

Significance
Phospholamban (PLN) can regulate Ca2+ uptake rates in the sarcoplasmic reticulum in cardiomyocytes. However, the mechanisms that control PLN levels are not fully understood. This study shows that PLN degradation depends on ubiquitinylation of its lysine 3 residue and p62-mediated selective autophagy. Metformin was shown to accelerate autophagy and to induce PLN degradation, resulting in increased Ca2+ uptake. These results suggest that changes in PLN degradation could account for the cardiac inotropic effects of metformin.
Keywords: selective autophagy, ubiquitinylation, protein degradation
Abstract
Phospholamban (PLN) is an effective inhibitor of the sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA). Here, we examined PLN stability and degradation in primary cultured mouse neonatal cardiomyocytes (CMNCs) and mouse hearts using immunoblotting, molecular imaging, and [35S]methionine pulse-chase experiments, together with lysosome (chloroquine and bafilomycin A1) and autophagic (3-methyladenine and Atg5 siRNA) antagonists. Inhibiting lysosomal and autophagic activities promoted endogenous PLN accumulation, whereas accelerating autophagy with metformin enhanced PLN degradation in CMNCs. This reduction in PLN levels was functionally correlated with an increased rate of SERCA2a activity, accounting for an inotropic effect of metformin. Metabolic labeling reaffirmed that metformin promoted wild-type and R9C PLN degradation. Immunofluorescence showed that PLN and the autophagy marker, microtubule light chain 3, became increasingly colocalized in response to chloroquine and bafilomycin treatments. Mechanistically, pentameric PLN was polyubiquitinylated at the K3 residue and this modification was required for p62-mediated selective autophagy trafficking. Consistently, attenuated autophagic flux in HECT domain and ankyrin repeat-containing E3 ubiquitin protein ligase 1-null mouse hearts was associated with increased PLN levels determined by immunoblots and immunofluorescence. Our study identifies a biological mechanism that traffics PLN to the lysosomes for degradation in mouse hearts.
Phospholamban (PLN) is a 52-amino acid peptide located in the sarcoplasmic reticulum (SR) membrane in cardiac, slow-twitch skeletal, and smooth muscle, where it exists as a monomer or pentamer. Whereas monomeric PLN physically interacts with sarco(endo)plasmic reticulum Ca2+ ATPase type 2a (SERCA2a) to antagonize its function, pentameric PLN complexes are thought to be a reservoir of inactive PLN (1–3). The physical interaction between SERCA2a and PLN reduces the apparent affinity of SERCA2a for Ca2+, thereby making SERCA2a less active in transporting Ca2+ from the cytoplasm to the lumen of the SR at the same concentration of cytoplasmic Ca2+. The physical interaction between the two proteins is regulated by phosphorylation of PLN at Ser16 by protein kinase A or at Thr17 by Ca2+/calmodulin-dependent protein kinase II (2). Phosphorylation of PLN reduces its affinity for SERCA2a, thereby increasing SERCA2a activity (2). Evidence from transgenic mice also supports the inhibitory function of PLN. Although targeted PLN deletion enhances baseline cardiac performance, cardiac-specific overexpression of superinhibitory forms of PLN leads to decreases in the affinity of SERCA2a for Ca2+ (2). These observations underscore the primary role of PLN as a regulator of SERCA2a activity and, therefore, as a crucial regulator of cardiac contractility. PLN inhibition of SERCA2a can be reversed by either external (i.e., activation of β-adrenergic receptors) or internal (i.e., increased intracellular Ca2+ concentration) stimuli.
Previous studies identified three PLN mutations in families of patients with hereditary dilated cardiomyopathy. These mutations, the substitution of Cys for Arg9 (R9C) (4), Arg14 deletion (RΔ14) (5), and the substitution of TGA for TAA in the Leu39 codon, creating a stop codon (L39stop) (6), also lead to dilated cardiomyopathy in transgenic mice. At the cellular level, ectopically expressed RΔ14 and L39stop PLN mutants localize at the plasma membrane in HEK-293T cells, cultured mouse neonatal cardiomyocytes, and cardiac fibroblasts, whereas wild-type and the R9C mutant reside within the endoplasmic reticulum (ER)/SR (6, 7). These data, together with a recent study by Sharma et al. (8), suggest a highly ordered trafficking of PLN, ultimately ensuring correct localization, and thus function, within the SR. However, PLN trafficking and degradation mechanisms in mammalian cardiomyocytes have not been clearly established.
Protein degradation and clearance of damaged organelles are critical for cellular physiology, and failure in proper clearance has been shown to have pathological repercussions (9). Autophagy is a major mechanism that mediates protein and organelle degradation in response to external and internal signals. External stimulation through pharmacological agonists, such as metformin and rapamycin, promotes autophagy via AMP-activated protein kinase (AMPK) and mammalian target of rapamycin signal pathways, whereas amino acid starvation and an increased intracellular AMP/ATP ratio serve as internal signals to promote autophagy via the Ca2+/Calmodulin-dependent kinase kinase-β (10). Steps in the autophagy pathway involve nucleation of targeted macromolecules on the ER membrane, trafficking of autophagosomes to lysosomes and, finally, fusion of the autophagosome-lysosome, resulting in targeted protein degradation (11). In the heart, autophagy plays a crucial role in response to insults, in part by relieving ER stress (12) and removing damaged mitochondria (13). Loss of autophagy could result in irreversible apoptosis and reduced cardiac functioning (14).
To characterize PLN degradation, we conducted a series of assays in cultured mouse neonatal cardiomyocytes (CMNCs) and the hearts of HECT domain and ankyrin repeat-containing E3 ubiquitin protein ligase 1 (Hace1)-null mice. Our results show that PLN degradation required both polyubiquitinylation and p62-mediated selective autophagy in CMNCs. Loss of HACE1 was associated with increased PLN levels, supporting the notion that selective autophagy modulates PLN degradation in vivo. Metformin promoted wild-type and R9C PLN degradation through autophagic pathways, resulting in metformin-induced inotropic enhancement.
Results
Endogenous PLN Is Degraded by Lysosomes in CMNCs.
CMNCs were cultured for 24 h in the presence of NH4Cl (20 mM) or chloroquine (CQ) (100 µM) to inhibit lysosomes, MG132 (10 µM), or Lac (5 µM) to inhibit proteasomes, or N-[N-(N-Acetyl-l-leucyl)-l-leucyl]-l-norleucine (ALLN; 100 µM) to inhibit the Ca2+-activated protease calpain (15–17). Fig. 1A shows that inhibiting lysosomal functions promoted endogenous PLN accumulation, whereas proteasome or calpain inhibitors did not alter PLN levels, but did increase the levels of connexin 43 (16) in CMNCs. Quantification of immunoblots revealed a significant 1.8-fold increase in PLN levels following CQ treatments (Fig. 1B). RT-PCR analyses confirmed that there was no change in PLN transcript levels in CMNCs in response to various inhibitors (Fig. 1C). The inhibition of lysosomal function by 100 µM CQ promoted accumulation of PLN monomers and pentamers (Fig. 1 D and E).
Fig. 1.

Endogenous PLN is degraded by lysosomes in CMNCs. (A) Western blots assessing PLN levels in response to different inhibitors. (B) Quantification of PLN protein levels in the presence of various inhibitors (n = 3). *P < 0.05. (C) RT-PCR for measuring pln transcript levels in response to inhibitor treatment. A pln cDNA plasmid was present and absent in positive and negative controls, respectively. (D) Western blot demonstrating the effect of CQ on the PLN pentamer and monomer levels. (E) Quantification of PLN pentamer and monomer levels in response to CQ. (F) Immunoblot assessing PLN protein levels in a CQ dosage-dependent fashion. Numbers represent quantification of PLN (6 kDa) protein levels (n = 3). (G) Immunoblot measuring endogenous PLN (6 kDa) levels in response to CQ treatment at different time points. Numbers represent quantification of PLN protein levels (n = 3). Loading controls, α-tubulin, 51 kDa. Lysosomal inhibitors, ammonium chloride (NH4Cl) and CQ; proteasomal inhibitors, lactacystin (Lac) and MG-132; calpain inhibitor, ALLN.
CMNCs were subjected to CQ treatment in a time- or dosage-dependent manner. Fig. 1F shows that PLN levels significantly increased as CQ concentration was increased to 100 μM (16), and that further increases in CQ concentration resulted in no greater increase in PLN levels. Hence, we used 100 µM CQ for CQ-related experiments throughout the study, unless otherwise indicated. In a time-dependent study, the elevation in PLN levels reached statistical significance within 12 h of CQ treatment and continued increasing until 24 h (Fig. 1G).
Autophagy Is Essential for PLN Degradation in Lysosomes.
Because the lysosomal inhibitor CQ promoted PLN accumulation, we determined whether PLN would colocalize with a lysosome marker, lysosome-associated membrane protein 1 (LAMP1), in the presence of CQ. Colocalization between PLN and LAMP1 did not appear to be affected qualitatively by CQ (Fig. 2A) and quantitative measurements of Pearson’s correlation for the effect of CQ on colocalization revealed a nearly neutral coefficient, indicating the absence of a relationship (Fig. 2B).
Fig. 2.

PLN is targeted to and degraded in lysosomes via autophagy in CMNCs. (A) Immunofluorescence assessing the colocalization of endogenous PLN and lysosome-specific protein LAMP1 in response to CQ treatment. (B) Quantification of PLN and LAMP1 colocalization in the presence of CQ (n = 25). (C) Immunofluorescence assessing colocalization signals between PLN and LC3 in response to 100 µM CQ and 100 nM bafilomycin. (D) Quantification of PLN and LC3 colocalization signals in the presence of CQ or bafilomycin (n = 25). *P < 0.05. (E) Immunoblot showing an increase in PLN protein levels in CMNCs in the presence of CQ or bafilomycin. (F) Immunoblots following 3-MA inhibition. Numbers represent quantification of PLN, LC3-II, and p62 levels. (G) Immunoblots following Atg5 silencing. Numbers represent quantification of PLN, ATG5, p62, and LC3-II levels in response to Atg5 knockdown. (Scale bars, 50 µm.)
Because CQ treatment did not increase the amount of PLN in the lysosomes, this may occur because CQ blocks the fusion of autophagosomes and lysosomes. Thus, we hypothesized that PLN could remain in autophagosomes and could colocalize with an autophagosome marker microtubule-associated protein 1A light chain 3 (LC3) in CQ-treated CMNCs. We incubated CMNCs with 100 µM CQ or with 100 nM bafilomycin (18), which is known to block autophagosome/lysosome fusion by specifically inhibiting the function of vacuolar-type H+-ATPase on lysosome membranes (19). Fig. 2C shows that CQ and bafilomycin promoted puncta formation for both PLN and LC3, whereas both proteins were more evenly distributed in DMSO controls. In addition, some PLN signals were found within the LC3+ puncta, suggesting the presence of PLN in autophagosomes (Fig. 2C). Orthogonal plane optical sectioning (XZ- and YZ-planes) showed colocalized signals between PLN and LC3 in response to both CQ and bafilomycin, but these signals were lower in the DMSO control. Finally, the average Pearson’s coefficient between PLN and LC3 was twofold higher in the CQ- and bafilomycin-treated CMNCs than in DMSO controls within 25 randomized images (Fig. 2D). Similarly, CQ and bafilomycin promoted PLN accumulation in CMNCs by more than twofold (Fig. 2E).
Increased colocalization between PLN and LC3 in the presence of lysosomal inhibitors implied that autophagy might modulate PLN trafficking in CMNCs. We inhibited autophagy with type III phosphatidylinositol 3-kinase antagonist 3-methyladenine (3-MA) and with Atg5 silencing in CMNCs. Treatment with 10 mM 3-MA significantly elevated PLN levels (Fig. 2F). Quantitative analyses showed more than a twofold increase in PLN levels in response to 3-MA inhibition in CMNCs (Fig. 2F). Inhibition was verified by LC3-II isoform depletion and p62/SQSTM1 protein accumulation, compared with controls. Similarly, Atg5 siRNA (100 nM) also promoted protein accumulation for PLN, p62/SQSTM1 and reduced the level of LC3-II in CMNCs (Fig. 2G).
Inhibition of PLN Ubiquitinylation Modulates PLN Degradation.
A proteomic search of protein ubiquitinylation in murine tissues revealed that PLN is likely to be ubiquitinylated (20), but this modification has not been verified directly. We hypothesized that ubiquitinylation of PLN would function as a marker for protein trafficking. Bioinformatic analyses showed that lysine residue 3 (K3) is conserved among species. Therefore, we hypothesized that K3 could be modified by ubiquitinylation. Whole-cell lysates from HEK-293T cells transfected with NF-PLN, HA-ubiquitin, or both plasmids revealed a polyubiquitinylation pattern associated with PLN pentamers, but not with monomeric PLN (Fig. 3A, Upper Right). In addition, several purified NF-PLN proteins separated in mass by ∼8.5 kDa could also be visualized by HA immunoblotting, but this pattern was not observed for NF-PLN or HA-ubiquitin transfected lysates alone, suggesting that PLN was polyubiquitinylated (Fig. 3A, Upper Left). We repeated this experiment with a PLN mutant in which alanine was substituted for lysine 3 (K3A) (21) to prevent any potential PLN ubiquitinylation. As predicted, the purified K3A NF-PLN mutant was not conjugated with HA-ubiquitin, whereas wild-type PLN was ubiquitinylated (Fig. 3B).
Fig. 3.

NF-PLN is ubiquitinylated in transfected HEK-293T cells and p62 interaction is required. (A, Left) In cotransfected cells, polyubiquitinylation assays with boiled lysates involved immunoprecipitation of NF-PLN followed by immunoblotting with HA-tagged ubiquitin. Asterisks denote potential PLN monomers attached to 1–4 Ub monomers (*1 Ub, mass 14.5 kDa; **2 Ub, mass 23 kDa; ***3 Ub, mass 31.5 kDa; and ****4 Ub, mass 40 kDa). (Left Lower) Immunoblotting with Flag shows that most of the PLN monomer was not ubiquitinylated. (Right) Control immunoblots with unboiled samples were performed to detect NF-PLN and HA-ubiquitin. In lane 3 of the lower panel, a unique HA-stained band of ∼40 kDa in lane 3 may indicate monoubiquitinylation of the PLN pentamer. Tubulin was used as a loading control. (B) (First panel, Left) Polyubiquitinylation assays using NF-PLN WT or NF-PLN K3A mutant with coexpressed HA-ubiquitin. IP was with Flag antibody and IB with HA antibody. Control experiments include IP/IB for Flag (second panel) and assessing total unboiled lysates for HA and Flag blots (Right). In lane 2, unique 23-, 40-, and 48.5-kDa bands indicate ubiquitinylation of PLN. (C, Left) Co-IPs were performed using unboiled lysates from cotransfections of NF-PLN WT plus HA-p62 WT or NF-PLN WT plus HA-p62 ΔUBA. Lane 2, p62 was co-IPed with ubiquitinylated NF-PLN. Lane 3, loss of the p62 ubiquitin-binding (UBA) domain abolished the PLN-p62 interaction. (Center) Control experiments include IP/IB for Flag (lane 2) demonstrating the presence in the co-IP of 40- and 48.5-kDa forms of ubiquitinylated NF-PLN. (Right) Total unboiled lysates and IB with Flag, HA, and tubulin.
Because several autophagic adaptor proteins may recognize ubiquitinylated proteins, including—but not limited to—Sequestosome 1 (p62/SQSTM1), for lysosome-dependent degradation (22), we investigated whether PLN might physically interact with p62. In Co-IP assays, HA-p62 was repeatedly coprecipitated with NF-PLN, but was never detected by immunoblotting in the absence of NF-PLN (Fig. 3C, Left). On the other hand, mutant HA-p62 without the UBA domain (HA-p62, ΔUBA) was not coimmunoprecipitated, even in the presence of NF-PLN, suggesting that the UBA domain of p62 was required for p62-PLN interaction. These results show that p62 interacts with polyubiquitinylated PLN via its UBA domain and that this interaction is critical for p62-mediated PLN trafficking in CMNCs.
Increased PLN Degradation by Metformin in CMNCs.
Metformin accelerates cardiac autophagy by promoting AMPK-mediated ULK1 phosphorylation (23) and also promotes p62-mediated selective autophagic flux in a dosage-dependent manner (24). Accordingly, we asked whether PLN degradation would be affected by metformin in CMNCs. Cells were incubated with 2.5 mM metformin for up to 24 h and cell lysates were subsequently collected for immunoblotting. Metformin induced an ∼30% decline in PLN levels within 24 h (Fig. 4 A and C), along with a large increase in AMPK phosphorylation that was observed as early as 12 h. In parallel, autophagy marker p62 decreased and LC3-II levels significantly increased following metformin treatment, suggesting that metformin enhanced autophagy activity in the treated CMNCs. Activated AMPK was associated with increased E3 ubiquitin ligase atrogin-1, although levels of another E3 ubiquitin ligase HACE1 were unchanged (Fig. 4A). Metformin did not affect the levels of other Ca2+ modulator proteins, including Na+/Ca2+exchanger, SERCA2a, and ryanodine receptor 2 (Fig. 4B).
Fig. 4.

Metformin induces PLN degradation. (A) Immunoblots assessing the protein levels of PLN, α-tubulin, total AMPK, phosphorylated AMPK at Threonine 172 (pAMPK-T172), p62, LC3, Atrogin-1, and HACE1 in response to metformin treatment for 24 h. (B) Immunoblots assessing the protein levels of Na+/Ca2+exchanger (NCX), SERCA2a, and ryanodine receptor 2 (RyR2) in the presence of metformin. α-Tubulin was included as a loading control. (C) Quantification of PLN protein levels in the presence of 2.5 mM metformin (n = 3). (D) Representative pulse-chase results for the [35S]-labeled PLN in the presence and absence of metformin. (E) Linear regression of PLN degradation based on the pulse-chase results (n = 3). (F) Representative Ca2+ transients acquired from CMNCs with PLN (wild-type and K3A) overexpression and metformin treatment (2.5 mM). Cells were pulsed at 1 Hz/5 V. (G) Quantification of time required from peak to 50% decay of the Ca2+ transient. CMNCs were transduced with empty vectors (null-transduction) or PLN cDNAs, and treated with DMSO or metformin; 100 µM milrinone was a positive control. *P < 0.05.
In parallel, we determined whether metformin treatment might affect PLN stability in CMNCs, measured by 35S-dependent metabolic labeling. Pulse-chase studies showed that the half-lives of wild-type PLN (τ = 9.5 ± 1.2 h) and R9C (τ = 8.8 ± 0.5 h) were similar in transduced CMNCs (Fig. 4 D and E) and that metformin significantly accelerated wild-type (τ = 6.5 ± 0.9 h) and R9C (τ = 6.1 ± 0.8 h) PLN degradation. This finding is consistent with immunoblotting results showing that metformin promoted PLN degradation in CMNCs (Fig. 4A).
Next, we determined if metformin could affect Ca2+ reuptake during electric field pulsing of Fura-2–loaded CMNCs to create a series of uniform Ca2+ transients (25, 26). CMNCs were transduced by lentiviral vectors harboring wild-type or K3A PLN, selected with 2 µM puromycin for 5 d, and then treated with 2.5 mM metformin overnight. A PDE3 inhibitor milrinone (100 µM), which promotes intracellular cAMP levels, was used as a positive control. The rates of Ca2+ reuptake were measured as the time from the peak of the Ca2+ transient to 50% decay (T50). Consistent with previous studies in HEK-293T cells (21, 27), overexpression of wild-type and K3A PLN resulted in reduced Ca2+ reuptake in CMNCs (Fig. 4 F and G). However, metformin increased Ca2+ reuptake in both nontransduced and wild-type PLN-transduced CMNCs, but not in CMNCs overexpressing K3A PLN, which could not be ubiquitinylated and, therefore, not degraded through the autophagy pathway. Thus, we observed a clear correlation between metformin-induced degradation of PLN and a metformin-related inotropic increase in Ca2+- reuptake.
Increased PLN Levels in Hace1-Null Mice.
Recent studies by Zhang et al. (22) and Rotblat et al. (28) showed that a homolog to the E6-AP carboxyl terminus domain and HACE1 is required for modulating stress responses in the heart and brain via autophagy. Deleting the Hace1 gene resulted in reduced autophagy flux in a p62-dependent manner in a severe transaortic constricted (sTAC) mouse heart, suggesting that HACE1 is critical in mediating p62-dependent autophagy flux in myocytes (22). We investigated PLN levels in Hace1-null mice. Immunoblots in Fig. 5A show that PLN levels were significantly higher in HACE1-null mouse hearts compared with wild-type controls, regardless of sTAC (Fig. 5B). Similarly, immunofluorescence showed that PLN levels were significantly higher in the hearts of HACE1-null mice, compared with wild-type counterparts using similar exposure time and laser intensity. Phalloidin was comparable between the different experimental conditions (Fig. 5C). These results support the view that PLN is trafficked by autophagy and any impairment in autophagy promotes PLN accumulation in cardiomyocytes (Fig. 6).
Fig. 5.

PLN degradation is affected by autophagic flux in vivo in adult mouse hearts. (A) PLN levels were higher in Hace1-null and Hace1-null/sTAC mouse heart compared with wild-type and wild-type/sTAC counterparts. (B) Quantification of PLN/GAPDH ratios in comparison with wild-type samples (n = 3). (C) Immunofluorescence of PLN in Hace1-null mouse hearts showed an increased PLN signal than that in wild-type mice under similar exposure time and laser source. Phalloidin staining was comparable between wild-type and Hace1-null mice. (Scale bar, 20 µm.) *P < 0.05.
Fig. 6.
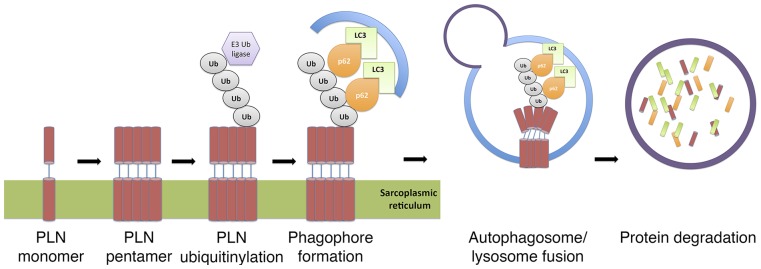
Schematic diagram illustrating p62-mediated selective autophagy for PLN trafficking to the lysosomes for degradation in cardiomyocytes. PLN is polyubiquitinylated by E3 ubiquitin ligases. This posttranslational modification bridges between p62 and PLN pentamers before phagaphore formations and protein trafficking to the lysosomes.
Discussion
In the present study we show that PLN degradation in cardiac lysosomes is mediated via p62-dependent autophagy. We show that pentameric PLN is polyubiquitinylated at the K3 residue and that K3A mutant proteins abrogate this modification. K3 ubiquitinylation is not required for PLN pentamerization, as both K3E and K3A PLN mutants could still form pentamers in transfected HEK-293 cells (29). Posttranslational modifications involving ubiquitin and ubiquitin-like modifiers play multifactorial roles in maintaining cell physiology, especially in maintaining protein and organelle homeostasis (30). The discovery of autophagy adaptor protein p62 and its physical association with LC3 provides more insight into selective autophagy-mediated degradation of ubiquitinylated proteins, pathogens, and organelles (31). In selective autophagy, adaptor proteins, including p62, interact with ubiquitinylated macromolecules via the UBA domain and with LC3 via the LC3-interacting region (32). Recruitment of LC3 to p62-centered aggregates facilitates lysosome-autophagosome fusion for protein degradation. Our study indicates that PLN pentamers are labeled with ubiquitins before protein clearance.
Successful macroautophagy requires a concerted effort of autophagosome biosynthesis and cargo protein trafficking to the lysosome for degradation. Our study demonstrates that selective autophagy via p62 is required for PLN degradation, but PLN translocation to the lysosomes has not been demonstrated directly. Stenoien et al. showed O6-alkylguanine-DNA alkyltransferase tagged-PLN was trafficked in a linear fashion between the SR and other organelles, including the lysosomes, in murine myoblasts and myotubes (33). p62 has been shown to physically interact with histone deacetylase-6 (HDAC6) (34) and dynein (35). HDAC6 is a cytosolic protein that plays a critical role in autophagosome-lysosome fusion; reduced HDAC6 levels are associated with stagnant autophagy and de novo protein aggregation (36). p62 interaction with HDAC6 and its recruitment to the microtubule organization center via dynein is important for dynein motor protein trafficking of ubiquitinylated proteins, as p62-null mouse embryonic fibroblasts become devoid of ubiquitin aggregates along microtubules (35). Taken together, these results can account for PLN translocation to the lysosomes after autophagosome biosynthesis.
Selective autophagy is a special arm of macroautophagy that removes ubiquitinylated macromolecules, organelles, and pathological agents (37). During the process, adaptor proteins, including at least p62 and optineurin, interact with ubiquitinylated macromolecules via UBA domains. These adaptor protein–ubiquitin complexes consequently depend on autophagy machinery for macromolecule clearance (37). Consistently, selective autophagy via p62 is enhanced via the autophagy agonist metformin in a dose-dependent manner, whereas silencing of autophagy with the modulator BECLIN1 attenuates the process (24). A recent study showed that HACE1 proteins play an intricate role in mediating autophagy flux by promoting optineurin-ubiquitin-p62 conjugation (38). Physical linkages between autophagy adaptor proteins via polyubiquitin chains are required for autophagy flux. Hace1 knockout in adult mouse hearts is associated with decreased autophagy flux, protein aggregation, and cardiac failure. In the present study, we demonstrated that Hace1 knockout was associated with a fourfold increase in PLN protein levels. PLN and LC3 colocalization at some puncta in neonatal cardiomyocytes further supports the notion that p62-mediated PLN degradation can be regulated by HACE1-mediated autophagy flux.
In this study we used both biochemistry and immunofluorescence to show that PLN is degraded in lysosomes, but is unresponsive to proteasome or calpain inhibitors. Similarly, an unconventional protease inhibitor, UCF-101, did not affect PLN levels in neonatal cardiomyocytes from diabetic mice (39). CQ has been shown to interfere with the autophagy pathway in many cell models (40). CQ in our system promoted PLN accumulation in a time- and dosage-dependent manner. We also showed that overlapping signals between PLN and LC3, an autophagosome resident protein, increased following CQ treatment in CMNCs. Bafilomycin prevents autophagosome-lysosome fusion (41). Here, bafilomycin promoted the accumulation of endogenous PLN in CMNCs. These results collectively demonstrate that PLN is trapped in autophagosomes when fusion between autophagosomes and lysosomes is interrupted.
Metformin is prescribed for the treatment of type-II diabetes. Clinically, the benefits of metformin in human heart failure remain unclear (42). Nevertheless, metformin administration is cardioprotective following transaortic ligation or occlusion of the left main descending coronary artery in experimental mammals, such as mice, rats, and canines (43–46). More importantly, the conferred protection was independent of obesity or type-II diabetes (46). Metformin accelerates autophagic mechanisms in many cells, including CMNCs (47), and is responsible for a 30% reduction of PLN in CMNCs in 24 h. Consistent with this observation, pulse-chase experiments carried out in CMNCs showed reduced protein stabilities of both wild-type PLN and R9C mutant. It is estimated that 40% of SERCA2a molecules are inhibited by PLN in mouse hearts (2). We propose that this physical interaction decreases following metformin treatment, thereby enhancing the activity of SERCA2a and manifesting as an inotropic effect in metformin-treated hearts. Finally, we propose that reduced PLN stability is an important feature in the protective effect of metformin in both CMNCs and mammalian smooth muscle.
Materials and Methods
Metabolic Labeling and Pulse-Chase Experiments.
The University of Toronto Animal Care and Use Committee approved all experiments using animals. Pulse-chase experiments with [35S]methionine and [35S]cysteine mixture were performed as described previously (48), with modifications. Transduced CMNCs were incubated in methionine- and cysteine-deficient medium (21013-024, Gibco) for 30 min, followed by 2 h of labeling with 0.2 mCi/mL l-[35S]methionine and l-[35S]cysteine mixture (NEG072, Perkin-Elmer). Cells were washed and maintained in DMEM/F12 medium for 0–8 h(s), followed by immunoprecipitation with M2 Flag antibody in RIPA buffer. Eluted proteins were subjected to SDS/PAGE, gels were dried, exposed to a phosphor-screen for 7 d, and analyzed using Storm 860 PhosphorImager (GE Healthcare).
Polyubiquitinylation Detection and Co-IP Assays.
HEK-293T cells were transfected with HA-ubiquitin and Flag-PLN (wild-type and K3A). Cells were harvested 48 h after transfection in 500 µL SDS lysis buffer [2% (wt/vol) SDS, 150 mM NaCl, 10 mM Tris⋅HCl; pH 8.0, supplemented with protease and phosphatase inhibitors], immediately boiled for 10 min, sonicated at 15% output for 10 s, diluted with 2 mL CHAPS IP buffer [0.3% (wt/vol) CHAPS, 40 mM Hepes; pH7.5, 120 mM NaCl, 1 mM EDTA, supplemented with protease and phosphatase inhibitors], and incubated at 4 °C for 30 min. Samples containing 500 µg of soluble protein were incubated with 1 µg M2 Flag antibody (Sigma) overnight at 4 °C, followed by immunoprecipitation with 20 µL BSA-blocked protein A/G agarose beads. Immobilized proteins were eluted with Laemmli buffer for immunoblotting.
For p62 and PLN interactions, coimmunoprecipitations were conducted as previously described (49). HEK-293T lysates were harvested in lysis buffer [100 mM NaCl, 20 mM Tris⋅HCl; pH8.0, 0.5 mM EDTA, and 0.1% (vol/vol) Nonidet P-40, 20% (vol/vol) glycerol], supplemented with proteasome and lysosome inhibitors. Samples containing 500 µg of soluble protein in 750-µL volume were incubated with 1 µg M2 Flag antibody (Sigma) overnight at 4 °C, followed by immunoprecipitation with 20 µL BSA-blocked protein A/G agarose beads. Immobilized proteins were subjected to immunoblotting.
Details for remaining experiments and materials can be found in SI Materials and Methods.
SI Materials and Methods
Tissue Culture.
HEK-293T were maintained in DMEM supplemented with 10% FBS, penicillin, and streptomycin. The procedures for lentiviral packaging and mouse neonatal cardiomyocytes culturing were described previously (25, 26). Small interfering RNA (siRNA) against mouse Atg5 transcripts (Invitrogen 161983) and scrambled sequence control (12935-300) were transfected in CMNCs with lipofectamine RNAiMAX transfection reagent (Invitrogen), as previously described (18).
Immunoblotting and Immunofluorescence.
Cells were washed twice with ice-cold PBS and lysates were harvested in RIPA buffer supplemented with protease and phosphatase inhibitors. Twenty micrograms of total protein were loaded for immunoblotting. Primary antibodies were prepared at the final concentration of 1 µg/mL in 5% (wt/vol) nonfat milk in Tris-buffered saline + 0.05% Tween-20, unless stated otherwise. Mouse PLN antibody (1D11) generation was described previously (29). Mouse M2 Flag antibody (F1804) was from Sigma-Aldrich; rabbit α-tubulin (2144), rabbit AMPKα (5831), rabbit Atg5 (2630), and rabbit Thr172 phospho-AMPKα (2535) were from Cell Signaling Technology. Mouse ryanodine receptor 2 antibody (C3-33) and SERCA2A antibody (2A7-A1) were from Thermo Scientific. Rabbit Na+/Ca2+exchanger (NB 300-127) antibody and LC3 antibody (NB 100-2220) were from Novus Biological. Anti-rabbit IgG-HRP (W4011) and anti-mouse IgG-HRP (W4021) were from Promega.
Immunofluorescence in CMNCs was carried out as previously described (1) and was visualized with a Zeiss spinning disk confocal microscope (Zeiss Observer Z1). Three-dimensional images were reconstructed and the Pearson product-moment correlation coefficient between two signals was analyzed by Imaris (BitPlane). Mouse monoclonal anti-LAMP1 antibody was from Developmental Studies Hybridoma Bank.
Immunohistochemistry on paraffin-embedded Hace1-null cardiac tissues was carried out as previously described (22).
Transcript Purification and RT-PCR.
Total RNA extraction was performed with TRIzol reagent (Invitrogen) according to the manufacturer’s instruction. cDNA synthesis was performed with SuperScript III First-strand Synthesis System (Life Technology). cDNA amplification was carried out with OneTaq polymerase (New England Biolabs) and was visualized on 1% (wt/vol) agarose gels.
Hace-1 Knockout Mice and sTAC.
Hace-1 knockout mice creation and sTAC procedures were carried out as previously described (22).
Calcium Transient.
Calcium transients were conducted as previously described (25, 26). Cardiac contractions were paced by electrical field pulse (ADInstructments, IE-210-V) at 5 V/1 Hz.
Plasmids and Reagents.
The construction of flag-tagged PLN (NF-PLN) has been described previously (5). To subclone NF-PLN (wild-type and K3A), human HA-p62 and HA-p62 (ΔUBA) in to pLex-MCS lentiviral plasmid (Open Biosystems), transgenes were amplified with OneTaq polymerase (New England Biolabs) with primers whose sequences and annealing temperatures are listed in Table S1. Sequences and reading frames were verified by sequencing (ACGT a). Ammonium chloride (NH4Cl, A9434), chloroquine diphosphate salt (CQ, C6628), Bafilomycin A1 (B1793), 3-methyladenine (M9281), and metformin hydrochloride (PHR1084) were from Sigma-Aldrich; Lactacystin (Lac, 426100), MG132 (474790) and N-Acetyl-Leu-Leu-Nle-CHO (ALLN, 208750) were from EMD. pRK5-HA-ubiquitin (plasmid 17608) was from Addgene.
Table S1.
A list of primers, their sequences, and PCR melting temperatures, used in this study
| Construct | Primer | Primer sequence (5′ → 3′) | Tm (°C) |
| NF-PLN | Sense | AGT GGA TCC GCC ATG GAC | 58 |
| Antisense | TCG CGT CGA CGA TCA GCA GGA GAC ATA TCT AGA TGA G | ||
| NF-PLN (K3A) |
Sense | CAT CAT GGA TCC ATG GAC TAC AAG GAC GAC GAT GAC AAG ATG GAG GCA GTT CCA | 65 |
| Antisense | CAT CAT CTC GAG TCA GAG AAG CAT GAC GAC GAT GAT GCA GAT CAG CAG GAG ACA TAT | ||
| HA-p62 | Sense | CAT CAT GAA TTC ATG GCG TCG CTC ACC GTG AAG | 57 |
| Antisense | ATG ATG CTC GAG TCA CAA CGG CGG GGG ATG CTT | ||
| HA-p62 (ΔUBA) |
Sense | CAT CAT GAA TTC ATG GCG TCG CTC ACC GTG AAG | 57 |
| Antisense | ATG ATG CTC GAG TCA CAT CTC CCG CCA GAG GCT GAG |
Statistical Analyses.
Experimental results were analyzed by Prism5 and data are shown as mean ± SEM. Experimental mean-fold protein intensities, relative to controls from triplicate assays, were considered different from controls at the P < 0.05 significance level using one-way ANOVA and Tukey post hoc test for each mean comparison.
Acknowledgments
This project was funded by the Heart and Stroke Foundation of Ontario Grants T-6281 and NS-6636 (to A.O.G.); Canadian Institutes of Health Research Grants MOP-106538 and GPG-102166 (to A.O.G.) and MOP-10254 (to D.H.M.); the Heart and Stroke Richard Lewar Centre of Excellence; the Ontario Research Fund–Global Leadership Round in Genomics and Life Sciences Grant GL2-01012 (to A.O.G. and P.P.L.); and a Research Fellowship from the Heart and Stroke/ Richard Lewar Centre of Excellence (to A.C.T.T.). A.O.G. is a Canada Research Chair in Cardiovascular Proteomics and Molecular Therapeutics.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508815112/-/DCSupplemental.
References
- 1.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110(12):1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacLennan DH, Kranias EG. Phospholamban: A crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4(7):566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 3.Simmerman HK, Jones LR. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol Rev. 1998;78(4):921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- 4.Schmitt JP, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299(5611):1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 5.Haghighi K, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA. 2006;103(5):1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haghighi K, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111(6):869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ha KN, et al. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc Natl Acad Sci USA. 2011;108(7):2735–2740. doi: 10.1073/pnas.1013987108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma P, et al. Endoplasmic reticulum protein targeting of phospholamban: A common role for an N-terminal di-arginine motif in ER retention? PLoS ONE. 2010;5(7):e11496. doi: 10.1371/journal.pone.0011496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaglia T, et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest. 2014;124(6):2410–2424. doi: 10.1172/JCI66339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woods A, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–3-3. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 12.Xie M, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129(10):1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maejima Y, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19(11):1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pedrozo Z, et al. Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc Res. 2013;98(2):277–285. doi: 10.1093/cvr/cvt029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laing JG, Tadros PN, Green K, Saffitz JE, Beyer EC. Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc Res. 1998;38(3):711–718. doi: 10.1016/s0008-6363(98)00060-1. [DOI] [PubMed] [Google Scholar]
- 17.Ishida M, et al. Regulated expression and role of c-Myb in the cardiovascular-directed differentiation of mouse embryonic stem cells. Circ Res. 2012;110(2):253–264. doi: 10.1161/CIRCRESAHA.111.259499. [DOI] [PubMed] [Google Scholar]
- 18.Sun X, et al. p27 protein protects metabolically stressed cardiomyocytes from apoptosis by promoting autophagy. J Biol Chem. 2014;289(24):16924–16935. doi: 10.1074/jbc.M113.542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner SA, et al. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics. 2012;11(12):1578–1585. doi: 10.1074/mcp.M112.017905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asahi M, McKenna E, Kurzydlowski K, Tada M, MacLennan DH. Physical interactions between phospholamban and sarco(endo)plasmic reticulum Ca2+-ATPases are dissociated by elevated Ca2+, but not by phospholamban phosphorylation, vanadate, or thapsigargin, and are enhanced by ATP. J Biol Chem. 2000;275(20):15034–15038. doi: 10.1074/jbc.275.20.15034. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat Commun. 2014;5:3430. doi: 10.1038/ncomms4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi A, et al. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014;14:53. doi: 10.1186/1475-2867-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bousette N, Abbasi C, Chis R, Gramolini AO. Calnexin silencing in mouse neonatal cardiomyocytes induces Ca2+ cycling defects, ER stress, and apoptosis. J Cell Physiol. 2014;229(3):374–383. doi: 10.1002/jcp.24459. [DOI] [PubMed] [Google Scholar]
- 26.Chis R, et al. α-Crystallin B prevents apoptosis after H2O2 exposure in mouse neonatal cardiomyocytes. Am J Physiol Heart Circ Physiol. 2012;303(8):H967–H978. doi: 10.1152/ajpheart.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toyofuku T, Kurzydlowski K, Tada M, MacLennan DH. Identification of regions in the Ca(2+)-ATPase of sarcoplasmic reticulum that affect functional association with phospholamban. J Biol Chem. 1993;268(4):2809–2815. [PubMed] [Google Scholar]
- 28.Rotblat B, et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci USA. 2014;111(8):3032–3037. doi: 10.1073/pnas.1314421111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toyofuku T, Kurzydlowski K, Tada M, MacLennan DH. Amino acids Glu2 to Ile18 in the cytoplasmic domain of phospholamban are essential for functional association with the Ca(2+)-ATPase of sarcoplasmic reticulum. J Biol Chem. 1994;269(4):3088–3094. [PubMed] [Google Scholar]
- 30.Nishida K, Taneike M, Otsu The role of autophagic degradation in the heart. J Mol Cell Cardiol. 2015;78:73–79. doi: 10.1016/j.yjmcc.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 31.Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci. 2014;127(Pt 1):3–9. doi: 10.1242/jcs.140426. [DOI] [PubMed] [Google Scholar]
- 32.Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 33.Stenoien DL, et al. Cellular trafficking of phospholamban and formation of functional sarcoplasmic reticulum during myocyte differentiation. Am J Physiol Cell Physiol. 2007;292(6):C2084–C2094. doi: 10.1152/ajpcell.00523.2006. [DOI] [PubMed] [Google Scholar]
- 34.Yan J, et al. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE. 2013;8(9):e76016. doi: 10.1371/journal.pone.0076016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calderilla-Barbosa L, et al. Interaction of SQSTM1 with the motor protein dynein—SQSTM1 is required for normal dynein function and trafficking. J Cell Sci. 2014;127(Pt 18):4052–4063. doi: 10.1242/jcs.152363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JY, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29(5):969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birgisdottir AB, Lamark T, Johansen T. The LIR motif—Crucial for selective autophagy. J Cell Sci. 2013;126(Pt 15):3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014;26(1):106–120. doi: 10.1016/j.ccr.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q, Hueckstaedt LK, Ren J. The protease inhibitor UCF-101 ameliorates streptozotocin-induced mouse cardiomyocyte contractile dysfunction in vitro: Role of AMP-activated protein kinase. Exp Physiol. 2009;94(9):984–994. doi: 10.1113/expphysiol.2009.049189. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Muthukrishnan P, Roukoz H, Grafton G, Jessurun J, Colvin-Adams M. Hydroxychloroquine-induced cardiomyopathy: A case report. Circ Heart Fail. 2011;4(2):e7–e8. doi: 10.1161/CIRCHEARTFAILURE.110.959916. [DOI] [PubMed] [Google Scholar]
- 41.Yoon YH, et al. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci. 2010;51(11):6030–6037. doi: 10.1167/iovs.10-5278. [DOI] [PubMed] [Google Scholar]
- 42.Aguilar D, Chan W, Bozkurt B, Ramasubbu K, Deswal A. Metformin use and mortality in ambulatory patients with diabetes and heart failure. Circ Heart Fail. 2011;4(1):53–58. doi: 10.1161/CIRCHEARTFAILURE.110.952556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calvert JW, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57(3):696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 44.Gundewar S, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104(3):403–411. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki H, et al. Metformin prevents progression of heart failure in dogs: Role of AMP-activated protein kinase. Circulation. 2009;119(19):2568–2577. doi: 10.1161/CIRCULATIONAHA.108.798561. [DOI] [PubMed] [Google Scholar]
- 46.Yin M, et al. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am J Physiol Heart Circ Physiol. 2011;301(2):H459–H468. doi: 10.1152/ajpheart.00054.2011. [DOI] [PubMed] [Google Scholar]
- 47.Quentin T, Steinmetz M, Poppe A, Thoms S. Metformin differentially activates ER stress signaling pathways without inducing apoptosis. Dis Model Mech. 2012;5(2):259–269. doi: 10.1242/dmm.008110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gramolini AO, et al. Sarcolipin retention in the endoplasmic reticulum depends on its C-terminal RSYQY sequence and its interaction with sarco(endo)plasmic Ca(2+)-ATPases. Proc Natl Acad Sci USA. 2004;101(48):16807–16812. doi: 10.1073/pnas.0407815101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duran A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]