

Abstract
Thirteen human interferon-α (IFNα) subtypes were expressed in E. coli and purified using an N-terminal affinity tag from the prodomain of subtilisn. IFNα subtypes were expressed in soluble form and purified from cell lysates or refolded and purified from inclusion bodies. Proteins produced by either protocol exhibited biological activities equal to or greater than commercially prepared IFNα preparations. The IFNαs were used to produce an anti-IFNα16 antibody (MAb-1B12) that specifically neutralized the biological activity of IFNα16, but not the 12 other IFNαs. Using MAb-1B12, and a previously generated IFNAR1/IFNAR2-FChk heterodimer, an assay was developed to determine total type I IFN biological activity and IFNα16-derived biological activity in an unknown sample.
Keywords: interferon, protein purification, interferon subtype, neutralizing antibodies, autoimmune disease
Introduction
Interferons (IFNs) are pleotropic cytokines that regulate resistance to viral infections, enhance immune responses and modulate cell survival and death [1, 2]. Three classes of IFNs have been discovered and classified as Type I, II and III IFNs that each signal through distinct receptor complexes [3]. In humans, the type I IFN family is comprised of thirteen IFNα subtypes, as well as IFN-β, -ω, -κ and -ε that signal through a common heterodimeric receptor complex of IFNAR1 and IFNAR2 [5, 9]. The IFNα subtypes share 75–95% amino acid sequence identity with each other and approximately 30% identity with IFNβ (Fig. 1). Upon receptor binding, the IFNs induce a signaling cascade that is initiated through the catalytic activation of Jak1 and Tyk2 tyrosine kinases, leading to phosphorylation of the receptor intracellular chains and recruitment of STAT1 and STAT2 transcription factors [4, 5]. STAT1 and STAT2 are subsequently phosphorylated and assemble with interferon regulatory factor 9 (IRF9) to form the ISGF3 complex. ISGF3 translocates to the nucleus and binds to interferon-stimulated response elements (ISREs) leading to the activation of interferon stimulated genes (ISGs).
Figure 1. Sequence analysis of IFNα subtypes.

Alignment of IFNα subtype protein sequences in ClustalW.
Despite significant sequence similarity, the IFNα subtypes exhibit distinct antiviral, antiproliferative and immunomodulatory activities [5–8]. The biological activities of the IFNs have led to the development of IFNα2 (IFNα2a and IFNα2b) for the treatment of Hepatitis C infections and certain types of cancers [9, 10]. The inclusion of other IFNαs might result in improved efficacy in different therapy regimens. Initial evidence supports this conclusion and an IFNα formulation consisting of 6 different IFNα subtypes (IFNα1, α2, α8, α10, α14 and α21) is now undergoing tests as a new anti-viral therapeutic [11–13]. In addition to a beneficial role as an antiviral and anticancer agent, the IFNαs have been linked to the initiation and progression of systemic lupus erythematosus (SLE), an autoimmune disease characterized by high levels of auto antibodies against nuclear antigens [14–17]. How increased levels of serum IFNα subtypes initiate or exacerbate SLE and other autoimmune diseases is currently unknown.
Large scale production of the IFNαs provides reagents for investigating the dual nature of the IFNαs, which are beneficial human therapeutics, but also play a negative role in autoimmune disease. Several expression systems including E. coli, yeast, baculovirus and mammalian cells have been used to produce and purify IFNαs [18–21]. However, most expression studies have focused on one or a limited number of IFNα subtypes [19, 20, 22, 23]. Here, we have individually expressed and purified all 13 human IFNα subtypes from E. coli and show they exhibit biological activities equivalent to commercially produced IFNαs. As a first step to understanding the role of individual IFNα subtypes in SLE patient serum, the produced IFNαs were used to identify a monoclonal antibody (MAb) that specifically neutralizes the biological activity of IFNα16. Using these proteins, an assay was designed that can be used to detect specific IFNα subtypes in unknown samples.
Materials and Methods
Cells, vectors and reagents
Escherichia coli DH5α was used for cloning IFN subtypes into the protein expression vector, pPAL7 (gift from Dr. Philip Bryan). The E. Coli BL21 (DE3) strain was used for protein expression. Restriction enzymes SpeI and EcoRI were obtained from New England Biolabs. The international standard for IFNα2a was obtained from National Institute of Biological Standards and Controls (NIBSC, Hertfordshire, UK) (Catalog no. Gxa01-901-535). Commercial IFNα1 (11125-1), IFNα8 (11115-1) and IFNα14 (11145-1) were purchased from PBL assay science (Piscataway, NJ).
Cloning
Synthetic cDNAs encoding IFNα subtypes (α1a, α2b, α4ab, α5, α6, α7, α8b, α10, α14c, α16, α17b, and α21b) were obtained from DNA 2.0 with SpeI and EcoRI restriction enzymes on their 5′ and 3′ ends, respectively. Plasmids containing the IFNα subtype cDNAs were digested with SpeI and EcoRI (New England Biolabs) to release the coding sequences that were subsequently ligated into the pPAL7. pPAL7-IFNα expression plasmids was transformed into BL21 (DE3) cells for expression.
IFNα protein expression
IFNα subtypes were expressed using the autoinduction method at 20°C (Studier, 2005). Briefly, cultures were inoculated into 1 ml of ZYP 0.8G media with 100 μg/ml ampicillin (ZY media with 0.8% glucose) and incubated for 4–5 hours at 37°C. This starter culture was inoculated into 50ml of ZY-0.8G media and incubated overnight at 37°C and 300 rpm. The next day, 5% of the culture was inoculated into ZYP-5052 rich media (0.5% glycerol, 0.05% glucose, 0.2% alpha-lactose). After approximately 4 hours (O.D. of 0.6), the cultures were transferred to a 20°C incubator and grown for 20 hours to induce protein expression.
IFNαs that were not soluble when expressed by autoinduction were induced using Isopropyl β-D-1-thiogalactopyranoside (IPTG). For these experiments, BL21 (DE3) starter cultures were grown overnight in 10 ml LB at 37°C. The next day, a 1% starter culture was inoculated into 500 ml of LB media and incubated in a 37°C shaker at 250 rpm for ~3 hours. Upon reaching an OD600 of 0.6–0.8, the cultures were induced with 1mM IPTG and incubated at 37°C for 5 hours.
Protein purification
E. coli cells, containing IFNαs expressed by autoinduction, were harvested by centrifuging at 6,000 rpm for 20 minutes. The culture pellet was resuspended in 50 ml of lysis buffer (100mM Tris acetate pH 8.0, 1mM EDTA and 100mM sodium acetate, pH 7.4) and sonicated for 5 min on ice at 40% amplitude (cycles of 9 seconds on and 9 seconds off). After sonication, the cell lysate was centrifuged at 48,000g for 45 minutes at 4° C and filtered through 0.2 μm PES syringe filter. The filtered supernatant was incubated with 5 ml of eXact™ beads (profinity eXact resin, Biorad) in batch for 30 minutes at 4°C. The resin was subsequently packed into Econo column (1.5cm diameter × 20 cm length; 35 ml volume) and washed with 15 column volumes (CVs) of lysis buffer and 3 CV of 1 M sodium acetate pH 7.4. The proteins were eluted from the beads by the addition of 7 CVs of elution buffer (100 mM Tris acetate pH 8.0, 100 mM sodium acetate pH 7.4 and 10 mM sodium azide). Each CV of elution buffer was incubated with the protein bound beads for 10 minutes to facilitate tag cleavage and then collected in tubes on ice. This collection step was repeated seven times until the entire elution buffer was collected. The eluates were pooled and dialyzed against 100 mM Tris acetate, pH 8.0 and 20 mM NaCl overnight at 4°C. The dialyzed protein was filtered and further purified by ion-exchange chromatography using Tricorn MonoQ 4.6/100PE, 1.7ml column (GE healthcare) with 5CV gradient ranging from 20mM to 200mM NaCl. The peak fractions from ion exchange chromatography were concentrated and further purified by preparative size exclusion chromatography using Superdex 200 column (24ml column volume with a flow rate of 0.3 ml/min.). Prior to injection, the Superdex 200 column was calibrated with gel filtration standards (Biorad, catalog no., 151-1901).
Each eXact column was regenerated by washing with 5 CV of 0.1M phosphoric acid followed by 10CV of lysis buffer. Following purification of each IFNα subtype, the ion exchange column was incubated overnight with 1mg/ml pepsin in 0.5M NaCl and 0.1M acetic acid at room temperature. Subsequently, the column was washed with 2 CV of distilled water, 2 CV of 2M NaCl, 4 CV of 1M NaOH, 2 CV of 2M NaCl and 2 CV of distilled water. After washing, the column was re-equilibrated with 4 CV of the running buffer (100mM Tris acetate and 20mM NaCl). The size exclusion column was cleaned after each purification run with 1 CV of 0.5M NaOH, 2 CV of distilled water and 2 CV of gel filtration running buffer (20mM Tris-HCl, 150mM NaCl).
Refolding
IFNα subtypes expressed in inclusion bodies (IB) were refolded prior to purification. Cell pellets obtained from 500ml of expression culture were resuspended in 25ml IB buffer (0.1M Tris-NaCl pH 8.0, 5mM EDTA, 0.5% Triton X 100, 0.1mM PMSF). The cell suspension was sonicated on ice for 2.5 minutes at 40% amplitude (10 second on and 5 second off) and centrifuged at 14,000 rpm for 20 minutes at 4°C. The IB pellet was washed 3X with IB buffer followed by a final wash in IB buffer without Triton X 100. The inclusion bodies were solubilized by the addition of a denaturing buffer (6M guanidine HCl, 0.1M Tris pH 8.0, 2.5mM EDTA, 5mM dithiothreitol), sonicated for 1 minute at 40% amplitude with 10 second burst and 5 second pause on ice; incubated for 10 minutes at room temperature. The mixture was sonicated again as described above followed by 1 hour incubation at RT. The mixture was centrifuged at 35,000 rpm for 1 hour at 4° C. The concentration of the denatured protein was measured by absorbance at 280 nm. The denatured protein was added to the refolding buffer (50mM NaCl, 100 mM Tris pH 8.0, 2.5 mM EDTA, 0.2 mM oxidized glutathione, 2 mM reduced glutathione, 0.8 M Arginine pH 9.3) rapidly stirred at 4° C to a final concentration of 0.5 mg/ml. The refolding mixture was transferred to a 10° C incubator for 48 hours. Following refolding, the proteins was dialyzed against 20 mM Tris acetate pH 8.0, 100mM urea with two changes of 8 hours each. The dialyzed protein was centrifuged at 10,000 rpm for 20 minutes and filtered using 0.45 μm filter. The protein was purified using eXact resin followed by ion-exchange and size exclusion chromatography as described for the soluble IFNs above.
Protein concentration
Protein concentration was determined by absorbance at 280nm. Extinction coefficients were determined using the EXPASY server (http://web.expasy.org/protparam). Theoretical molecular weights and pI values for each IFNα were obtained using the EXPASY server.
Mass spectrometry
IFNα molecular weights were determined by MALDI-TOF mass spectrometry. Briefly, samples were analyzed in the positive mode on a Voyager Elite mass spectrometer with delayed extraction technology (PerSeptive Biosystems, Framingham MA). The acceleration voltage was set at 25kV and 100 laser shots were summed. Experiments were performed using sinapinic acid (Aldrich, D13, 460-0) at 5mg/mL dissolved in acetonitrile: 0.1% TFA (1:1) as the matrix. The mass spectrometer was calibrated using apomyoglobin (Sigma Aldrich). Samples were diluted 1:10 with matrix, and 1 μL was pipetted onto a stainless steel 96 spot plate for analysis. Data was smoothed and processed using Data Explorer Software (AB Sciex).
Endotoxin measurement
Endotoxin contamination levels were assessed using Limulus Ameobocyte Lysate (LAL) QCL-1000™ Endotoxin assay kit (Lonza, catalog no. 50-647U). IFNα samples were diluted (1:500) and the endotoxin concentration in the samples was measured using a standard curve. Endotoxin levels are reported as endotoxin units (EU) per μg of protein.
Luciferase activity assay
HL116 cells were plated in white opaque plates (Corning) at 4 × 105 cells/ml (100μL/well) and incubated overnight at 37°C. Serial dilutions of the IFNα subtypes were prepared in the DMEM-glutamax media (Invitrogen, Catalog no. 10566-016) and incubated for 30 minutes at 37°C prior to addition to the cells. Diluted IFNαs were added to the cells and incubated at 37°C for 5 hours. Post incubation, the microplate and all the reagents were equilibrated to room temperature. Steady-Glo (Promega) Luciferase assay reagent (50 μL) was added to each well and luminescence was measured using a Biotek Synergy2 plate reader according to manufacturer’s instructions. Experiments were performed in duplicates and repeated on at least three independent occasions. Sigmoidal dose response curves were analyzed using the PRISM software (Graphpad Inc.) to derive 50% effective concentrations (EC50).
Statistical analysis
One-way analysis of variance (ANOVA) was used to compare IFNα EC50 values. The Tukey-Kramer multiple comparisons test was then used to determine which pairs of means were significantly different. All statistical tests were two-sided and were performed using a significance level of 5%. SAS software (version 9.3; SAS Institute, Cary, NC) was used for the statistical analyses.
ELISA
Interferons were plated in duplicate at 2μg/mL in PBS, pH 7.4 (100 μL/well) and incubated overnight at 4°C. All subsequent steps were performed at room temperature. Following IFN absorption, the buffer was discarded and 100μl of blocking reagent (PBS, 0.5% Proclin 300, 0.05% Tween 20, 1%BSA) was added to the wells for 1 hour. Subsequently, the plates were washed 3X with 200μL of wash buffer (PBS, 0.05% Tween 20). Antibody supernatants (100μL) were added to the plates and incubated for 1 hour. Following 3 wash steps, anti-mouse IgG-HRP conjugate (1:4000 dilution, Southern Biotech) was added to the wells for 1 hour. Following 3 additional washes, the plates were incubated with 100μL of TMB (3,3′,5,5′-Tetramethylbenzidine) for 15 minutes, followed by the addition of 100μl of stop solution (0.16M sulfuric acid). Absorbance was measured at 450nm and 670nm using the Biotek Synergy2 plate reader. A background corrected absorbance was obtained by subtracting the OD measured at 670nm from the OD measured at 450nm.
Monoclonal antibody production
For antibody production, BALB/C mice were immunized with 100 μg of IFNα16 in complete Freund’s adjuvant injected subcutaneously into the base of the tail and the thigh area of the hind legs. Mice received booster injections of 50μg of IFNα16 in PBS on days 7, 14 and 20. Mice were sacrificed on day 21 and cells from lymph nodes and spleen were fused to the PU31 hybridoma line (ATTC). Approximately 10 days after fusion, cell supernatants were screened for IFNα16 binding by ELISA. Positive hybridomas were subcloned by limiting dilution and re-evaluated by ELISA. The selected Ab, MAB-1B12, was purified from 1 liter of supernatant using Gammabind Plus Sepharose beads (GE healthcare). Bound antibody was eluted from the beads in 1ml fractions with low pH elution buffer (1M Glycine, pH 2.8). The pH of the fractions was adjusted with 1M Tris-HCl pH 9.0.
Results
Design of an IFNα expression construct for purification
To optimize the speed and efficiency of purifying 13 IFNα subtypes, an engineered subtilisin prodomain (SPD) was fused to the N-terminus of each IFNα subtype [24]. The SPD functions as a high affinity purification tag that is efficiently removed by an auto-cleavage reaction during elution from subtilisin eXact affinity chromatography resin. To optimize SPD cleavage, a threonine-serine (TS) linker was inserted between the SPD tag and N-terminus of each IFNα. As a result, each purified IFNα retains two additional amino acids (Thr-Ser) on its N-termini.
We previously determined that an SPD-IFNα2a fusion protein could be expressed in E. coli in soluble form [25]. To extend this finding, Incyte plasmids encoding human IFNα1 and IFNα21 genes were sub-cloned into the pPAL7 vector, which encodes the N-terminal SPD, for expression testing. However, no detectable protein production was observed for either SPD-IFNα1 or SPD-IFNα21 fusion proteins. To enhance protein expression, cDNAs encoding IFNα1a, IFNα2b, IFNα4ab, IFNα5, IFNα6, IFNα7a, IFNα8b, IFNα10, IFNα14c, IFNα16, IFNα17b and IFNα21b were synthesized using codons optimized for high-level expression in E. coli. IFNα subtype variants (e.g. IFNα8a vs. IFNα8b) were chosen based on their prevalence in the human population, when data was available [26–29]. The synthetic IFNα cDNAs were sub-cloned into the pPAL7 plasmid and transformed into BL21 (DE3) for expression studies.
Expression and purification of IFNα subtypes
The 13 SPD-IFNαs were expressed using the auto induction method at 20°C in ZY-5052 media (ref. [30], 0.5 liter scale). As shown in Figure 2A for IFNα1, autoinduction resulted in the accumulation of SPD-IFNα1 with a molecular weight of ~27 kDa, which corresponds to the ~7 kDa SPD and the ~20 kDa IFNα1. A three-step purification strategy was developed to isolate pure IFNα subtypes. First, the fusion proteins were captured by eXact bead affinity chromatography, followed by on- column cleavage of the SPD tag by the addition of elution buffer. Second, low levels of uncut SPD-IFNαs were removed by anion exchange chromatography. Third, size exclusion chromatography was performed on each IFNα. The purity of IFNα1 at each step in the purification process is shown in Figure 2B. To prevent cross contamination of different IFNα’s during purification, each subtype was purified on its own eXact bead affinity resin. The ion exchange and gel filtration columns were cleaned and considered suitable for a new IFNα subtype purification when final fractions from column washes exhibited no detectable IFN bioactivity. Bioactivity was evaluated using an IFN reporter cell line (HL116 cells), which contain a stable IFN-inducible firefly luciferase reporter gene [31]. A total of six IFNαs (IFNα1, IFNα2a, IFNα2b, IFNα4, IFNα5 and IFNα14) were expressed and purified as soluble SPD-IFNα fusion proteins, resulting in 3 to 20 milligrams of pure protein per 0.5 liter culture.
Figure 2. Expression and purification of IFNαs.
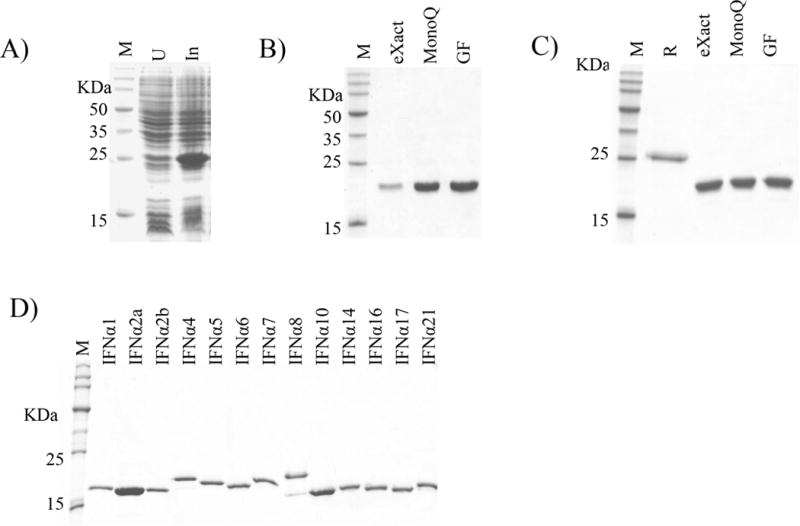
(A) Codon optimized expression of soluble SPD-IFNα1. Molecular weight markers (M), uninduced (U) and induced (In) samples are shown. (B) SDS-PAGE gel of soluble SPD-IFNα1 purification steps (C) SDS-PAGE gel of refolded SPD-IFNα14 purification steps. Lane markers on the gels are R, refolded protein; ‘eXact’, eXact affinity purified; ‘MonoQ’, purified by MonoQ ion exchange chromatography; ‘GF’, purified by gel filtration chromatography. (D) Final SDS-PAGE of the 13 purified IFNα subtypes.
The remaining seven SPD-IFNα fusion proteins (IFNα6, IFNα7, IFNα8, IFNα10, IFNα16, IFNα17 and IFNα21) were not soluble when expressed using auto-induction conditions. For these IFNα subtypes, the protein induction method was changed to 1mM IPTG for 5 hours and the SPD-IFNαs were refolded from insoluble inclusion bodies (IB). Refolded SPD-IFNαs were purified as described for the soluble SPD-IFNα fusion proteins (Figure 2C). SPD-IFNα refolding experiments resulted in protein yields (~8 to 20 mg) that were similar to the SPD-IFNαs expressed by the auto-induction method.
Amino acid sequence comparisons between soluble and insoluble IFNαs did not identify unique amino acids that would predict solubility. Consistent with this result, expression of the soluble SPD-IFNα14 fusion protein using the IPTG induction method resulted in SPD-IFNα14 IB formation. This suggests SPD-IFNα solubility is at least partially due to differences in protein expression rate and/or overall expression level. Importantly, IFNα14 produced in soluble or refolded form exhibited essentially identical gel filtration and biological activity profiles (Table 1). The biological potency of refolded SPD-IFNα14, prior to removal of the SPD tag, was ~5–fold lower than the biological potency of IFNα14 after SPD removal.
Table 1.
Biochemical and Biological Activity of purified IFNα subtypes.
| IFNα Subtype | Molecular Weight Determination
|
Biological Activity
|
||||
|---|---|---|---|---|---|---|
| Theoretical | Mass spectrometry | Δ (Da.) | Gel filtration | EC50 (pM) | Specific activity (IU/mg) | |
| IFNα1a | 19,574 | 19,536 | 38 | 20,423 | 222.4 ± 19.5 | 3.0 ± 0.2×107† |
| IFNα2a | 19,429 | 19,431 | 2 | 21,649 | 8.4 ± 1 | 4.3 ± 0.4×108† |
| IFNα2b | 19,457 | 19,437 | 20 | 24,469 | 7.8 ± 1.4 | 5.1×108 |
| IFNα4 | 19,567 | 19,534 | 33 | 18,389 | 40 ± 4.3 | 9.9 ± 0.6×107† |
| IFNα5 | 19,712 | 19,756 | 44 | 18,713 | 8.2 ± 1.2 | 4.8×108 |
| IFNα6 | 19,934 | 19,899 | 34 | 25,637 | 7.8 ± 0.74 | 5.1×108 |
| IFNα7 | 19,795 | 19,772 | 23 | 17,348 | 29.7 ± 4.7 | 1.3×108 |
| IFNα8 | 19,672 | 19,642 | 30 | 23,628 | 3.1 ± 0.6 | 1.3×109 |
| IFNα10 | 19,594 | 19,562 | 32 | 15,711 | 1.8 ± 0.3 | 2.2×109 |
| S-IFNα14 | 19,896 | 19,854 | 42 | 18,710 | 1.4 ± 0.2 | 3.1 ± 0.3×109† |
| R-IFNα14 | 19,896 | N.D. | N.D. | 18,701 | 1 ± 0.1 | 4.0×109 |
| IFNα16 | 19,470 | 19,466 | 4 | 21,523 | 8.5 ± 1 | 4.7×108 |
| IFNα17 | 19,487 | 19,482 | 5 | 15,803 | 4.7 ± 1 | 8.4×108 |
| IFNα21 | 19,500 | 19,496 | 4 | 15,260 | 10.9 ± 1.1 | 3.6×108 |
| IFNα1a* | N.D. | N.D. | N.D. | N.D. | 110 ± 7 | 3.6×107 |
| IFNα8* | N.D. | N.D. | N.D. | N.D. | 3.7 ± 0.5 | 1.1×109 |
| IFNα14* | ND | ND. | N.D. | N.D. | 5.4 ± 0.6 | 7.3×108 |
S-IFNα14, obtained from soluble SPD-IFNα14; R-IFNα14, obtained from refolded SPD-IFNα14;
mean specific activity derived from 3 different expression/purification experiments;
IFNα’s purchased from commercial source; N.D., Not Determined; Specific activity was calculated using IFNα2a NIH standard.
Chemical Purity (SDS-PAGE/Mass Spectrometry) of the IFNα Subtypes
A final SDS-PAGE gel of all 13 purified IFNαs is shown in Figure 1D. Although the molecular weights of the IFNα subtypes differ by no more than 500Da (Table 1), they exhibit significant molecular weight variability in the gel. The distinct electrophoretic mobility of IFNα8 in SDS-PAGE gels was previously shown to be caused by charged amino acids Glu84 and Asp90 [32]. To further validate the chemical composition of each IFNα, MALDI-TOF mass spectrometry was performed and the resulting experimentally determined molecular weights were compared against the predicted molecular weights (Table 1). The deviation between experimental and theoretical molecular masses ranged from 44 Da for IFNα5 to 2 Da for IFNα2a. In additional to protein purity, endotoxin levels of the purified IFNαs were determined. Endotoxin levels ranged from 0–1 EU/μg of protein, which is within the range of values observed in IFNα preparations obtained from commercial sources.
Size exclusion chromatography of the IFNαs
Size exclusion chromatography (SEC) of the IFNα subtypes was performed as a purification and diagnostic step to confirm proper folding of the molecules. All 13 IFNαs exhibited SEC chromatograms consistent with properly folded monomeric proteins (Fig. 3A). Estimated molecular weights of the IFNαs, determined by SEC, ranged from 25,637 kDa for IFNα6 to 15,260 kDa for IFNα21 (Table 1). The 3 IFNαs exhibiting the highest molecular weights by SEC are IFNα6, IFNα2b and IFNα8b, while the smallest are IFNα10, IFNα17, and IFNα21. No correlation was observed between the subtle differences in IFNα size, monitored by SEC, and the IFNα biological activity profiles (Table 1).
Figure 3. Biochemical and Functional Analysis of IFNα subtypes.
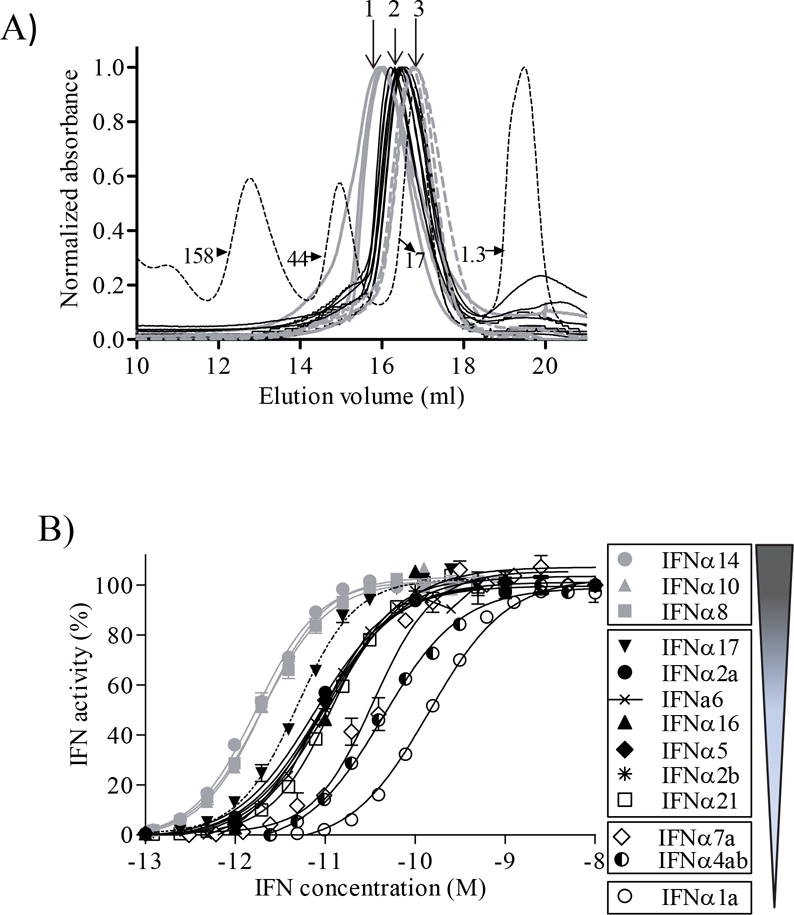
(A) Gel filtration chromatograms of the purified IFNα subtypes. Molecular weight standards are shown as a dashed line. The molecular weights of the peaks are labeled in kDa. IFNα chromatograms exhibiting similar profiles (3 different profiles, 1, 2, 3) are represented with the same line color/type study (1 = grey, 2 = black and 3 = dashed grey lines). (B) Average dose response curves for each IFNα subtype on HL116 cells. Graphical representation of the IFNα subtype potency groupings with lowest potency IFNαs at the bottom of the Figure.
Biological activity of IFNα subtypes
The bioactivity of each IFNα subtype was evaluated using an HL116 reporter cell line, which contains an IFN-inducible firefly luciferase reporter gene [31]. Dose response curves were generated for each IFNα subtype (Fig. 3B), from at least 6 independent measurements, to derive half-maximal effective concentrations (EC50). IFNα EC50 values ranged from 1.4 ± 0.2 pM for IFNα14 to 222 ± 19.5 pM for IFNα1 (Table 1), which corresponds to a 159-fold difference in biological potency among the subtypes. A statistical analysis of the EC50 values revealed the IFNαs cluster into four distinct activity groups (p<0.0001, Fig 3B). The most potent IFNαs (group 1) are IFNα14, IFNα10 and IFNα8 with EC50 values that range from 1.4 ± 0.2 pM to 3.1 ± 0.6 pM. Group 2 consists of 7 IFNαs (IFNα17 > IFNα2a, IFNα2b, IFNα5, IFNα6, IFNα16, > IFNα21) that exhibit EC50 values between 4.7 ± 1 pM (IFNα17) and 10.9 ± 1.1 pM (IFNα21). Group 3 consists of IFNα4 and IFNα7 with EC50 values of 29.7 ± 4.7 pM and 40 ± 4.3 pM. Group 4 consists of IFNα1 with an EC50 of 222 ± 19.5 pM.
The specific activity of each subtype was determined using the world health organization NIH standard for IFNα2a [33]. All IFNs, except IFNα1, exhibited specific activities ranging from 108–109 IU/mg. Similar specific activities have been previously reported in purification studies of IFNα2a and IFNα8 [22, 23, 34]. The bioactivity of IFNα1, IFNα8 and IFNα14 produced from SPD fusion proteins was also compared against commercial protein preparations. IFNα1 and IFNα8 exhibited EC50 values and specific activities equivalent to the commercial preparations, while IFNα14 exhibited 5-fold higher specific activity compared to commercially obtained IFNα14 (Table 1). Specific activity was also used to estimate the reproducibility of the protein expression/purification protocols. For these studies, four IFNs (IFNα1, IFNα2a, IFNα4 and IFNα14) were expressed and purified three separate times resulting in 3 distinct preparations of the four subtypes. Specific activity measurements for these IFNα preparations were essentially identical (Table 1), suggesting the protein expression/purification protocols reproducibly generate biologically active IFNαs.
Production of a neutralizing antibody specific for IFNα16
The purified IFNαs were used to generate an antibody that could specifically neutralize the biological activity of IFNα16, but not the other 12 IFNα subtypes. Anti-IFNα16 antibodies were generated by immunizing BALB/c mice with IFNα16. A total of 768 fusion supernatants were evaluated by ELISA, of which 98 exhibited strong signals consistent with IFNα16 binding. A secondary ELISA screen was performed on the 98 positive clones that identified one MAb (MAb-1B12) that bound tightly to IFNα16, but exhibited essentially no affinity to the other 12 IFNα subtypes. MAb-1B12 supernatant also neutralized IFNα16 biological activity in HL116 luciferase assays, suggesting MAb-1B12 is an IFNα16-specific neutralizing antibody. To confirm these results, MAb-1B12 was purified using protein G affinity chromatography. Purified MAb-1B12 was able to neutralize the biological activity of 10pM and 100pM IFNα16 on HL116 cells with IC50 (50% inhibitory concentration) values of 2.1±0.5 nM and 20±0.5 nM, respectively (Fig 4A).
Figure 4. Characterization of MAb-1B12 and detection of IFNα16.
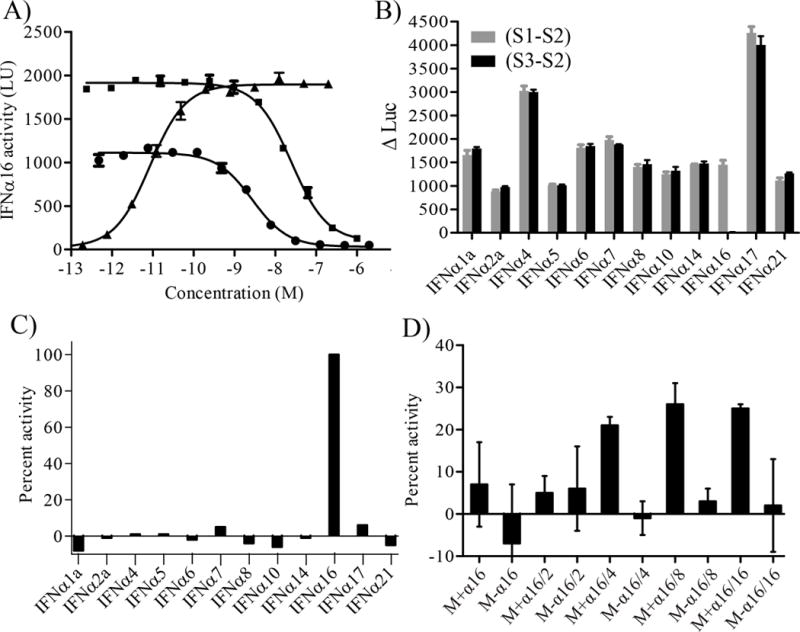
(A) Neutralization potential of MAb-1B12. MAb-1B12 IC50 values were derived in the presence of constant IFNα16 values of 10pM (●) and 100pM (■). A dose response curve for IFNα16 (▲) is shown for comparison. (B) MAb-1B12 specificity was determined by plotting S1-S2 (total IFN activity) and S3-S2 values (IFNα16 activity) for samples that contain the IFNα subtypes denoted on the x-axis. (C) Data in Figure 4B were re-plotted to calculate the percent IFNα16 activity in each sample using the equation {(S1-S2)−(S3-S2)/(S1-S2)}*100. (D) Detection of IFNα16 in a mixture of the eleven other IFNα subtypes (IFNα2b was not included in the assay). A mixture (M) of all IFNα subtypes except IFNα16 was made. Assayed samples contained the mixture plus IFNα16 (M+α16) or the mixture without IFNα16 (M-α16). The samples were measured at different dilutions as noted on the x-axis. For example, M+α16/2 and M+α16/4 correspond to twofold and fourfold diluted samples, respectively. Data are plotted as described in Figure 4C.
Assay to Elucidate total IFNα and IFNα16 Activity
A strategy for measuring IFNα levels in media samples was established to further characterize the specificity of MAb-1B12 and determine the feasibility of evaluating IFNα16 levels in samples containing different IFNα subtypes. The assay is performed by incubating HL116 cells with an IFNα sample that has been prepared in 3 ways (S1, S2 and S3). Sample 1 (S1) contains IFNα and corresponds to the unknown. Sample 2 (S2) contains the same IFNα solution plus the addition of an IFNAR1/IFNAR2-FChk heterodimer. Sample 3 (S3) contains IFNα incubated with MAb-1B12. IFNAR1/IFNAR2-FChk is a soluble type I IFN receptor heterodimer that functions as a potent antagonist of all IFNα subtypes [25]. Thus, the difference in luciferase counts obtained between S1 and S2 (e.g. S1-S2) corresponds to the total type I IFN activity in the sample. The difference in luciferase counts between sample S3 (e.g. IFNα+MAb-1B12) and S2 (S3-S2) corresponds to the amount of IFNα activity neutralized by MAb-1B12 in the sample.
To characterize the specificity of MAb-1B12, HL116 cells were incubated with each IFNα subtype (S1), IFNα + 10nM IFNAR1/IFNAR2-FChk (S2) and IFNα + 100nM MAb-1B12 (S3) and luciferase levels were measured. Total IFNα subtype activity (S1-S2) and neutralized IFNα16 (S3-S2) are plotted in Figure 4B. This experiment confirmed that MAb-1B12 specifically neutralizes IFNα16 bioactivity, since S1-S3 luciferase levels are essentially identical to total IFN levels (S1-S2), except for the sample containing the IFNα16 subtype. The S3-S2 measurement records no luciferase counts for the IFNα16 containing sample since all IFNα16 was neutralized in the experiment by MAb-1B12. The S3-S2 value provides a good method for evaluating the neutralization efficiency/specificity of MAb-1B12 for IFNα16. However, it is easier to interpret IFNα16 levels as a fraction of the total type I IFN activity neutralized in the S3-S2 sample, which is determined by plotting [(S1-S2)−(S3-S2)]/(S1-S2). Re-plotting the same data according to this equation clearly identifies the IFNα16 containing sample, but not samples with other IFNα subtypes, as containing ~100% IFNα16 (Fig. 4C).
To evaluate a sample containing multiple IFNαs, 11 IFNα subtypes, each at a final concentration of 1pM, were mixed with 12pM IFNα16 (Mixα16) or no IFNα16 (Mix). S1, S2, and S3 data were collected, using HL116 reporter cells, for Mix and Mixα16 samples at five different dilutions. As observed in the prior experiment with single IFNα subtypes, MAb-1B12 selectively neutralized IFNα16 activity in the Mixα16 samples diluted 4-fold, 8-fold and 16-fold (.e.g. S3-S2 data). At higher IFN concentrations (undiluted and 2-fold diluted) IFNα16 levels in Mixα16 and Mix samples were essentially identical and inaccurately suggested the samples do not contain IFNα16. This errant result is presumably due to competition of IFNα16 with the other 11 IFNα subtypes for receptor binding/activation. Thus, even though IFNα16 is “neutralized” by MAb-1B12 in these samples, luciferase production/IFNα activity is dominated by other IFNα subtypes. A second problem observed at high IFNα concentrations is saturation of the cellular dose response curve, where small changes in the levels of one IFNα are not efficiently transduced into a change in luciferase counts. In contrast to the results at high IFN concentration, the 4-fold, 8-fold and 16-fold diluted Mixα16 samples return consistent IFNα16 fraction levels of 21% (±2%), 26% (±6%), and 25% (±1%), respectively (Figure 4D). Measurement of the 4-fold, 8-fold, and 16-fold dilutions of the Mix samples, which do not contain IFNα16, returned IFNα16 fraction values of −1% (±4%), 3% (±3%) and 2% (±12%), respectively.
Discussion
This report describes protocols suitable for large scale expression and purification of 13 human IFNα subtypes in E. coli. The purification strategy takes advantage of the SPD affinity tag, which allows high affinity SPD-IFNα fusion protein capture and efficient tag removal during elution from the column. Thus, IFNα subtype purification by this method is rapid and avoids disrupted IFNα activity caused by six histidine tags [35] and/or the need for a secondary protease cleavage step in the purification process [21]. The affinity purified IFNαs were subjected to two additional purification steps, anion exchange chromatography and gel filtration, to ensure all SPD-IFNα fusion protein was removed from the preparations. Using this expression and purification strategy, we obtained between 3 and 20 milligrams of each IFNα subtype from 0.5 liter culture.
Almost all reports of IFNα expression in E. coli or yeast expression systems have focused on IFNα2 variants (.e.g. IFNα2a,b,c) or IFNα8 [20, 23, 34–36]. To our knowledge, this is the first report demonstrating the expression and purification of all 13 IFNα subtypes, which share between 75% and 95% amino acid sequence identity. For this reason, it was necessary to identify a rapid purification protocol that could be standardized for all 13 proteins. An added complexity of purifying all IFNα subtypes was to ensure that each protein preparation was not cross-contaminated with another IFNα subtype, which could lead to inaccurate interpretations of their biological activities. To overcome this problem, we used separate eXact affinity media and columns for each IFNα subtype. In addition, anion exchange and gel filtration columns used in the experiments were extensively cleaned and column effluent was tested for lack of IFN biological activity using the HL116 luciferase reporter cell line. To our knowledge, potential contamination issues have not been addressed in prior IFNα expression reports. However, IFNα contamination can be a problem, as was recently reported for an anti-IFNα antibody prepared using columns also used to purify IFNαs [37, 38].
The biological potency of IFNα subtypes have been evaluated previously on primary fibroblasts, endothelial cells and several cell lines using proteins from the same vendor [8, 39]. For most IFNs, our analysis of IFNα subtype potency agrees with these reports. In particular, IFNα1 is the least active IFNα subtype, while IFNα8, IFNα10 and IFNα14 are generally the most active. However, Moll et al. observed IFNα2b, IFNα6 and IFNα7 exhibited the highest potency in assays performed with primary endothelial cells [8]. This observation differs from our studies and assays performed in other cell lines [39] where these subtypes consistently exhibit middle to low activities. One possibility for these activity differences may be a fundamental difference in primary and immortalized cells, although the mechanism for such a difference has not been determined. A secondary explanation for these differences could be the assay format and measurement methods (RT-PCR vs. luciferase production) used to determine IFNα biological activity.
The production of purified IFNα subtypes is a first step in obtaining useful reagents to monitor IFNα subtype levels in human sera. Towards this goal, a murine monoclonal antibody, MAb-1B12, which specifically binds and neutralizes the biological activity of IFNα16 was developed. For MAb-1B12 selection and testing, it was essential to produce IFNα16, as well as the other 12 IFNα subtypes. IFNα16 shares the greatest sequence identity (87%) with IFNα4 and the lowest with IFNα1 (78%). Similar amino acid sequence identities are observed between IFNα1, IFNα2a/b, IFNα5, IFNα6, IFNα8, IFNα14 and IFNα21 suggesting subtype-specific neutralizing Abs may be made for these IFNαs. Consistent with this hypothesis, an IFNα1-specific neutralizing MAb has been generated as a potential therapeutic in lupus [40]. However, it may be more challenging to generate Abs that discriminate IFNα4, IFNα7, IFNα10 and IFNα17 subtypes, which share ~95% sequence identity with one another.
The IFNα subtypes, MAb-1B12 and IFNAR1/IFNAR2-FChk were used to develop an assay to determine total type I IFN biological activity and IFNα16-specific activity in an unknown biological sample. Such an assay may be applicable to monitoring serum type I IFN levels in a variety of clinical settings. Monitoring differences in total type I IFN and IFNα subtypes may provide unique indicators of disease status and/or provide insight on treatment response to novel reagents under development for autoimmune disease. The studies performed here demonstrate how IFNAR1/IFNAR2-FChk can be used to provide reproducible measurements of total type I IFN activity levels that exclude type III IFN activity that may be present in a sample. Measuring IFNα16 levels was more difficult since IFNα16 activity was highly dependent on the amounts of other IFNα subtypes present in the sample, which compete with IFNα16 for receptor binding and activation of the luciferase reporter gene. This problem was overcome, at least partially, by diluting the starting sample to limit competitive receptor binding from other IFNαs. Interestingly, by mass IFNα16 represented ~50% of the total IFNα in the test sample. However, the measured IFNα16 activity fraction was ~25%, which is presumed to be due to signaling background of the other 11 IFNαs in the mixture. Obtaining an measurement of IFNα16 activity amongst the other IFNαs will require the production of more subtype-specific Abs that selectively inhibit the other IFNαs in the sample to prevent receptor binding competition. Despite this current limitation, the assay can clearly distinguish samples that contain IFNα16 from those that do not. Ultimately, these studies may elucidate unique roles of different IFNα subtypes during normal immune function or their contribution to the initiation and/or maintenance of autoimmune disease.
Acknowledgments
This work was supported by a grant from Lupus Research Institute to M.R.W. The authors thank Dr. Robert A. Oster at UAB for assisting with statistical analysis. The purchase of the Applied Biosystems (now AB Sciex) Voyager DE-Pro MALDI-TOF Mass Spectrometer in the Targeted Metabolomics and Proteomics Laboratory came from funds provided by NIH Shared Instrumentation grant (S10 RR13795) plus UAB Health Services Foundation General Endowment Fund. The UAB Epitope Recognition Immunoreagent Core is partially supported by the UAB Rheumatic Disease Core Center (P30 ARO48311).
References
- 1.Gonzalez-Navajas JM, et al. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12(2):125–35. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):258–67. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 3.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 4.Stark GR. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev. 2007;18(5–6):419–23. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borden EC, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6(12):975–90. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baig E, Fish EN. Distinct signature type I interferon responses are determined by the infecting virus and the target cell. Antivir Ther. 2008;13(3):409–22. [PubMed] [Google Scholar]
- 7.Gibbert K, Dittmer U. Distinct antiviral activities of IFN-alpha subtypes. Immunotherapy. 2011;3(7):813–6. doi: 10.2217/imt.11.74. [DOI] [PubMed] [Google Scholar]
- 8.Moll HP, et al. The differential activity of interferon-alpha subtypes is consistent among distinct target genes and cell types. Cytokine. 2011;53(1):52–9. doi: 10.1016/j.cyto.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem. 2007;282(28):20047–51. doi: 10.1074/jbc.R700004200. [DOI] [PubMed] [Google Scholar]
- 10.Pestka S. Purification and cloning of interferon alpha. Curr Top Microbiol Immunol. 2007;316:23–37. doi: 10.1007/978-3-540-71329-6_3. [DOI] [PubMed] [Google Scholar]
- 11.Robertson C, et al. Methods and uses of antibodies in the purification of interferon. 2006. Google Patents. [Google Scholar]
- 12.Munoz-Espinosa LE, et al. Re-treatment with highly purified nIFNalpha in Mexican nonresponder patients with chronic genotype 1 hepatitis C. Arch Med Res. 2013;44(6):444–8. doi: 10.1016/j.arcmed.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Musch E, et al. Successful application of highly purified natural interferon alpha (multiferon) in a chronic hepatitis C patient resistant to preceding treatment approaches. Hepatogastroenterology. 2004;51(59):1476–9. [PubMed] [Google Scholar]
- 14.Niewold TB, et al. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8(6):492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knight JS, Kaplan MJ. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol. 2012;24(5):441–50. doi: 10.1097/BOR.0b013e3283546703. [DOI] [PubMed] [Google Scholar]
- 16.Ronnblom L, Alm GV, Eloranta ML. The type I interferon system in the development of lupus. Semin Immunol. 2011;23(2):113–21. doi: 10.1016/j.smim.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Crow MK. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther. 2010;12(Suppl 1):S5. doi: 10.1186/ar2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lefevre F, et al. Production, purification and biological properties of an Escherichia coli-derived recombinant porcine alpha interferon. J Gen Virol. 1990;71(Pt 5):1057–63. doi: 10.1099/0022-1317-71-5-1057. [DOI] [PubMed] [Google Scholar]
- 19.Kalie E, et al. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J Biol Chem. 2007;282(15):11602–11. doi: 10.1074/jbc.M610115200. [DOI] [PubMed] [Google Scholar]
- 20.Shi L, et al. Efficient expression and purification of human interferon alpha2b in the methylotrophic yeast, Pichia pastoris. Protein Expr Purif. 2007;54(2):220–6. doi: 10.1016/j.pep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Stifter SA, et al. Purification and biological characterization of soluble, recombinant mouse IFNbeta expressed in insect cells. Protein Expr Purif. 2014;94:7–14. doi: 10.1016/j.pep.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Srivastava P, et al. Overexpression and purification of recombinant human interferon alpha2b in Escherichia coli. Protein Expr Purif. 2005;41(2):313–22. doi: 10.1016/j.pep.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Rabhi-Essafi I, et al. A strategy for high-level expression of soluble and functional human interferon alpha as a GST-fusion protein in E. coli. Protein Eng Des Sel. 2007;20(5):201–9. doi: 10.1093/protein/gzm012. [DOI] [PubMed] [Google Scholar]
- 24.Ruan B, et al. Engineering subtilisin into a fluoride-triggered processing protease useful for one-step protein purification. Biochemistry. 2004;43(46):14539–46. doi: 10.1021/bi048177j. [DOI] [PubMed] [Google Scholar]
- 25.Deshpande A, et al. Kinetic analysis of cytokine-mediated receptor assembly using engineered FC heterodimers. Protein Sci. 2013 doi: 10.1002/pro.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hussain M, Gill DS, Liao MJ. Identification of interferon-alpha 7, -alpha 14, and -alpha 21 variants in the genome of a large human population. J Interferon Cytokine Res. 1996;16(10):853–9. doi: 10.1089/jir.1996.16.853. [DOI] [PubMed] [Google Scholar]
- 27.Hussain M, Gill DS, Liao MJ. Both variant forms of interferon-alpha4 gene (IFNA4a and IFNA4b) are present in the human population. J Interferon Cytokine Res. 1997;17(9):559–66. doi: 10.1089/jir.1997.17.559. [DOI] [PubMed] [Google Scholar]
- 28.Hussain M, et al. Interferon-alpha 8b is the only variant of interferon-alpha 8 identified in a large human population. J Interferon Cytokine Res. 1996;16(7):523–9. doi: 10.1089/jir.1996.16.523. [DOI] [PubMed] [Google Scholar]
- 29.Hussain M, et al. A new allele of interferon-alpha17 gene encoding IFN-alpha17b is the major variant in human population. J Interferon Cytokine Res. 1998;18(7):469–77. doi: 10.1089/jir.1998.18.469. [DOI] [PubMed] [Google Scholar]
- 30.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Uze G, et al. Domains of interaction between alpha interferon and its receptor components. J Mol Biol. 1994;243(2):245–57. doi: 10.1006/jmbi.1994.1651. [DOI] [PubMed] [Google Scholar]
- 32.Meister A, et al. Biological activities and receptor binding of two human recombinant interferons and their hybrids. J Gen Virol. 1986;67(Pt 8):1633–43. doi: 10.1099/0022-1317-67-8-1633. [DOI] [PubMed] [Google Scholar]
- 33.Meager A, et al. Establishment of new and replacement World Health Organization International Biological Standards for human interferon alpha and omega. J Immunol Methods. 2001;257(1–2):17–33. doi: 10.1016/s0022-1759(01)00460-4. [DOI] [PubMed] [Google Scholar]
- 34.Platis D, Foster GR. High yield expression, refolding, and characterization of recombinant interferon alpha2/alpha8 hybrids in Escherichia coli. Protein Expr Purif. 2003;31(2):222–30. doi: 10.1016/s1046-5928(03)00187-6. [DOI] [PubMed] [Google Scholar]
- 35.Schmeisser H, et al. Binding Characteristics of IFN-alpha Subvariants to IFNAR2-EC and Influence of the 6-Histidine Tag. J Interferon Cytokine Res. 2006;26(12):866–76. doi: 10.1089/jir.2006.26.866. [DOI] [PubMed] [Google Scholar]
- 36.Piehler J, Schreiber G. Biophysical analysis of the interaction of human ifnar2 expressed in E. coli with IFNalpha2. J Mol Biol. 1999;289(1):57–67. doi: 10.1006/jmbi.1999.2726. [DOI] [PubMed] [Google Scholar]
- 37.Moll HP, et al. Contamination with recombinant IFN accounts for the unexpected stimulatory properties of commonly used IFN-blocking antibodies. Eur J Immunol. 2011;41(1):252–4. doi: 10.1002/eji.200940145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moll HP, et al. Neutralizing type I IFN antibodies trigger an IFN-like response in endothelial cells. J Immunol. 2008;180(8):5250–6. doi: 10.4049/jimmunol.180.8.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavoie TB, et al. Binding and activity of all human alpha interferon subtypes. Cytokine. 2011;56(2):282–9. doi: 10.1016/j.cyto.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 40.Ouyang S, et al. Structural insights into a human anti-IFN antibody exerting therapeutic potential for systemic lupus erythematosus. J Mol Med (Berl) 2012;90(7):837–46. doi: 10.1007/s00109-012-0866-3. [DOI] [PubMed] [Google Scholar]