

Abstract
A common deletion polymorphism within B-cell chronic lymphocytic leukemia-lymphoma like 11 gene (BIM) was deemed to be a genetic cause leading to compromised kinase inhibitor therapeutic efficacy in cancer individuals. However, the results reported were not consistent. Thus, a comprehensive meta-analysis containing 12 eligible studies including 1,532 Asian patients was conducted to investigate a steady and reliable conclusion. The results showed that BIM deletion polymorphism was significantly associated with tyrosine kinase inhibitor (TKI) clinical efficacy in term of response rate (Ph = 0.349, HR = 0.438, 95%CI = 0.274–0.699) and disease control rate (Ph = 0.941, HR = 0.370, 95%CI = 0.202–0.678) in EGFR-mutated NSCLC population, not in CML and HCC subgroups. Additionally, EGFR-mutated NSCLC patient harbored BIM deletion polymorphism was associated with a shorter progression-free survival (PFS) than those with BIM wild polymorphism (Ph = 0.580, adjusted HR = 2.194, 95%CI = 1.710–2.814). However, no significant association was examined between BIM deletion polymorphism and overall survival (OS) and toxic adverse events in EGFR-mutated NSCLC population and it was not associated with PFS and OS in HCC subgroup. These findings revealed that BIM deletion polymorphism might be a genetic cause of intrinsic resistance to TKI therapy and it could be emerged as an independent predictor to identify patients who would benefit from TKI targeted therapy in EGFR-mutated NSCLC.
Gefitinib, erlotinib and imatinib are a therapeutic class of tyrosine kinase inhibitors (TKIs) that significantly reduce tyrosine kinase activity by inhibiting its phosphorylation level and are important treatment options in patients with tyrosine kinase-driven malignancies, including epidermal growth factor receptor (EGFR) mutated non-small-cell lung cancer (NSCLC) and breakpoint cluster region-Abelson oncogene 1(Bcr-Abl1), non-receptor tyrosine kinase-driven chronic myeloid leukemia (CML)1,2,3. However, approximately primary resistance invariably emerged in 20% of CML patients to imatinib and 30% of EGFR-mutated NSCLC patients to EGFR-TKI therapy4,5,6, revealing that personalized difference in genetic background might influence treatment efficacy of TKI in cancer patients. EGFR T790M mutation and mesenchymal-epithelial transition (MET) amplification have been demonstrated to be genetic causes of acquired resistance to TKI7,8, However, genetic cause of intrinsic resistance to TKI in kinase-driven cancer patients remains unknown. Results of several recent studies showed that Kirsten rat sarcoma viral oncogene (KRAS) mutation, phosphatase and tensin gene (PTEN) loss were significantly associated with primary resistance to TKI therapy in kinase-driven malignancies9,10. However, these findings only account for a small proportion of cases. Thus, investigations are needed to further understand and overcome these possible primary resistant cancer patients with treatment of TKI.
B-cell chronic lymphocytic leukemia-lymphoma like11 (BCL2L11 also known as BIM), which is located in 2q12-q13, is a member of the B-cell CLL/lymphoma 2 (Bcl-2) family genes that encodes protein BIM including BCL2-homology domain 3 (BH3)-only domain11,12. BIM has emerged as a crucial mediator of apoptotic signal pathway that triggered by TKI13. It can directly activate the pro-apoptotic function, oppose to all members of the prosurvival Bcl-2 subfamily and bind to all members of the pro-apoptotic Bcl-2 family to promote cell apoptosis14. A common 2,903 bp deletion polymorphism was observed in intron 2 of BIM recently, and it had been demonstrated that it might affect RNA alternative splicing, leading to decreased generation of BIM spliced isoforms without essential BH3 domain15. Since BH3 domain plays an important role in cell apoptosis and apoptosis is one of the pivotal pathways for cancer cell death induced by TKI16,17. We hypothesized that the deletion polymorphism within BIM would mediate the treatment efficacy and survival of cancer patient with TKI therapy.
Recently, accumulating evidences showed that the BIM deletion polymorphism was associated with inferior responses to TKI and a shorter progression-free survival (PFS) in TKI treated cancer patients15,18,19. Others were suggested that BIM deletion polymorphism was not significantly correlated with the kinase inhibitor efficacy for EGFR-mutated NSCLC, CML and hepatocellular cancer (HCC) patients20,21,22,23. In order to obtain an objective and consistent conclusion, we therefore conducted this comprehensive systematic review and meta-analysis of the association between BIM deletion polymorphism and clinical response and survival outcome of kinase inhibitor treated cancer patients.
Materials and methods
Literature search
A comprehensive literature search was conducted in databases of Web of Science, PUBMED and CNKI using the following keywords and search terms: “BIM or BCL2L11 or Bcl-2-Like Protein 11”, “tyrosine kinase inhibitor or TKI”, “polymorphism” as well as “gefitinib or erlotinib or imatinib or sorafenib” dating up to 1 December 2014. Meanwhile, hand search was performed to obtain substantial relevant study by reviewing all references within all eligible articles. All selected literatures were journal articles in Chinese and English. The methods used for this study were selected in accordance with the preferred reporting items for systematic review and meta-analyses (PRISMA) statement24. This study was approved by the Institution Ethics Commission of Southeast University, and the methods were carried out in accordance with the approved guidelines.
Inclusion and exclusion criteria
Relevant article was obtained by identification of title and abstract of each articles searched from the databases and reference list of eligible studies and eligible literatures were identified by screening the full-text of relevant study fulfilling the following eligibility criteria: 1) retrospective or prospective study investigated the association between BIM deletion polymorphism and kinase inhibitor efficacy or survival of cancer patient, 2) eligible study provided sufficient data concerning BIM polymorphism and TKI response status, toxic adverse events, survival (PFS and overall survival (OS)), or sufficient information for such data to be calculated or provided by author; 3) response and toxicity assessments were in accordance with the international guidelines. On the contrary, studies with duplicated or without sufficient data, study investigating susceptibility, review, view, letter, reply were excluded from the study.
Data extraction
The following data were gathered from each included eligible article: study design, name of the first author, year of publication, cancer type, sample size, sex, ethnicity, TKI information, definition of response and non-response, genotype distributions, response rate (RR), disease control rate (DCR), toxic adverse events, OS and PFS data. These data were extracted by two independent reviewers (Hou-Qun Ying and Jie Chen), and any discrepancies between them were resolved to reach consensus by discussion.
Statistical analysis
Statistical analysis of the extracted data was conducted using Stata software (Version11.0, Stata Corporation, College Station, TX). The odds ratio (OR), hazard ratio (HR) and corresponding 95% confidential interval (CI) were used as common measurements to assess the strength of association between BIM deletion polymorphism and clinical outcome of cancer patients with TKI therapy. The pooled OR, HR and corresponding 95%CI were calculated using the random or fixed model according to the results of heterogeneity analyses. Subgroup analysis was performed by cancer type. Q test and I2 were used to evaluate the heterogeneity across the included studies25. Ph < 0.1 or I2 > 50% suggested a significant statistical heterogeneity across studies and the random model was selected to pool data, otherwise, results from the fixed-model were reported. Sensitivity analysis was performed to evaluate the robustness of primary results by successively omitting an eligible study or changing the evaluation model. Begg’s funnel plot and egger’s test were calculated to test for publication bias and obvious asymmetry of begg’s funnel plot and Pe < 0.05 were considered statistical significance26.
Results
Eligible study
Using the keyword and search term, a total of 288 articles were found from the above databases and 16 articles were obtained by manual retrieval the reference from eligible articles. However, 81 duplicated articles, 197 unrelated articles, 4 reviews, 4 studies investigating susceptibility, 2 studies concerning non-deletion polymorphism, one study regarding non-kinase inhibitor therapy, one study concerning mechanism, one study without data, one communication, one view and one reply were excluded from this meta-analysis. Therefore, ultimately, only 10 articles including 12 studies15,18,19,20,21,22,23,27,28,29, which met the inclusion criteria, were enrolled in the study to investigate association between BIM deletion polymorphism and clinical efficacy and survival of cancer patients with kinase inhibitor therapy. The flow chart for the study search and screen process was depicted in Fig. 1.
Figure 1. Flow chart of eligible study selection.

Study characteristics
A total of 10 articles including 2 prospective studies and 10 retrospective studies with 1,532 cases were included in this meta-analysis. Among them, 6 articles including 6 studies with 839 patients, 4 articles containing 5 studies with 604 patients and one article containing 1 studies with 89 patients investigating the association between BIM polymorphism and clinical outcome of EGFR-TKI, imatinib as well sorafenib for EGFR-mutated NSCLC, CML and HCC, respectively. All included studies were all conducted in East and Southeast Asian population. In 6 studies, the individuals were advanced, recurrent EGFR-mutated NSCLC patients. The patients in 4 and one studies were Bcl-Abl fusion gene positive CML cases and II-IV stage HCC, respectively. Among the studies, clinical response of all solid cancers, CML and toxic adverse event were evaluated in accordance with RESCIST version 1.1 and ELN criteria as well as CTC3.0, respectively. However, only 7 and 3 eligible studies respective reported PFS and OS in solid cancer. The baseline characteristics of eligible studies were described in Table 1.
Table 1. The baseline characteristics of the study included in this study.
| Author and year | Country or Region | Race | Cancer | Sample size | Males (%) | TNM stage | Kinase inhibitor | Criteria | Study design | Definition of response | Definition of non-response | Clinical outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ng 2012[1] | Singapore, Malaysia | Asian | Bcr-Abl1 positive CML | 138 | 58.70% | — | Imatinib | ELN | Retrospective | Optimal response | Suboptimal response or failure | Sensitive, resistant |
| Ng 2012[2] | Japan | Asian | Bcr-Abl1 positive CML | 65 | 56.90% | — | Imatinib | ELN | Retrospective | Optimal response | Suboptimal response or failure | Sensitive, resistant |
| Ng 2012[3] | Singapore, Japan | Asian | Mutant EGFR-NSCLC | 141 | 33.30% | III-IV, relapse | Gefitinib, erlotinib | — | Retrospective | — | — | PFS |
| Katagiri 2013 | Japan | Asian | Bcr-Abl1 positive CML | 37 | — | — | Imatinib | ELN | Retrospective | Sustained CMR for >24 months | Fluctuating CMR for >24 months | Sustained or fluctuating CMR for >24 months |
| Lee 2013 | Korea | Asian | Mutant EGFR-NSCLC | 193 | 37.10% | IIIB-IV, relapse | Gefitinib, erlotinib | RESCIST 1.1 | Retrospective | CR/PR | SD/PD | RR, DCR, PFS |
| Shao 2013 | Taiwan | Asian | HCC | 89 | 89.90% | II-IV | Sorafenib | RESCIST 1.1 | Retrospective | CR/PR | SD/PD | RR, DCR, PFS, OS, |
| Shinohara 2013 | Japan | Asian | Bcr-Abl1 positive CML | 144 | 65.80% | — | Imatinib | ELN | Prospective | CMR | Non-CMR | CMR, non-CMR |
| Zheng 2013 | China | Asian | Mutant EGFR-NSCLC | 123 | 49.60% | IIIB-IV | Gefitinib, erlotinib | RESCIST 1.1, CTC3.0 | Retrospective | CR/PR | SD/PD | RR, DCR, PFS, adverse events |
| Chen 2014 | China | Asian | Bcr-Abl1 positive CML | 220 | 50.90% | — | Imatinib | ELN | Retrospective | Optimal response | Suboptimal response or failure | Sensitive, resistant |
| Isobe 2014 | Japan | Asian | Mutant EGFR-NSCLC | 70 | 27.10% | IV, relapse | Gefitinib, erlotinib | RESCIST 1.1, CTC3.0 | Retrospective | CR/PR | SD/PD | RR, DCR, PFS, OS, adverse events |
| Lee 2014[1] | Korea, Taiwan | Asian | Mutant EGFR-NSCLC | 146 | 39.20% | IIIB-IV | Gefitinib, erlotinib, afatinib | RESCIST 1.1 | Prospective | CR/PR | SD/PD | RR, PFS, OS |
| Zhao 2014 | China | Asian | Mutant EGFR-NSCLC | 166 | 48.20% | IIIB- IV | Gefitinib, erlotinib | RESCIST 1.1 | Retrospective | CR/PR | SD/PD | RR, DCR, PFS |
Abbreviation: CML: chronic myeloid leukemia; NSCLC: non-small cell lung cancer; HCC: hepatocellular cancer; ELN: European leukemiaNet criteria; RSCST: response evaluation criteria in solid tumors; CTC: national cancer institute common terminology criteria; CMR: complete molecular response; CR: complete response; PR: partial response; PD: progressive disease; SD: stable disease; RR: response rate; DCR: disease control rate; PFS: progression-free survival; OS: Overall survival.
Efficacy of TKI
The pooled results of the meta-analysis were listed in Table 2 and Fig. 2. The clinical RR in patient with TKI therapy who harbored BIM deletion polymorphism was inferior to the patients with BIM wild polymorphism in EGFR-mutated NSCLC population (Ph = 0.349, OR = 0.438, 95%CI = 0.274–0.699). Furthermore, there was an inverse association of BIM deletion polymorphism with DCR in EGFR-mutated NSCLC cancer patients (Ph = 0.941, OR = 0.370, 95%CI = 0.202–0.678). However, BIM deletion polymorphism wasn’t correlated with RR in neither CML (Ph = 0.143, OR = 0.888, 95%CI = 0.537-1.470) nor HCC (OR = 0.791, 95%CI = 0.197–3.174) subgroups. Additionally, no significant association was observed between BIM deletion polymorphism and DCR in HCC individuals (OR = 0.791, 95%CI = 0.197–3.174).
Table 2. Meta-analysis results of the association between BIM deletion polymorphism and response rate and disease control rate.
| Group |
Response rate |
Disease control rate |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study(cases) | Ph (I2) | Pe |
OR (95%CI) |
Study(cases) | Ph (I2) | Pe |
OR (95%CI) |
|||
| Fixed | Random | Fixed | Random | |||||||
| NSCLC | 5 (698) | 0.349 (10.1%) | 0.769 | 0.438 (0.274–0.699) | 0.450 (0.269–0.753) | 4 (552) | 0.941(0.0%) | 0.136 | 0.370 (0.202–0.678) | 0.364 (0.200–0.664) |
| CML | 5 (604) | 0.143 (41.8%) | 0.393 | 0.888 (0.537–1.470) | 0.797 (0.373–1.703) | — | — | — | — | — |
| HCC | 1 (89) | — | — | 1.875 (0.194–18.102) | 1.875 (0.194–18.102) | 1 (89) | — | — | 0.791 (0.197–3.174) | 0.791 (0.197–3.174) |
Abbreviation: NSCLC: non-small cell lung cancer; CML: chronic myeloid leukemia; HCC: hepatocellular cancer; Ph: p-value of heterogeneity test; Pe: p-value of egger’s test; OR: odds ratio; 95%CI: 95% confidential interval.
Figure 2. The results of meta-analysis of association between BIM deletion polymorphism and response rate and disease control rate of kinase inhibitor therapy in malignancy.
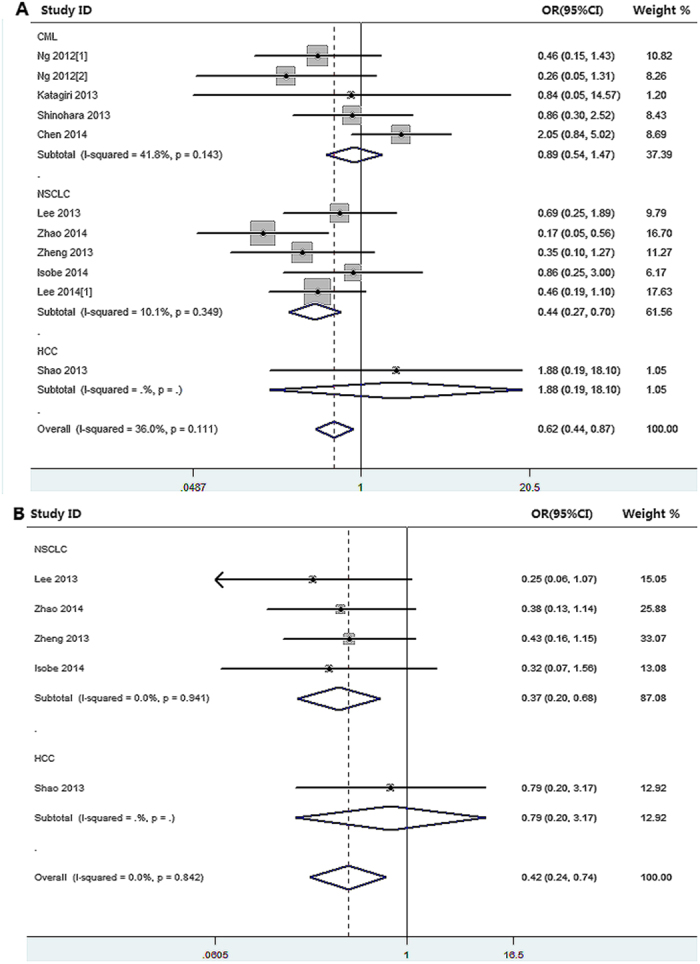
(A): BIM deletion polymorphism and response rate; (B): BIM deletion polymorphism and disease control rate.
PFS and OS
7 eligible studies reported association between BIM deletion polymorphism and PFS in TKI treated solid cancer patients were enrolled in our study and the pooled results showed a significant association between them only in EGFR-mutated NSCLC patients (univariate analysis: Ph = 0.164, HR = 2.000, 95%CI = 1.629–2.455; multivariate analysis: Ph = 0.580, HR = 2.194, 95%CI = 1.710–2.814), not in HCC subgroup (univariate analysis: HR = 0.720, 95%CI = 0.364–1.422; multivariate analysis: HR = 0.866, 95%CI = 0.408–1.837) (Table 3 and Fig. 3). However, there was no significant correlation between BIM deletion polymorphism and OS in EGFR-mutated NSCLC (univariate analysis: Ph = 0.057, HR = 1.361, 95%CI = 0.559–3.315) and HCC (univariate analysis: HR = 1.170, 95%CI = 0.740–1.850; multivariate analysis: HR = 0.668, 95%CI = 0.300–1.500), respectively (Table 3 and Fig. 3).
Table 3. Meta-analysis results of the association between BIM deletion polymorphism and progression-free and overall survivals.
| Group |
Progression-free survival |
Overall survival |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| study(case) |
HR(95%CI) |
Adjusted HR (95%CI) |
study(case) |
HR(95%CI) |
Adjusted HR (95%CI) |
|||||
| Fixed | Random | Fixed | Random | Fixed | Random | Fixed | Random | |||
| NSCLC | 6 (839) | 2.000 (1.629–2.455) | 2.067 (1.591–2.685) | 2.194 (1.710–2.814) | 2.194 (1.710–2.814) | 2 (216) | 1.419 (0.893–2.256) | 1.361 (0.559–3.315) | — | — |
| HCC | 1 (89) | 0.720 (0.364–1.422) | 0.720 (0.364–1.422) | 0.866 (0.408–1.837) | 0.866 (0.408–1.837) | 1 (89) | 1.170 (0.740–1.850) | 1.170 (0.740–1.850) | 0.668 (0.300–1.500) | 0.668 (0.300–1.500) |
Abbreviation: NSCLC: non-small cell lung cancer; HCC: hepatocellular cancer; HR: hazard ratio; 95%CI: 95% confidential interval.
Figure 3. The results of meta-analysis of association between BIM deletion polymorphism and progression-free, overall survival in malignancy with kinase inhibitor therapy.
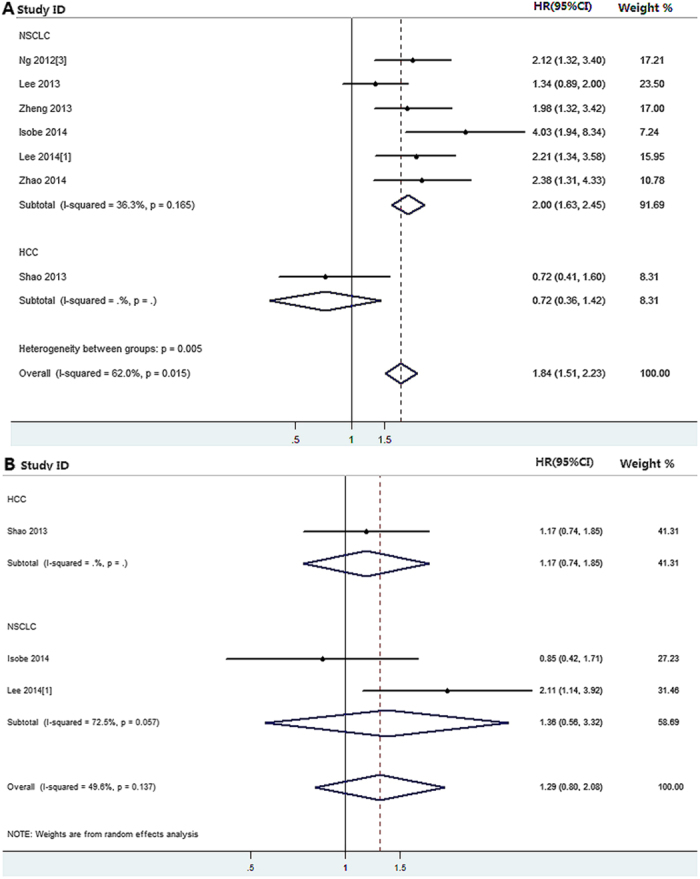
(A) BIM deletion polymorphism and progression-free survival; (B) BIM deletion polymorphism and overall survival.
Adverse events
The association between BIM deletion polymorphism and toxicity in individuals triggered by TKI was evaluated in two eligible studies including 193 EGFR-mutated NSCLC patients. The overall effect of meta-analysis showed no association between BIM deletion polymorphism and rash (Ph = 0.361, OR = 1.134, 95%CI = 0.536–2.399), diarrhea (Ph = 0.153, OR = 1.000, 95%CI = 0.446–2.239), interstitial pneumonia (Ph = 0.77, OR = 0.467, 95%CI = 0.057–3.808) and liver function damage (Ph = 0.406, OR = 0.470, 95%CI = 0.085–2.612), respectively.
Sensitivity analysis and publication bias
Results of sensitivity analysis showed that the pooled OR and HR were not significant alternated by omitting each eligible article successively or changing evaluation model. Visual assessment of begg’s funnel plot symmetry and egger’s test did not suggest evidence of substantial publication or small-study bias (Table 2 and Fig. 4).
Figure 4. Begg’s funnel plots of BIM deletion polymorphism and response rate, disease control rate and progression-free survival in malignancy with kinase inhibitor therapy.

(A) BIM deletion polymorphism and response rate; (B) BIM deletion polymorphism and disease control rate; (C) BIM deletion polymorphism and progression-free survival.
Discussion
This study, to the best of our knowledge, is the first synopsis of the literature on the effect of BIM deletion polymorphism on intrinsic resistance and clinical outcome of cancer patient with kinase inhibitor therapy. Upon systematic review and meta-analysis of the data from 12 eligible studies, we found that mutant EGFR NSCLC patient harbored BIM wild polymorphism with TKI therapy out-performed the patients with BIM deletion polymorphism in term of RR, DCR and PFS. However, there was no evidence that kinase inhibitor treated EGFR-mutated NSCLC and HCC individual harbored BIM deletion polymorphism improved OS in comparison with those with BIM wild polymorphism. In addition, BIM deletion polymorphism wasn’t associated with toxic adverse events in EGFR-mutated NSCLC cases triggered by TKI therapy. These robust findings revealed that clinical outcome of TKI treated mutant EGFR NSCLC individuals who harbored BIM deletion polymorphism was inferior to those carrying BIM wild polymorphism in Asian population.
Kinase inhibitor therapy can be effective for EGFR-mutated NSCLC, and Bcl-Abl CML as well as HCC30. However, only approximately 70–80% and 3.3% of EGFR-mutated NSCLC and CML, HCC patients exhibited treatment response to TKIs and sorafenib, respectively29,31,32. BIM is an essential mediator in cell apoptosis that induced by kinase inhibitor33,34. Germline variation within BIM may result in alternative expression of BIM isoforms lacking BH3 domain, leading to intrinsic resistance in kinase inhibitor therapy15,35. Therefore, a common deletion polymorphism within BIM was deemed as a candidate genetic cause of intrinsic resistance to kinase inhibitor therapy in these malignancies.
In this meta-analysis, we found that individuals with mutant EGFR NSCLC harbored BIM deletion polymorphism were inferior response to TKI than did patients with BIM wild polymorphism, suggesting that BIM deletion polymorphism was inverse correlated with clinical efficacy of TKI therapy and it might be a genetic cause mediating intrinsic resistance to TKI treatment in mutant EGFR NSCLC individuals. Furthermore, DCR for mutant EGFR NSCLC cases carrying BIM deletion polymorphism were significantly decreased in comparison with those with BIM wild polymorphism, suggesting that individuals with BIM deletion polymorphism could not obtain benefit more from TKI therapy and it could be used as a genetic biomarker for predicting TKI efficacy in EGFR-mutated NSCLC. Additionally, we also found that TKI therapy in mutated-EGFR NSCLC showed inferiority of patients carrying BIM deletion polymorphism over BIM wild polymorphism in term of PFS in both univariate and multivariate analyses, indicating that BIM deletion polymorphism was an independent prognostic factor for advanced EGFR-mutated NSCLC with TKI therapy. However, the pooled results showed no statistically significant association between BIM deletion polymorphism and OS in univariate analysis, revealing that it could not be emerged as a genetic biomarker to predict OS in EGFR-mutated NSCLC patient with TKI therapy in Asian population. The deletion polymorphism is a 2,903 bp fragment deletion locus which is located in intron 2 and its frequency is only 13% in Asian population, but absent in African and Caucasian populations15,20,36,37. The deletion polymorphism region contains cis elements that suppresses BIM exon 3 splicing and leads to preferential splicing of exon 3 over exon 415,38, resulting in impaired expression of BH3 domain. Since BH3 is a crucial domain in BIM that acts as an apoptosis facilitator in response to stress signals like DNA damage, and its functions irreplaceably in the apoptosis pathway39,40. Inhibition BIM activity resulted in failure of TKI therapy and high expression of BIM was necessary in success of TKI targeted therapy in levels of vivo and vitro13,15,35,41,42,43. All participants of eligible studies concerning NSCLC were advanced stage patients, and EGFR of all included cases were mutated (T790M, exon 19 deletion, L858R mutation, and so on). This may be the reason why BIM deletion polymorphism was associated with poor clinical outcome in mutant EGFR NSCLC with TKI targeted therapy.
However, we did not find any significant association between BIM deletion polymorphism and clinical outcome of Bcl-Abl tyrosine kinase-driven CML and HCC in terms of RR, DCR, PFS and OS. The mechanism for this remains poor understand. Only an eligible study including 89 cases concerning HCC in present study attenuated the statistical power22,44. However, cell apoptosis induced by TKI in CML might be not completely depended on BIM pathway and cancer response to TKI in patients with BIM deletion polymorphism might depend on other proapopotic regulators 45,46. We also did not observed the significant association between BIM deletion polymorphism and incidences of rash, diarrhea, interstitial pneumonia and liver function damage due to TKI therapy among mutated-EGFR NSCLC cases, indicating that BIM deletion polymorphism could not be a predictor to evaluate toxic adverse event inducing by TKI in EGFR-mutated NSCLC patients. Although knockdown of BIM could inhibit overproduction of reactive oxygen species and apoptosis mediating by FOXO347,48, the occurred toxicity of TKI might be not only affected by BIM deletion polymorphism. Therefore, prospective cohort studies are warrant to investigate the useful biomarker to predict TKI related toxic adverse events.
To the best of our knowledge, this study is the first and the largest sample size synopsis of the literature on the effect of BIM deletion polymorphism on intrinsic resistance and clinical efficacy and survival of cancer patient with kinase inhibitor targeted therapy. The results of publication bias and sensitivity analysis showed no publication bias and the pooled results were robust, suggesting that the results were reliable and steady. However, several limitations of the present study should be addressed as follow. Eligible study was only searched and screened in databases of Web of Science, PUBMED and CNKI and manual retrieval in English and Chinese, which might lose other language-published studies and consequently result in selection bias. Sample sizes of each cancer group was not large enough to get more precise results.
In summary, BIM deletion polymorphism might be a genetic cause of intrinsic resistance to TKI therapy in EGFR-mutated NSCLC and it could be emerged as an independent predictive biomarker to identify patients who would benefit from TKI targeted therapy in EGFR-mutated NSCLC.
Additional Information
How to cite this article: Ying, H.-Q. et al. The effect of BIM deletion polymorphism on intrinsic resistance and clinical outcome of cancer patient with kinase inhibitor therapy. Sci. Rep. 5, 11348; doi: 10.1038/srep11348 (2015).
Acknowledgments
This study was supported by the Fundamental Research Funds for the Central Universities, University Graduate Student Scientific Innovation Project of Jiangsu (No.KYLX_0201), National Natural Science Foundation of China (No. 81172141), Nanjing Health Young Talent Project, Nanjing Medical Science and Technique Development Foundation to Y.Q.P. (No. QRX11255) and B.S.H. (No. QRX11254).
Footnotes
Author Contributions H.Q.Y. extracted the data and wrote the manuscript. J.C. extracted the data and reviesed the manuscript. B.S.H. and Y.Q.P. searched and screened the eligible studies. F.W. and Q.W.D. conducted the statistics. H.L.S. and X.L. prepared Tables and Figures. S.K.W. designed the study, reviewed and approved the manuscript.
References
- Lin Y., Wang X. & Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res 4, 411–35 (2014). [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A., Kantarjian H. & Cortes J. Imatinib and beyond--exploring the full potential of targeted therapy for CML. Nat Rev Clin Oncol 6, 535–43 (2009). [DOI] [PubMed] [Google Scholar]
- Fausel C. Targeted chronic myeloid leukemia therapy: Seeking a cure. Am J Health Syst Pharm 64, S9–15 (2007). [DOI] [PubMed] [Google Scholar]
- Keedy V. L. et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 29, 2121–7 (2011). [DOI] [PubMed] [Google Scholar]
- Mok T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361, 947–57 (2009). [DOI] [PubMed] [Google Scholar]
- Rosell R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13, 239–46 (2012). [DOI] [PubMed] [Google Scholar]
- Rho J. K. et al. Combined treatment with silibinin and epidermal growth factor receptor tyrosine kinase inhibitors overcomes drug resistance caused by T790M mutation. Mol Cancer Ther 9, 3233–43 (2010). [DOI] [PubMed] [Google Scholar]
- Hammerman P. S., Janne P. A. & Johnson B.E. Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Clin Cancer Res 15, 7502–7509 (2009). [DOI] [PubMed] [Google Scholar]
- Takeda M. et al. De novo resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutation-positive patients with non-small cell lung cancer. J Thorac Oncol 5, 399–400 (2010). [DOI] [PubMed] [Google Scholar]
- Sos M. L. et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 69, 3256–61 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar P. E., Lessene G., Strasser A. & Adams J. M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15, 49–63 (2014). [DOI] [PubMed] [Google Scholar]
- Kivits R. A. & Furneaux C. BIM: enabling sustainability and asset management through knowledge management. ScientificWorldJournal 2013, 983721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa D. B. et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 4, 1669–79; discussion 1680 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle R. J. & Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9, 47–59 (2008). [DOI] [PubMed] [Google Scholar]
- Ng K. P. et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 18, 521–8 (2012). [DOI] [PubMed] [Google Scholar]
- Kelekar A. & Thompson C. B. Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol 8, 324–30 (1998). [DOI] [PubMed] [Google Scholar]
- Lutz R. J. Role of the BH3 (Bcl-2 homology 3) domain in the regulation of apoptosis and Bcl-2-related proteins. Biochem Soc Trans 28, 51–6 (2000). [DOI] [PubMed] [Google Scholar]
- Zhao M. et al. The Bim deletion polymorphism clinical profile and its relation with tyrosine kinase inhibitor resistance in Chinese patients with non-small cell lung cancer. Cancer 120, 2299–307 (2014). [DOI] [PubMed] [Google Scholar]
- Isobe K. et al. Clinical significance of BIM deletion polymorphism in non-small-cell lung cancer with epidermal growth factor receptor mutation. J Thorac Oncol 9, 483–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Liu H., Xing H., Sun H. & Zhu P. The BIM deletion polymorphism cannot account for intrinsic TKI resistance of Chinese individuals with chronic myeloid leukemia. Nat Med 20, 1090 (2014). [DOI] [PubMed] [Google Scholar]
- Lee J. K. et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol 24, 2080–7 (2013). [DOI] [PubMed] [Google Scholar]
- Shao Y. Y., Chang Y. L., Huang C. Y., Hsu C. H. & Cheng A. L. The germline BIM deletion polymorphism is not associated with the treatment efficacy of sorafenib in patients with advanced hepatocellular carcinoma. Oncology 85, 312–6 (2013). [DOI] [PubMed] [Google Scholar]
- Shinohara Y. et al. A multicenter clinical study evaluating the confirmed complete molecular response rate in imatinib-treated patients with chronic phase chronic myeloid leukemia by using the international scale of real-time quantitative polymerase chain reaction. Haematologica 98, 1407–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch V. et al. PRISMA-Equity 2012 extension: reporting guidelines for systematic reviews with a focus on health equity. PLoS Med 9, e1001333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zintzaras E. & Ioannidis J. P. Heterogeneity testing in meta-analysis of genome searches. Genet Epidemiol 28, 123–37 (2005). [DOI] [PubMed] [Google Scholar]
- Peters J. L., Sutton A. J., Jones D. R., Abrams K. R. & Rushton L. Comparison of two methods to detect publication bias in meta-analysis. JAMA 295, 676–80 (2006). [DOI] [PubMed] [Google Scholar]
- Zheng L. et al. Relationship between BIM gene polymorphism and therapeutic efficacy in the retreatment of advanced non-small cell lung cancer with tyrosine kinase inhibitor. Chin J Lung Cancer 16, 632–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H. et al. Bcl-2-like protein 11 deletion polymorphism predicts survival in advanced non-small-cell lung cancer. J Thorac Oncol 9, 1385–92 (2014). [DOI] [PubMed] [Google Scholar]
- Katagiri S., Umezu T., Ohyashiki J. H. & Ohyashiki K. The BCL2L11 (BIM) deletion polymorphism is a possible criterion for discontinuation of imatinib in chronic myeloid leukaemia patients. Br J Haematol 160, 269–71 (2013). [DOI] [PubMed] [Google Scholar]
- Takeuchi S. & Yano S. Clinical significance of epidermal growth factor receptor tyrosine kinase inhibitors: Sensitivity and resistance. Respir Investig 52, 348–356 (2014). [DOI] [PubMed] [Google Scholar]
- Mitsudomi T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 11, 121–8 (2010). [DOI] [PubMed] [Google Scholar]
- Cheng A. L. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10, 25–34 (2009). [DOI] [PubMed] [Google Scholar]
- Yoshida T. & Haura E. B. The potential benefits of BIM in the further pursuit of biomarker discovery in cancer therapeutics. Cancer Discov 1, 289–90 (2011). [DOI] [PubMed] [Google Scholar]
- Akiyama T., Dass C. R. & Choong P. F. Bim-targeted cancer therapy: a link between drug action and underlying molecular changes. Mol Cancer Ther 8, 3173–80 (2009). [DOI] [PubMed] [Google Scholar]
- Nakagawa T. et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res 73, 2428–34 (2013). [DOI] [PubMed] [Google Scholar]
- Soh S. & Ong S. T. A novel BIM deletion polymorphism: implications and lessons for cancer targeted therapies. Rinsho Ketsueki 54, 1714–9 (2013). [PubMed] [Google Scholar]
- Ong S. T., Chuah C. T., Ko T. K., Hillmer A. M. & Lim W. T. Reply: the BIM deletion polymorphism cannot account for intrinsic TKI resistance of Chinese individuals with chronic myeloid leukemia. Nat Med 20, 1090–1 (2014). [DOI] [PubMed] [Google Scholar]
- Juan W. C., Roca X. & Ong S. T. Identification of cis-acting elements and splicing factors involved in the regulation of BIM Pre-mRNA splicing. PLoS One 9, e95210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happo L. et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 116, 5256–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordigarden A. et al. BH3-only protein Bim more critical than Puma in tyrosine kinase inhibitor-induced apoptosis of human leukemic cells and transduced hematopoietic progenitors carrying oncogenic FLT3. Blood 113, 2302–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber A. C. et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov 1, 352–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa K., Okamoto I., Nishio K., Janne P. A. & Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res 17, 2140–8 (2011). [DOI] [PubMed] [Google Scholar]
- Simasi J., Oelkrug C., Schubert A., Nieber K. & Gillissen A. The role of BIM-EL and BCL2-alpha on the efficacy of erlotinib and gefitinib in lung cancer. Respir Physiol Neurobiol (2014). [DOI] [PubMed] [Google Scholar]
- Hofmeister E. H., King J., Read M. R. & Budsberg S. C. Sample size and statistical power in the small-animal analgesia literature. J Small Anim Pract 48, 76–9 (2007). [DOI] [PubMed] [Google Scholar]
- Pellicano F. et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells 32, 2324–37 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai H. et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res 11, 759–67 (2013). [DOI] [PubMed] [Google Scholar]
- Hagenbuchner J. et al. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J Cell Sci 125, 1191–203 (2012). [DOI] [PubMed] [Google Scholar]
- Sade H. & Sarin A. Reactive oxygen species regulate quiescent T-cell apoptosis via the BH3-only proapoptotic protein BIM. Cell Death Differ 11, 416–23 (2004). [DOI] [PubMed] [Google Scholar]