

Abstract
The pleural space, a sterile secluded environment in the thoracic cavity, represents an attractive metastatic site for various cancers of lung, breast and gastrointestinal origins. Whereas lung and breast adenocarcinomas could invade the pleural space because of their anatomic proximity, “distant” cancers like ovarian or gastrointestinal tract adenocarcinomas may employ more active mechanisms to the same end. A pleural metastasis is often accompanied by a malignant pleural effusion (MPE), an unfavorable complication that severely restricts the quality of life and expectancy of the cancer patient. MPE is the net “product” of three different processes, namely inflammation, enhanced angiogenesis and vascular leakage. Current efforts are focusing on the identification of cancer cell autocrine (specific mutation spectra and biochemical pathways) and paracrine (cytokine and chemokine signals) characteristics as well as host features (immunological or other) that underlie the MPE phenotype. Herein we examine the pleural histology, cytology and molecular characteristics that make the pleural cavity an attractive metastasis destination for lung adenocarcinoma. Mesothelial and tumor features that may account for the tumor’s ability to invade the pleural space are highlighted. Finally, possible therapeutic interventions specifically targeting MPE are discussed.
Keywords: Lung cancer, pleural membranes, visceral pleural invasion (VPI), malignant pleural effusion (MPE), EGFR mutations
Introduction
The pleural membranes—visceral and parietal—are mesothelial structures lining the lungs and the inside of the thoracic cavity, respectively. These layers define the pleural space, a sterile and protected environment equipped with lymphatics designed to recycle the normal pleural fluid. The fluid allows the sliding of the lung surface against the thoracic wall during breathing. Healthy mesothelial membranes are plastic in nature and perform multiple functions in the embryo and the adult. For example, during organogenesis mesothelial cells undergo epithelial to mesenchymal transition, whereas during adulthood they generate nearly all vasculogenic cells and also a diverse array of stromal cells to populate the internal organs (1). These membranes, which are of themselves innocuous and once thought to be biologically inert, may give rise to a particularly aggressive primary cancer, malignant pleural mesothelioma (MPM), after asbestos exposure (2). Furthermore, the pleural cavity is a frequent metastasis site of “proximal” lung and breast or “distal” gastrointestinal and ovary cancers as well as lymphoma (3). Metastasis is singlehandedly the gravest outcome for a tumor in terms of patient survival. Additionally, metastasis to the pleura may lead to a troublesome complication, malignant pleural effusion (MPE), which hallmarks stage IV disease. MPE severely restricts the patients’ survival to an average of 3-4 months and diminishes their life quality by imposing a number of adverse symptoms such as dyspnea and chest pain. Currently, no specific cure exists for MPE and palliative interventions are the only existing option. Not all tumors metastasizing to the pleura cause MPE (4,5). Autopsy findings show that ~60% of patients with pleural metastasis have effusions (5). Apart from lung adenocarcinoma, neoplasms including MPM, lymphoma as well as breast, colon, gastric and ovary adenocarcinomas may result in MPE (4,5). This suggests a potentially identifiable common MPE “signature” and biological pathways among these diverse tumor types. Furthermore, it suggests that there must be more than the anatomic proximity that defines lung adenocarcinoma expansion and metastasis to the pleural space. Here we will review the current literature by examining the features rendering the pleural space an attractive metastasis site for lung cancer, discuss the mechanisms that could account for pleural metastasis and the generation of MPE and present possible targets for pharmacological and therapeutic interventions.
Biological and histological attributes of lung cancer
Lung cancer, the major killer cancer accounting for millions of deaths every year world-wide (http://seer.cancer.gov/statfacts/html/lungb.html) is a heterogeneous disease group, divided into two major categories, non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC). This distinction is based on the histologic features, response to conventional therapies and the biology of the cancer genome. More specifically, SCLC is generally associated with smoking-elaborated RB1 point mutations, whereas NSCLC includes three cell types (adenocarcinoma, squamous cell carcinoma, and large cell carcinoma), further divided into various subtypes or variants. Lung adenocarcinoma is the most frequent type accounting for 40% of the cases (6). The molecular trends of lung adenocarcinoma include a high mutation rate of 8.87 mutations per megabase DNA (predominantly C to A transversions) and tumors bearing 46% TP53, 33% KRAS and 14% EGFR mutations (7). Transversion-high groups correlate with a positive smoking history and mutations in the KRAS gene.
Epidemiologically, gender differences exist because female non-smokers appear prone to transversion-low EGFR mutations (7,8). Additionally, lung cancer signatures in Asiatic populations are similar to that of Caucasian origin with transversions being predominant in smokers. However, in Asian populations the percentages of mutated EGFR and KRAS appear to be reversed when compared to Caucasians, with EGFR mutations accounting for 30-40% and KRAS for 10% or less of NSCLC cases (9). Although the basis for this demographic difference is not known, genetic factors rather than smoking, second hand smoking or environmental carcinogens causing oxidative DNA damage probably account for the increased EGFR mutation percentages in Asian ethnicities (10).
KRAS and EGFR are key features in a receptor tyrosine kinase pathway (RTK, namely the EGFR signaling pathway) which is active in virtually all mammalian epithelia. EGFR (alternatively ERBB1 or HER1) encodes for a transmembrane receptor tyrosine kinase that, upon binding to a variety of extracellular ligands (TGF-α, amphiregulin, epigen, betacellulin, epiregulin and heparin-binding EGF), signals through two major axes in the cell cytoplasm (i.e., RAS/RAF/MEK/ERK/MAPK pathway and PIK3CA/AKT/mTOR pathways) and thus controls every aspect of cellular life and death such as survival, growth, proliferation, and apoptosis. A staggering 75% of lung adenocarcinomas are caused by mutations appearing in the RAS/RAF/MEK/ERK/MAPK pathway, whereas a minor 25% are attributed to PIK3CA/AKT/mTOR pathway mutations (7). Evidently, autocrine EGFR signaling contributes to tumor cell proliferation, apoptosis evasion and metastasis whereas paracrine signaling contributes to cancer associated inflammation, angiogenesis and metastasis. Both mutations and deletions, as well as perturbations of gene copy numbers affecting mostly ten genes of the EGFR signaling pathway confer growth and evolutionary advantage to lung adenocarcinoma cells. These are the “driver” oncogenes, namely the RTKs, EGFR and HER2, the RAS family members KRAS and NRAS and the kinases ALK, BRAF, PIK3CA, AKT, ROS, and MAP2K1. Usually, a single overactive oncogene underlies the proliferation and survival of lung adenocarcinoma through a mechanism known as “oncogene addiction” (11). With the exception of PIK3CA co-existing with mutations in EGFR and KRAS, other driver gene mutations are mutually exclusive in lung cancer. This is particularly exemplified between mutations of EGFR (~12-14%) and KRAS genes (~33-35%) that together account for the 47% of NSCLC cases in western countries. However, exceptions to this have been recently identified (12,13). The conceptual breakthrough of driver oncogene identification, oncogene addiction and the exclusivity of oncogenic mutations in lung cancer (in both cancer research and treatment) lead to the application of RTK inhibitors (TKI) targeting mutant EFGR, along with conventional chemotherapy in the treatment of lung adenocarcinoma.
TKIs gefitinib and erlotinib are approved for NSCLC patients worldwide. Erlotinib significantly prolongs the survival time of patients with advanced NSCLC (14). In recent years many studies reported that TKIs specifically benefit NSCLC patients bearing EGFR mutations. However, despite cytotoxic chemotherapy, which does not alter EGFR mutation status, TKI treatment confers improvement in the overall survival of patients bearing EGFR mutations, but on the other hand—shortly after initial lung cancer remission—frequently results in cancer resistance (15). Cancers that initially benefit from EGFR targeted therapies subsequently become refractory by a plethora of mechanisms and the task to combat them becomes ever more difficult (16). Additionally, due to evolutionary pressure posed by EGFR targeted therapies, tumors may select mutation spectra that predispose to new more aggressive forms that metastasize to the pleura or distant organs [(17), and see below]. KRAS is downstream of EGFR in their common signaling pathway and mutations in KRAS would lead to cancer growth regardless of EGFR signal modulation via TKIs or antibodies against mutant EGFR. Indeed, KRAS mutations are negative predictive biomarkers of TKI therapies (18). Wild type EGFR status and the presence of KRAS mutations should not necessarily be used to exclude cancer patients for treatment with TKIs as minor (but significant) benefits can be exerted to these patients predominantly by erlotinib (19-21).
Malignant pleural effusion (MPE)
Anatomy & histology
MPE is traditionally attributed to occlusion of parietal pleural stomata or obstruction of lymph vessels in mediastinal lymph nodes (1,22). Lymphatic block, however, cannot fully explain MPE which is underlined by inflammation, enhanced angiogenesis and vascular leakage. Recent findings that exploit newly developed murine MPE models (22-24) point out that a vicious cycle of tumor-host interactions take place in the pleural cavity. This phenomenon extensively involves the host innate immune system and vasculature abetted by enhanced plasma extravasation into the pleural space (5). Vascular endothelial growth factor-A (VEGF-A) is a critical cytokine elaborating extravasation and the formation of MPEs that could explain the high rate fluid accumulation observed during thoracoscopy of patients with MPE (24-26). Elevated levels of VEGF produced by both tumor cells of variable origin and infiltrating immune cells, result in increased vascular permeability, cancer cell transmigration, and angiogenesis (26). However, VEGF is not the only vasoactive cytokine in MPE. Other MPE promoting molecules are pro-inflammatory cytokines such as tumor necrosis factor (TNF), C-C-motif chemokine ligand 2 (CCL2) and osteopontin (OPN) (5). In particular, host and tumor elaborated OPN exerts distinct effects: the former recruits macrophages and promotes angiogenesis whereas the latter causes evasion of cancer cell apoptosis (27). These data imply that a strategy aimed at alleviating fluid extravasation in MPE that specifically targets VEGF alone may be hampered as a pharmacological intervention because of the broad spectrum of vasoactive molecules operating in MPE. This is particularly important to take into consideration as a great number of current MPE-relevant clinical trials use bevacizumab, an antibody targeting VEGF (see below).
On the other hand, both TNF and CCL2 are key cytokines with central roles in inflammation which also participate in MPE formation. Notwithstanding, CCL2 mediates the recruitment of macrophages, memory T and dendritic cells upon inflammation. Several lines of evidence suggest that CCL2 is playing key roles in MPE by—among others—recruiting a monomyelocytic/macrophage cellular infiltrate in experimental and human MPE. The administration of anti-CCL2 neutralizing antibodies alleviated MPE in mice, by annihilating immune cell infiltrates and by restricting angiogenesis and fluid extravasation (28). These results suggest that tumor and or host elaborated CCL2 is epistatic to the tumor paracrine pathways that ultimately lead to pleural fluid accumulation and MPE and, as such, comprises a very attractive target for therapeutic interventions.
MPE associates with a smoldering inflammation but a detailed phenotyping of immune cells participating in MPE is not yet available. Which immune cell types participate in MPE? More than 80% of MPE feature elevated lymphocyte counts. However, increased lymphocyte counts do not specifically characterize MPE, but also other pleurisies (tuberculous, chronic rheumatoid pleurisy and chylothorax). Studies in immunocompetent mice suggest that certain T lymphocytes (Tregs or Th17) and their associated cytokines play important roles in MPE pathogenesis (29,30). Furthermore, the host elaborated T-helper 2 cytokine IL-5 is essential for the recruitment of eosinophils in MPE. Deletion of the IL-5 gene protects mice from MPE (23). Accordingly, increased percentages of eosinophils (12-24%) were identified in the pleura during MPE manifestation.
By using an MPE model that accurately recapitulates the hallmarks of human MPE our group was able to identify at least three immunologically distinct myelogenous cell populations that are actually recruited to the pleural cavity of MPE-ridden animals (28,31). Among these cell types, a rather rare and previously underappreciated minority cell population, mast cells, was characterized as a cardinal culprit of MPE and associated phenomena of inflammation, angiogenesis and vascular leakage. This rare cell is recruited to the pleural cavity upon CCL2 signaling elaborated from cancer cells and release vasoactive factors and cytokines that support the survival of cancer cells in the pleural cavity (31). Our data suggest that complex immune cell interactions underlie MPE. However, the ordered recruitment of these cell types to the pleural cavity is following a cause-and-effect linear fashion that holds promise for targeted pharmacologic interventions.
Mutation spectra associated with MPE
In general, MPE is associated with particular mutation spectrums within the tumor cells and not with tumor growth rates (32,33). In fact, EGFR mutations are more common in the pleural metastases and fluid as compared with primary tumors (32-42). EGFR mutations are enriched (26.5%) in patients with MPE whereas the frequency of KRAS mutations in MPE patients is lower than that in NSCLC in general (18.8% vs. 33.3% respectively) (33,42). This trend was maintained in a study conducted in Asiatic population where paired MPE and primary tumor samples from 386 lung adenocarcinoma patients were analyzed. In total 244 samples screened positive (66.3%) and 116 (31.5%) screened negative for EGFR mutations. Of the 116 EGFR negative mutations 39 samples (33.6%) screened positive for the EML4-ALK fusion gene whereas a minor 6.5% screened positive for KRAS mutations (43). From these data the EML4-ALK fusion gene emerges as a possible MPE-associated mutation. In other studies, the presence of EGFR mutations in patients with MPE is significantly higher than in those without, thus indicating that EGFR mutations may facilitate the migration of cancer cells to the pleural cavity (43,44).
Evidently, whereas KRAS is the most frequent mutation in NSCLC and may predispose to visceral pleural invasion (VPI), MPE associates better with mutant EGFR and the EML4-ALK fusion gene in both Caucasian and Asiatic populations. Whereas EGFR pathway perturbations are by far the most common cancer drivers in the lung, KRAS mutations are common denominators in other types of cancers. One way to explain this difference is to suppose that lung, breast cancer and MPM are cancers invading the pleura because of local proximity and not through the bloodstream, whereas distant adenocarcinomas may employ specific biochemical pathways directing them to invade the pleura. Thus, KRAS mutations underlying distant cancers in the colon, stomach or ovary, may elaborate autocrine and paracrine signaling mechanisms directly facilitating pleural homing. In addition, MPE might be a secondary effect to active (but not necessarily mutant) KRAS signaling mediated downstream of mutant EGFR and/or EML4-ALK kinase genes.
The proteomic “signature” of lung cancer signaling offers complementarities to the transcriptional profiling and could potentially identify novel biomarkers and therapeutic targets. Recently, lung cancer-specific plasma proteomes were defined (45). Microvesicles (or exosomes) are released from cancer cells and host immune cells in the pleural cavities of MPE patients. Although their function(s) remain unclear, their proteomic signature after purification and mass spectrometry revealed that EGFR pathway proteins (epiregulin, amphiregulin, TGF-β, KRAS, and RAB5) are overrepresented in MPE associated exosomes. This finding underscores the importance of this pathway in MPE pathogenesis (46).
Sampling from the primary tumor and the disseminated tumor foci in the pleura and/or MPE suggests that different cancer cell mutations are present between these two sites. The concomitant sampling of primary and metastatic tumors revealed discordance in the mutational spectra present in tumor cells in these two sites, a phenomenon that adds to the complexity of MPE pathogenesis (44,47,48). Mutational discordance may arise from tumor evolution under pharmacological pressure. For example, this is the case of EGFR acquired resistance and disease relapse after treatment with TKIs. Alternatively, however, mutational discordance may indicate a tumor adaptive strategy to invade “new lands” and may suggest that EGFR mutations predispose pleural metastasis. These two possibilities are not mutually exclusive (44). Nevertheless, this relatively recent assumption is not yet adequately substantiated. More studies examining paired MPE and primary tumor samples as well as mechanistic studies in mouse models will help towards the clarification of these issues.
Mesothelial attributes facilitating metastasis
Pleural membranes, due to their anatomic site and their visceral versus parietal topology, may present specific gene expressions, different paracrine signals, differential permeability, elasticity and tissue plasticity. All of these characteristics may positively impact cancer cell dissemination in the pleural space. Despite the general appreciation of the pleural role in lung and breast cancer dissemination, very little is known about the molecular characteristics, paracrine signaling and other properties of the pleural membranes that may attract cancer cells to the pleural space. Which mesothelial attribute(s) could render the pleural space a desired metastatic destination? Tissue specific metastasis is thought to be mediated by active mechanisms based on tumor “homing” to the target organ by ligand-receptor interactions. Such a mechanism should involve complementary molecules, featured on tumor and the target organ cells. For example, tumor cells expressing a specific cytokine receptor are “sensing” its cytokine ligand that is expressed by cells in a distant organ. The tumor cells upon breaching the endothelial barrier and entering the bloodstream disseminate to this distant site. This type of mechanism is used predominantly by the immune system where inflammatory or immune surveillance cells populate distant sites upon inflammation or organ surveillance. A notorious example is based on the interaction of CXCL12 chemokine (previously known as stromal cell derived factor-1, SCDF1) and the G-coupled receptor CXCR4. This interaction has important ramifications for physiologic embryogenesis and tumor invasion. CXCR4 is commonly expressed by virtually all mammalian cell types ranging from hematopoetic, endothelial and neuronal origin. Furthermore, CXCR4 is overexpressed in more than 23 human cancers. CXCR4-expressing cells respond to CXCL12 gradients in target organs during embryonic hematopoiesis, and additionally cancer cells—via the same mechanism—escape primary tumors and home to organs that release CXCL12 (49). In NSCLC, disease prognosis correlates with localization of CXCR4 to the nuclear and/or the cytoplasmic membrane compartment. Higher CXCR4 expression in the cytoplasmic membrane correlates with a higher tendency to locally invade neighboring tissues and with increased propensity of tumor cells to form distant metastases (49).
Another pathway that could account for pleural metastasis is the lysophosphatidic acid (LPA) pathway. LPA controls motility, migration, cancer adhesion invasion as well as differentiation of many cell types (50). LPA biosynthesis is controlled by autotaxin (ATX), a secreted lysophospholipase converting lysophosphatidylcholine to LPA. ATX is a molecule highly expressed in the omentum, a visceral membrane of mesothelial origin (like the pleural membranes), which is a frequent metastasis target for gastrointestinal invasive adenocarcinomas that subsequently produce ascites (51), a terminal disease hallmark resembling MPE. Interestingly, pleural membrane-originated MPM overtly express ATX, whereas bone marrow platelet-derived ATX, through binding to integrin alpha V/beta3 expressed by breast cancer cells, drives metastasis to the bone marrow (52). Furthermore, integrin B3 was shown to be important for EGFR acquired resistance and its upregulated expression increases cell “stemness”. Upon treatment with TKIs, cells expressing integrin B3 are enriched in the tumor cellular population. These cells (comprising the alleged cancer stem cells) acquire resistance to TKIs whereby integrin B3-Galectin engages the alternative KRAS-RalB signaling complex downstream of EGFR (17). Under the light that integrin B3 is upregulated in EGFR positive lung cancer cell lines because of TKI therapy, and that KRAS and RalB are enriched in MPE- associated exosomes, it will be interesting to examine ATX’s role in pleural metastasis.
Visceral pleural invasion (VPI)
Anatomy vs. biology
Lung and breast tumors, due to their anatomical proximity, colonize the pleura in a function called VPI. VPI is usually defined as tumor cells invading beyond the elastic layer (monitored by elastin stain) of the visceral pleura. The VPI is a poor prognostic factor in both breast cancer and NSCLC (53,54). In the TNM staging system, VPI associates with extensive N2 involvement (55), independently of the primary tumor size (56). This suggests that a specific tumor cell pathway predisposes the tumor for VPI through the subpleural lymphatics and hilar lymph nodes, and into the mediastinal lymph nodes (57). The nature of this pathway is not yet known. However, one study points out that G12C or G12D mutant KRAS-bearing lung cancers are more likely prone to VPI (57). The definite lack of suitable animal models to study the molecular basis of VPI makes it difficult to examine important molecular aspects of extravasation-related VPI, involving crosstalk between tumor cells, endothelial cells, basement membrane and macrophages.
VPI detection
The worldwide incidence of lung adenocarcinoma is steadily increasing. In 2012, lung adenocarcinoma accounted for 40% of NSCLC cases, while in 2014 the percentage is estimated to be 48.7% or more. Accordingly, minimal pleural invasion due to direct mechanisms (VPI)—with or without effusion—is expected to become more common in the future. Recently, 13.2% of more than 2,000 patients with NSCLC presented metastatic minimal effusions and this was shown to negatively impact survival even when detected in the first stages of the disease (54). In the recent TNM classification, the MPE status was changed from T4 to M1a (58,59). Despite its importance and adverse prognosis for survival, however, minimal invasion is not thoroughly sought for when staging NSCLC (54). This is probably due to its problematic clinical assessment. Nevertheless, minimal invasion assessment is shown to be of prognostic significance in determining accurate staging (especially in early disease stages). The pleural fluid biopsy obtained with minimally invasive procedures [pleural lavage cytology (PLC)] could lead to the definitive identification of malignant cells, a finding hallmarking MPE. However, when VPI measures 10 mm or less (as is often the case of early stage disease), the minimally invasive procedures will not confer enough sensitivity (54) and video-assisted thoracoscopic surgery (VATS) can be alternatively considered for the identification of malignant cells. VATS is a method that requires anesthesia and may be of risk for patients with poor performance status. VATS will help to accurately stage the disease and understand the nature of minimal invasion towards MPE development and the mechanisms underlying it.
Based on Light’s criteria the majority of MPEs are categorized as exudates (60). PLC is a common method for the detection of malignant cells in pleural fluid. Why some tumors colonizing the pleura cause MPE and some others do not, is still unclear but the answer to this question can be more mundane than expected.
The presence of malignant cells in the pleura can be identified either before or after lung surgery in patients with resectable lung cancer. This distinction can be of paramount importance for the prognostic significance of PLC since pleural invasion before resection could signify the natural disease process with severe consequences for patients’ survival. However, another possibility for a positive PLC, (although admittedly not substantiated enough), is the dispersion of malignant cells during fine needle aspiration conducted for diagnostic purposes (60,61). Thus, a major cause of a positive PLC can be either the metastatic pleural dissemination of tumor cells through direct mechanisms (VPI, subpleural lymphatics mediated metastasis and infiltrated lymph nodes) (60-62) or the results of iatrogenic cancer dissemination during lung resection. This could potentially represent an important issue to be resolved because as stated before, a naturally occurring positive PLC may be considered as an early sign of pleural invasion and would signify a negative prognosis and a local and/or systemic cancer recurrence, especially for patients with early stage disease (63). It is uncertain whether a positive PLC after lung resection has the same prognostic significance (62).
Potential for therapeutic interventions
With respect to the PLC sensitivity, it is difficult to distinguish the cytologic morphology of malignant cells originated from different primary tumors; thus, the sensitivity of PLC ranges from 40-87% (1). Since morphology is not enough, the systematic analysis of the mutation spectrum of a positive PLC would be able to guide subsequent approaches such as intraoperative intrapleural application of chemotherapy, adjuvant chemotherapy or cytotoxic chemotherapy. A method of choice to assess the mutation spectrum of a positive PLC would be PNA-clamping due to the increased sensitivity of this method compared to more traditional methods based on Sanger sequencing (64,65).
Given the prevalence of EGFR mutations in MPE, administration of TKIs like gefitinib or erlotinib may be beneficial for MPE patients, but the pharmacodynamics of these agents in MPE are not known. Gefitinib was administered to NSCLC patients with MPE along with chest draining with good results (37-39,66,67). However, during gefitinib treatment the drug-sensitive L858R EGFR cells underwent apoptosis in the pleural cavity, whereas previously undetected cancer cell clones bearing the drug-resistant T790M EGFR mutation expanded (66,67). Thus, gefitinib administration was associated with tumor evolution towards cells bearing EGFR resistance-conferring mutations. Furthermore, erlotinib therapy was shown to increase the 1-year rate survival of NSCLC patients bearing EGFR mutations and MPE (68). In fact, erlotinib is beneficial to different degrees for patients with both wild-type and mutant EGFR (69). Furthermore, erlotinib is reportedly stabilizing the disease in patients with mutant KRAS (19,20), despite KRAS mutations emerged as negative predictive factors for TKI therapy response (21). Thus, a larger-scale systematic assessment of erlotinib efficacy against MPE irrespective of pleural metastasis mutation status may be of significance.
Currently, there are more than 70 reported clinical studies using a variety of therapies ranging from biological, genetic and drug interventions for MPE (Figure 1). Out of those studies, three currently being conducted are specifically administering bevacizumab, a monoclonal antibody targeting VEGF, a cardinal factor relevant to angiogenesis and enhanced vascular leakiness leading to MPE. It is reasonable to assume that VEGF blockade will diminish MPE. Notwithstanding, the broad spectrum of vasoactive molecules underscoring MPE points out that a strategy against MPE that is specifically targeting VEGF may be not as efficient and additional complementary therapies must be considered.
Figure 1.
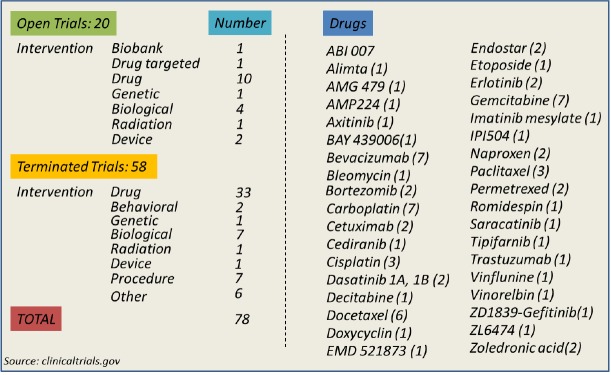
(Left) Open and terminated clinical trials on MPE, categorized according to intervention; (Right) used drugs in MPE related clinical trials. In parenthesis the number of trials that use the specific drug is presented. MPE, malignant pleural effusion.
Conclusions
Lung adenocarcinoma incidence increases worldwide and the reason for that is not known. Whereas traditional and new generation TKIs represent a major breakthrough in lung cancer therapeutics, the use of these molecules exerts a selection pressure on tumors, by pushing them to survival limits, ultimately driving them to evade “new lands” and metastasize.
Evidence based on recently developed mouse models accurately modeling human MPE, suggests that this phenomenon is underlined by traceable molecular mechanisms and pathways commonly employed by normal immune system function. Full-blown MPE is characterized by smoldering inflammation where a plethora of immunological cell types recruited to combat the enemy become instead trapped in the “vicious cycle” of supporting the survival and growth of cancer cells. However, certain immunological cell types initiate the MPE avalanche and these are provoked by specific epistatic chemokine signals. This, in turn holds promise towards the identification of several potential targets aiming towards MPE-specific therapeutic applications.
Both cancer mutation spectra and mesothelial attributes affect VPI and MPE. The exploitation of the existing MPE mouse models and the development of more sophisticated genetic tools to explore phenomena like VPI and minimal pleural effusion is mandatory towards a complete understanding of the pathways and the genetic lesions leading to pleural metastasis and MPE formation. This will bring us closer to more effective, specific, targeted and personalized therapies to this currently untreated disease.
Notwithstanding, the management of VPI, minimal pleural effusion, MPE, and true positive PLC results are essential for accurate staging and proper treatment of the disease. Based on their prognostic significance, and provided that the existing caveats will be addressed thoroughly and appropriately, it may be of value to include standards evaluating VPI, MPE and a positive PLC status into the TNM staging system.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- 1.Light RW. editor. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013. [Google Scholar]
- 2.Assis LV, Isoldi MC. Overview of the biochemical and genetic processes in malignant mesothelioma. J Bras Pneumol 2014;40:429-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts ME, Neville E, Berrisford RG, et al. Management of a malignant pleural effusion: British Thoracic Society Pleural Disease Guideline 2010. Thorax 2010;65 Suppl 2:ii32-40. [DOI] [PubMed] [Google Scholar]
- 4.Egan AM, McPhillips D, Sarkar S, et al. Malignant pleural effusion. QJM 2014;107:179-84. [DOI] [PubMed] [Google Scholar]
- 5.Stathopoulos GT, Kalomenidis I. Malignant pleural effusion: tumor-host interactions unleashed. Am J Respir Crit Care Med 2012;186:487-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travis WD, Brambilla E, Riely GJ. New pathologic classification of lung cancer: Relevance for clinical practice and clinical trials. J Clin Oncol 2013;31:992-1001. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dacic S, Shuai Y, Yousem S, et al. Clinicopathological predictors of EGFR/KRAS mutational status in primary lung adenocarcinomas. Mod Pathol 2010;23:159-68. [DOI] [PubMed] [Google Scholar]
- 9.Mitsudomi T, Yatabe Y. Epidermal growth factor mutations in Lung Cancer Pathol Int 2007;57:233-44. [DOI] [PubMed]
- 10.Krishnan VG, Ebert PJ, Ting JC, et al. Whole-genome sequencing of asian lung cancers: second-hand smoke unlikely to be responsible for higher incidence of lung cancer among asian never-smokers. Cancer Res 2014;74:6071-81. [DOI] [PubMed] [Google Scholar]
- 11.Weinstein IB, Joe A. Oncogene addiction. Cancer Res 2008;68:3077-80. [DOI] [PubMed] [Google Scholar]
- 12.Sun JM, Hwang DW, Ahn JS, et al. Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS ONE 2013;8: e64816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choughule A, Sharma R, Trivedi V, et al. Coexistance of KRAS mutation with mutant but not wild type EGFR predicts to tyrosine kinase inhibitors in humal lung cancer. BJC 2014;1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shepherd FA, Rodrigues Pereira J, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123-32. [DOI] [PubMed] [Google Scholar]
- 15.Cortot AB, Jänne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev 2014;23:356-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013;19:1389-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seguin L, Kato S, Franovic A, et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol 2014;16:457-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riely GJ, Ladanyi M. KRAS mutations: an old oncogene becomes a new predictive biomarker. J Mol Diagn 2008;10:493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu CQ, da Cunha Santos G, Ding K, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada clinical trials group study. J Clin Oncol 2008;26:4268-75. [DOI] [PubMed] [Google Scholar]
- 20.Schneider CP, Heigener D, Schott-von-Römer K, et al. Epidermal growth factor receptor-related tumor markers and clinical outcomes with erlotinib in non-small cell lung cancer: an analysis of patients from german centers in the TRUST study. J Thorac Oncol 2008;3:1446-53. [DOI] [PubMed] [Google Scholar]
- 21.Linardou H, Dahabre H, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 2008;9:962-72. [DOI] [PubMed] [Google Scholar]
- 22.Stathopoulos GT, Zhu Z, Everhart MB, et al. Nuclear Factor-kB affects tumor progression in a mouse model of malignant pleural effusion. Am J Respir Cell Mol Biol 2006;34:142-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stathopoulos GT, Sherrill TP, Karabela SP, et al. Host-derived interleukin-5 promotes adenocarcinoma-induced malignant pleural effusion. Am J Respir Crit Care Med 2010;182:1273-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yano S, Shinohara H, Herbst RS, et al. Production of experimental malignant pleural effusions is dependent on invasion of the pleura and expression of vascular endothelial growth factor/vascular permeability factor by human lung cancer cells. Am J Pathol 2000;157:1893-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zebrowski BK, Yano S, Liu W, et al. Vascular endothelial growth factor levels and induction of permeability in malignant pleural effusions. Clin Cancer Res 1999;5:3364-8. [PubMed] [Google Scholar]
- 26.Bradshaw M, Mansfield A, Peikert T, et al. The role of vascular endothelial growth factor in the pathogenesis, diagnosis and treatment of malignant pleural effusion. Curr Oncol Rep 2013;15:207-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Psallidas I, Stathopoulos GT, Maniatis NA, et al. Secreted phosphoprotein-1 directly provokes vascular leakage to foster malignant pleural effusion. Oncogene 2013;32:528-35. [DOI] [PubMed] [Google Scholar]
- 28.Marazioti A, Kairi CA, Spella M, et al. Beneficial impact of CCL2 and CCL12 neutralization on experimental malignant pleural effusion. PLoS One 2013;8:e71207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin H, Tong ZH, Xu QQ, et al. Interplay of Th1 and Th17 cells in murine models of malignant pleural effusion. Am J Respir Crit Care Med 2014;189:697-706. [DOI] [PubMed] [Google Scholar]
- 30.Marazioti A, Blackwell TS, Stathopoulos GT. The Lymphatic System in Malignant Pleural Effusion. Drain or immune switch? Am J Respir Crit Care Med 2014;189:626-7. [DOI] [PubMed] [Google Scholar]
- 31.Giannou AD, Marazioti A, Spella M, et al. Mast cells mediate malignant pleural effusion formation. J Clin Invest 2015;125:2317-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou J, Bella AE, Chen Z, et al. Frequency of EGFR mutations in lung adenocarcinoma with malignant pleural effusion: Implication of cancer biological behaviour regulated by EGFR mutation. J Int Med Res 2014;42:1110-7. [DOI] [PubMed] [Google Scholar]
- 33.Smits AJ, Kummer JA, Hinrichs JW, et al. EGFR and KRAS mutations in lung carcinomas in the Dutch population:increased EGFR mutation frequency in malignant pleural effusion in lung adenocarcinoma. Cell Oncol (Dordr) 2012;35:189-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asano H, Toyooka S, Tokumo M, et al. Detection of EGFR gene mutation in lung cancer by mutant-enriched polymerase chain reaction assay. Clin Cancer Res 2006;12:43-8. [DOI] [PubMed] [Google Scholar]
- 35.Hung MS, Lin CK, Lew SW, et al. Epidermal growth factor receptor mutations in cells from non-small cell lung cancer malignant pleural effusion. Chang Gung Med J 2006;29:373-9. [PubMed] [Google Scholar]
- 36.Wu SG, Gow CH, Yu CJ, et al. Frequent epidermal growth factor receptor gene mutation in malignant pleural effusion of lung adenocarcinoma. Eur Respir J 2008;32:924-30. [DOI] [PubMed] [Google Scholar]
- 37.Jian G, Songwen Z, Ling Z, et al. Prediction of epidermal growth factor receptor mutations in the plasma/pleural effusion to efficacy of gefitinib treatment in advanced non-small cell lung cancer. J Cancer Res Clin Oncol 2010;136:1341-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimura H, Fujiwara Y, Sone T, et al. EGFR mutation status in tumor derived from pleural effusion fluid is a practical basis for predicting the response to gefitinib. Br J Cancer 2006;95:1390-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soh J, Toyooka S, Aoe K, et al. Usefullness of EGFR mutation screening in pleural fluid to predict the clinical outcome of gefitinib treted patients with lung cancer. Int J Cancer 2006;119:2353-8. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Zhao Y, Wang M, et al. Detection and comparison of epidermal growth factor receptor mutations in cells and fluid of malignant pleural effusion in non small cell lung cancer. Lung Cancer 2008;60:175-82. [DOI] [PubMed] [Google Scholar]
- 41.Tsai TH, Su KY, Wu SG, et al. RNA is favourable for analyzing EGFR mutations in malignant pleural effusion of lung cancer. Eur Respir J 2012;39:677-84. [DOI] [PubMed] [Google Scholar]
- 42.Froudarakis ME. Pleural effusion in lung cancer: more questions than answers. Respiration 2012;83:367-76. [DOI] [PubMed] [Google Scholar]
- 43.Wu SG, Kuo YW, Chang YL, et al. EML4-ALK translocation predicts better outcome in lung adenocarcinoma patients with wild-type EGFR. J Thorac Oncol 2012;7:98-104. [DOI] [PubMed] [Google Scholar]
- 44.Han HS, Eom DW, Kim JH, et al. EGFR mutation status in primary lung adenocarcinomas and corresponding metastatic lessions: discordance in pleural metastasis. Clin Lung Cancer 2011;12:380-6. [DOI] [PubMed] [Google Scholar]
- 45.Taguchi A, Politi K, Pitteri SJ, et al. Lung cancer signatures in plasma based on proteome profiling of mouse tumor models. Cancer Cell 2011;20:289-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park JO, Choi DY, Choi DS, et al. Identification and characterization of proteins isolated from microvesicles derived from human lung cancer pleural effusions Proteomics 2013;13:2125-34. [DOI] [PubMed] [Google Scholar]
- 47.Gow CH, Chang YL, Hsu YC, et al. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol 2009;20:696-702. [DOI] [PubMed] [Google Scholar]
- 48.Park S, Holmes-Tisch AJ, Cho EY, et al. Discordance of molecular biomarkers associated with epidermal growth factor receptor pathway between primary tumors and lymph node metastasis in non-small cell lung cancer. J Thorac Oncol 2009;4:809-15. [DOI] [PubMed] [Google Scholar]
- 49.Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res 2014;124:31-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willier S, Butt E, Grunewald TG. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: a focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol Cell 2013;105:317-33. [DOI] [PubMed] [Google Scholar]
- 51.Shelton EL, Galindo CL, Williams CH, et al. Autotaxin signaling governs phenotypic heterogeneity in visceral and parietal mesothelia. Plos One 2013;8:e69712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leblanc R, Lee SC, David M, et al. Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone Blood 2014;124:3141-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.David E, Thall PF, Kalhor N, et al. Visceral pleural invasion is not predictive of survival in patients with lung cancer and smaller tumor size. Ann Thorac Surg 2013;95:1872-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryu JS, Ryu HJ, Lee SN, et al. Prognostic impact of minimal pleural effusion in non-small-cell lung cancer. J Clin Oncol 2014;32:960-7. [DOI] [PubMed] [Google Scholar]
- 55.Yoshida K, Sugiura T, Takifuji N, et al. Randomized phase II trial of three intrapleural regimens of malignant pleural effusion in previously untreated non-small cell lung cancer: JCOG 9515. Lung Cancer 2007;58:362-8. [DOI] [PubMed] [Google Scholar]
- 56.Manac’h D, Riquet M, Medioni J, et al. Visceral pleura invasion by non-small cell lung cancer: an underrated bad prognostic factor. Ann Thorac Surg 2001;71:1088-93. [DOI] [PubMed] [Google Scholar]
- 57.Raparia K, Villa C, Raj R, et al. Peripheral lung adenocarcinoma with KRAS mutations are more likely to invade visceral pleura. Arch Pathol Lab Med 2015;139:189-93. [DOI] [PubMed] [Google Scholar]
- 58.Goldstraw P, Crowley J, Chansky K, et al. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol 2007;2:706-14. [DOI] [PubMed] [Google Scholar]
- 59.Nam HS. Malignant pleural effusion: medical approaches for diagnosis and management. Tuberc Respir Dis (Seoul) 2014;76:211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sawabata N, Maeda H, Ohta M, et al. Operable non-small cell lung cancer diagnosed by transpleural techniques: do they affect relapse and prognosis? Chest 2001;120:1595-8. [DOI] [PubMed] [Google Scholar]
- 61.Riquet M, Badoual C, Le Pimpec Barthes F, et al. Visceral pleura invasion and pleural lavage tumor cytology by lung cancer: a prospective appraisal. Ann Thorac Surg 2003;75:353-5. [DOI] [PubMed] [Google Scholar]
- 62.Toufektzian L, Sepsas E, Drossos V, et al. Pleural lavage cytology: where do we stand? Lung Cancer 2014;83:14-22. [DOI] [PubMed] [Google Scholar]
- 63.Nakagawa T, Okumura N, Kokado Y, et al. Clinical relevance of intraoperative pleural lavage cytology in non-small cell lung cancer. Ann Thorac Surg 2007;83:204-8. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka T, Nagai Y, Miyazawa H, et al. Reliability of the peptide nucleic acid-locked nucleic acid polymerase chain reaction clamp-based test for epidermal growth factor receptor mutations integrated into the clinical practice for non-small cell lung cancers. Cancer Sci 2007;98:246-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang JY, Park CU, Yeo CD, et al. Comparison of PNA clamping and direct sequencing for detecting KRAS mutations in matched tumour tissue, cell block, pleural effusion and serum from patients with malignant pleural effusion. Respirology 2015;20:138-46. [DOI] [PubMed] [Google Scholar]
- 66.Kubo A, Koh Y, Kawaguchi T, et al. Makignant pleural effusion from lung adenocarcinoma treated by gefitinib. Intern Med 2011;50:745-8. [DOI] [PubMed] [Google Scholar]
- 67.Honda Y, Takigawa N, Fushimi S, et al. Disappearance of an activated EGFR mutation after treatment with EGFR tyrosine kinase inhibitors. Lung Cancer 2012;78:121-4. [DOI] [PubMed] [Google Scholar]
- 68.Guo R, Chen X, Wang T, et al. Subsequent chemotherapy reverses acquired tyrosine kinase inhibitor resistance and restores response to tyrosine kinase inhibitor in advanced non-small-cell lung cancer. BMC Cancer 2011;11:90-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brugger W, Triller N, Blasinska-Morawiec M, et al. Prospective molecular marker analyses of EGFR and KRAS from a randomized, placebo-controlled study of erlotinib maintenance therapy in advanced non-small-cell lung cancer. J Clin Oncol 2011;29:4113-20. [DOI] [PubMed] [Google Scholar]