

Abstract
Bradykinin (BK) and its receptors (B1 and B2 receptors) play important roles in inflammatory nociception. However, the patterns of expression and physiological/pathological functions of B1 and B2 receptors in trigeminal ganglion (TG) neurons remain to be fully elucidated. We investigated the functional expression of BK receptors in rat TG neurons. We observed intense immunoreactivity of B2 receptors in TG neurons, while B1 receptors showed weak immunoreactivity. Expression of the B2 receptor colocalized with immunoreactivities against the pan-neuronal marker, neurofilament H, substance P, isolectin B4, and tropomyosin receptor kinase A antibodies. Both in the presence and absence of extracellular Ca2+ ([Ca2+]o), BK application increased the concentration of intracellular free Ca2+ ([Ca2+]i). The amplitudes of BK-induced [Ca2+]i increase in the absence of [Ca2+]o were significantly smaller than those in the presence of Ca2+. In the absence of [Ca2+]o, BK-induced [Ca2+]i increases were sensitive to B2 receptor antagonists, but not to a B1 receptor antagonist. However, B1 receptor agonist, Lys-[Des-Arg9]BK, transiently increased [Ca2+]i in primary cultured TG neurons, and these increases were sensitive to a B1 receptor antagonist in the presence of [Ca2+]o. These results indicated that B2 receptors were constitutively expressed and their activation induced the mobilization of [Ca2+]i from intracellular stores with partial Ca2+ influx by BK. Although constitutive B1 receptor expression could not be clearly observed immunohistochemically in the TG cryosection, cultured TG neurons functionally expressed B1 receptors, suggesting that both B1 and B2 receptors involve pathological and physiological nociceptive functions.
Keywords: bradykinin, B1 receptor, B2 receptor, neuropathic pain, pain, trigeminal ganglion neuron, Ca2+ signaling
Introduction
Tissue damage results in an accumulation of endogenous chemical substances, such as bradykinin (BK), which are released by nociceptive afferents and/or non-neural cells in the injured area of the tissue (Julius and Basbaum, 2001; Basbaum et al., 2009). BK receptors, which are divided into two subtypes (B1 and B2), are plasma membrane G-protein-coupled receptors of the seven-transmembrane-domain family. The existence of B1 and B2 receptors has been confirmed by pharmacological and radioligand-binding studies, as well as by mRNA expression analyses, in a wide variety of cells (Hess et al., 1994; Pesquero et al., 1996; Hall, 1997). Previous studies have indicated that B2 receptors couple with the Gq protein. Activation of the Gq protein activates phospholipase C, which induces a number of intracellular second messenger systems, including 1, 2-diacylglycerol and inositol 1, 4, 5-trisphosphate, which activates protein kinase C and mobilizes intracellular Ca2+, respectively (Walker et al., 1995; Tiwari et al., 2005).
BK-induced changes in the chemical environment surrounding axons cause peripheral sensitization, which is associated with inflammatory responses (Basbaum et al., 2009). Neuropathic pain is also involved in peripheral and central sensitization, which increases chronic pain states (Cervero and Laird, 1996; Scholz and Woolf, 2002; Ochoa, 2009). Injury to trigeminal ganglion (TG) neurons, which occasionally induces neuropathic pain, has been reported to be mediated by both B1 and B2 receptors in the orofacial area. Formalin-induced orofacial pain responses in rats are reduced by B2 receptor inhibition (Chichorro et al., 2004). In addition, administration of B1 and B2 receptor antagonists delays the development of thermal hyperalgesia in the orofacial area, which is induced by constriction of the infraorbital nerve in rats and mice (Luiz et al., 2010). Thus, the functional role of BK receptors in TG neurons in physiological and pathological nociception has been well described by behavioral studies. However, the basic expression patterns of B1 and B2 receptors in TG neurons are still unclear and remain to be fully elucidated.
In the present study, we investigated the expression and localization, as well as physiological and pharmacological properties, of B1 and B2 receptors in primary cultured rat TG neurons.
Materials and Methods
Ethical Approval
All the animals used in our study were treated in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences, which was approved by the Council of the Physiological Society of Japan and the American Physiological Society. In addition, the study followed the guidelines that were established by the National Institutes of Health (USA) regarding the care and use of animals for experimental procedures. This study was approved by the Animal Research Ethics Committee of Tokyo Dental College (approval No. 252502).
Cell Culture
TG cells were isolated from neonatal Wistar rats (7 days old) (Kawaguchi et al., 2015) that were under pentobarbital sodium anesthesia (50 mg/kg) following the administration of isoflurane (3.0 Vol%). TG cells were dissociated by enzymatic treatment with Hank’s balanced salt solution (Life Technologies, Grand Island, NY, USA) containing 20 U/mL papain (Worthington Biochemical Corporation, Lakewood, NJ, USA) for 20 min at 37°C, which was followed by dissociation by trituration. After dissociation, the TG cells were plated on 35 mm-diameter dishes (Corning Incorporated Life Sciences, Tewksbury, MA, USA) and cultured for 48 h at 37°C (95% air and 5% CO2). The primary cells were cultured in Leibovitz’s L-15 medium (Life Technologies) containing 10% fetal bovine serum, 1% penicillin-streptomycin (Life Technologies), 1% fungizone (Life Technologies), 26 mM NaHCO3, and 30 mM glucose (pH 7.4). For the immunocytochemistry, TG cells were subjected to primary culture on poly-L-lysine-coated cover glasses (Matsunami Glass Ind., Ltd., Osaka, Japan).
Immunofluorescence Analysis
TGs isolated from neonatal Wistar rats (7 days old) were fixed in optimal cutting temperature compound and rapidly frozen in liquid nitrogen. Frozen tissues were cut at a thickness of 10 μm and placed on slides. After fixation by 50% ethanol and 50% acetone at −20°C for 30 min, primary cultured TG cells and cryosections were treated with 10% donkey serum at room temperature for 20 min and then incubated overnight at 4°C with primary antibodies (Kuroda et al., 2013). A cocktail of primary antibodies (Neuro-ChromTM Pan Neuronal Marker, EMD Millipore, Billerica, MA, USA; 1:50 dilution), including mouse anti-Neuronal nuclei (NeuN), anti-microtubule-associated protein 2 (MAP2), anti-βIII tubulin, and anti-neurofilament H (NF-H) antibodies, was used as a neuronal marker. TG cells were also incubated with either mouse anti-NF-H (SantaCruz, CA, USA; 1:200 dilution) as an A-neuron marker, mouse anti-substance P (SP; R&D Systems, Minneapolis, MN, USA; 2.5 μg/100 μl dilution) as a peptidergic C-neuron marker, FITC-conjugated anti-isolectin B4 (IB4; Vector laboratories, CA, USA; 1:200 dilution) as a non-peptidergic C-neuron marker, goat anti-high-affinity nerve growth factor (NGF) receptor (a tropomyosin receptor kinase A (TrkA); R&D Systems; 1.5 μg/100 μl dilution) as an NGF-responsive nociceptor marker (Mantyh et al., 2011), and rabbit anti-B1 receptor (Alomone Labs, Jerusalem, Israel; 1:50 dilution) and rabbit anti-B2 receptor (Alomone Labs; 1:50 dilution) (Duehrkop et al., 2013; Dutra et al., 2013) antibodies. For negative controls, the sections were incubated with non-immune IgGs (Abcam, Cambridge, UK; 1:50; N = 4 from four rats) (Figure 2M). The cells and tissues were then washed and incubated with a secondary antibody at room temperature for 30 min. The secondary antibodies were Alexa Fluor 488 donkey anti-rabbit IgG, Alexa Fluor 568 donkey anti-mouse IgG, Alexa Fluor 568 donkey anti-rabbit IgG, and Alexa Fluor 568 donkey anti-goat IgG (1:50 dilution; Life Technologies) for the fluorescence staining and 4′, 6-diamino 2-phenylindole dihydrochloride (Life Technologies) for the nuclear staining (room temperature for 5 min). The cells and tissues were examined under fluorescence microscopes (Carl Zeiss AG, Jena, Germany; Keyence Corporation, Osaka, Japan).
Figure 1.

Immunolocalization of B1 and B2 receptors in primary cultured trigeminal ganglion (TG) neurons and TG cryosections. (A,D,G,J) Cells positive for the pan neuronal marker in primary cultured TG neurons (A,D) and TG cryosections (G,J). (B,H) Immunoreactivity to the B1 receptor antibody (green) in primary cultured TG neurons (B) and TG cryosections (H). (C,I) Triple immunofluorescence staining with antibodies against the pan neuronal marker (red) and B1 receptor (green) in primary cultured TG neurons (C) and TG cryosections (I). Nuclei are shown in blue. (E,K) Positive immunoreactivity to the B2 receptor antibody (green) in primary cultured TG neurons (E) and TG cryosections (K). (F,L) Triple immunofluorescence staining with antibodies against the pan neuronal marker (red) and B2 receptor (green) in primary cultured TG neurons (F) and TG cryosections (L). Nuclei are shown in blue. Scale bars are 50 μm in (A–F), and 20 μm in (G–L). Each set of images showing representative immunolocalization of B1 (A–C) and B2 receptors (D–F) in primary cultured TG neurons was obtained from six different rats, while that showing immunolocalization of B1 (G–I) and B2 receptors (J–L) in TG cryosections was obtained from five different rats.
Figure 2.

Immunolocalization of B2 receptors in the soma of TG neurons in cryosections. (A) Positive immunoreactivity to NF-H as an A-neuron marker in TG neurons (arrowhead). (B,E,H,K) B2 receptor immunoreactivity (arrowheads). (C) Triple immunofluorescence staining with antibodies against B2 receptors (green) and NF-H (red). Nuclei are shown in blue. (D) Positive immunoreactivity to SP as a peptidergic C-neuron marker in TG neurons (arrowheads). (F) Triple staining with antibodies against B2 receptors (green) and SP (red). Nuclei are shown in blue. (G) Positive immunoreactivity to IB4 as a non-peptidergic C-neuron marker in TG neurons (arrowheads). (I) Triple staining with antibodies against B2 receptors (red) and IB4 (green). Nuclei are shown in blue. (J) Positive immunoreactivity to TrkA as an nerve growth factor (NGF)-responsive nociceptor marker in TG neurons (arrowheads). (L) Triple staining with antibodies against B2 receptors (green) and TrkA (red). Nuclei are shown in blue. (M) No fluorescence was detected in the negative control. Scale bars: 20 μm. Each set of photos showing representative colocalization of B2 receptors with NF-H (A–C) and SP (D–F) was obtained from six different rats. Each set of photos showing representative colocalization of B2 receptors with IB4 (G–I) and TrkA (J–L) was obtained from four different rats.
Solutions and Reagents
A standard solution containing (in mM) 137 NaCl, 5.0 KCl, 2.0 CaCl2, 0.5 MgCl2, 0.44 KH2PO4, 0.34 Na2HPO4, 4.17 NaHCO3, and 5.55 glucose (pH 7.4) was used as an extracellular solution. A high-K+ solution containing (in mM) 91 NaCl, 50 KCl, 2.0 CaCl2, 0.5 MgCl2, 0.44 KH2PO4, 0.34 Na2HPO4, 4.17 NaHCO3, and 5.55 glucose (pH 7.4) was used to discern TG neurons from glial cells by activation of depolarization-induced increases in intracellular free Ca2+ concentration ([Ca2+]i) in neurons. BK, a selective B2 receptor antagonist (HOE140), a selective B1 receptor antagonist (R715) and a highly selective B1 receptor agonist (Lys-[Des-Arg9]BK) were obtained from Tocris Bioscience (Bristol, UK). All the other reagents were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO, USA), except where indicated.
Measurement of [Ca2+]i
Primary cultured TG cells were loaded for 90 min at 37°C in Hank’s solution containing 10 μM of fura-2 acetoxymethyl ester (Dojindo Laboratories, Kumamoto Japan) and 0.1% (w/v) pluronic acid F-127 (Life Technologies). Cultured TG cells were then rinsed with fresh Hank’s solution and mounted on a microscope stage (Olympus Corporation, Tokyo, Japan). Fura-2 fluorescence emission was measured at 510 nm in response to alternating excitation wavelengths of 340 nm (F340) and 380 nm (F380) with an Aquacosmos system and software (Hamamatsu Photonics K.K., Shizuoka, Japan), which controls the excitation wavelength selector and intensified charge-coupled device camera system (Hamamatsu Photonics K.K.). [Ca2+]i was measured as the fluorescence ratio of F340 and F380 (RF340/F380) and expressed as F/F0 units. The RF340/F380 value (F) was normalized to the resting value (F0).
Statistical and Offline Analysis
The data were expressed as the mean ± standard error (S.E.) or standard deviation of the mean of N observations, where N represents the number of independent experiments or cells, respectively. The Kruskal–Wallis test, Dunn’s posthoc test, or Mann–Whitney U-test was used to determine the nonparametric statistical significance. P values less than 0.05 were considered significant. The statistical analysis was performed with GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
The dependence of the changes in [Ca2+]i on each pharmacological agent was determined by fitting the data to the following function with Origin 8.5 (OriginLab Corporation, Northampton, MA, USA):
| (1) |
where K is the equilibrium binding constant, [x]o indicates the applied concentration of the pharmacological agents, and F/F0int and F/F0fin are the initial and final F/F0 responses, respectively.
Results
Immunolocalization of BK Receptors in TG Neurons
The cultured TG neurons showed positive immunoreactivity to a neuronal marker cocktail (Neuro-ChromTM pan-neuronal marker), which contained mouse anti-NeuN, anti-MAP2, and anti-βIII tubulin antibodies (Figures 1A,D). Intense B2 receptor immunoreactivity was observed in primary cultured TG neurons (Figure 1E), and it showed colocalization with the pan neuronal marker (Figure 1F) in somata, dendrites, axons, and perinuclear regions. Weak but positive B1 receptor immunoreactivity was also observed in primary cultured TG cells (Figure 1B), and the immunoreactivity colocalized with the pan neuronal marker (Figure 1C).
In the TG cryosections, we could observe positive immunoreactivity against the neuronal marker cocktail (Figures 1G,J). These TG neurons in the cryosections showed positive immunoreactivity to the B2 receptor antibody (Figure 1K), showing colocalization with the pan neuronal marker (Figure 1L) in somata, dendrites, axons, and perinuclear regions. However, the TG cryosections did not show B1 receptor immunoreactivity (Figures 1H,I). Positive immunoreactivity was also observed with NF-H (an A-neuron marker; Figure 2A), SP (a peptidergic C-neuron marker; Figure 2D), IB4 (a non-peptidergic C-neuron marker; Figure 2G), and high-affinity NGF receptor (TrkA; an NGF-responsive nociceptor marker; Figure 2J) antibodies. These immunoreactivities against NF-H, SP, IB4, and TrkA antibodies showed colocalization with those against the B2 receptor antibodies (Figures 2B,C,E,F,H,I,K,L).
BK-Induced [Ca2+]i Increases in TG Neurons
We observed rapid and transient [Ca2+]i increases in TG neurons following the administration of five different concentrations of BK (0.01, 0.1, 1.0, 10, and 100 nM) in the presence of external Ca2+ (2.0 mM; Figure 3A). A semilogarithmic plot (Figure 3B) illustrates F/F0 values as a function of the applied BK concentrations, and the equilibrium-binding constant was the half-maximal 50% effective concentration (EC50) of 1.0 nM.
Figure 3.
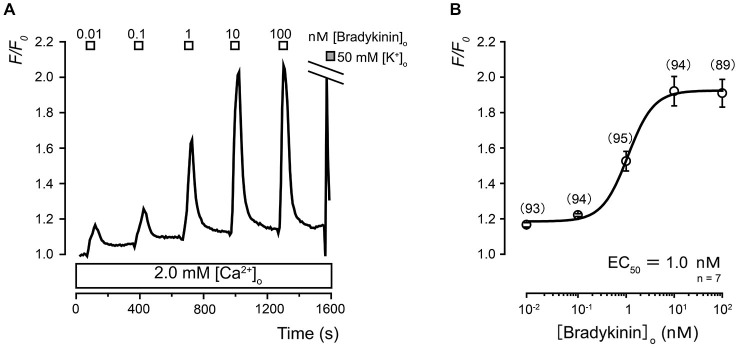
Ca2+ dependence of extracellular bradykinin (BK)-induced [Ca2+]i responses in TG neurons. (A) Examples of transient [Ca2+]i increases following the application of a series of BK concentrations. In the presence of extracellular Ca2+ (2.0 mM; lower white box), the application of BK induced transient [Ca2+]i increases in a concentration-dependent manner. The concentrations of BK (0.01–100 nM) that were administered are shown in the uppermost white boxes. (B) The data points illustrate the F/F0 values as a function of the applied BK concentration. Each data point represents the mean ± standard error (S.E.) of seven experiments (the numbers in parentheses represent the number of tested cells). The curve on the semilogarithmic scale was fitted according to Equation 1, which is described in the text. The upper gray box in (A) indicates the timing of the application of the 50 mM KCl solution. The equilibrium binding constant of BK was 1.0 nM.
HOE140, a B2 Receptor Antagonist, Inhibited the BK-Induced [Ca2+]i Increases in TG Neurons
We examined the BK-induced [Ca2+]i responses in both the presence and absence of external Ca2+. The application of BK (1.0 nM) rapidly increased [Ca2+]i to a peak F/F0 value of 1.7 ± 0.03 F/F0 units in the presence (2.0 mM) of external Ca2+ and 1.4 ± 0.03 F/F0 units in the absence (0 mM) of external Ca2+ (Figures 4A,D). The amplitudes of the BK-induced [Ca2+]i increases significantly differed between those in the presence and absence of extracellular Ca2+. In the absence of extracellular Ca2+, BK (1.0 nM)-induced [Ca2+]i increases were significantly inhibited by a B2 receptor antagonist (100 nM of HOE140) (Figures 4C,D) but not by a B1 receptor antagonist (1.0 μM of R715) (Figures 4B,D).
Figure 4.

Pharmacological identification of BK receptors in TG neurons. (A) Examples of transient [Ca2+]i increases following the administration of 1.0 nM of BK (upper white boxes) with (lower gray boxes) or without (lower white box) extracellular Ca2+ (2.0 mM). (B) Examples of BK-induced (1.0 nM; upper white boxes) [Ca2+]i increases with (upper black box) or without R715 in the absence (lower white box) or presence (lower gray boxes) of external Ca2+. (C) Examples of BK-induced (1.0 nM; upper white boxes) [Ca2+]i increases with (upper black box) or without HOE140 in the absence (lower white box) or presence (lower gray boxes) of external Ca2+. (A,B,C) The upper gray boxes indicate the timing of the application of the 50 mM of KCl solution. (D) The summary bar graph indicates [Ca2+]i increases during the first (upper column) and second (second upper column) application of 1.0 nM of BK with external Ca2+ (2.0 mM) (gray boxes on the right side). The mean values of the increase in [Ca2+]i following the first (third upper column) and second (fourth upper column) application of 1.0 nM of BK in the absence of external Ca2+ as well as the application of 1.0 nM of BK with 1.0 μM of R715 (fifth upper column) and 100 nM of HOE140 (sixth upper column) in the absence of external Ca2+ (white boxes on the right side) are shown. Each column denotes the mean ± S.E. of the indicated (in parentheses) number of experiments. The statistical significance between the columns (shown by solid lines) is indicated by asterisks: *p < 0.05.
The B1 Receptor Antagonist R715 did not Affect the BK-Induced [Ca2+]i Increases
The BK-induced (1.0 nM) increases in [Ca2+]i were not significantly inhibited in TG neurons by the administration of four different concentrations of the B1 receptor antagonist (0.001, 0.01, 0.1, and 1.0 μM of R715) in the presence (2.0 mM) of external Ca2+ (Figures 5A,B).
Figure 5.

Pharmacological identification of B1 receptors in TG neurons. (A) Examples of BK-induced (1.0 nM; upper white boxes) [Ca2+]i increases showing insensitivity to R715 (0.001–1.0 μM; upper black boxes) in the presence of external Ca2+ (2.0 mM; lower white box). (B) The summary bar graph indicates [Ca2+]i increases following the first (upper column) application of 1.0 nM of BK with external Ca2+ (2.0 mM). The mean values for the increases in [Ca2+]i following the application of 1.0 nM of BK with 0.001 μM (second upper column), 0.01 μM (third upper column), 0.1 μM (fourth upper column), or 1.0 μM (fifth upper column) of R715 in the presence of external Ca2+ (white boxes on the right side) are shown. Each column denotes the mean ± S.E. of the indicated (in parentheses) number of experiments. There is no statistical significance between the columns. (C) Examples of transient [Ca2+]i increases following the application of a series of concentrations of Lys-[Des-Arg9]BK (0.01–100 nM; upper white boxes) in the presence of extracellular Ca2+ (2.0 mM; lower white box). (D) The data points illustrate the F/F0 values as a function of the applied concentrations of Lys-[Des-Arg9]BK. Each data point represents the mean ± S.E. of six independent experiments (the numbers in parentheses represent the number of tested cells). The curve on the semilogarithmic scale was fitted according to Equation 1, which is described in the text. The equilibrium-binding constant for Lys-[Des-Arg9]BK was 0.4 nM. (E) Examples of Lys-[Des-Arg9]BK-induced (10 nM; upper white boxes) [Ca2+]i increases that were significantly inhibited by 1.0 μM of R715 (upper black box) in the presence of external Ca2+ (2.0 mM; lower white box). (A,C,E) The application of 50 mM of the KCl solution is shown in the gray boxes. (F) Summary bar graph of the [Ca2+]i increases following 10 nM of Lys-[Des-Arg9]BK with (lower black column) or without (upper white column) 1.0 μM of R715. Each column denotes the mean ± S.E. of the indicated number (in parentheses) of independent experiments. The statistical significance between the columns (shown by solid lines) is indicated by asterisks: *p < 0.05.
Pharmacological Identification of B1 Receptors in TG Neurons
We investigated the [Ca2+]i increases during the administration of Lys-[Des-Arg9]BK, which is an endogenous, potent, and highly selective B1 receptor agonist (Talbot et al., 2009; More et al., 2014). The increases in [Ca2+]i in the TG neurons were induced by the administration of five different concentrations of Lys-[Des-Arg9]BK (0.01, 0.1, 1, 10, and 100 nM) in the presence of extracellular Ca2+ (2.0 mM) (Figure 5C). A semilogarithmic plot (Figure 5D) illustrates the F/F0 values as a function of the applied concentration of Lys-[Des-Arg9]BK with an equilibrium-binding constant of 0.4 nM. In the presence of extracellular Ca2+, the Lys-[Des-Arg9]BK-induced increase in [Ca2+]i was significantly inhibited by a B1 receptor antagonist (1.0 μM of R715) (Figures 5E,F).
Discussion
The present study demonstrated the functional expression of BK receptors (B1 and B2) in TG neurons. B2 receptors were present on axons and dendrites in A-neurons, non-peptidergic C-neurons, peptidergic C-neurons, and NGF-responsive nociceptors. While the localization pattern of the B1 receptor was not clear in the TG cryosections, weak immunoreactivity for B1 receptors was observed in the primary cultured TG neurons. The application of BK activated B2 receptors and Lys-[Des-Arg9]BK activated the B1 receptors. B2 receptor activation mobilized [Ca2+]i by releasing Ca2+ from internal Ca2+ stores with partial Ca2+ influx from the extracellular medium.
B2 receptors, which are expressed ubiquitously and constitutively in healthy tissues, are essential in the early stages of general pain generation (Hall, 1992). The constitutive expression of B2 receptors in TG neurons has been studied by reverse transcription-polymerase chain reaction (RT-PCR) analyses (Ceruti et al., 2011) and immunocytochemical analyses in cultured TG neurons (Patwardhan et al., 2005). Although BK-induced [Ca2+]i increases have also been reported in TG neurons (Ceruti et al., 2008, 2011), precise functional expression patterns of B1 and B2 receptors in TG neurons remained unclear. The results of the present study showing the functional expression and localization of B2 receptors in TG neurons were in line with the previous results. The results of this study were also in line with the pharmacological properties of BK, which is a potent and endogenous agonist for B2 receptors and not B1 receptors in the sympathetic neurons of the rat superior cervical ganglion (Babbedge et al., 1995) and in Chinese hamster ovary (CHO) cells stably expressing recombinant human B1 or B2 receptors (Simpson et al., 2000). Furthermore, BK has an affinity for B2 receptors that is 500 times that for B1 receptors (Simpson et al., 2000). Therefore, B2 receptors are histologically and functionally expressed, and endogenous BK preferentially activates B2 receptors in rat TG neurons.
The expression of the B1 receptor, which is induced as a result of tissue damage and inflammation, is involved in chronic inflammation or tissue injury (Hall, 1992). The observations of the constitutive B1 receptor expression in TG and dorsal root ganglion (DRG) neurons have been inconsistent. In DRG neurons, some immunohistochemical studies have reported constitutive B1 receptor expression (Ma et al., 2000; Wotherspoon and Winter, 2000). In contrast, other studies have described that B1 receptor activation-induced [Ca2+]i responses could not be observed in DRG neurons (Brand et al., 2001). In TG neurons, an immunohistochemical study has shown the constitutive expression of B1 receptors (Ma et al., 2000). In contrast, RT-PCR analyses have demonstrated that B1 receptor mRNA was barely expressed in intact tissue, while it was weakly expressed in primary cultured TG neurons. In primary cultured TG neurons, the levels of expression of B1 receptor mRNA have been reported to depend on the length of the culture period (Ceruti et al., 2011). The present immunohistochemical and immunocytochemical results were similar to the previous RT-PCR results; B1 receptor immunoreactivity was weakly positive in cultured TG neurons and could not be detected in intact TG tissue. Although few report concerning B1 receptor-induced [Ca2+]i response in TG neurons exist, in the [Ca2+]i imaging in the present study, the B1 receptor agonist, Lys-[Des-Arg9]BK which is a metabolite of endogenous BK in peripheral tissues (Regoli et al., 2001), dose-dependently increased [Ca2+]i in the presence of extracellular Ca2+, and this increase was suppressed by a B1 receptor-specific antagonist (Figures 5C–F). These results of B1 receptor expression in primary cultured TG neurons suggest that the expression of B1 receptors is induced in TG neurons by tissue damage and/or inflammation. However, further studies are required to evaluate the expression patterns of B1 receptors in native TG neurons.
BK-induced [Ca2+]i increases were observed in both the presence and absence of extracellular Ca2+. However, the amplitudes of the [Ca2+]i increases in the absence of extracellular Ca2+ were significantly smaller (84.9 ± 11.3%, N = 161) than those in the presence of Ca2+ (100%; Figures 4A,D). This indicated that the BK-induced [Ca2+]i mobilization (by B2 receptor activation) was mainly composed of Ca2+ release from internal stores with partial Ca2+ influx from the extracellular medium. Notably, BK has been reported to activate voltage-dependent Ca2+ channels in rat submucosal plexus neurons (Avemary and Diener, 2010; Rehn et al., 2013), and transient receptor potential cation channel subfamily-V member-1 channels in rat DRG neurons (Ferreira et al., 2004; Mistry et al., 2014). However, BK-induced Ca2+ currents could not be recorded in TG neurons (Kitakoga and Kuba, 1993). Although further studies are needed to clarify which Ca2+ influx pathways contribute to the BK-induced Ca2+ influx in TG neurons, the present results clearly indicate that BK mobilizes [Ca2+]i through both intracellular Ca2+ release and Ca2+ influx.
In addition, NGF-TrkA signaling plays important roles in not only the developmental processes of peptidergic nociceptive afferents, but also in the generation of acute and chronic pain state in adults. The signaling also up-regulates B2 receptor expression in peptidergic nociceptors (Mantyh et al., 2011). Thus, the results showing colocalization of B2 receptor and TrkA immunoreactivity in TG neurons strongly support reports describing that the B2 receptor mediates inflammatory/neuropathic pain induced by peripheral sensitization in the orofacial region (Chichorro et al., 2004; Luiz et al., 2010); however, the present results obtained from neonatal rat may not reflect the situation in adults.
In conclusion, B2 receptors were expressed constitutively, and their activation induced the mobilization of [Ca2+]i by releasing Ca2+ from intracellular stores with partial Ca2+ influx. In contrast, B1 receptor expression was faint in cultured TG neurons and absent in neurons in TG cryosections, although a metabolite of endogenous BK elicited [Ca2+]i increases. These results indicated that both BK and its metabolites activated [Ca2+]i mobilization in TG neurons through B2 and B1 receptor activation, respectively.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a Grant-in-Aid (#23592751/26861559/25861762) for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- Avemary J., Diener M. (2010). Bradykinin-induced depolarisation and Ca2+ influx through voltage-gated Ca2+ channels in rat submucosal neurons. Eur. J. Pharmacol. 635, 87–95. 10.1016/j.ejphar.2010.03.009 [DOI] [PubMed] [Google Scholar]
- Babbedge R., Dray A., Urban L. (1995). Bradykinin depolarises the rat isolated superior cervical ganglion via B2 receptor activation. Neurosci. Lett. 193, 161–164. 10.1016/0304-3940(95)11690-x [DOI] [PubMed] [Google Scholar]
- Basbaum A. I., Bautista D. M., Scherrer G., Julius D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. 10.1016/j.cell.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M., Klusch A., Kurzai O., Valdeolmillos M., Schmidt R. F., Petersen M. (2001). No evidence for bradykinin B1 receptors in rat dorsal root ganglion neurons. Neuroreport 12, 3165–3168. 10.1097/00001756-200110080-00036 [DOI] [PubMed] [Google Scholar]
- Ceruti S., Fumagalli M., Villa G., Verderio C., Abbracchio M. P. (2008). Purinoceptor-mediated calcium signaling in primary neuron-glia trigeminal cultures. Cell Calcium 43, 576–590. 10.1016/j.ceca.2007.10.003 [DOI] [PubMed] [Google Scholar]
- Ceruti S., Villa G., Fumagalli M., Colombo L., Magni G., Zanardelli M., et al. (2011). Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 Knock-in mice: implications for basic mechanisms of migraine pain. J. Neurosci. 31, 3638–3649. 10.1523/JNEUROSCI.6440-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervero F., Laird J. M. (1996). Mechanisms of touch-evoked pain (allodynia): a new model. Pain 68, 13–23. 10.1016/s0304-3959(96)03165-x [DOI] [PubMed] [Google Scholar]
- Chichorro J. G., Lorenzetti B. B., Zampronio A. R. (2004). Involvement of bradykinin, cytokines, sympathetic amines and prostaglandins in formalin-induced orofacial nociception in rats. Br. J. Pharmacol. 141, 1175–1184. 10.1038/sj.bjp.0705724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duehrkop C., Banz Y., Spirig R., Miescher S., Nolte M. W., Spycher M., et al. (2013). C1 esterase inhibitor reduces lower extremity ischemia/reperfusion injury and associated lung damage. PLoS One 8:e72059. 10.1371/journal.pone.0072059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutra R. C., Bento A. F., Leite D. F. P., Manjavachi M. N., Marcon R., Bicca M. A., et al. (2013). The role of kinin B1 and B2 receptors in the persistent pain induced by experimental autoimmune encephalomyelitis (EAE) in mice: evidence for the involvement of astrocytes. Neurobiol. Dis. 54, 82–93. 10.1016/j.nbd.2013.02.007 [DOI] [PubMed] [Google Scholar]
- Ferreira J., da Silva G. L., Calixto J. B. (2004). Contribution of vanilloid receptors to the overt nociception induced by B2 kinin receptor activation in mice. Br. J. Pharmacol. 141, 787–794. 10.1038/sj.bjp.0705546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. M. (1992). Bradykinin receptors: pharmacological properties and biological roles. Pharmacol. Ther. 56, 131–190. 10.1016/0163-7258(92)90016-s [DOI] [PubMed] [Google Scholar]
- Hall J. M. (1997). Bradykinin receptors. Gen. Pharmacol. 28, 1–6. 10.1016/S0306-3623(96)00174-7 [DOI] [PubMed] [Google Scholar]
- Hess J. F., Borkowski J. A., Stonesifer G. Y., MacNeil T., Strader C. D., Ransom R. W. (1994). Cloning and pharmacological characterization of bradykinin receptors. Braz. J. Med. Biol. Res. 27, 1725–1731. [PubMed] [Google Scholar]
- Julius D., Basbaum A. I. (2001). Molecular mechanisms of nociception. Nature 413, 203–210. 10.1038/35093019 [DOI] [PubMed] [Google Scholar]
- Kawaguchi A., Sato M., Kimura M., Ichinohe T., Tazaki M., Shibukawa Y. (2015). Expression and function of purinergic P2Y12 receptors in rat trigeminal ganglion neurons. Neurosci. Res. [Epub ahead of print]. 10.1016/j.neures.2015.04.008 [DOI] [PubMed] [Google Scholar]
- Kitakoga O., Kuba K. (1993). Bradykinin-induced ion currents in cultured rat trigeminal ganglion cells. Neurosci. Res. 16, 79–93. 10.1016/0168-0102(93)90075-2 [DOI] [PubMed] [Google Scholar]
- Kuroda H., Sobhan U., Sato M., Tsumura M., Ichinohe T., Tazaki M., et al. (2013). Sodium-calcium exchangers in rat trigeminal ganglion neurons. Mol. Pain 9:22. 10.1186/1744-8069-9-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiz A. P., Schroeder S. D., Chichorro J. G., Calixto J. B., Zampronio A. R., Rae G. A. (2010). Kinin B1 and B2 receptors contribute to orofacial heat hyperalgesia induced by infraorbital nerve constriction injury in mice and rats. Neuropeptides 44, 87–92. 10.1016/j.npep.2009.10.005 [DOI] [PubMed] [Google Scholar]
- Ma Q. P., Hill R., Sirinathsinghji D. (2000). Basal expression of bradykinin B1 receptor in peripheral sensory ganglia in the rat. Neuroreport 11, 4003–4005. 10.1097/00001756-200012180-00020 [DOI] [PubMed] [Google Scholar]
- Mantyh P. W., Koltzenburg M., Mendell L. M., Tive L., Shelton D. L. (2011). Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology 115, 189–204. 10.1097/ALN.0b013e31821b1ac5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry S., Paule C. C., Varga A., Photiou A., Jenes A., Avelino A., et al. (2014). Prolonged exposure to bradykinin and prostaglandin E2 increases TRPV1 mRNA but does not alter TRPV1 and TRPV1b protein expression in cultured rat primary sensory neurons. Neurosci. Lett. 564, 89–93. 10.1016/j.neulet.2014.02.006 [DOI] [PubMed] [Google Scholar]
- More A. S., Kim H. M., Khang G., Hildebrandt T., Bernlöhr C., Doods H., et al. (2014). Des-Arg9-bradykinin causes kinin B1 receptor mediated endothelium-independent contractions in endotoxin-treated porcine coronary arteries. Pharmacol. Res. 90, 18–24. 10.1016/j.phrs.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Ochoa J. L. (2009). Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 72, 1282–1283. 10.1212/01.wnl.0000346325.50431.5f [DOI] [PubMed] [Google Scholar]
- Patwardhan A. M., Berg K. A., Akopain A. N., Jeske N. A., Gamper N., Clarke W. P., et al. (2005). Bradykinin-induced functional competence and trafficking of the δ-opioid receptor in trigeminal nociceptors. J. Neurosci. 25, 8825–8832. 10.1523/jneurosci.0160-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesquero J. B., Pesquero J. L., Oliveira S. M., Roscher A. A., Metzger R., Ganten D., et al. (1996). Molecular cloning and functional characterization of a mouse bradykinin b1 receptor gene. Biochem. Biophys. Res. Commun. 220, 219–225. 10.1006/bbrc.1996.0384 [DOI] [PubMed] [Google Scholar]
- Regoli D., Rizzi A., Perron S. I., Gobeil F. (2001). Classification of kinin receptors. Biol. Chem. 382, 31–35. 10.1515/bc.2001.005 [DOI] [PubMed] [Google Scholar]
- Rehn M., Bader S., Bell A., Diener M. (2013). Distribution of voltage-dependent and intracellular Ca2+ channels in submucosal neurons from rat distal colon. Cell Tissue Res. 353, 355–366. 10.1007/s00441-013-1643-5 [DOI] [PubMed] [Google Scholar]
- Scholz J., Woolf C. J. (2002). Can we conquer pain? Nat. Neurosci. 5(Suppl.), 1062–1067. 10.1038/nn942 [DOI] [PubMed] [Google Scholar]
- Simpson P. B., Woollacott A. J., Hill R. G., Seabrook G. R. (2000). Functional characterization of bradykinin analogues on recombinant human bradykinin B1 and B2 receptors. Eur. J. Pharmacol. 392, 1–9. 10.1016/s0014-2999(00)00046-7 [DOI] [PubMed] [Google Scholar]
- Talbot S., Théberge-Turmel P., Liazoghli D., Sénécal J., Gaudreau P., Couture R. (2009). Cellular localization of kinin B1 receptor in the spinal cord of streptozotocin-diabetic rats with a fluorescent [Nα-Bodipy]-des-Arg9-bradykinin. J. Neuroinflammation 6:11. 10.1186/1742-2094-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari M. M., Prather P. L., Mayeux P. R. (2005). Mechanism of bradykinin-induced Ca2+ mobilization in murine proximal tubule epithelial cells. J. Pharmacol. Exp. Ther. 313, 798–805. 10.1124/jpet.104.080408 [DOI] [PubMed] [Google Scholar]
- Walker K., Perkins M., Dray A. (1995). Kinins and kinin receptors in the nervous system. Neurochem. Int. 26, 1–16; discussion 17–26. 10.1016/0197-0186(94)00115-b [DOI] [PubMed] [Google Scholar]
- Wotherspoon G., Winter J. (2000). Bradykinin B1 receptor is constitutively expressed in the rat sensory nervous system. Neurosci. Lett. 294, 175–178. 10.1016/s0304-3940(00)01561-5 [DOI] [PubMed] [Google Scholar]