an atheroprotective role for SR-BI in liver and peripheral tissues. J Clin Invest. 2006;116(10):2767–2776. doi: 10.1172/JCI26893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iqbal AJ, McNeill E, Kapellos TS, et al. Human CD68 promoter GFP transgenic mice allow analysis of monocyte to macrophage differentiation in vivo. Blood. 2014;124(15):e33–e44. doi: 10.1182/blood-2014-04-568691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill BG, Higdon AN, Dranka BP, Darley-Usmar VM. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim Biophys Acta. 2010;1797(2):285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoneman V, Braganza D, Figg N, et al. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100(6):884–893. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasir K, Guallar E, Navas-Acien A, Criqui MH, Lima JA. Relationship of monocyte count and peripheral arterial disease: Results from the national health and nutrition examination survey 1999–2002. Arterioscler Thromb Vasc Biol. 2005;25(9):1966–1971. doi: 10.1161/01.ATV.0000175296.02550.e4. [DOI] [PubMed] [Google Scholar]
- 35.Johnsen SH, Fosse E, Joakimsen O, et al. Monocyte count is a predictor of novel plaque formation: A 7-year follow-up study of 2610 persons without carotid plaque at baseline the tromso study. Stroke. 2005;36(4):715–719. doi: 10.1161/01.STR.0000158909.07634.83. [DOI] [PubMed] [Google Scholar]
- 36.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ullevig S, Kim HS, Asmis R. S-glutathionylation in monocyte and macrophage (dys)function. Int J Mol Sci. 2013;14(8):15212–15232. doi: 10.3390/ijms140815212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci U S A. 2012;109(41):E2803–E2812. doi: 10.1073/pnas.1212596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim HS, Ullevig SL, Nguyen HN, Vanegas D, Asmis R. Redox regulation of 14-3-3zeta controls monocyte migration. Arterioscler Thromb Vasc Biol. 2014;34(7):1514–1521. doi: 10.1161/ATVBAHA.114.303746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen EM, Mieyal JJ. Protein-thiol oxidation and cell death: Regulatory role of glutaredoxins. Antioxid Redox Signal. 2012;17(12):1748–1763. doi: 10.1089/ars.2012.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin F, Sancheti H, Cadenas E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid Redox Signal. 2012;17(12):1714–1727. doi: 10.1089/ars.2012.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mailloux RJ, Xuan JY, Beauchamp B, Jui L, Lou M, Harper ME. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J Biol Chem. 2013;288(12):8365–8379. doi: 10.1074/jbc.M112.442905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pastore A, Piemonte F. S-glutathionylation signaling in cell biology: Progress and prospects. Eur J Pharm Sci. 2012;46(5):279–292. doi: 10.1016/j.ejps.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. S-glutathionylation in protein redox regulation. Free Radic Biol Med. 2007;43(6):883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 45.Kim SJ, Jung HJ, Choi H, Lim CJ. Glutaredoxin 2a, a mitochondrial isoform, plays a protective role in a human cell line under serum deprivation. Mol Biol Rep. 2012;39(4):3755–3765. doi: 10.1007/s11033-011-1152-0. [DOI] [PubMed] [Google Scholar]
- 46.Diotte NM, Xiong Y, Gao J, Chua BH, Ho YS. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim Biophys Acta. 2009;1793(2):427–438. doi: 10.1016/j.bbamcr.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 47.Wu H, Lin L, Giblin F, Ho YS, Lou MF. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic Biol Med. 2011;51(11):2108–2117. doi: 10.1016/j.freeradbiomed.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ullevig S, Zhao Q, Lee CF, Seok Kim H, Zamora D, Asmis R. NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler Thromb Vasc Biol. 2012;32(2):415–426. doi: 10.1161/ATVBAHA.111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez-Prados JC, Traves PG, Cuenca J, et al. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 50.Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem. 2011;286(24):21865–21875. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Retraction notice to "overexpression of glutaredoxin-2 reduces myocardial cell death by preventing both apoptosis and necrosis". J Mol Cell Cardiol. 2012;53(5):744. doi: 10.1016/j.yjmcc.2012.08.016. [J. mol. cell. cardiol. 44 (2008) 252–260. [DOI] [PubMed] [Google Scholar]
- 52.Retraction notice to "role of glutaredoxin-1 in cardioprotection: An insight with Glrx1 transgenic and knockout animals". J Mol Cell Cardiol. 2012;53(5):745. doi: 10.1016/j.yjmcc.2012.08.015. [J. mol. cell. cardiol. 44 (2008) 261–269. [DOI] [PubMed] [Google Scholar]
- 53.Lillig CH, Berndt C. Glutaredoxins in thiol/disulfide exchange. Antioxid Redox Signal. 2013;18(13):1654–1665. doi: 10.1089/ars.2012.5007. [DOI] [PubMed] [Google Scholar]
- 54.Schutte LD, Baumeister S, Weis B, Hudemann C, Hanschmann EM, Lillig CH. Identification of potential protein dithiol-disulfide substrates of mammalian Grx2. Biochim Biophys Acta. 2013;1830(11):4999–5005. doi: 10.1016/j.bbagen.2013.07.009. [DOI] [PubMed] [Google Scholar]