

Abstract
Most plant viruses that seriously damage agricultural crops are transmitted by insects. However, the mechanisms enabling virus transmission by insect vectors are poorly understood. The brown planthopper (Nilaparvata lugens) is one of the most serious rice pests, causing extensive damage to rice plants by sucking the phloem sap and transmitting viruses, including Rice ragged stunt virus (RRSV). In this study, we investigated the mechanisms of RRSV transmission from its insect vector to the rice plant in vivo using the terminal deoxynucleotidyl transferase dUTP nick-end labeling assay and RNA interference technology. RRSV induced apoptosis in the salivary gland cells of its insect vector, N. lugens. The RRSV-induced apoptosis was regulated through a caspase-dependent manner, and inhibition of the expression of N. lugens caspase-1 genes significantly interfered with virus transmission. Our findings establish a link between virus-associated apoptosis and virus transmission from the insect vector to the host plant.
Introduction
Insect pests and plant viruses that are transmitted by insect vectors are major biological threats to crop production. The interactions between insect vectors and plant viruses have received extensive attention worldwide because of their importance in the agricultural sector. The most economically devastating insect vectors are restricted to a few Hemiptera and Thysanoptera taxa, such as planthoppers, aphids, whiteflies and thrips. The long-distance migrating brown planthopper, Nilaparvata lugens Stål (Hemiptera: Delphacidae), is considered one of the most destructive pests of rice throughout Asia, causing severe damage to rice plants by sucking the rice phloem sap and transmitting plant viruses, including rice grassy stunt virus (RGSV) and rice ragged stunt virus (RRSV)1,2.
RRSV is a member of the genus Oryzavirus in the family Reoviridae and causes rice ragged stunt disease. The disease was first discovered in 1976–1977 in Indonesia and the Philippines3, and then became prevalent in most rice-growing regions in South East Asia and southern China4,5. Since its discovery, RRSV has become one of the most important rice pathogens in these regions. RRSV is known to be horizontally transmitted by the brown planthopper in a persistent propagative manner but is not vertically transmitted via eggs or rice seeds6. Nilaparvata lugens acquires the virus by sucking the sap of infected rice plants. The virus first enters the epithelial cells of the midgut, where it proceeds to the visceral muscles surrounding the midgut, then disperses throughout the visceral muscles of the midgut and hindgut, and finally reaches the salivary glands2. During subsequent sap feeding, the virus within the saliva is transmitted to the host rice plant7. Despite our understanding of the invasion route within the insect body, the mechanisms underlying the transmission of this virus from its insect vector to the rice plant remain largely unknown.
The salivary gland is vital to the biological success of insects via its principal secretory function, e.g., arbovirus transmission in mosquito depends on the function of salivary gland during blood feeding8. We are interested in investigating the responses of the salivary gland to RRSV infection because this organ provides the essential link for understanding virus transmission from the insect vector to the rice plant. There are several intriguing observations indicating that arboviruses induce apoptosis in the midgut and salivary glands of mosquitoes8,9. The apoptosis associated with arbovirus infections can affect vector competence in mosquitoes10. In insect tissues, cytopathologic changes such as virus-activated apoptosis could have potential effects on virus dissemination or release. However, until now there has been no solid evidence showing a causal relationship between virus-induced apoptosis and virus transmission.
The molecular pathways that regulate apoptosis in insects have been most clearly studied in Drosophila melanogaster and Aedes aegypti by far11,12,13,14. The core components in the apoptosis pathway are initiator and effector caspases, which are a family of cysteine proteases that are ubiquitously expressed as inactive zymogens15,16. Apoptotic signals trigger initiator caspases, which function to activate downstream effector caspases that lack long prodomains and the ability to self-activate, and lead to programmed cell death14,17. We identified five caspase homolog genes through searching the N. lugens genome and transcriptome datasets18,19,20,21,22. Three genes are highly homologous to each other and share significant sequence similarities with insect caspase-1 genes11,23,24,25,26,27. Caspase-1, the first insect caspase to be discovered, was identified in Spodoptera frugiperda28. Insect caspase-1 is most closely related to the mammalian apoptotic effectors caspase-3 and caspase-729. We also identified two long prodomain-carrying caspase homolog genes in N. lugens. One shares similar characteristics with insect caspase-Nc genes that feature caspase activation and recruitment domains (CARDs). The other homolog has significant sequence identities with caspase-8 genes of several insect species, which contain the long prodomain regions with no homology to known characterized motifs. In this study, we focused on these caspases to determine their association with RRSV-induced apoptosis in N. lugens salivary glands. The silencing of caspase-1 or caspase-Nc expression inhibited RRSV-induced apoptosis in the salivary glands. However, the silencing of caspase-8 expression did not inhibit the occurrence of apoptosis in the RRSV-infected salivary glands. Our findings revealed that the interference of caspase-1 expression significantly reduced RRSV transmission from N. lugens to the rice plant. This study provides new insights, in that virus transmission was accompanied by caspase-dependent apoptosis in the salivary gland, which addresses a so far unknown step of virus transmission in a monophagous sap-sucking arthropod herbivore and is important for better understanding of insect vector–virus–plant host interactions.
Results
RRSV proliferation in the salivary glands of N. lugens
To understand the characteristics of RRSV invasion, we monitored the RRSV proliferation in the salivary glands after infection of the host, N. lugens. Quantitative real-time PCR analysis showed that specific amplification was achieved using one pair of primers against the RRSV major capsid protein P8 gene, for which no nonspecific amplification or primer-dimer artifacts were observed. The relative transcript levels of the P8 gene were barely detectable during 2–4 d p.i. (Fig. 1). As the infection progressed, viral proliferation increased rapidly, with a relative transcript level of 1.0 × 103 at 6 d p.i. and a peak of 9.2 × 103 at 12 d p.i., followed by a decrease to 3.3 × 103 at 16 d p.i. These results reveal that the maximum accumulation of the virions in the salivary glands was at approximately 12 d p.i. The RRSV proliferation process was clearly visualized using immunofluorescence staining (Fig. 2). The virions were captured in the cytoplasmic region in a portion of the cells of the salivary gland tissues at 6 d p.i. and were then observed in more than half of the cells in these tissues at 8 d p.i. The rapid proliferation of the virus resulted in its occupying all salivary gland cells at 10 d p.i. The virions were not detectable during 2–4 d p.i. Further evidence of RRSV invasion was obtained by examining virus-infected salivary glands using TEM at 8 d p.i. (Fig. 3). RRSV has an icosahedral capsid of approximately 65–70 nm in diameter, which includes electron-dense cores of approximately 45–50 nm in diameter that are surrounded by outer shells of approximately 10 nm in width2,30,31. TEM scanning showed that virions of approximately the same size were distributed in the cytoplasm of the RRSV-infected salivary gland cells in clusters (Fig. 3b,c). Such virions were not detectable in uninfected salivary glands (Fig. 3a). Some virion-containing salivary gland cells show structural changes. Highly condensed chromatin in large compact granular masses was observed in the nuclear regions (Fig. 3b,c). In these cells, the nuclear membrane disappeared and/or invagination of the cell membrane formed. The structural changes indicate that these salivary gland cells were undergoing apoptosis. In contrast, uninfected salivary gland cells displayed a large nucleus with limited condensation of the chromatin localized around the nuclear membrane (Fig. 3a). To confirm whether the virions observed by TEM were RRSVs, we used IEM to localize the viruses in the salivary gland cells. There was 5-nm immunogold labeling against a RRSV P8 antigen detected in the cytoplasm of the salivary gland cells (Fig. 3d), consistent with the TEM results. These results clearly demonstrate the presence of RRSV in the cytoplasm of N. lugens salivary gland cells after virus infection. More importantly, TEM showed that RRSV infection induced apoptosis-related morphological changes in N. lugens salivary gland cells.
Figure 1. Relative quantification analysis of RRSV P8 transcript levels in the salivary glands of N. lugens.
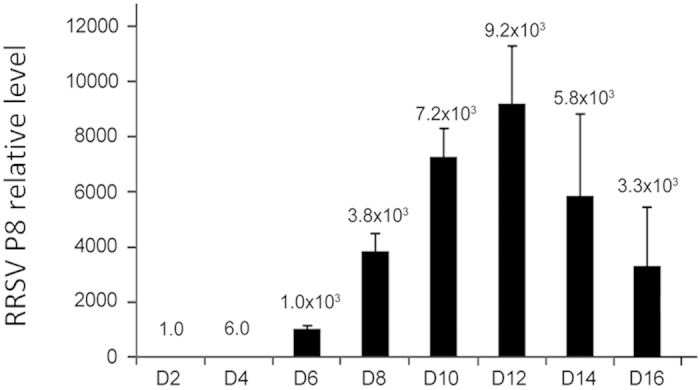
Total RNA was extracted from salivary glands at the indicated times following RRSV infection and subjected to quantitative real-time PCR analysis using a pair of primers to specifically amplify the RRSV major capsid protein gene, P8. The relative P8 transcript levels were calculated using N. lugens 18S rRNA as an internal control. A mixture of 100 salivary glands was used as one biological sample due to the very low quantity of salivary glands. Samples from each time point were tested in three biological replicates, and the mean value used to analyze the relative transcript levels.
Figure 2. Confirmation of RRSV proliferation by immunofluorescence staining analysis.
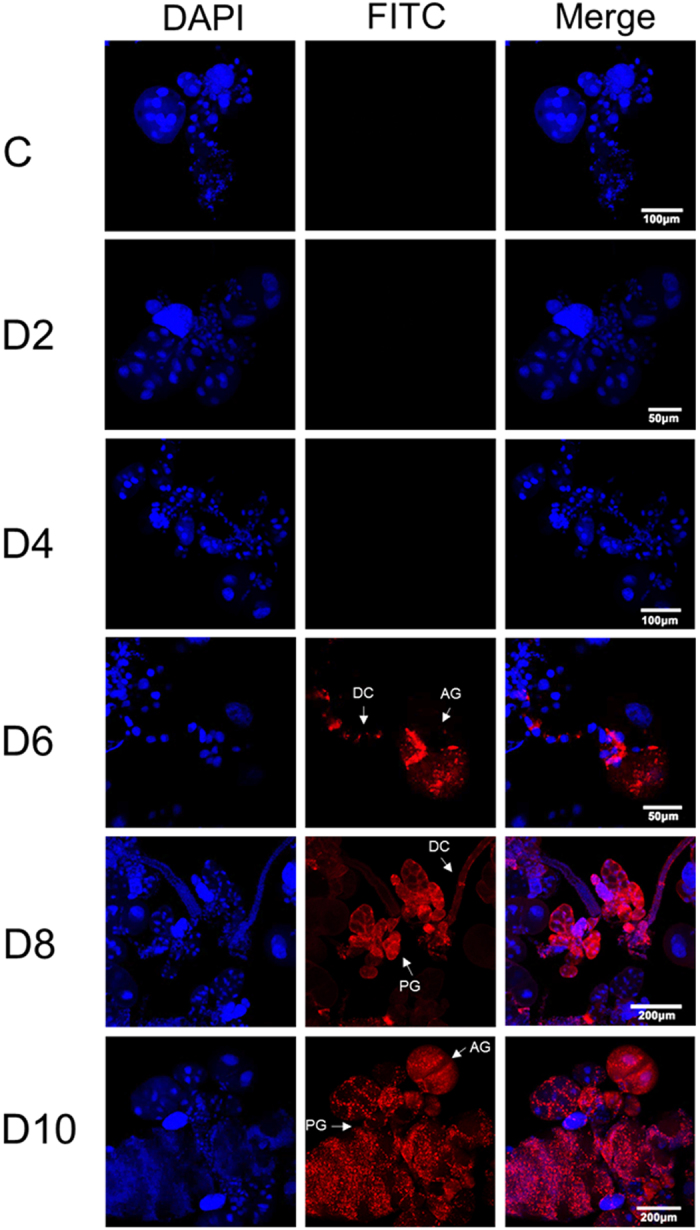
The salivary glands were dissected from RRSV-infected nymphs at the indicated times, and RRSV virions were immunolabeled with virus–FITC. Red (FITC) and blue (DAPI) fluorescence corresponding to the RRSV virions and the nuclei in the salivary gland cells, respectively. PG, principal glandular lobules; AG, accessory glands; DC, duct cells.
Figure 3. Identification of the location of the RRSV virions in the salivary gland cells at 8 d p.i. using transmission electron microscopy and immunoelectron microscopy.

(a) The salivary glands of uninfected N. lugens nymphs. (b and c) The electron micrographs show RRSV virions distributed in the cytoplasm of the salivary gland cells. (d) Immunoelectron micrograph shows the immunogold labeling of RRSV in the cytoplasm of the salivary gland cells. The red arrows indicate the RRSV virions. The blue arrows show the mitochondria. The green arrows indicate 5-nm gold-conjugated goat-anti-mouse IgG against the RRSV major capsid protein P8 antigens. The inset shows the enlargement of the boxed areas. Nc, nucleus; Mc, mitochondria.
RRSV induces apoptosis in salivary glands
To investigate RRSV-associated apoptosis in the salivary gland, we examined virus-infected and uninfected tissues from 2–8 d p.i. using the TUNEL assay (Fig. 4A). Apoptosis was not detectable in salivary gland cells of uninfected individuals. Apoptosis was also not observed in salivary glands at 2 or 4 d p.i., possibly because RRSV had not yet spread to the salivary gland cells during this infection stage. However, apoptotic cells displaying significant greenish-yellow fluorescence signals, as visualized in the nuclear regions using a FITC filter, were found in clusters at 6 or 8 d p.i. Apoptosis occurred in the RRSV-infected cells that were indicated by red fluorescence when using the viral antibody conjugated to Cy3 to detect the virus. The N. lugens salivary glands are paired, sac-like, glandular lobules that anteriorly connect to a common duct that opens into the pharynx (Fig. 4B), and they consist of the principal glandular lobules, accessory gland and duct regions. We dissected the salivary glands from five pools of 200 RRSV-infected N. lugens nymphs at each time point. In the viruliferous insects, a portion of the salivary gland cells that included the principal gland cells and the duct epithelium cells were undergoing apoptosis at 6 and 8 d p.i. (Fig. 4A), while the accessory gland cells were not undergoing apoptosis. These results suggest that RRSV infection induced apoptosis in the salivary glands, but this was limited to a portion of the principal gland cells and the duct epithelium cells.
Figure 4. RRSV-induced apoptosis in the salivary glands of N. lugens.

(A) The salivary glands were dissected from RRSV-infected N. lugens nymphs at the indicated times. Uninfected individuals were used as controls. The salivary glands were analyzed using a TUNEL assay to detect the apoptotic cells. Greenish-yellow fluorescence signals (FITC) indicate the nuclear regions of the principal glandular lobule cells and the duct epithelium cells undergoing apoptosis at 6 and 8 d p.i., respectively. Red fluorescence signals indicate the immunolabeled RRSV virions with virus-Cy3. Blue fluorescence (DAPI) shows the nuclei of the salivary gland cells. (B) Structure of the N. lugens salivary gland. The salivary glands were dissected and observed using a Leica S8AP0 stereomicroscope.
Possible apoptosis-activating factors in RRSV-infected salivary glands
Caspases are central components of the machinery responsible for apoptosis in animal cells. However, significantly less is known about insect caspases other than those described in Drosophila. To understand the regulation mechanism involved in RRSV-induced apoptosis, we focused on caspase genes to investigate their roles in RRSV-associated apoptosis in N. lugens salivary glands. We identified five caspase homologs, as shown in a supplementary file 2, by searching the N. lugens genome and transcriptome sequences obtained from N. lugens salivary gland, fat body, gut, ovary, testis and integument tissues in our recent studies. Three genes were highly homologous to each other and shared significant sequence similarities with insect caspase-1s, the effector death proteases that are thought to execute apoptosis in insects. Gene structure analysis revealed that these caspase-1 genes were located at different scaffolds with single or multiple (4–5) exons (Fig. 5A). Their predicted protein products (288, 271 and 258 amino acid residues, individually) possessed characteristic caspase domains (Pfam domain PF00656) that nearly spanned the entire coding regions of the putative proteases (Fig. 5B). We designated these genes as Nlcaspase-1a, Nlcaspase-1b and Nlcaspase-1c. Phylogenetic analysis showed that the caspase-1 genes of Lepidopteran, Hymenopteran and Dipteran insects formed three major clusters (Fig. 5C). The homologous genes from two Hemiptera insect species, N. lugens and Acyrthosiphon pisum, formed an independent cluster and were closely related to each other, suggesting that they had the closest phylogenetic relationship among the compared insect species. Two other N. lugens caspase homologs encoded 686 and 604 amino acid residues containing the characteristic caspase domains and showed significant sequence similarities with insect Nedd2-like caspases (Nc) and caspases-8, respectively. We designated these two genes as Nlcaspase-Nc and Nlcaspase-8. Nlcaspase-Nc included a long amino-terminal CARD (Pfam domain PF00619), a specific protein–protein interaction motif, which is commonly considered to be the primary apoptotic initiator in the cell death pathway. A comparison of insect Nc-like proteins revealed that they are composed of a CARD sequence with 63–89 amino acid residues at the amino-terminus and a caspase sequence of 231–310 amino acid residues at the carboxyl terminus (Fig. 5D). The phylogenetic tree showed that Lepidopteran and Dipteran Nc-like genes formed a cluster, while the N. lugens and A. pisum Nc-like genes were in another cluster and were closely related to the homologous genes of Hymenopteran insects. Nlcaspase-8 shared high similarities with insect caspase-8/dredd genes. Their deduced amino acid sequences contained a caspase domain with 235–277 amino acid residues and a long prodomain but lacking protein–protein interaction motifs that mark them as initiator caspases, unlike mammalian caspase-8 with the death effector domains (DED) (Fig. 5E). Nlcaspase-8 is phylogenetically most closely related to Tribolium castaneum caspase-8 among the compared insect species (Fig. 5E).
Figure 5. Identification of N. lugens caspase genes.

(A) The gene organization of N. lugens caspase on scaffolds. The green arrows in the upper panel indicate the gene sizes and transcription orientations on the scaffolds. The transcript sequences of the caspase genes were matched to N. lugens genomic sequences to identify the exons and introns using the online tool Spidey (http://www.ncbi.nlm.nih.gov/spidey/spideyweb.cgi). The exons are indicated by orange boxes. The schematic representation of the deduced protein structures is shown under the gene structures. The caspase domains are indicated by pink pentagons. The size bar indicates the amino acid residues of the deduced proteins. (B) Sequence alignment of caspase domains of the deduced N. lugens caspase-1 proteins. The ClustalX program was used for alignments. The GenBank accession numbers for the sequences are as follows: N. lugens caspase-1a (KF956388), caspase-1b (KF956389), and caspase-1c (KF956390). The active-site histidine and cysteine residues required for caspase activity are marked by asterisks. The amino acids shaded in black and gray indicate the conserved and type-conserved residues, respectively. (C) Phylogenetic analysis of insect caspase-1 genes. The phylogenetic tree was constructed by Maximum Likelihood using the program Mega 5.05 (http://www.megasoftware.net/). The Jones–Taylor–Thornton (JTT) for amino acid substitution model was used, while a test of phylogeny was carried out using the bootstrap method with 1000 replications; bootstrap values >50% are shown on each node of the tree. (D) Phylogenetic analysis and the predicted domains of the insect Nc-like genes. (E) Phylogenetic analysis and the predicted domains of the insect caspase-8 genes. The CARD and caspase domains are indicated by blue box and pink pentagon, respectively. H.a, Helicoverpa armigera; T.n, Trichoplusia ni; S.l, Spodoptera litura; M.s, Manduca sexta; B.m, Bombyx mori; N.v, Nasonia vitripennis; M.r, Megachile rotundata; A.m, Apis mellifera; C.q, Culex quinquefasciatus; M.d, Musca domestica; D.m, Drosophila melanogaster; P.h.c, Pediculus humanus corporis; A.p, Acyrthosiphon pisum; N.l, N. lugens. A.a, Aedes aegypti; C.f, Camponotus floridanus; B.i, Bombus impatiens; A.e, Acromyrmex echinatior; A.f, Apis florea; D.p, Danaus plexippus; L.d, Lymantria dispar; S.f, Spodoptera frugiperda. T.c, Tribolium castaneum; Z.n, Zootermopsis nevadensis; C.c, Ceratitis capitata; A.d, Anopheles darling.
RRSV-induced apoptosis occurred at 6–8 d p.i., and so we focused on 8 d p.i. to observe the RNAi effect. We first conducted RNAi experiments by microinjecting N. lugens nymphs with a mixture of dsRNAs specific to the three Nlcaspase-1 genes at a 1:1:1 quality ratio in order to maximally inhibit the expression of all Nlcaspase-1 genes. Interference notably decreased the expression levels of the three Nlcaspase-1 genes in N. lugens during 2–8 d p.i. compared with dsGFP-injected individuals (Fig. 6A). The relative transcript levels of the three Nlcaspase-1 genes were reduced by approximately 70–90% upon dsRNA injections throughout the tested period, indicating that RNAi sufficiently down-regulated the expression of the target genes. RRSV infection did not cause apoptosis in the salivary glands of dscaspase-1 mixture-treated nymphs but induced apoptosis in the salivary glands of dsGFP-injected nymphs at 8 d p.i. (Fig. 6B). The greenish-yellow fluorescence signals were clearly observed in the nuclear regions of principal gland cells of the RRSV-infected salivary glands. These results suggest that silencing of the three Nlcaspase-1 genes effectively inhibited apoptosis in the salivary glands of RRSV-infected nymphs, while dsGFP treatment did not affect the occurrence of apoptosis at 8 d p.i. Subsequently, we focused on each Nlcaspase-1 gene to investigate the RNAi effect on RRSV-induced apoptosis in the salivary glands. In each case, the targeted mRNA levels of Nlcaspase-1a, Nlcaspase-1b or Nlcaspase-1c were greatly reduced by approximately 90, 90 and 80%, respectively; while there were no apparent reductions in transcript levels for non-target genes, indicating that the dsRNA-mediated silencing was sequence specific (Fig. 6A). Examining three pools of 200 nymphs each showed that expression inhibition of Nlcaspase-1a, Nlcaspase-1b or Nlcaspase-1c genes sufficiently impeded apoptosis in RRSV-infected salivary glands, suggesting that these Nlcaspase-1s were indispensable in RRSV-induced apoptosis (Fig. 6B). These results enabled us to determine whether silencing the expression of the Nlcaspase-1 genes affected the viral proliferation or accumulation in N. lugens, which would result in less or no apoptosis in salivary glands. We tested this by knockdown of expression of all Nlcaspase-1 genes using microinjection of a mixture of dsRNAs into N. lugens nymphs to ensure sufficient inhibition. RRSV was then inoculated into the nymphs, and the resultant viral loads in the whole body and salivary glands of N. lugens nymphs were determined using quantitative real-time PCR analysis at 8 d p.i. Among 70 dscaspase-1- and 70 dsGFP-treated nymphs, 16 (23%) and 15 (21%) individuals were infected by RRSV (Fig. 6C), respectively, indicating a similar infection rate for insects exposed to the different treatments. Viral loads in dsGFP- or dscaspase-1-injected nymph individuals varied greatly but did not differ significantly between the two groups. Furthermore, we compared the viral loads in the salivary glands of dscaspase-1- and dsGFP-treated nymphs. As the quantity of salivary glands was very low, we prepared a mixture including 100 salivary glands as one biological sample, and three biological repeats were analyzed. The viral loads in the salivary glands of the dscaspase-1-injected nymphs were almost the same as those in the dsGFP-infected nymphs, suggesting that RRSV proliferation or accumulation was not significantly correlated with dsRNA treatment (Fig. 6C). To verify the data from the quantitative real-time PCR, western blotting was conducted to determine RRSV capsid P8 protein levels in dsGFP- and dscaspase-1-treated nymphs. For each treatment, 10 whole bodies or 100 salivary glands were mixed as a sample for analysis. The blotting showed the capsid P8 protein bands were detected in the whole bodies and the salivary glands of the dsGFP- and dscaspase-1-treated nymphs (Fig. 6C). There were similar signal strengths of the capsid P8 proteins in the whole bodies or the salivary glands between the different treatments. We also investigated the roles of Nlcaspase-Nc and Nlcaspase-8 genes in RRSV-induced apoptosis. Microinjection with dscaspase-Nc or dscaspase-8 led to significant inhibition of expression of target genes. The transcript levels of Nlcaspase-Nc and Nlcaspase-8 were greatly reduced by approximately 92 and 93%, respectively (Fig. 6D). Apoptosis was not observed in RRSV-infected salivary gland cells from the dscaspase-Nc-treated nymphs, but was seen in dscaspase-8-treated nymphs after examining three pools of 200 nymphs each, suggesting that Nlcaspase-Nc was an important factor in RRSV-induced apoptosis; while Nlcaspse-8 may not have been an indispensable component in the apoptotic response to RRSV infection (Fig. 6B). Although the detailed functional roles of N. lugens caspases are yet to be discovered, our findings clearly indicate that the RRSV-associated apoptosis that occurred in the N. lugens salivary gland was regulated in a caspase-dependent manner.
Figure 6. Analysis of the apoptosis-activating factors in RRSV-infected salivary glands.

(A) The silencing of Nlcaspase-1 gene expression in N. lugens. Second-instar nymphs were microinjected with the dscaspase-1 mixture or each dscaspase-1 individually. The relative transcript levels of each Nlcaspase-1 gene were analyzed as described in Fig. 1. The expression silencing of the three Nlcaspase-1 genes by microinjection with the dscaspase-1 mixture is shown in the upper left panel. Black, grey and white boxes indicate the transcript levels of Nlcaspase-1a, -1b and -1c genes at the indicated time points after RNAi. The transcript levels of Nlcaspase-1a, -1b or -1c genes in dsGFP- and each dscaspase-1-treated nymphs are shown in the upper right, the lower left and right panels, respectively. (B) The RNAi effect on RRSV-induced apoptosis in N. lugens salivary glands. The nymphs were microinjected with the dsRNAs specific to the target genes and infected with RRSV. The salivary glands were dissected from the insects at 8 d p.i. The tissues were stained using a TUNEL assay to detect the apoptotic signals (green) and counterstained with DAPI to show the nuclei (blue) of the salivary gland cells. RRSV virions (red) were immunolabeled with virus-Cy3. (C) Determination of the viral loads in the whole body and the salivary glands. The nymphs were microinjected with the dscaspase-1 mixture and infected with RRSV. The RRSV P8 transcript levels in each nymph (the upper left, one dot represents one insect) and the salivary glands (the upper right) at 8 d p.i. were measured as described in Fig. 1. Western blotting analysis of RRSV capsid P8 protein in the whole bodies (left) and the salivary glands (right) of dsGFP- and dscaspase-1 mixture-treated nymphs is shown in the lower panel. For each treatment, 10 whole bodies or 100 salivary glands were mixed as a sample for analysis. Data represent one of three biological replicate experiments. β-Actin was used to show equal protein loading. (D) The silencing of Nlcaspase-Nc and Nlcaspase-8 gene expressions in N. lugens. The relative transcript levels of Nlcaspase-Nc gene (left) and Nlcaspase-8 gene (right) after RNAi were analyzed as described above.
Transmission of RRSV from the insect vector to the rice plant
To understand whether the observed RRSV-induced apoptosis was linked to viral transmission from the insect vector to the rice plant, we silenced the expressions of three Nlcaspase-1 genes because they were the essential effectors leading to apoptosis. We also silenced the expression of Nlcaspase-8 gene to understand its relevance to the virus transmission. The dscaspase-1-, dscaspase-8- or dsGFP-microinjected nymphs were inoculated with RRSV and then individually propagated on a series of fresh rice seedlings at 24-h intervals for 7 d (Fig. 7A). In dscaspase-1-, dscaspase-8- or dsGFP-treated group, more than 210 rice seedlings infected with at least 30 viruliferous nymphs were analyzed using reverse transcription-PCR combined with quantitative real-time PCR to detect the virus. In the dsGFP-treated group, all tested rice seedlings were virus-negative at 1–4 d p.i., suggesting that virus transmission did not occur from the nymphs to the rice seedlings during this infection stage (Fig. 7A). The virus was detectable in rice seedlings as early as 5 d p.i. The transmission rate from the viruliferous nymphs to the rice seedlings was 53.4% at 5 d p.i. and then increased to 74 and 88% at 6 and 7 d p.i., respectively, implying that the RRSV proliferation in the nymphs promoted viral transmission. In the dscaspse-8-treated group, the virus was detectable in the rice seedlings at 5 d p.i. The transmission rate was 50% at 5 d p.i. and then increased to 67 and 83% at 6 and 7 d p.i., respectively. There were no significant differences of the transmission rates between dsGFP- and dscaspase-8-treated groups during the tested period. In the dscaspase-1-treated group, RRSV was not transmitted by the N. lugens nymphs to the fresh rice seedlings at 1–5 d p.i. The earliest transmission appeared at 6 d p.i. and, in this case, the transmission rate (15%) was significantly lower than that in the dsGFP- and dscaspse-8-treated nymphs. Despite the transmission rate increasing to 52% at 7 d p.i., it was still much lower than for controls. We determined the virus loads in the salivary glands of N. lugens nymphs after microinjection with dscaspase-1, dscaspse-8 or dsGFP. Quantitative real-time PCR analysis showed that P8 gene expression was not detectable in the salivary glands of nymphs at 1–4 d p.i. but became detectable at 5 d p.i. (Fig. 7B, upper panel). The relative transcript levels were low at 5 d p.i. in dsGFP-, dscaspase-8- and dscaspase-1-treated samples, then quickly increased at 6–7 d p.i. Reverse transcription-PCR analysis detected the weak bands of the amplified P8 gene produced at 5 d p.i. and the strong bands at 6–7 d p.i. in dscaspase-1-, dscaspase-8- and dsGFP-treated samples (Fig. 7B, lower panel). Although the virus loads were very low in the salivary glands of the nymphs at 5 d p.i., RRSV was effectively transmitted to the rice plant from dsGFP- and dscaspse-8-treated nymphs, but could not be transmitted from dscaspase-1-treated nymphs. With the progress of proliferation, viral loads in the salivary glands of dscaspse-1-, dscaspase-8- and dsGFP-treated nymphs increased rapidly, but transmission efficiency was much lower in dscaspase-1-treated compared with dsGFP- and dscaspase-8-treated nymphs at 6–7 d p.i. These results clearly suggest that the silencing of Nlcaspase-1 genes caused a significant reduction of RRSV transmission from the insect vector to the rice plants; while the silencing of Nlcaspase-8 gene did not significantly affect the virus transmission.
Figure 7. Transmission of RRSV from viruliferous N. lugens insects to rice seedlings.

(A) Second-instar nymphs were microinjected with the dscaspase-1 mixture or dscaspase-8 and inoculated on viruliferous rice seedlings, after which they were transferred to fresh rice seedlings as shown in the schematic representation. The dsGFP-injected individuals were used as controls. The day on which the insects were transferred to healthy rice seedlings was regarded as the first day and is shown on the x-axis. The transmission rate of RRSV from the viruliferous individuals to the fresh rice seedlings is shown on the y-axis. Error bars indicate the standard deviations of three independent experiments. The asterisks indicate statistical significance at p < 0.05 (*) by Student’s t-test compared to the dsGFP-treated control. (B) Determination of the virus loads after dsRNA treatments in N. lugens salivary glands. The N. lugens nymphs were treated with dscaspase-1 mixture, dscaspase-8 or dsGFP and infected with RRSV as described in this figure (A). Total RNA was extracted from the salivary glands at the indicated times and subjected to quantitative real-time PCR and reverse transcription-PCR analysis. For quantitative real-time PCR, samples from each time point were tested in three biological replicates, and the mean value used to analyze the relative transcript levels as described in Fig. 1. Reverse transcription-PCR analysis is shown in the lower panel. Total RNA (1 μg) was used as a template. The dscas-1 mix, dscas-8 and dsGFP refer to dscaspase-1 mixture, dscaspase-8 and dsGFP-treated samples, respectively.
Discussion
The mechanisms underlying virus transmission from the insect vector to the host plant remain largely unknown. In the present study, we examined the biological characteristics of plant virus transmission via investigating insect vector–virus–host plant interactions. Quantitative real-time PCR analysis revealed that RRSV continually propagated in the N. lugens salivary glands and reached a maximal accumulation at 12 d p.i. Tracking of the virions through immunofluorescence staining further confirmed the progressive accumulation of RRSV in the salivary glands. TEM and IEM scanning clearly showed that the virions were abundant in the cytoplasm of salivary gland cells, suggesting the proliferation of RRSV in the tissue. The fact that RRSV was not detectable by immunofluorescence staining and quantitative real-time PCR at 2–4 d p.i. was most likely because the virus had not yet spread to the salivary glands during this infection stage. This result is consistent with a report from Wei et al.2, which showed RRSV infection route in various tissues of N. lugens.
Our study revealed that RRSV infection induced apoptosis in N. lugens salivary glands. The occurrence of apoptosis was limited to the viruliferous insects, indicating that RRSV accumulation was required to induce apoptosis in the salivary gland cells. Only a portion of the principal gland cells and of the duct epithelium cells of the salivary glands underwent apoptosis upon RRSV infection. Interestingly, the accessory gland cells did not undergo apoptosis, suggesting that virus-associated apoptosis was restricted to a subset of cells within this tissue. Thus the apoptosis that occurred only in principal gland cells and the duct epithelium cells of the salivary glands may be an evolutionary trade-off between the need for RRSV to ensure its release from the insect and the need to ensure its survival in the insect.
Caspases play an important role in triggering apoptosis in animal cells. In this study, we provide direct evidence that caspases were necessary for RRSV-induced apoptosis in N. lugens salivary glands. Due to the accomplishments in the N. lugens genome project by our research team, we were able to perform a deep search of the genome to identify caspase genes. Five caspase genes homologous to insect caspase-1, caspase-Nc and caspase-8 were identified in the N. lugens genome. Our experimental results indicated that the expression inhibition of N. lugens caspase genes through in vivo RNAi did not lead to phenotypic defects in morphological characters and lethality. However, the silencing of Nlcaspase-1 and Nlcaspase-Nc expression caused the failure of apoptosis in the salivary glands of viruliferous insects. In contrast, RRSV dramatically induced apoptosis in dscaspase-8- and dsGFP-treated insects. These findings reveal that the molecular mechanisms by which RRSV induced apoptosis in N. lugens acted in a caspase-dependent manner. Nlcaspase-1 and Nlcaspase-Nc were critical in RRSV-induced apoptosis pathways, while Nlcaspase-8 did not seem to be an essential factor during the apoptosis process. The detailed functions of these Nlcaspase genes and their interactions in the caspase activation cascades require further elucidation.
As an insect vector, N. lugens can effectively transmit RRSV to the rice plant. Successful transmission was achieved by the majority of viruliferous insects that were treated with dsGFP. A minority of individuals appeared to exhibit no ability to transmit the virus, most likely due to individual differences, e.g., in the virus loads in the viruliferous insects, in the feeding behaviors of the insect vectors and in the microphysiological or microecological systems of the individual insects. Silencing of Nlcaspase-8 expression generated the similar transmission rates at 5–7 d p.i. when compared to the dsGFP-treated group, suggesting that Nlcaspase-8 seemed not to be involved in RRSV transmission. However, silencing of all three Nlcaspase-1 genes significantly interfered with RRSV transmission from the viruliferous insects to the rice plants at 5–6 d p.i., indicating that Nlcaspase-1s were associated with virus transmission from N. lugens to rice seedlings. The fact that the transmission rate increased to 50% at 7 d p.i. implied that Nlcaspase-1s might not have been unique factors in the mechanism of virus transmission. A recent study reported that mosquito saliva serine protease enhances dissemination of dengue virus from vector to the mammalian host32. We hypothesize that certain salivary proteins may be conducive to promoting the virus transmission in N. lugens. The discovery of RRSV-induced salivary proteins will enable us to better understand the virus transmission mechanism from its insect vector to the plant host.
In this study, our findings established a link between virus-induced apoptosis and virus transmission. This is possibly a strategy of the adaptive evolution of the insect vector with the virus. The connection of RRSV transmission with virus-induced apoptosis in salivary gland cells provides new insights into the specific interactions between the insect vector, virus and plant host. Understanding the characteristics of RRSV transmissibility by N. lugens will contribute to the control of diseases caused by insect-mediated plant viruses.
Methods
Insects, viruses and rice plants
The N. lugens strain employed in this study was originally collected from a rice field on the Huajiachi Campus of Zhejiang University, Hangzhou, China. The insects used in this experiment were the offspring of a single female and were reared at 26 ± 0.5 °C with 50 ± 5% humidity on rice seedlings under a 16:8 h light:dark photoperiod. Newly molted, second-instar N. lugens nymphs were used for these experiments. RRSV was kindly provided by Prof. Tai-Yun Wei of the Institute of Plant Virology of Fujian Agricultural and Forestry University and Prof. Xu-Dong Zhu of the China National Rice Research Institute and was maintained at the Molecular Biology Laboratory of the Institute of Insect Sciences of Zhejiang University. The rice TN1 strain, which is susceptible to RRSV infection, was used in assays involving insect-mediated virus infection.
Analysis of apoptosis
Second-instar N. lugens nymphs were inoculated using RRSV-infected rice seedlings for 48 h and then transferred to healthy rice seedlings. The day on which the insects were transferred to the healthy rice seedlings was regarded as the first day of the infection. The nymphs were collected at 48-h intervals during 2–8 d post infection (p.i.); uninfected nymphs were used as the control. For tissue extraction, the nymphs were anesthetized on ice, and their salivary glands were dissected and washed in a phosphate-buffered saline (PBS) solution (137 mM NaCl, 2.68 mM KCl, 8.1 mM Na2HPO4 and 1.47 mM KH2PO4 at pH 7.4). Apoptosis was analyzed using the DeadEndTM Fluorometric TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) system (Promega, Madison, WI, USA). Briefly, the salivary glands were fixed in 4% paraformaldehyde in PBS (v/v) overnight at 4 °C and washed in PBS at room temperature, followed by incubation with 2% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) for 1 h. Then, the salivary glands were permeabilized in 500 μl of 20 μg/ml proteinase K for 5 min and washed in PBS for 5 min. After fixation in 4% paraformaldehyde for 5 min, followed by washing in PBS for 5 min, the tissues were equilibrated in 100 μl of equilibration buffer [200 mM potassium cacodylate (pH6.6), 25 mM Tris-HCl (pH6.6), 0.2 mM DTT, 0.25 mg/ml BSA and 2.5 mM cobalt chloride] for 10 min. Next, 50 μl of a recombinant terminal deoxynucleotidyl transferase reaction mixture was added to the tissues to catalytically incorporate fluorescein-12-dUTP at 3¢-OH DNA ends, followed by incubation for 1 h at 37 °C. After incubation, 500 μl of 2 × SSC was added to the tissues for 10 min, followed by washing three times in PBS (5 min each). Then, the tissues were blocked with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) at room temperature for 1 h. The monoclonal antibody against a major RRSV capsid P8 protein, which was produced in our laboratory, was added at a dilution of 1:200 for 1.5 h and visualized with an Cy3-labeled secondary goat anti-mouse IgG (Abbkine, Redlands, CA, USA), which was diluted at 1:200. For nucleus (DNA)-specific staining, the salivary glands were stained with 100 nM of 4¢,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) for 2 min, after which they were washed three times with PBS. Fluorescence images were examined using a Zeiss LSM780 confocal laser-scanning microscope (Zeiss, Göttingen, Germany).
Investigation of RRSV proliferation in N. lugens salivary glands using quantitative real-time PCR
Second-instar N. lugens nymphs were infected with RRSV as described in the previous sections. Total RNA was extracted from the salivary glands of nymphs at 48-h intervals using RNAiso plus (TaKaRa, Dalian, China). The total RNA samples extracted from 100 salivary glands were used as an individual template for quantitative real-time PCR analysis (a total of 300 salivary glands per time point for three biological replicates). The concentration of each RNA sample was adjusted with DEPC-treated H2O to 1 μg/μl, and 1 μg of RNA was reverse-transcribed in a 10-μl reaction using ReverTra Ace® qPCR RT Master Mix with the gDNA Remover Kit (ToYoBo, Osaka, Japan) to remove any contaminating genomic DNA. RNA with no-reverse-transcriptase was used as the no-template control (NTC). Quantitative real-time PCR was performed using the BIO-RAD CFX96TM Real-Time System (Bio-Rad, Hercules, CA, USA) and the iQTM SYBR Green® Supermix Kit (Bio-Rad). The first-strand cDNA and a no-reverse-transcription control were used as templates for three biological replication assays under the following conditions: denaturation at 95 °C for 2 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. Fluorescence of PCR products was detected by adding a heat-dissociation protocol (temperature range 65–95 °C) during the last step of each cycle. Following amplification, melting curves were constructed and data analysis was performed on Bio-Rad CFX Manager 2.1 software. One pair of primers (sense: 5¢-GAGATAACGCTTGGAGGACA-3¢ and antisense: 5¢-GGATTGAATTACTCGCAGGA-3¢), that specifically amplify the RRSV P8 gene encoding the major capsid protein were designed based on the gene sequence (GenBank accession no. HM125546). As an internal control, expression of N. lugens 18S rRNA gene (GenBank accession no. JN662398) was analyzed using the following primers: 5¢-CGCTACTACCGATTGAA-3¢ (sense) and 5¢-GGAAACCTTGTTACGACTT-3¢ (antisense). In our previous studies, the utility of the N. lugens 18S rRNA gene was validated for its stable expression in N. lugens tissues, developmental stages and immune-induced individuals18,20,21. In the present study, the results were standardized to the expression level of N. lugens 18S rRNA. The relative transcript levels of RRSV P8 were calculated using the ∆∆Ct method and the following equation: ∆Ct = (Ct of RRSV P8 gene) – (Ct of N. lugens 18S rRNA gene). The biological repeats were analyzed and the average threshold cycle (Ct) value was used to quantify the relative transcript levels of RRSV P8.
Identification of caspase genes from the N. lugens genomic and transcriptomic databases
The brown planthopper genome assemblies have been deposited at GenBank under accession number AOSB00000000 (BioProject PRJNA177647). The caspase genes were searched against the N. lugens genome sequence based on the KEGG (ftp://ftp.uniprot.org/pub/databases/uniprot/, v58), Swissprot (ftp://ftp.uniprot.org/pub/databases/uniprot/, release-2012_03) and Trembl (ftp://ftp.uniprot.org/pub/databases/uniprot/, release-2012_03) annotations. Predicted coding sequences of caspase genes were used as reference sequences to match the N. lugens transcriptome in the Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra, SRX023419), which were obtained through high-throughput Illumina technology in our previous studies18,21, using the tBLASTX algorithm with a cut-off E-value of 10−10. The deduced CARD and caspase domains were determined by using Pfam (http://www/sanger.ac.uk/Software/Pfam/), Simple Modular Architecture Research Tool (SMART) (http://smart.embl.de/), InterProScan (http://www.ebi.ac.uk/Tools/pfa/iprscan/) and Conserved Domains of the National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
Phylogenetic analysis
The N. lugens caspase-1a, caspase-1b, caspase-1c, caspase-Nc and caspase-8 sequences were aligned with the best-matched homologs of other insect species using the ClustalX program33. The phylogenetic trees were constructed by the maximum likelihood method using the program Mega 5.05 (http://www.megasoftware.net/)34. Homologous relationships were determined using bootstrap analysis with 1000 replications.
Preparation of double-stranded RNA (dsRNA)
Nucleotide sequences specific to the N. lugens caspase-1a, caspase-1b, caspase-1c, caspase-Nc and caspase-8 were individually cloned into the pGEM-T Easy vector (Promega). Aequorea victoria green fluorescent protein (GFP) was used as a control. Specific dsRNAs were synthesized via in vitro transcription using PCR-generated DNA templates that contained the T7 promoter sequence at both ends. The specific primers used to generate these DNA templates are shown in a supplementary file 1. The specific dsRNAs for each gene were synthesized using the MEGAscript T7 Transcription Kit (Ambion, Austin, TX) according to the manufacturer’s instructions. Following transcription, the DNA template was removed using TURBO DNase (Ambion) and the dsRNAs were purified with the RNAqueous Kit (Ambion). The size of the dsRNA products was confirmed by electrophoresis on a 1% agarose gel that was run in Tris-Acetate-EDTA buffer.
RNA interference (RNAi) via microinjection
Second-instar N. lugens nymphs were used in the RNAi experiments. Each virus-free nymph was anesthetized with carbon dioxide (CO2) for 5–10 s at PCO2 = 5 mPa. Approximately 250 ng of dsRNA was microinjected into the thorax of each nymph between the mesocoxa and the hind coxa using the FemtoJet Microinjection System (Eppendorf, North America). The treated nymphs were reared at 26 ± 0.5 °C with 50 ± 5% humidity on fresh, healthy rice seedlings under a 16:8 h light:dark photoperiod for 24 h and then transferred to the RRSV-infected rice seedlings for 48 h. The morphological phenotypes and mortality rates of the insects were observed and determined using a stereomicroscope (Leica S8AP0, Germany) every 24 h following the dsRNA treatments.
Confirmation of RRSV infection in the salivary glands using immunofluorescence staining
Second-instar N. lugens nymphs were inoculated on RRSV-infected rice seedlings for 48 h and then transferred to healthy rice seedlings. The salivary glands were dissected from the N. lugens nymphs at 48-h intervals, and uninfected nymphs were used as controls. The salivary gland samples were fixed using 4% paraformaldehyde in PBS for 6 h at 4 °C and then blocked with 10% fetal bovine serum (Gibco) at room temperature for 2 h. A monoclonal antibody against a major RRSV capsid P8 protein was diluted 1:200, followed by overnight incubation at 4 °C and visualization using a FITC-labeled secondary goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA), diluted 1:100. After the subsequent washes, the tissues were stained using 100 nM DAPI (Sigma-Aldrich) for 2 min at room temperature. Following three washes with PBS, each for 10 min, fluorescence images were observed using a Zeiss LSM 780 confocal microscope (Zeiss).
Transmission electron microscopy (TEM) observations
Second-instar N. lugens nymphs were infected with RRSV as described in the previous sections. The nymphs were collected at 8 d p.i.; uninfected nymphs were used as a control. The salivary glands were dissected from the nymphs and fixed in 2.5% (v/v) glutaraldehyde in PBS at 4 °C overnight. Following fixation, the samples were washed three times in 0.1 M PBS (pH 7.0) and then post-fixed with 1% (v/v) osmium tetroxide for 1 h at room temperature. The fixed salivary glands were dehydrated through incubation in a graded series of ethanol (50, 70, 80, 90, 95 and 100%, v/v) for 10 min each and then soaked in acetone for 20 min. At room temperature, the samples were placed in a 1:1 mixture of acetone and Spurr resin for 1 h, then in a 1:3 mixture for 3 h and in Spurr resin alone overnight. Finally, the samples were polymerized at 70 °C for 16 h. Semi-thin sections (2 μm) were cut using glass knives on an LKB Bromma 11800 pyramitome (LKB, Bromma, Sweden) and stained with methylene blue, and ultra-thin sections were cut with a diamond knife using PowerTome-PC (RMC, Boeckeler Instruments, Tucson, AZ, USA). The sections were stained with 3% uranyl acetate and alkaline lead citrate and observed using TEM with a model JEM-1230 (JEOL, Tokyo Japan) at an accelerating voltage of 80 kV.
Immunoelectron microscopy (IEM)
As described in the previous sections, RRSV-infected N. lugens nymphs were collected at 8 d p.i.; and uninfected nymphs were used as a control. Salivary glands were dissected and fixed in 4% paraformaldehyde (v/v), 0.3% glutaraldehyde and 4% sucrose in 0.1 M sodium cacodylate buffer (pH 7.4) for 3 h. After dehydration in serially graded methanol (30, 50, 70, 80, 90, 95 and 100%, v/v), the tissues were embedded in Lowicryl K4M (Polysciences, Inc., Warrington, PA, USA). Polymerization was performed in an ultraviolet irradiator at −20 °C for 2 d and then at room temperature for 2 d. Ultra-thin sections were cut as described for the TEM observations. The sections were blocked with 1% BSA for 5 min and then incubated with the anti-RRSV capsid P8 protein mouse serum (1:50) at room temperature for 2 h, followed by incubation with 5-nm gold-conjugated goat-anti-mouse IgG (1:200, Sigma-Aldrich) for 2 h. The sections were stained in 3% uranyl acetate (w/v in distilled water) and observed using TEM with a model JEM-1230 at an accelerating voltage of 80 kV.
Western blotting analysis
Salivary glands and whole bodies were collected from N. lugens nymphs and homogenized in RIPA Lysis Buffer (Beyotime, Shanghai, China), respectively. The protein concentrations were quantified using Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA) following the manufacturer’s instructions. After adding 6 × SDS loading buffer, lysates were boiled for 10 min. The proteins were separated by SDS-PAGE and transferred to PVDF membrane. The blot was probed with a RRSV capsid P8-specific mouse primary antibody (1:5,000 dilution) and detection was achieved using a goat anti-mouse IgG-conjugated horseradish peroxidase (HRP) antibody (Jackson ImmunoResearch, West Grove, PA, USA) at a dilution of 1:10,000. Western blots were imaged using a Chemiluminescence Detection Kit (Bio-Rad, Hercules, CA, USA) with the Molecular Imager® ChemiDoc™ XRS+ System (Bio-Rad, Hercules, CA, USA). The β-actin polyclonal rabbit serum was prepared as in our previous report35, and used to monitor equal protein loading.
Transmission efficiency of RRSV from the insect vector to the rice plants
Second-instar N. lugens nymphs were microinjected with a mixture of dscaspase-1 at a 1:1:1 quality ratio or dscaspase-8 and maintained on healthy rice seedlings for 24 h. The dsRNA-treated nymphs were placed on RRSV-infected plants for virus acquisition over an acquisition access period (AAP) of 48 h. Each nymph was individually transferred to a series of fresh, virus-free rice seedlings at the 1–2 leaf stage in culture bottles (one nymph per bottle) and reared at 26 ± 0.5 °C with 50 ± 5% humidity under a 16:8 h light:dark photoperiod. Two weeks later, the transmission of RRSV from the insects to the rice seedlings was determined using quantitative real-time PCR combined with reverse transcription-PCR. Briefly, total RNA was extracted from the insects and the leaf tissue of the rice seedlings using RNAiso plus (TaKaRa) and employed as the templates for reverse transcription. PCR analysis was performed using the specific primers to amplify the RRSV P8 gene encoding the major capsid protein. The transmission rates of RRSV to the rice seedlings by the viruliferous individuals of N. lugens were calculated for 1–7 d p.i. based on the PCR results. The dsGFP-microinjected individuals were used as controls. RRSV accumulation after dsRNA treatments in N. lugens salivary glands was investigated using quantitative real-time PCR and reverse transcription-PCR. Total RNA was extracted from the salivary glands of dscaspase-1-, dscaspase-8- or dsGFP-treated N. lugens nymphs at 24-h intervals during 1–7 d p.i. Reverse transcription was conducted using 1 μg of genomic DNA-removed RNA sample. The procedure for quantitative real-time PCR analysis is described in Materials and Methods: ‘Investigation of RRSV proliferation in N. lugens salivary glands using quantitative real-time PCR’. Reverse transcription-PCR was carried out using a pair of primers for amplifying the RRSV P8 gene (sense: 5¢-GAGCAAACTTGAGGCGTAT-3¢ and antisense: 5¢-TTGGTCGTGTTGTATCTGG-3¢). The N. lugens β-actin gene (GenBank accession no. EU179846) was analyzed as a control using the following primers: 5¢-TGGACTTCGAGCAGGAAATGG-3¢ (sense) and 5¢-ACGTCGCACTTCATGATCGAG-3¢ (antisense). The PCR reaction was performed under the following conditions: denaturation at 94 °C for 4 min followed by 35 cycles of 94 °C for 30 s, 60 °C for 30 s and then extension at 72 °C for 10 min.
Additional Information
How to cite this article: Huang, H.-J. et al. Rice ragged stunt virus-induced apoptosis affects virus transmission from its insect vector, the brown planthopper to the rice plant. Sci. Rep. 5, 11413; doi: 10.1038/srep11413 (2015).
Supplementary Material
Acknowledgments
We thank Prof. Tai-Yun Wei (Institute of Plant Virology of Fujian Agricultural and Forestry University) and Prof. Xu-Dong Zhu (China National Rice Research Institute) for kindly providing RRSV. We thank Prof. Jian-Guo Chen (School of Life Sciences, Peking University) and Prof. Li Yu (School of Life Sciences, Tsinghua University) for helping us to interpret the TEM pictures. This work was supported by the National Basic Research Program of China (973 Program, No. 2014CB138402) and the National Natural Science Foundation of China (Grant No. 31371934).
Footnotes
Author Contributions Y.B. designed experiments and wrote the manuscript. Y.B. analyzed the N. lugens genomic and transcriptomic data. H.H., S.L., X.H. and Y.Y. performed the experiments. C.Z, X.Z, H.X. and J.W. provided valuable suggestions and helped to revise the manuscript. All authors discussed the results and commented on the manuscript.
References
- Zheng L., Mao Q., Xie L. & Wei T. Infection route of rice grassy stunt virus, a tenuivirus, in the body of its brown planthopper vector, Nilaparvata lugens (Hemiptera: Delphacidae) after ingestion of virus. Virus Res. 188, 170–173 (2014). [DOI] [PubMed] [Google Scholar]
- Jia D. et al. Assembly of the viroplasm by viral non-structural protein Pns10 is essential for persistent infection of rice ragged stunt virus in its insect vector. J. Gen. Virol. 93, 2299–2309 (2012). [DOI] [PubMed] [Google Scholar]
- Ling K., Tiongco E. & Aguiero V. Rice ragged stunt, a new virus disease. Plant Dis. Rep 62, 701–705 (1978). [Google Scholar]
- Wu J. et al. Identification of Pns6, a putative movement protein of RRSV, as a silencing suppressor. J. Virol 7, 335 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Wu J., Adkins S., Xie L. & Li W. Rice ragged stunt virus segment S6-encoded nonstructural protein Pns6 complements cell-to-cell movement of Tobacco mosaic virus-based chimeric virus. Virus Res. 152, 176–179 (2010). [DOI] [PubMed] [Google Scholar]
- Cabauatan P. Q., Cabunagan R. C. & Choi I.-R. Rice viruses transmitted by the brown planthopper Nilaparvata lugens Stål in Planthoppers: New Threats to the Sustainability of Intensive Rice Production Systems in Asia (eds Heong K.L. & Hardy B. ) 357–368 (International Rice Research Institute, Asian Development Bank, Australian Government, Australian Centre for International Agricultural Research, 2009). [Google Scholar]
- Hogenhout S. A., Ammar E.-D., Whitfield A. E. & Redinbaugh M. G. Insect vector interactions with persistently transmitted viruses*. Annu. Rev. Phytopathol. 46, 327–359 (2008). [DOI] [PubMed] [Google Scholar]
- Kelly E. M., Moon D. C. & Bowers D. F. Apoptosis in mosquito salivary glands: Sindbis virus-associated and tissue homeostasis. J. Gen. Virol. 93, 2419–2424 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan R. & Scott T. W. Apoptosis in mosquito midgut epithelia associated with West Nile virus infection. Apoptosis 11, 1643–1651 (2006). [DOI] [PubMed] [Google Scholar]
- Wang H., Gort T., Boyle D. L. & Clem R. J. Effects of manipulating apoptosis on Sindbis virus infection of Aedes aegypti mosquitoes. J. Virol. 86, 6546–6554 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. & Doumanis J. The fly caspases. Cell Death Differ. 7, 1039–1044 (2000). [DOI] [PubMed] [Google Scholar]
- Xu D. et al. Genetic control of programmed cell death (apoptosis) in Drosophila. Fly 38, 43 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant B., Blair C. D., Olson K. E. & Clem R. J. Annotation and expression profiling of apoptosis-related genes in the yellow fever mosquito, Aedes aegypti. Insect Biochem. Mol. Biol. 38, 331–345 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. & Clem R. J. Defining the core apoptosis pathway in the mosquito disease vector Aedes aegypti: the roles of iap1, ark, dronc, and effector caspases. Apoptosis 16, 105–113 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner M. O. The biochemistry of apoptosis. Nature 407, 770–776 (2000). [DOI] [PubMed] [Google Scholar]
- Thornberry N. A. & Lazebnik Y. Caspases: enemies within. Science 281, 1312–1316 (1998). [DOI] [PubMed] [Google Scholar]
- Cooper D. M., Granville D. J. & Lowenberger C. The insect caspases. Apoptosis 14, 247–256 (2009). [DOI] [PubMed] [Google Scholar]
- Xue J. et al. Transcriptome analysis of the brown planthopper Nilaparvata lugens. PLoS One 5, e14233 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y.-Y. et al. Genomic insights into the serine protease gene family and expression profile analysis in the planthopper, Nilaparvata lugens. BMC Genomics 15, 507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y.-Y. et al. The genome-and transcriptome-wide analysis of innate immunity in the brown planthopper, Nilaparvata lugens. BMC Genomics 14, 160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y.-Y. et al. De novo intestine-specific transcriptome of the brown planthopper Nilaparvata lugens revealed potential functions in digestion, detoxification and immune response. Genomics 99, 256–264 (2012). [DOI] [PubMed] [Google Scholar]
- Xue J. et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 15, 521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Chai L., Wang J. & Zhao X. Molecular cloning and characterization of Hearm caspase-1 from Helicoverpa armigera. Mol. Biol. Rep. 35, 405–412 (2008). [DOI] [PubMed] [Google Scholar]
- Courtiade J., Pauchet Y., Vogel H. & Heckel D. G. A comprehensive characterization of the caspase gene family in insects from the order Lepidoptera. BMC Genomics 12, 357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoa D., Trang L. & Takeda M. Expression analyses of caspase‐1 and related activities in the midgut of Galleria mellonella during metamorphosis. Insect Mol. Biol. 21, 247–256 (2012). [DOI] [PubMed] [Google Scholar]
- Liu Q., Qi Y. & Chejanovsky N. Spodoptera littoralis caspase-1, a Lepidopteran effector caspase inducible by apoptotic signaling. Apoptosis 10, 787–795 (2005). [DOI] [PubMed] [Google Scholar]
- Hebert C. G., Valdes J. J. & Bentley W. E. Investigating apoptosis: Characterization and analysis of Trichoplusia ni-caspase-1 through overexpression and RNAi mediated silencing. Insect Biochem. Mol. Biol. 39, 113–124 (2009). [DOI] [PubMed] [Google Scholar]
- Ahmad M. et al. Spodoptera frugiperda caspase-1, a novel insect death protease that cleaves the nuclear immunophilin FKBP46, is the target of the baculovirus antiapoptotic protein p35. J. Biol. Chem. 272, 1421–1424 (1997). [DOI] [PubMed] [Google Scholar]
- Fuchs Y. & Steller H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H., Saleh N. & Roechan M. Reovirus-like particles associated with rice ragged stunt diseased rice and insect vector cells. Ann. Phytopath. Soc. Japan 45, 229 (1979). [Google Scholar]
- Miyazaki N. et al. Structural evolution of reoviridae revealed by oryzavirus in acquiring the second capsid shell. J. Virol. 82, 11344–11353 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway M. J. et al. Mosquito saliva serine protease enhances dissemination of dengue virus into the mammalian host. J. Virol. 88, 164–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F. & Higgins D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.-J. et al. Two insulin receptors determine alternative wing morphs in planthoppers. Nature 519, 464–467 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.