

Abstract
Hypophosphatasia (HPP) is an inborn-error-of-metabolism disorder characterized by deficient bone and tooth mineralization due to loss-of function mutations in the gene (Alpl) encoding tissue-nonspecific alkaline phosphatase (TNAP). Alpl−/− mice exhibit many characteristics seen in infantile HPP including long bone and tooth defects, vitamin B6 responsive seizures and craniosynostosis. Previous reports demonstrated that a mineral-targeted form of TNAP rescues long bone, verterbral and tooth mineralization defects in Alpl−/− mice. Here we report that enzyme replacement with mineral-targeted TNAP (asfotase-alfa) also prevents craniosynostosis (the premature fusion of cranial bones) and additional craniofacial skeletal abnormalities in Alpl−/− mice. Craniosynostosis, cranial bone volume and density, and craniofacial shape abnormalities were assessed by microsocopy, histology, digital caliper measurements and micro CT. We found that craniofacial shape defects, cranial bone mineralization and craniosynostosis were corrected in Alpl−/− mice injected daily subcutaneously starting at birth with recombinant enzyme. Analysis of Alpl−/− calvarial cells indicates that TNAP deficiency leads to aberrant osteoblastic gene expression and diminished proliferation. Some but not all of these cellular abnormalities were rescued by treatment with inorganic phosphate. These results confirm an essential role for TNAP in craniofacial skeletal development and demonstrate the efficacy of early postnatal mineral-targeted enzyme replacement for preventing craniofacial abnormalities including craniosynostosis in murine infantile HPP.
Keywords: craniofacial; craniosynostosis; mineralization; disorders of calcium/phosphate; animal models; micro computed tomography (micro CT, µCT)
1.1 Introduction
Tissue-nonspecific alkaline phosphatase (TNAP) is a well-established and essential enhancer of tissue mineralization [1–3]. TNAP is GPI-anchored to the surface of bone forming cells [4], where it functions to promote hydroxyapatite crystal growth by increasing inorganic phosphate (Pi) to pyrophosphate (PPi) concentration ratios, and dephosphorylating osteopontin [5–8]. Hypophosphatasia (HPP) is the metabolic disorder that occurs due to loss-of function mutations in the gene (Alpl) encoding TNAP [9–11]. Clinical manifestations of HPP are heterogenous in terms of severity and age of onset, with more severe forms of the disorder initiating in utero or infancy [3,12]. Infants affected with HPP can exhibit defective bone and tooth mineralization, epileptic seizures due to diminished hydrolysis of pyridoxal-5’-phosphate (PLP) leading to vitamin B6 deficiency, and craniosynostosis.
During skull growth, cranial bones ultimately come into close proximal relationships but continue to increase in size via new bone formation along outer edges, with maintenance of undifferentiated tissue (cranial suture tissue) between the growing bones. When suture tissue is prematurely lost, adjacent cranial bones become prematurely fused (craniosynostosis) [13]. This can lead to limited growth in the area of fusion, abnormal skull and facial shapes, and high intracranial pressure requiring cranial surgery [14–16]. Craniosynostosis is an established attribute of infantile HPP [3, 10, 12, 17,18]. Craniosynostosis in a rachitic disorder such as hypophosphatasia may seem counterintuitive, yet craniosynostosis is also associated with other forms of rachitic disease [19–24]. We recently reported that Alpl−/− mice exhibit craniofacial skeletal abnormalities and craniosynostosis similar to that seen in infants with HPP [25]. Alpl−/− mice therefore serve as an excellent model to further understand the pathogenesis of rachitic craniosynostosis. In an open label phase 1 clinical trial, children suffering from HPP were treated daily with mineral-targeted TNAP enzyme (asfotase-alfa) starting months to years after birth. This treatment was highly efficacious for rescuing long bone defects seen in these patients, yet many children still manifested craniosynostosis [26]. Because craniosynostosis develops during fetal or early postnatal life and cannot be reversed without surgery after it has occurred [27–29], we hypothesize that treatment must be initiated at earlier developmental time points to affect craniosynostosis.
The Alpl−/− mouse model of infantile HPP was previously used to investigate abnormalities that occur in association with HPP [1,25,30,31], and to establish that treatment with a mineral-targeted form of TNAP can rescue rib, vertebral, long bone and tooth mineralization defects in murine HPP [32–34]. Here, we show that TNAP enzyme replacement can also diminish craniofacial skeletal abnormalities and prevent craniosynostosis in Alpl−/− mice when initiated shortly after birth.
1.2 Materials and Methods
1.2.1 Animals
Preparation and genotyping of Alpl+/− mice were previously reported [1,6,33]. Alpl+/− mice were backcrossed against 50% C57Bl/6 – 50% 129SF2/J wild type (Alpl+/+) mice for ten generations prior to crossing Alpl+/− × Alpl+/− mice for generating progeny used in this study. This mixed genetic background was maintained intentionally as it results Alpl−/− mice with variable phenotype severity, similar to the phenotype variability observed in human infantile HPP. Mice were given free access to modified rodent diet containing increased levels of pyridoxine (vitamin B6, 325 ppm) to suppress seizures and extend life span in Alpl−/− mice. Animal use followed federal guidelines for the care and use of animals in research.
Alpl−/− mice on this background are variable in terms of severity of their hypophosphatasia phenotype. To take phenotype variability into account, severity of the long bone ossification defect in Alpl−/− mice was scored using mouse paw radiographs (Supplemental Information, Figs. 1 and 2), as follows. A normal paw phenotype shows all secondary ossification centers present and mineralized digits. A mild phenotype shows some secondary ossification centers are absent and mineralized digits. A moderate phenotype shows all secondary ossification centers absent and mineralized digits. A severe phenotype shows profound dysmorphology, with absent ossification centers and no mineralization of digits.
1.2.2 Enzyme Replacement
Recombinant TNAP enzyme (asfotase-alfa) is composed of soluble human TNAP fused to the constant region of human IgG1 (Fc), and a C-terminal deca-aspartate motif that confers targeting to hydroxyapatite [26,35]. Alpl−/− mice were injected daily subcutaneously with vehicle, 2.0 or 8.2 mg/kg of protein starting at birth (4 mL/kg), according to previously established protocols for rescue of murine HPP. Efficacy of this dose and delivery method for this recombinant form of TNAP was previously shown to increase serum alkaline phosphatase levels; and prevent long bone, rib, vertebral and tooth defects in murine HPP [32–34]. Craniofacial skeletal assessments were performed on treated Alpl−/− mice, as compared to vehicle treated Alpl−/− mice and wild type littermates. Animal genotype and treatment group was blinded for analyses then unblinded for statistics.
Previous studies showed that dose levels of mineral-targeted TNAP required for rescue of mineralization defects in Alpl−/− mice is dependent upon body site [33]. Craniosynostosis occurs in Alpl−/− mice between two and three weeks after birth. To compare dose levels required for preventing craniosynostosis, Alpl−/− mice were injected with 2.0 (n=8) or 8.2 mg/kg/day (n=6) of recombinant enzyme, and compared to control Alpl−/− (n=24) and Alpl+/+ mice (n=24) three to six weeks after birth. To establish craniofacial skeletal shape and cranial bone mineralization changes which occur prior to the onset of craniosynostosis, and to determine if treatment influences these independently of craniosynostosis, Alpl−/− mice were injected with 8.2 mg/kg/day (n=45) of recombinant enzyme and compared to untreated Alpl−/− (n=46) and Alpl+/+ mice (n=46) and at approximately two weeks after birth.
1.2.3 Micro computed tomography
Whole skulls were scanned at an 18 µm isotropic voxel resolution using the eXplore Locus SP micro-computed tomography imaging system (GE Healthcare Pre-Clinical Imaging, London, ON, Canada), as previously described [25,38]. Density, volume and mineral content of cranial bones from P15 mice were measured using previously established methods and regions of interest using Microview version 2.2 software (GE Healthcare Pre-Clinical Imaging, London, ON) and established algorithms [38–40].
1.2.4 Craniosynostosis Assessment
Fusion of the coronal suture (fusion between frontal and parietal bones) was identified on micro CT scans of skulls dissected from control Alpl−/− mice, 2.0 mg/kg/day treated Alpl−/− mice, 8.2 mg/kg/day treated Alpl−/− mice, and Alpl+/+ mice. Cranial sutures were viewed using the two-dimensional micro-CT slices in an orthogonal view across the entire length of the coronal suture, as previously described [25,37,38]. Bone fusions identified on micro CT images were verified by visualization of specimens under a dissecting microscope (Leica M60 TL5000; Leica Microsystems Inc., Buffalo Groves, IL).
1.2.5 Linear Craniofacial Measurements
Digital calipers were used to conduct craniofacial linear skeletal measurements because identification of some skeletal landmarks on micro CT scans can be difficult due to craniofacial bone hypomineralization in untreated Alpl−/− mice. P15 (approximately two week-old) vehicle treated Alpl−/−, 8.2 mg/kg/day treated Alpl−/− and control Alpl+/+ mouse skulls were carefully dissected and fixed. Linear measurements were calculated using previously reported craniofacial skeletal landmarks [25,36–38], including five standard measurements currently in use by the Craniofacial Mutant Mouse Resource of Jackson Laboratory (Bar Harbor, ME). Linear measurements were normalized to total skull length (measured from nasale to opisthion) to account for potential size differences between Alpl−/− and Alpl+/+ mice. No significant difference between genders was found therefore genders were combined for comparison of genotype and treatment groups.
1.2.6 Craniofacial Morphometric Analysis
A subset of thirty-three previously established craniofacial landmarks were placed on micro CT scans of the P15 skulls, and included only those landmarks that were readily visualized on micro CT scans of all samples (Supplemental Information, Fig. 3) [36,38]. Landmarks were placed and landmark coordinate data was generated using Dolphin Imaging 11.0 software (Dolphin Imaging and Management Solutions, Chatsworth, CA), as previously reported [38]. Landmark coordinate data was imported into WinEDMA 1.0.1 software (Department of Basic Medical Science, School of Medicine, University of Missouri, Kansas City, MO) for comparison of form, size and shape by Euclidean Distance Matrix Analysis (EDMA) [38,41]. Briefly, EDMA is a morphometric analysis that uses landmark x,y,z coordinate data without using a fixed coordinate axis. The analysis calculates all the linear distances between all possible pairs of landmarks in each individual and compares these distances as ratios between groups.
1.2.7 Histology
For microscopic visualization of mineralized tissues; dissected and fixed skulls were serially dehydrated, washed in isopropanol, incubated in xylene and then embedded in methyl methacrylate. This method does not remove mineral and therefore allows for assessment of bone fusion. Methacrylate blocks were trimmed in the sagittal plane to within 4 mm of the sagittal suture. 4 µm sections perpendicular to the coronal suture were prepared with a Leica RM2255 microtome equipped with a tungsten carbide blade (Leica Microsystems Inc., Buffalo Groves, IL). Sections were transferred to slides and dried overnight. Sections were stained for mineral by incubation in 5% AgNO3 (Von Kossa stain) followed by counter-staining in a 1% toludine blue 1% sodium borate solution.
To assess proliferation in mouse calvarial tissues; fixed and decalcified tissues from 5 dayold mice were paraffin embedded. 8 µm sections perpendicular to the coronal suture were sectioned and stained using anti-Ki67 primary antibody (Spring Bioscience), a horse radish peroxidase-conjugated secondary antibody, colorimetric detection with chromogen diaminobenzidine (DAB) and counter staining with hematoxylin and eosin.
1.2.8 Cell preparation and culture
Primary cells were isolated from dissected calvaria by collagenase digestion, as previously described [38,42]. Briefly, bones were rinsed with media then serially digested in a solution containing 2 mg/ml collagenase P and 1 mg/ml trypsin. Cells from the third digestion were utilized for experiments. TNAP specific shRNA and non-target shRNA expressing MC3T3E1(C4) pre-osteoblastic calvarial cells were previously characterized [25]. Cells were induced to differentiate by culture in custom formulated aMEM media containing no phosphate, supplemented with 50 ug/ml ascorbate, 10% fetal bovine serum (FBS), and 10,000 ug/ml penicillin/streptomycin (P/S). Where indicated, sodium phosphate was added at a final concentration of 5 mM. RNA was isolated using Trizol reagent (Invitrogen) following manufacturer protocols. mRNA levels were assayed by reverse transcription and real time PCR. Real time PCR was performed utilizing the murine glyceraldehye 3-phosphate dehydrogenase (GAPDH) primer/probe set Mm01545399_m1, the murine osteocalcin (OCN) Mm03413826_mH primer/probe set, the murine bone sialoprotein (BSP) primer/probe set Mm00492555_m1, the murine tissue non-specific alkaline phosphatase (TNAP) primer/probe set Mm00475834_m1, the murine Runx2 primer/probe set Mm00501578_m1 and Taqman Universal PCR Master Mix (Applied Biosystems). Real-time PCR was performed on a GeneAmp 7700 thermocyler (Applied Biosystems) and quantified by comparison to a standard curve. mRNA levels are reported after normalization to GAPDH mRNA levels. Alkaline phosphatase enzyme activity was assayed using the colorimetric substrate, NBT/ BCIP (Sigma). For quantification, wells were scanned and densitometry was measured using NIH Image software. To assay cellular proliferation, cells were seeded and grown in media containing 10% fetal bovine serum for five days. Cells were stained with trypan blue and counted in quintuplet at each time point.
1.3 Results
1.3.1 Mineral-targeted enzyme replacement prevents craniosynostosis in Alpl−/− mice
Bony fusion between frontal and parietal bones (fusion of the coronal suture) occurs in severely affected Alpl−/− mice (approximately one third of all Alpl−/− mice) between two and three weeks after birth [25]. Subcutaneous daily injection of a mineral-targeted soluble recombinant form of TNAP (asfotase-alfa) at 2.0 mg/kg and 8.2 mg/kg dose levels was previously shown to increase serum alkaline phosphatase levels, extend life and rescue long bone, vertebral and dental defects in Alpl−/− mice [31–34]. Here we show that TNAP enzyme replacement prevents craniosynostosis. Craniosynostosis in the form of coronal suture fusion is evident in vehicle treated Alpl−/− mice, but not in Alpl−/− mice treated with mineral-targeted TNAP enzyme; as demonstrated by micro CT, microscopic visualization of dissected skulls and histology (Fig. 1). Craniosynostosis was evident in 29% of control Alpl−/− mice. Craniosynostosis was not seen in any Alpl−/− mice treated with low or high dose recombinant TNAP, and the incidence of coronal suture fusion was significantly higher in vehicle treated compared to asfotase-alfa treated Alpl−/− mice (p<.05, two tailed Fisher’s exact test).
Figure 1. Mineral-targeted TNAP enzyme replacement prevents craniosynostosis in murine HPP.
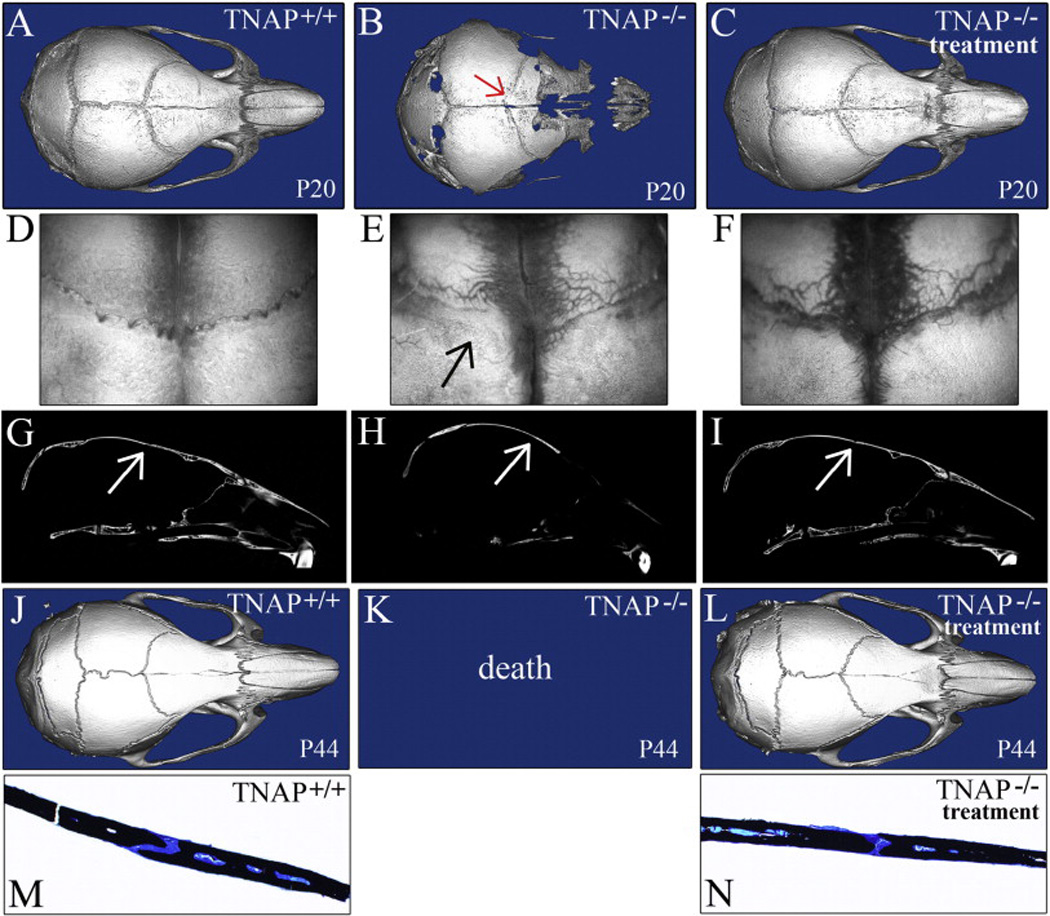
A,B,C) Isosurface micro CT images show coronal suture fusion present in vehicle treated P20 Alpl−/− mouse with severe HPP phenotype (B), but not in 2.0 mg/kg/day treated Alpl−/− (C) or in Alpl+/+ (A) mice (red arrow points to fused coronal suture in vehicle treated P20 Alpl−/− mouse). D,E,F) Dissected skulls of P20 mice were visualized through a dissection microscope. Fusion of the coronal suture is apparent in vehicle treated Alpl−/− (E) but not in treated Alpl−/− (F) or in Alpl+/+ (D) mice (black arrow points to fused coronal suture in untreated P20 Alpl−/− mice). G,H,I) Micro CT lateral slice images show coronal suture fusion present in vehicle treated P20 Alpl−/− mice (B), but not in 2.0 mg/kg/day treated Alpl−/− (C) or in Alpl+/+ (A) mice (arrows points to the coronal suture). J,K,L) Isosurface micro-CT images show that treated P44 Alpl−/− mice show no evidence of craniosynostosis. Skulls of P44 8.2 mg/kg/day treated mice (I) appear normal, as compared to Alpl+/+ (G) mice (H). Because vehicle treated Alpl−/− mice do not survive past three weeks post-birth, vehicle treated P44 Alpl−/− cannot be shown. M,N) Histologic staining of non-decalcified bone surrounding the coronal suture of P44 skulls is shown (black stain is mineralized tissue (32×). The coronal suture was fused in 29% of untreated Alpl−/− mice, but not in Alpl+/+ or asfotase-alfa treated Alpl−/− mice, regardless of dose.
1.3.2 Mineral-targeted enzyme replacement rescues mineralization defects in the craniofacial skeleton of Alpl−/−mice
To further establish abnormalities prior to the onset of craniosynostosis and determine if recombinant TNAP enzyme therapy is efficacious for preventing these defects, we next investigated treatment effects in two week-old mice. Micro CT isosurface images of 15 day-old Alpl−/− mice treated with 8.2 mg/kg/day of recombinant enzyme (Fig. 1E,F) show apparent rescue of craniofacial bone mineralization defects, when compared to vehicle treated Alpl−/− (Fig. 1C,D) and wild type mice (Fig. 1A,B). Consistent with the typical HPP phenotype, multiple cranial vault and facial bones of untreated Alpl−/− mice lack adequate mineralization to appear on micro CT scans constrained to a bone tissue threshold. This hypomineralization phenotype is not evident in asfotase-alfa treated Alpl−/− mice. Interestingly, micro CT based quantification of individual cranial bones revealed a surprising phenotype involving increased frontal bone mineral volume, density and mineral content in vehicle treated Alpl−/− mice, as compared to wild type mice at post-natal day 15 (Table 1). This phenotype was not seen in the treated Alpl−/− mice. Tissue mineral content of the parietal bone was also increased in vehicle treated P15 Alpl−/− mice but not in asfotase-alfa treated Alpl−/− mice. Together, these results indicate that treatment with mineral-targeted TNAP normalizes both hypomineralization (eg: maxilla, zygoma and anterior aspects of frontal bone) and hypermineralization (eg: parietal bone and posterior aspects of frontal bone) defects seen in the craniofacial skeleton of Alpl−/− mice.
Table I.
Cranial Bone Volume, Density and Mineral Content in untreated vs. treated TNAP−/− and TNAP+/+ Mice.
| Bone Volume Fraction |
Bone Mineral Content (mg) |
Bone Mineral Density (mg/cc) |
Tissue Mineral Content (mg) |
Tissue Mineral Density (mg/cc) |
|
|---|---|---|---|---|---|
| TNAP−/− vehicle frontal | 0.31 +/− .16# | .007 +/− .002*# | 441 +/− 67*# | .003 +/− .002# | 602 +/− 31*# |
| TNAP−/− asfotase-alfa frontal | 0.23 +/− .10 | .006 +/− .001 | 401 +/− 34 | .002 +/− .001 | 580 +/− 26 |
| TNAP+/+ frontal | 0.25 +/− .15 | .006 +/− .002 | 411 +/− 49 | .002 +/− .002 | 569 +/− 28 |
| TNAP−/− vehicle parietal | 0.32 +/− .13 | .007 +/− .001 | 432 +/− 39 | .003 +/− .002 | 594 +/− 27* |
| TNAP−/− asfotase-alfa parietal | 0.29 +/− .12 | .006 +/− .001 | 415 +/− 42 | .003 +/− .001 | 579 +/− 32 |
| TNAP+/+ none parietal | 0.37 +/− .12 | .007 +/− .001 | 443 +/− 37 | .003 +/− .002 | 579 +/− 27 |
Indicates statistical significance between genotypes.
indicates statistical significance between treatment groups.
1.3.3 Mineral-targeted enzyme replacement diminishes craniofacial shape abnormalities in Alpl−/− mice
Alpl−/− mice exhibit craniofacial shape abnormalities within two weeks after birth [25]. Daily treatment with 8.2 mg/kg of mineral-targeted TNAP rescued these abnormalities to a large extent. Digital caliper linear measurements normalized to skull length demonstrate that nose length, nasal bone length and frontal bone length are shorter in P15 vehicle treated Alpl−/− mice, when compared to treated Alpl−/− or Alpl+/+ mice (Fig. 3A). Because severity of the HPP phenotype is variable in Alpl−/− mice on this mixed genetic background, we also compared craniofacial shape differences by long bone phenotype severity. These results reveal more extreme craniofacial skeletal defects in P15 Alpl−/− mice that are dependent both upon severity of the HPP phenotype and TNAP enzyme supplementation (Fig. 3B). Cranial height, cranial width and inner canthal distance are significantly larger; while nose length, nasal bone length and frontal bone length are significantly smaller in Alpl−/− mice of the severe long bone ossification phenotype category, when compared to treated Alpl−/− and Alpl+/+ mice. All Alpl+/+ and 44 of 46 treated Alpl−/− mice were scored as having normal long bone ossification phenotypes and all of these mice also had a normal craniofacial shape. Two asfotase-alfa treated Alpl−/− mice were scored as having mild long bone ossification phenotypes. These mice also had a normal craniofacial shape and were excluded from analyses due to limited sample numbers.
Figure 3. TNAP enzyme replacement normalizes craniofacial shape abnormalities in murine HPP.

A) When grouped by genotype and treatment, linear craniofacial measurements of P15 mice show that vehicle treated Alpl−/− mice exhibit increased cranial height and decreased nose length, nasal bone length and frontal bone length when compared to Alpl+/+ mice or treated Alpl−/− mice. Measurements of asfotase-alfa treated Alpl−/− mice are not different than those of wild type mice. Grey = vehicle treated Alpl−/− (n=45), white = Alpl+/+ (n=46), black = 8.2 mg/kg/day treated Alpl−/− (n=46). B) Severity of the HPP phenotype is variable in Alpl−/− mice on this mixed genetic background. When separated by genotype, treatment and severity of the long bone ossification defect; greater craniofacial measurement differences become apparent between vehicle treated, treated and wild type mice. Vehicle treated Alpl−/− mice with a severe long bone ossification defect exhibit increased cranial height, cranial width and inner canthal distance; with decreased nose length, nasal bone length and frontal bone length when compared to Alpl+/+ mice or treated Alpl−/− mice. All Alpl+/+ and 44 of 46 treated Alpl−/− were of the normal bone ossification phenotype, therefore only the normal phenotype of these groups is presented and analyzed. Data is presented as means +/− standard deviations. *p <.05 vs. Alpl+/+, #p <.05 vs. treated Alpl−/− mice. C) Representative craniofacial landmark distance EDMA mean ratios calculated using x,y,z coordinate data from micro CT scans, are shown on a mouse skull sagittal micro CT section. Yellow lines indicate those distances that are significantly larger in P15 vehicle treated Alpl−/− mice as compared to 8.2 mg/kg/day treated Alpl−/− mice. White lines indicate those distances that are significantly smaller in vehicle treated d Alpl−/− mice as compared to treated Alpl−/− mice. Note that smaller distances in the vehicle treated Alpl−/− mouse skull involve anterior-posterior dimensions while larger distances involve vertical cranial vault dimensions. Cranial vault width dimensions were also larger in vehicle treated when compared to treated Alpl−/− skulls, as assessed by EDMA.
To substantiate these results, three-dimensional coordinate data from landmarks placed on micro CT scans of mouse skulls were also used to compare craniofacial form and shape by Euclidean Distance Matrix Analysis (EDMA). Results show that the overall craniofacial skeletal form (form includes size and shape) of vehicle treated Alpl−/− mice is different than that of Alpl−/− mice treated with 8.2 mg/kg/day of recombinant TNAP (T = 1.10, p <.001) by two weeks after birth. Shape analysis using geometric mean as a scaling factor confirmed that vehicle treated vs. mineral-targeted TNAP treated skulls are also statistically different in shape by two weeks after birth (Z = −0.052, α = 0.01). EDMA shape confidence intervals for individual linear measurements were consistent with results of our digital caliper analysis. Anterior-posterior skull length distances were smaller while skull height distances were larger in vehicle treated, when compared to treated Alpl−/− mice (Fig. 3C). Together these results support our subjective assessment that TNAP enzyme replacement minimizes the skull and facial shape defects seen in affected Alpl−/− mice. Overall vehicle treated Alpl−/− mice have skulls are acrocephalic (tall) and brachycephalic (wide), when compared to those of treated of Alpl−/− mice and wild type littermates.
1.3.4 Role of inorganic phosphate (Pi) in TNAP deficiency induced cellular abnormalities
We previously showed that calvarial MC3T3E1(C4) cells in which TNAP expression was suppressed by shRNA exhibit cellular abnormalities including aberrant osteoblastic gene expression and diminished proliferation [25]. Consistent with this data, primary cells isolated from Alpl−/− calvaria express higher levels of Runx2, osteocalcin (OCN) and bone sialoprotein (BSP) mRNA before osteoblast differentiation in culture (Fig. 4C–E) and lower levels upon differentiation (Fig. 4F–H), when compared to wild type cells. Alpl−/− cells are also less proliferative than Alpl+/+ cells (Fig. 4I). Diminished proliferation of bone lining and suture cells is also apparent in Alpl−/− calvaria (Fig. 4K) as compared to Alpl+/+ (Fig. 4J). Regions of high proliferation are evident along the leading edge of both frontal and parietal bones adjacent to the coronal suture in wild type mice. Less proliferation is evident in these and other regions of Alpl−/− calvarial tissues.
Figure 4. Gene expression and proliferation changes in Alpl−/− calvarial cells.
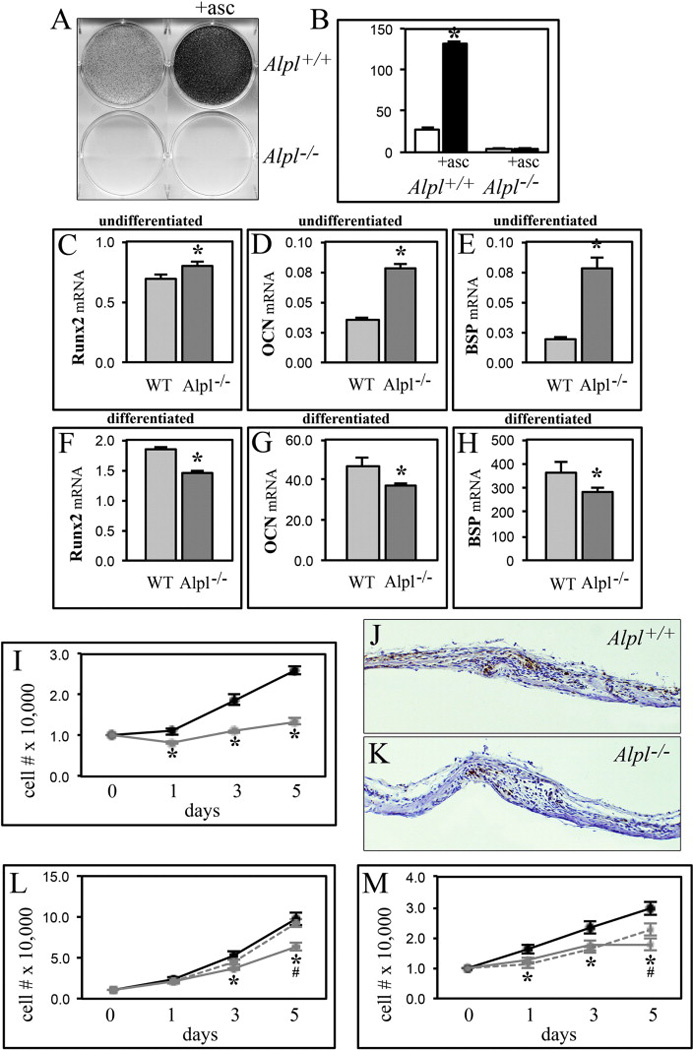
(A,B) Primary calvarial cells were cultured with ascorbate to induce osteoblast differentiation. Alkaline phosphatase enzyme activity was visualized by incubation of cells with a colorimetric substrate and quantified by densitometry. (C–H) RNA was isolated from cells after culture with or without ascorbate to induce osteoblast differentiation. Runx2, osteocalcin (OCN) and bone sialoprotein (BSP) mRNA levels were measured by real time PCR. Results are presented as normalized to GAPDH. Light grey = wild type (WT), dark grey = Alpl−/−. (I) Cells were stained with trypan blue and counted at indicated time points after plating to assay for proliferation. (J,K) Calvarial tissue sections were stained with Ki67 antibody as a marker of proliferation. 10× magnification of parietal and frontal bones surrounding the coronal suture from P5 Alpl+/+ (J) and Alpl−/− (K) mice are shown. Note the diminished number of proliferative cells in Alpl−/− calvaria. (L) MC3T3E1(C4) cells stably transduced with TNAP specific or non-target shRNA were cultured in media supplemented with/without 5mM sodium phosphate. Cells were stained with trypan blue and counted at indicated time points after plating to assay for proliferation. Black = nontarget shRNA, solid grey = TNAP shRNA, dashed grey = TNAP shRNA with sodium phosphate. (M) Primary cavarial cells were cultured in media supplemented with/without 5mM sodium phosphate. Cells were stained with trypan blue and counted at indicated time points after plating to assay for proliferation. Black = Alpl+/+, solid grey = Alpl−/−, dashed grey = Alpl−/− with sodium phosphate. *p<.05 vs. wild type cells for all panels.
Alpl−/− mice do not exhibit systemic hypophosphatemia but TNAP does function to generate inorganic phosphate (Pi) at the extracellular membrane [5,6]. Extracellular Pi influences matrix mineralization, and can also stimulate changes in gene expression and proliferation [43–46]. Therefore, we next investigated the potential for Pi to rescue proliferation and gene expression abnormalities seen in TNAP deficient cells. Results show that addition of Pi to culture media increased the proliferation of TNAP deficient MC3T3E1(C4) cells to the levels seen in control cells (Fig. 4L). Consistently, Pi also increased the proliferation of Alpl−/− cells (Fig. 4M). Treatment with Pi did not rescue gene expression differences between TNAP deficient and control MC3T3E1(C4) cells, or between Alpl−/− and Alpl+/+ cells (data not shown). Together these results suggest that diminished extracellular Pi mediates some but not all of the cellular changes induced by TNAP deficiency.
1.4 Discussion
Alpl−/− mice exhibit metabolic, long bone, tooth and craniofacial skeletal abnormalities similar to those seen in human infantile HPP [1,25,30,31], and these mice were previously used to demonstrate that daily subcutaneous injections of the mineral-targeted form of TNAP (asfotase-alfa) can increase serum alkaline phosphatase levels, diminish seizures, extend life, and rescue long bone, vertebral and rib defects in a murine model of infantile HPP [32–34]. Success of this work supported testing of this form of TNAP in infants and children severely affected with HPP. Published results from a phase 1 open label study demonstrated that TNAP enzyme replacement diminishes plasma levels of the TNAP substrates inorganic pyrophosphate (PPi) and pyridoxal 5′-phosphate (PLP), improves skeletal mineralization, respiratory function, motor skills and cognitive development in patients with early and severe onset of HPP [26]. Notably though, the incidence of craniosynostosis was unchanged by treatment. Because craniosynostosis cannot be reversed after it has occurred, this finding suggests either that the treatment is not efficacious for preventing craniosynostosis or that the treatment was initiated after the onset of craniosynostosis in these patients. Results presented here demonstrate that daily subcutaneous injections of mineral-targeted TNAP initiated shortly after birth prevents craniosynostosis in murine infantile HPP. Approximately one third of untreated Alpl−/− mice develop craniosynostosis in the form of bony coronal suture fusion by three weeks after birth [25]. No Alpl−/− mice treated daily with injections of mineral-targeted TNAP showed any evidence of craniosynostosis at the low or high dose level. Infantile hypophosphatasia is a progressive disorder and craniosynostosis in HPP and other rachitic disorders tends to occur later than that seen in other forms of craniosynostosis [28]. Given early diagnosis, these results therefore support testing of recombinant TNAP enzymes for preventing or diminishing the severity of craniosynostosis in humans. In utero delivery systems will be needed to prevent prenatal onset craniosynostosis in humans [47]. Studies investigating the long-term impact of enzyme replacement on the skull are also warranted.
Craniosynostosis only occurs in untreated Alpl−/− mice with a severe long bone (paw) ossification defect. Skulls of mice scored as having a severe paw ossification phenotype also present with a more severe craniofacial hypomineralization phenotype, in that only Alpl−/− mice with a severe paw phenotype have craniofacial bones that are hypomineralized to the extent that they do not appear in micro CT scans constrained to a bone tissue threshold (Fig. 1C,D). The fact that craniosynostosis only occurs in Alpl−/− mice with a severe bone mineralization phenotype could indicate that craniosynostosis occurs indirectly, as a result of cellular changes that occur in response to severely abnormal cranial bone mineralization. Alternatively, this finding could indicate that compensators for TNAP exist that function to influence both bone mineralization and craniosynostosis. A previous study showed that the skulls of Alpl−/− mice treated with 16 mg/kg/day of recombinant soluble alkaline phosphatase appeared similar to those of Alp+/+ mice [48]. Future studies will be needed to determine if mineral-targeting of TNAP alters efficacy for preventing craniosynostosis, as compared directly to treatment with soluble forms of alkaline phosphatase. Future studies comparing phenotype severity and craniosynostosis incidence with systemic biomarker levels, such as inorganic pyrophosphate, will be important for illuminating mechanisms of HPP-associated craniosynostosis and modifiers of HPP phenotype severity.
We next assessed pathogenic changes prior to the onset of craniosynostosis +/− treatment because we are interested in understanding how TNAP deficiency causes craniosynostosis and the potential for enzyme replacement therapy to influence these changes. As expected, TNAP enzyme replacement prevented the cranial bone hypomineralization seen in Alpl−/− mice by two weeks after birth. Multiple facial bones of untreated Alpl−/− mice, for example, are so hypomineralized that they do not appear on micro CT scans constrained to a bone tissue threshold. This is not the case in asfotase-alfa treated Alpl−/− mice. These results are consistent with previously published data showing that TNAP enzyme replacement increases vertebral, rib and long bone hypomineralization in Alpl−/− mice [32–34] and HPP patients [26]. TNAP enzyme replacement also prevented a localized cranial bone hypermineralization phenotype seen in untreated Alpl−/− mice at two weeks after birth. Frontal bones of Alpl−/− mice are hypomineralized by three weeks after birth [25], indicating that the hypermineralization seen at two weeks after birth is transient. Localized hypermineralization of long bone metaphyses has been reported in children with HPP, and theorized to occur as a compensatory response to otherwise mechanically incompetent bone structure [49,50]. Cranial bones are not weight bearing but do experience forces from mastication [51]. Pediatric cranial bones also deform to a significantly greater extent than adult cranial bones in response to mechanical force [52]. It is possible that the transient hypermineralization of cranial bones in two week-old Alpl−/− mice is a compensatory response to weakened overall cranial bone structure. Alternatively, the hypermineralization could be due to abnormal expression of other enzymes essential to mineralization, such ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase 1) and/or PHOSPHO-1 (phosphatase, orphan 1), in response to TNAP deficiency.
Treatment with mineral-targeted TNAP also diminished craniofacial skeletal shape abnormalities in murine HPP. Vehicle treated Alpl−/− mice exhibit an acrocephalic and brachycephalic skull shape (tall, wide and shorter in anterior-posterior length), similar to that seen in some infants affected with HPP [12,25]. Asfotase-alfa treated Alpl−/− mice have skulls that are similar to those of wild type mice. Skulls of treated Alpl−/− mice are less tall, less wide and longer in anterior-posterior length than those of vehicle treated Alpl−/− mice, as assessed using digital calipers or landmark coordinate data from micro CT scans. Because the rescue in craniofacial shape shown here occurred prior to the onset of coronal suture fusion (skulls were analyzed two weeks after birth for shape; coronal suture fusion is not yet seen at this time point), it seems likely that TNAP enzyme replacement normalizes craniofacial skeletal shape through mechanisms in addition to the prevention of craniosynostosis. If mineral-targeted TNAP is able to diminish skull and facial skeletal shape abnormalities in patients, this treatment could be an important augmentation to surgeries used to separate fused cranial bones. Facial shapes influence perceived intelligence and dominance, and improvement in craniofacial deformity enhances quality of life [53–55].
It is worth noting that frontal bone length but not parietal bone length is different between Alpl−/− and wild type mice. Parietal bone length is also not altered by treatment. Micro CT data reported here and in our previous publication showed that mineralization abnormalities are also less severe in parietal than in frontal bone [25]. This data indicates that TNAP plays a more essential role in frontal than parietal bone development. That the influence of TNAP on craniofacial bones is bone specific is also well exhibited in the micro CT images of these mice (Fig. 1). Comparative investigation of more severely affected vs. less severely affected craniofacial bones of Alpl−/− mice could yield information on modifiers of the HPP ossification phenotype.
We previously showed that suppression of TNAP expression by shRNA in calvarial MC3T3E1(C4) cells led to changes in cell behavior independent of matrix mineralization, which suggested a potential direct effect of TNAP on cell behavior [25]. Here we show that calvarial cells isolated from Alpl−/− mice show changes similar to those seen in TNAP deficient MC3T3E1(C4) cells. Expression of Runx2, osteocalcin (OCN) and bone sialoprotein (BSP) were higher in Alpl−/− than in Alpl+/+ primary calvarial cells prior to differentiation, and lower in Alpl−/− than Alpl+/+ cells at later stages of differentiation. Alpl−/− calvarial cells are also less proliferative than Alpl+/+ cells when assessed in vitro and in vivo. TNAP is an ectoenzyme that hydrolyzes ATP and pyrophosphate (PPi) to inorganic phosphate (Pi) and adenosine. This process is essential for bone mineralization even in the presence of systemic normal phosphate, suggesting that local changes in PPi and Pi mediate hydroxyapatite crystal deposition [56] Previous studies also showed that Pi can stimulate signaling to influence osteoblast differentiation and proliferation [44–46,57]. We show here that treatment with Pi rescues the diminished proliferation of TNAP deficient MC3T3E1(C4) and Alpl−/− primary calvarial cells, suggesting that local changes in Pi may mediate proliferative changes induced by TNAP deficiency. Pi treatment did not rescue differences in gene expression between TNAP deficient and control cells (data not shown). Together, these results indicate that TNAP has direct influences on cell behavior that may be mediated in part by extracellular Pi.
Approximately 40% of infants affected with HPP develop craniosynostosis but it is unknown how premature cranial bone fusion occurs in this rachitic disorder [17]. Our results implicate multiple potential mechanisms by which TNAP deficiency could lead to craniosynostosis. TNAP deficiency could promote premature osteoblast differentiation of cranial suture cells, even while inhibiting the full functionality of mature osteoblasts. TNAP deficiency could also diminish proliferation by cranial suture and/or bone lining cells. These types of differentiation and proliferation changes could diminish available undifferentiated cranial suture tissue over time, potentiating the tendency for craniosynostosis [13]. Alternatively, cells within the cranial bone and suture environment could respond to TNAP deficiency by upregulating other promineralization factors. This latter possibility is supported by the fact that transient hypermineralization is evident in Alpl−/− frontal bones prior to the onset of craniosynostosis in these mice. Distinguishing between these potential mechanisms will be important for understanding how craniosynostosis occurs in HPP and other rachitic disorders.
Supplementary Material
Figure 2. Qualitative rescue of craniofacial hypomineralization and shape abnormalities by mineral-targeted TNAP enzyme replacement prior to craniosynostosis, in murine HPP.

Micro CT based, isosurface images of 15-day post-natal Alpl+/+ (A,B), vehicle treated Alpl−/− (C,D) and treated Alpl−/− mouse skulls (E,F) are shown. The shown untreated Alpl−/− mouse has the severe HPP phenotype. Treated mice received daily injections of 8.2 mg/kg of mineral-targeted recombinant TNAP enzyme starting as newborns. Multiple cranial vault and facial bones of vehicle treated Alpl−/− mice are severely hypo-mineralized when compared to those of mineral-targeted TNAP treated Alpl−/− and Alpl+/+ skulls. Notably, the parietal and intraparietal bones appear less affected while the anterior aspect of the frontal and facial bones are too deficient in mineralization to appear on micro CT scans constrained to a bone tissue threshold. The vehicle treated Alpl−/− skull also presents decreased anterior-posterior length but increased height and width when compared to the treated Alpl−/− and Alpl+/+ skulls, and is more dome-shaped in overall appearance.
Highlights.
Treatment with a mineral-targeted form of soluble TNAP significantly diminishes the incidence of craniosynostosis in murine infantile hypophosphatasia
Treatment with a mineral-targeted form of soluble TNAP normalizes craniofacial shape abnormalities in murine infantile hypophosphatasia
Treatment with a mineral-targeted form of soluble TNAP rescues craniofacial bone hypomineralization and hypermineralization in murine infantile hypophosphatasia
Inorganic phosphate rescues the diminished proliferation seen in TNAP deficient calvarial cells
Acknowledgements
This work was supported by a National Institute of Health collaborative supplement to grant R01 DE012889 (to J.L.M.) and by a research grant (to N.E.H.) from Alexion Pharmaceuticals Inc. (Chesire, CT), the licensed manufactures of asfotase-alfa (the mineral-targeted form of TNAP enzyme utilized in this study). Alexandre Caron was employed by Alexion Pharmaceuticals Inc. during the time of data collection for this study.
Abbreviations used are
- TNAP
tissue-nonspecific alkaline phosphatase
- HPP
hypophosphatasia
- Pi
inorganic phosphate
- PPi
inorganic pyrophosphate
- micro CT
micro computed tomography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
The authors have no other potential conflicts of interest to report.
References
- 1.Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432–446. doi: 10.1002/(SICI)1097-0177(199703)208:3<432::AID-AJA13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 2.Murshed M, Harmery D, Millan JL, McKee MD, Karsenty G. Unique coexpression I osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes and Dev. 2009;19:1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.OnlineMendelian Inheritance in Man and OMIM, MIM Number 171760. Baltimore, Md, USA: Johns Hopkins University; 2009. http://omim.org/entry/171760. [Google Scholar]
- 4.Low MG, Saltiel AR. Structural and functional roles of glycosyl-phosphatidylinositol in membranes. Science. 1988;239(4837):268–275. doi: 10.1126/science.3276003. [DOI] [PubMed] [Google Scholar]
- 5.Johnson KA, Hessle L, Vaingankar S, Wennberg C, Mauro S, Narisawa S, et al. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1365–R1377. doi: 10.1152/ajpregu.2000.279.4.R1365. [DOI] [PubMed] [Google Scholar]
- 6.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harmey D, Johnson KA, Zelken J, Camacho NP, Hoylaerts MF, Noda M, et al. Elevated skeletal osteopontin levels contribute to the hypophosphatasia phenotype in Akp2(−/−) mice. J Bone Miner Res. 2006;21(9):1377–1386. doi: 10.1359/jbmr.060619. [DOI] [PubMed] [Google Scholar]
- 8.Narisawa S, Yadav MC, Millán JL. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J Bone Miner Res. 2013;28(7):1587–1598. doi: 10.1002/jbmr.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007;2:40. doi: 10.1186/1750-1172-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- 11.Millan JL, editor. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Wiley-VCH; 2006. [Google Scholar]
- 12.Fraser D. Hypophosphatasia. Am J Med. 1957;22(5):730–746. doi: 10.1016/0002-9343(57)90124-9. [DOI] [PubMed] [Google Scholar]
- 13.Rice DP. Developmental Anatomy of Craniofacial Sutures. In: Rice DP, editor. Craniofacial Sutures Development, Disease and Treatment. Karger, Switzerland: 2008. [Google Scholar]
- 14.Cohen MM., Jr Sutural biology and the correlates of craniosynostosis. Am J Med Genet. 1993;47:581–616. doi: 10.1002/ajmg.1320470507. [DOI] [PubMed] [Google Scholar]
- 15.Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207(5):637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen SA, Yazdy MM, Frías JL, Honein MA. Priorities for public health research on craniosynostosis: summary and recommendations from a Centers for Disease Control and Prevention-sponsored meeting. Am J Med Genet A. 2008;146A(2):149–158. doi: 10.1002/ajmg.a.32106. [DOI] [PubMed] [Google Scholar]
- 17.Collmann H, Mornet E, Gattenlöhner S, Beck C, Girschick H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst. 2009;25(2):217–223. doi: 10.1007/s00381-008-0708-3. [DOI] [PubMed] [Google Scholar]
- 18.Wenkert D, Benigno M, Mack K, McAlister W, Mumm S, Whyte M. Hypophosphatasia: prevalence of clinical problems in 175 pediatric patients; Proceedings of the American Society for Bone and Mineral Research 31st Annual Meeting; Denver, Colo, USA. 2009. Abstract A09001674. [Google Scholar]
- 19.Reilly BJ, Leeming JM, Fraser D. Craniosynostosis in the rachitic spectrum. J Pediatr. 1964;64:396–405. doi: 10.1016/s0022-3476(64)80192-x. [DOI] [PubMed] [Google Scholar]
- 20.Willis FR(1), Beattie TJ. Craniosynostosis in X-linked hypophosphataemic rickets. J Paediatr Child Health. 1997;33(1):78–79. doi: 10.1111/j.1440-1754.1997.tb00997.x. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy JG, Reid CA. Craniofacial synostosis in association with vitamin D--resistant rickets. Ann Plast Surg. 1980;4(2):149–153. doi: 10.1097/00000637-198002000-00013. [DOI] [PubMed] [Google Scholar]
- 22.Inman PC, Mukundan S, Jr, Fuchs HE, Marcus JR. Craniosynostosis and rickets. Plast Reconstr Surg. 2008;121(4):217e–218e. doi: 10.1097/01.prs.0000305381.61117.2f. [DOI] [PubMed] [Google Scholar]
- 23.Murthy AS. X-linked hypophosphatemic rickets and craniosynostosis. J Craniofac Surg. 2009;20(2):439–442. doi: 10.1097/SCS.0b013e31819b9868. [DOI] [PubMed] [Google Scholar]
- 24.Roy WA, Iorio RJ, Meyer GA. Craniosynostosis in vitamin D-resistant rickets. A mouse model. J Neurosurg. 1981;55(2):265–271. doi: 10.3171/jns.1981.55.2.0265. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Nam HK, Campbell C, Gasque KC, Millán JL, Hatch NE. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia. Bone. 2014;67:81–94. doi: 10.1016/j.bone.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366(10):904–913. doi: 10.1056/NEJMoa1106173. [DOI] [PubMed] [Google Scholar]
- 27.Connolly JP, Gruss J, Seto ML, Whelan MF, Ellenbogen R, Weiss A, et al. Progressive postnatal craniosynostosis and increased intracranial pressure. Plast Reconstr Surg. 2004;113(5):1313–1323. doi: 10.1097/01.prs.0000111593.96440.30. [DOI] [PubMed] [Google Scholar]
- 28.Seruya M, Oh AK, Boyajian MJ, Myseros JS, Yaun AL, Keating RF, Rogers GF. Age at initial consultation for craniosynostosis: comparison across different patient characteristics. J Craniofac Surg. 2013;24(1):96–98. doi: 10.1097/SCS.0b013e318270fb83. [DOI] [PubMed] [Google Scholar]
- 29.Weber B, Schwabegger AH, Oberaigner W, Rumer-Moser A, Steiner H. Incidence of perinatal complications in children with premature craniosynostosis. J Perinat Med. 2010;38(3):319–325. doi: 10.1515/jpm.2010.028. [DOI] [PubMed] [Google Scholar]
- 30.Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14(12):2015–2026. doi: 10.1359/jbmr.1999.14.12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster BL, Nagatomo KJ, Tso HW, Tran AB, Nociti FH, Jr, Narisawa S, et al. Tooth root dentin mineralization defects in a mouse model of hypophosphatasia. J Bone Miner Res. 2013;28(2):271–282. doi: 10.1002/jbmr.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millan JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777–787. doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yadav MC, Lemire I, Leonard P, Boileau G, Blond L, Beliveau M, et al. Dose response of bone-targeted enzyme replacement for murine hypophosphatasia. Bone. 2011;49(2):250–256. doi: 10.1016/j.bone.2011.03.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKee MD, Nakano Y, Masica DL, Gray JJ, Lemire I, Heft R, et al. Enzyme replacement prevents dental defects in a mouse model of hypophosphatasia. J Dent Res. 2011;90:470–476. doi: 10.1177/0022034510393517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishioka T, Tomatsu S, Gutierrez MA, Miyamoto K, Trandafirescu GG, Lopez PL, et al. Enhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab. 2006;88(3):244–255. doi: 10.1016/j.ymgme.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217(2):137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 37.Perlyn CA, DeLeon VB, Babbs C, Govier D, Burell L, Darvann T, et al. The craniofacial phenotype of the Crouzon mouse: analysis of a model for syndromic craniosynostosis using three-dimensional MicroCT. Cleft Palate Craniofac J. 2006;43(6):740–748. doi: 10.1597/05-212. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Nam HK, Wang E, Hatch NE. Further Analysis of the Crouzon Mouse, Effects of the FGFR2C342Y Mutation are Cranial Bone Dependent. Calc Tissue Int. 2013;92(5):451–466. doi: 10.1007/s00223-013-9701-2. PMCID: 3631296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meganck JA, Kozloff KM, Thornton MM, Broski SM, Goldstein SA. Beam hardening artifacts in micro-computed tomography scanning can be reduced by X-ray beam filtration and the resulting images can be used to accurately measure BMD. Bone. 2009;45(6):1104–1116. doi: 10.1016/j.bone.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Umoh JU, Sampaio AV, Welch I, Pitelka V, Goldberg HA, Underhill TM, et al. In vivo micro-CT analysis of bone remodeling in a rat calvarial defect model. Phys Med Biol. 2009;54(7):2147–2161. doi: 10.1088/0031-9155/54/7/020. [DOI] [PubMed] [Google Scholar]
- 41.Lele S, Richtsmeier JT. An invariant approach to statistical analysis of shapes. Boca Raton, FL: Chapman & Hall/CRC; 2001. [Google Scholar]
- 42.Hatch NE, Li Y, Franceschi RT. FGF2 stimulated expression of the pyrophosphate generating enzyme, PC-1, is mediated by Runx2. J Bone Min Res. 2009;24(4):652–662. doi: 10.1359/JBMR.081213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nam HK, Liu J, Li Y, Kragor A, Hatch NE. Ectonucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1) protein regulates osteoblast differentiation. J Biol Chem. 2011;286(45):39059–39071. doi: 10.1074/jbc.M111.221689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conrads KA, Yi M, Simpson KA, Lucas DA, Camalier CE, Yu LR, et al. A combined proteome and microarray investigation of inorganic phosphate-induced pre-osteoblast cells. Mol Cell Proteomics. 2005;4(9):1284–1296. doi: 10.1074/mcp.M500082-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Khoshniat S, Bourgine A, Julien M, Weiss P, Guicheux J, Beck L. The emergence of phosphate as a specific signaling molecule in bone and other cell types in mammals. Cell Mol Life Sci. 2011;68(2):205–218. doi: 10.1007/s00018-010-0527-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camalier CE, Yi M, Yu LR, Hood BL, Conrads KA, Lee YJ, et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J Cell Physiol. 2013;228(7):1536–1550. doi: 10.1002/jcp.24312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugano H, Matsumoto T, Miyake K, Watanabe A, Iijima O, et al. Successful gene therapy in utero for lethal murine hypophosphatasia. Hum. Gene Ther. 2012;23(4):399–406. doi: 10.1089/hum.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gasque KC, Foster BL, Kuss P, Yadav MC, Liu J, et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl−/− mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015;72:137–147. doi: 10.1016/j.bone.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Girschick HJ, Schneider P, Kruse K, Huppertz HI. Bone metabolism and bone mineral density in childhood hypophosphatasia. Bone. 1999;25(3):361–367. doi: 10.1016/s8756-3282(99)00164-7. [DOI] [PubMed] [Google Scholar]
- 50.Girschick HJ, Haubitz I, Hiort O, Schneider P. Long-term follow-up of bone mineral density in childhood hypophosphatasia. Joint Bone Spine. 2007;74(3):263–269. doi: 10.1016/j.jbspin.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 51.Shibazaki-Yorozuya R, Wang Q, Dechow PC, Maki K, Opperman LA. Changes in biomechanical strain and morphology of rat calvarial sutures and bone after Tgf-β3 inhibition of posterior interfrontal suture fusion. Anat Rec. 2012;295(6):928–938. doi: 10.1002/ar.22474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coats B, Margulies SS. Material properties of human infant skull and suture at high rates. J Neurotrauma. 2006;23(8):1222–1232. doi: 10.1089/neu.2006.23.1222. [DOI] [PubMed] [Google Scholar]
- 53.Kleisner K, Chvátalová V, Flegr J. Perceived intelligence is associated with measured intelligence in men but not women. PLoS One. 2014;9(3):e81237. doi: 10.1371/journal.pone.0081237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torrance JS, Wincenciak J, Hahn AC, DeBruine LM, Jones BC. The relative contributions of facial shape and surface information to perceptions of attractiveness and dominance. PLoS One. 2014;9(10):e104415. doi: 10.1371/journal.pone.0104415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi WS, Lee S, McGrath C, Samman N. Change in quality of life after combined orthodontic-surgical treatment of dentofacial deformities. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109(1):46–51. doi: 10.1016/j.tripleo.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 56.Millán JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. 2013;93(4):299–306. doi: 10.1007/s00223-012-9672-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergwitz C, Jüppner H. Phosphate sensing. Adv Chronic Kidney Dis. 2011;18(2):132–144. doi: 10.1053/j.ackd.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.