

Abstract
Renal cell carcinoma (RCC) accounts for approximately 3% of all new cancer cases. Although the classification of RCC is based mainly on histology, this method is not always accurate. We applied comparative genomic hybridization (CGH) to determine genomic alterations in 46 cases of different RCC histological subtypes [10 cases of clear cell RCC (CCRCC), 13 cases of papillary RCC (PRCC), 12 cases of chromophobe RCC (CRCC), 9 cases of Xp11.2 translocation RCC (Xp11.2RCC), 2 cases of undifferentiated RCC (unRCC)], and investigated the relationships between clinical parameters and genomic aberrations. Changes involving one or more regions of the genome were seen in all RCC patients; DNA sequence gains were most frequently (>30%) seen in chromosomes 7q, 16p, and 20q; losses from 1p, 3p, 13q, 14q, and 8p. We conclude CGH is a useful complementary method for differential diagnosis of RCC. Loss of 3p21-25, 15q, and gain of 16p11-13 are relatively particular to CCRCC vs. other types of RCC. Gain of 7p13-22, 8q21-24, and loss of 18q12-ter, 14q13-24, and Xp11-q13/Y are more apparent in PRCC, and gain of 8q21-24 is characteristic of type 2 PRCC vs. type 1 PRCC. Loss of 2q12-32, 10p12-15, and 11p11-15, 13p are characteristic of CRCC, and gain of 3p and loss of 11p11-15 and 13p are significant differentiators between common CRCC and CRCC accompanied by sarcomatous change groups. Gain of Xp11-12 is characteristic of the Xp11.2RCC group. Based on Multivariate Cox regression analysis, aberration in 5 chromosome regions were poor prognostic markers of RCC, and include the gain of chromosome 12p12-ter (P = 0.034, RR = 3.502, 95% CI 1.097-11.182), 12q14-ter (P = 0.002, RR = 5.115, 95% CI 1.847-14.170), 16q21-24 (P = 0.044, RR = 2.629, 95% CI 1.027-6.731), 17p12-ter (P = 0.017, RR = 3.643, 95% CI 1.262-10.512) and the loss of 18q12-23 (P = 0.049, RR = 2.911, 95% CI 1.006-8.425), which may provide clues of new genes involved in RCC tumorigenesis.
Keywords: Renal cell carcinoma, comparative genomic hybridization, chromosomal change
Introduction
Renal cell carcinoma (RCC) accounts for approximately 3% of all new cancer cases and incidence rates have been steadily grown over the last 3 decades [1-3]. About one-third of patients present with metastatic disease at initial diagnosis and surgical resection remain the only curative therapy for RCC; however, up to 50% of patients undergoing nephrectomy for clinically localized RCC will develop local recurrence or distant metastasis. Over all stages, nearly 50% of patients die within 5 years of diagnosis [4-6]. As in most cancers, clinical variables play a major role in prognosis of localized RCC, but accumulation of genetic aberrations is central to the initiation and prognosis of RCC and other cancers. Therefore, integration of genetic markers into traditional approaches may allow a more accurate prediction of prognosis [7,8].
RCC consists of a heterogeneous group of epithelial tumors with different histological features. There are 4 main subtypes of renal cell carcinoma associated with distinct clinical outcomes and classified according to their histopathology: clear cell RCC (CCRCC), papillary RCC (PRCC), chromophobe RCC (CRCC), and the recently recognized rare renal carcinoma associated with Xp11.2 translocation/TFE3 gene fusions (Xp11.2RCC). Although the classification of RCC is based mainly on histology, the morphology of RCC subtypes is sometimes similar, so the method is not always accurate. The WHO classification has introduced genetic alterations as a hallmark corresponding to the histologic subtypes of RCC. We applied comparative genomic hybridization (CGH) to identify the genomic alterations in 46 cases of different histological subtypes of renal cell carcinoma. We analyzed correlations between chromosome aberrations and clinicopathological variables, including tumor stage and nuclear grade, and validated the use of CGH for differential diagnosis of RCCs.
Materials and methods
Ethics statement
Ethical approval was obtained from Institutional Ethics Review Board (IERB), The First Affiliated Hospital, Shihezi University School of Medicine and all participants provided written informed consent for themselves.
Primary tumor
Forty-six methanol-fixed, paraffin-embedded primary renal cell carcinoma samples were retrieved from the archives of the Department of Pathology, Shihezi University School of Medicine of Xinjiang, China. There were 5 subtypes of RCC: 10 cases of CCRCC, 13 cases of PRCC (6 cases of type 1 PRCC, 7 cases of type 2 PRCC), 12 cases of chrRCC (8 cases of common CRCC, CRCC C; 4 cases of CRCC accompanied with sarcomatous change, CRCC S), 9 cases of Xp11.2 RCC, 2 cases of undifferentiated RCC (unRCC). All original slides including hematoxylin-eosin and immunohistochemical staining from each case were reviewed and assessed in accordance with current diagnostic criteria by senior pathologists [9]. TNM stage was determined in all cases using 2009 staging criteria [10,11] and followed up. Each paraffin block was reviewed to assure at least 70% tumor content before sectioning and DNA extraction.
DNA extraction
Total DNA was extracted from the samples using the standard phenol/chloroform extraction method, and peripheral blood cells were used as controls. DNA quality was checked on a 1% agarose gel and the amount of extracted DNA was measured spectrophotometrically at 260 nm (impurity and ratio of DNA to non-DNA were also cross-checked at 280 nm). Extractions were stored at -80°C prior to labeling by nick translation.
Comparative genomic hybridization
Comparative genomic hybridization was performed according to the manufacturer’s protocol (Vysis Inc., U.S). Briefly, labeling reactions were performed with 1 μg DNA and the nick translation labeling kit (Vysis) in a volume of 50 μl with 0.1 mmol/L dNTP pool containing 0.3 mmol/L each dATP, dGTP, and dCTP; 0.1 mmol/L dTTP; 0.2 mmol/L fluorescein isothiocyanate (FITC)-dUTP (for the experimental sample) or cyanine 3 (Cy3)-dUTP (for the 46, XY male reference); nick translation buffer and nick translation enzyme. Probe size was determined by separation on 1% agarose gel. Metaphase slides were denatured at 73 ± 1°C for 5 min in 70% methanamide/2× SSC and dehydrated in an ethanol series (70%, 85%, and 100%). The hybridization mixture consisted of approximately 200 ng Spectrum Red total genomic reference DNA coprecipitated with 10 µg human Co-1 DNA (Invitrogen, USA) and dissolved in hybridization buffer before hybridization to metaphase chromosomes. The probe mixtures were denatured at 73°C for 5 min then competitively hybridized to the denatured normal metaphase chromosomes in a humid chamber at 37°C for 3 days. After washing, chromosomes were counterstained with 4,6-diamidino-2-phenylindole-2 HCl (DAPI II; Vysis) and embedded in an antifading agent to reduce photobleaching.
Microscopy and digital image analysis
A fluorescence microscope equipped with appropriate filters (DAPI, FITC, and Cy3) was used to visualize the signals. For each hybridization panel, raw images from at least 5 metaphases were captured through a computer-driven CCD camera and analyzed with ISIS image software (Carl Zeiss Inc., Germany). Chromosomes were indentified by their DAPI banding patterns. Threshold levels of 1.25 and 0.8 were used to score gains and losses, respectively. High-level amplification was indicated by a ratio greater than 1.5. All centromeres, as well as chromosome p35-36 and the heterochromatic regions of chromosome 22, were excluded from further analysis, because these regions can yield unreliable hybridization data due to incompletely suppressed repetitive DNA sequences. Positive and negative controls provided comparisons for evaluation and interpretation of the data. Normal female DNA (labeled green) was used as negative control and normal male DNA was used for reference (labeled red). The intensity profiles for this experiment should be within the threshold values as determined by image analysis. DNA from MPE600 cells (with known genetic aberrations that are easy to detect by comparative genomic hybridization) was used as a positive control (labeled green) and normal male DNA was used as reference.
Statistical analysis
Pearson’s χ2 test or Fisher’s exact test were used to compare differences between different groups. Multivariate cox regression analysis was used to analyze the risk factors of RCC prognosis. All data were analyzed using SPSS 17.0 statistical software. A P value < 0.05 was considered statistically significant.
Results
Subject characteristics
A total of 46 RCC tumors were included in the analysis. Table 1 lists host and tumor characteristics. The mean age was 53.6 years. Men were overrepresented in the group (1.87:1). Using the 2009 TNM classification for renal cell carcinoma [10,11], 8 patients had stage I, 15 had stage II, 13 had stage III, and 10 had stage IV tumors (17.4%, 32.6%, 28.3%, and 21.7%).
Table 1.
Characteristics and follow-up data of 46 cases of RCC
| No | Type | Gender/age | pTNM | Stage | Follow up | Survival time (years) |
|---|---|---|---|---|---|---|
| 1 | CCRCC | F/31 | T2M0N0 | 2 | survive | 14 |
| 2 | CCRCC | F/25 | T1M0N0 | 1 | Death | 21 |
| 3 | CCRCC | M/52 | T2M0N0 | 2 | Death | 20 |
| 4 | CCRCC | M/39 | T2M0N0 | 2 | Survive | 18 |
| 5 | CCRCC | M/56 | T1M0N0 | 1 | Survive | 17 |
| 6 | CCRCC | M/55 | T3M0N0 | 3 | Death | 1 |
| 7 | CCRCC | M/39 | T3M0N0 | 3 | Survive | 10 |
| 8 | CCRCC | F/74 | T2M0N0 | 2 | Survive | 9 |
| 9 | CCRCC | M/70 | T2M0N0 | 2 | Survival | 8 |
| 10 | CCRCC | M/70 | T2M1NO | 4 | Death | 16 |
| 11 | Type 1 PRCC | M/62 | T1M0N0 | 1 | Survival | 18 |
| 12 | Type 1 PRCC | M/49 | T2M0N0 | 2 | Death | 13 |
| 13 | Type 1 PRCC | F/61 | T2M0N0 | 2 | Survival | 13 |
| 14 | Type 1 PRCC | M/36 | T1M0N0 | 1 | Survival | 10 |
| 15 | Type 1 PRCC | F/48 | T2M0N0 | 2 | Survival | 6 |
| 16 | Type 1 PRCC | M/52 | T2M0N0 | 2 | Survival | 3 |
| 17 | Type 2 PRCC | M/58 | T3M0N0 | 3 | Death | 29 |
| 18 | Type 2 PRCC | F/56 | T2M0N0 | 2 | Death | 29 |
| 19 | Type 2 PRCC | M/70 | T3M0N0 | 3 | Death | 2 |
| 20 | Type 2 PRCC | M/53 | T2M0N0 | 2 | Death | 5 |
| 21 | Type 2 PRCC | F/55 | T2M0N1 | 3 | Death | 7 |
| 22 | Type 2 PRCC | M/61 | T2M1N0 | 4 | Death | 4 |
| 23 | Type 2 PRCC | M/88 | T2M1N0 | 4 | Death | 4 |
| 24 | CRCC C | F/52 | T2M0N0 | 2 | Death | 34 |
| 25 | CRCC C | F/36 | T1M0N0 | 1 | Survival | 19 |
| 26 | CRCC C | M/42 | T1M0N0 | 1 | Survival | 16 |
| 27 | CRCC C | F/72 | T3M0N0 | 3 | Death | 15 |
| 28 | CRCC C | M/32 | T1M0N0 | 1 | Survival | 13 |
| 29 | CRCC C | F/74 | T2M0N0 | 2 | Survival | 9 |
| 30 | CRCC C | F/30 | T1M0N1 | 3 | Death | 5 |
| 31 | CRCC C | M/36 | T2M0N0 | 2 | Survival | 7 |
| 32 | CRCC S | M/25 | T3M0N1 | 3 | Death | 34 |
| 33 | CRCC S | F/32 | T4M0N0 | 4 | Death | 13 |
| 34 | CRCC S | M/75 | T2M1NO | 4 | Death | 11 |
| 35 | CRCC S | M/64 | T2M1N1 | 4 | Death | 10 |
| 36 | Xp11.2RCC | M/26 | T2M0N0 | 2 | Death | 37 |
| 37 | Xp11.2RCC | M/47 | T3MONO | 3 | Death | 36 |
| 38 | Xp11.2RCC | F/47 | T2M0N0 | 2 | Death | 26 |
| 39 | Xp11.2RCC | M/85 | T3MON0 | 3 | Survival | 9 |
| 40 | Xp11.2RCC | M/43 | T1M0N1 | 4 | Death | 8 |
| 41 | Xp11.2RCC | M/64 | T1M0N0 | 1 | Death | 8 |
| 42 | Xp11.2RCC | M/72 | T2M1NI | 4 | Death | 5 |
| 43 | Xp11.2RCC | F/63 | T2M1NO | 4 | Survival | 4 |
| 44 | Xp11.2RCC | F/72 | T2M0N1 | 4 | Survival | 5 |
| 45 | UnRCC | M/56 | T3MONO | 3 | Death | 2 |
| 46 | UnRCC | M/60 | T3M1NO | 3 | Death | 3 |
Comparative genomic hybridization profiles
Comparative genomic hybridization revealed DNA sequence gains and losses in all 46 primary renal cell carcinoma cases, with 260 gains and 282 losses. The mean numbers of aberrations per tumor sample were 5.7 gains and 6.1 losses. Gains were most frequently detected on chromosomes 7q, 16p, and 20q, common large regions of chromosome gains in RCC cases were most frequently detected on chromosomes 7q21-22, 7q31-ter, 12q14-ter, 16q21-24, and 20q12-ter. Frequently occurring losses involved 1p, 3p, 13q, 14q, and 8p, involving 1p31-ter, 3p20-ter, 4q, 6q, 8p, 9q12-33, 13q12-22, and 14q (Figure 1; Table 2).
Figure 1.
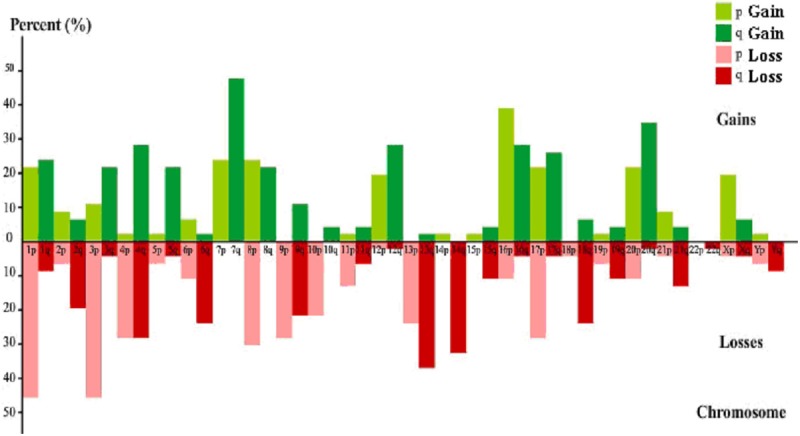
Frequency of chromosome gains and losses in renal cell carcinoma. Green histograms are gains, red are losses.
Table 2.
Common large region of chromosome aberrations in renal cell carcinoma cases (>10%)
| Chromosome number | Gain | Number (n=46) | RR | 95% CI | P | Loss | Number (n=46) | RR | 95% CI | P |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1p11-13 | 8 | 0.850 | 0.328-2.206 | 0.738 | 1p11-22 | 19 | 1.170 | 0.529-2.589 | 0.698 |
| 1p20-32 | 9 | 0.824 | 0.329-2.063 | 0.680 | 1p31-ter | 13 | 0.700 | 0.259-1.887 | 0.480 | |
| 3 | 3p11-ter | 7 | 0.556 | 0.191-1.678 | 0.304 | 3p11-14 | 7 | 2.681 | 0.888-8.100 | 0.080 |
| 3q13-29 | 8 | 1.019 | 0.366-2.841 | 0.971 | 3p20-ter | 15 | 0.720 | 0.245-2.121 | 0.552 | |
| 4 | 4q21-32 | 6 | 0.466 | 0.130-1.665 | 0.240 | 4p12-ter | 9 | 1.478 | 0.508-4.300 | 0.473 |
| 4q | 14 | 2.360 | 0.854-6.519 | 0.098 | ||||||
| 5 | 5q21-ter | 9 | 1.830 | 0.6.9-5.497 | 0.561 | |||||
| 6 | 6q | 13 | 1.389 | 0.469-4.111 | 0.553 | |||||
| 7 | 7p13-ter | 9 | 0.350 | 0.109-1.121 | 0.077 | |||||
| 7q11 | 7 | 1.757 | 0.605-5.103 | 0.300 | ||||||
| 7q21-22 | 18 | 1.950 | 0.743-5.118 | 0.175 | ||||||
| 7q31-ter | 16 | 1.680 | 0.517-5.457 | 0.388 | ||||||
| 8 | 8p12-ter | 9 | 1.575 | 0.546-4.544 | 0.401 | 8p- | 12 | 0.529 | 0.209-1.340 | 0.179 |
| 8q20-ter | 10 | 1.392 | 0.562-3.448 | 0.475 | ||||||
| 9 | 9q12-31 | 5 | 0.532 | 0.130-2.181 | 0.380 | 9p | 10 | 0.796 | 0.282-2.252 | 0.668 |
| 9q12-33 | 13 | 0.851 | 0.345-2.094 | 0.725 | ||||||
| 10 | 10p12-ter | 10 | 1.665 | 0.603-4.598 | 0.325 | |||||
| 12 | 12p11-ter | 7 | 3.502 | 1.097-11.182 | 0.034 | |||||
| 12q14-ter | 14 | 5.115 | 1.847-14.170 | 0.002 | ||||||
| 13 | 13p- | 12 | 1.137 | 0.418-3.097 | 0.801 | |||||
| 13q12-22 | 19 | 1.992 | 0.761-5.214 | 0.160 | ||||||
| 14 | 14q- | 14 | 1.442 | 0.576-3.612 | 0.434 | |||||
| 16 | 16p12-13 | 7 | 0.557 | 0.148-2.100 | 0.387 | |||||
| 16q21-24 | 11 | 2.629 | 1.027-6.731 | 0.044 | ||||||
| 17 | 17p12-ter | 6 | 3.643 | 1.262-10.512 | 0.017 | 17p12-ter | 11 | 1.221 | 0.378-3.942 | 0.739 |
| 17q12-ter | 10 | 1.330 | 0.464-3.810 | 0.595 | ||||||
| 18 | 18q12-23 | 11 | 2.911 | 1.006-8.425 | 0.049 | |||||
| 19 | 19q- | 6 | 1.842 | 0.540-6.282 | 0.329 | |||||
| 20 | 20p | 8 | 1.141 | 0.241-5.404 | 0.868 | |||||
| 20q12-ter | 17 | 1.044 | 0.446-2.444 | 0.920 |
Chromosomal changes in different renal cell carcinoma subtypes
Comparative genomic hybridization profiles showed that chromosomal changes varied among the 4 renal cell carcinoma subtypes (10 CCRCC, 13 PRCC, 12CRCC, 9 Xp11.2RCC). Representative analyses are shown in Figures 2 and 3. There are relative characteristic chromosomal changes in different subtypes of RCC (Table 3).
Figure 2.
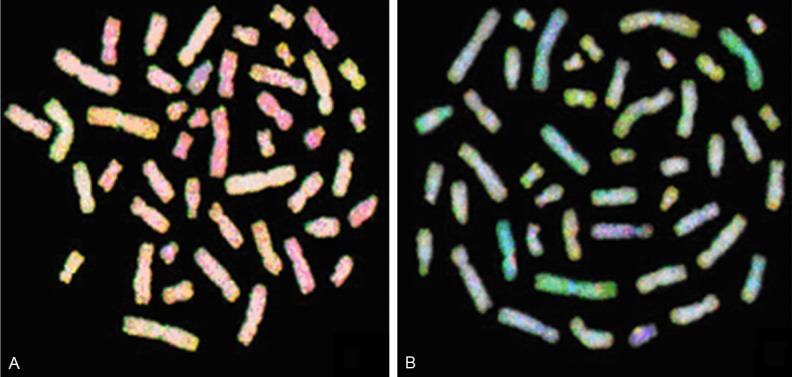
Comparative genomic hybridization metaphase spreads of RCC 20 (A) and RCC 25 (B). Green areas are gains, red areas are losses, yellow/yellowish areas are normal, and blue areas are heterochromatin. Hybridization to repetitive sequences/heterochromatin were blocked by unlabeled human Cot-1 DNA and stained blue with 4,6-diamidino-2 phenylindole-2 HCL (DAPI).
Figure 3.
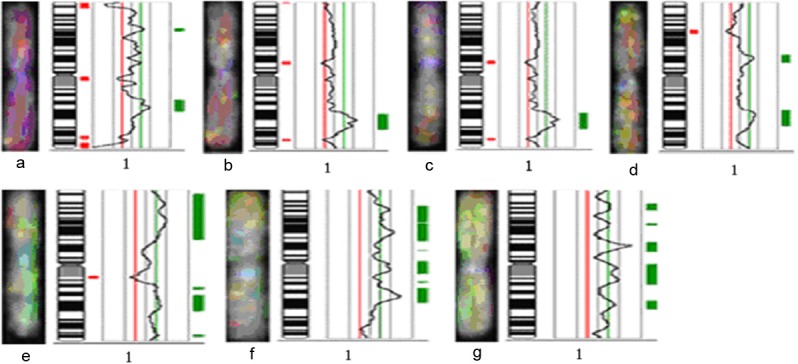
Comparative genomic hybridization profiles of chromosome 1. Green to red fluorescence thresholds (represented by the green/red line) are 0.8 and 1.25, respectively. The curve shows DNA copy status. Curves to the left of the red line indicate losses, curves to the right indicate gains. a, b, c, d, e, f, g represent RCC cases 3, 20, 32, 4, 24, 25, and 27, respectively.
Table 3.
Characteristic chromosomal change in different subtype of renal cell carcinoma
| N | Characteristic chromosomal change | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||
| -3p21-25 | P | -15q | P | +16p11-13 | P | ||||||
| CCRCC | 10 | 8 | 4 | 6 | |||||||
| *Non-CCRCC | 34 | 5 | 0.0003 | 1 | 0.0130 | 1 | 0.0001 | ||||
| **PRCC | 13 | 5 | 0.0903 | 1 | 0.1269 | 1 | 0.0186 | ||||
| **CRCC | 12 | 0 | 0.0001 | 0 | 0.0287 | 0 | 0.0281 | ||||
| **Xp11.2RCC | 9 | 0 | 0.0007 | 0 | 0.0867 | 0 | 0.0108 | ||||
| +7p13-22 | P | +8q21-24 | P | -18q12-ter | P | -14q13-24 | P | -Xp11-q13/-Y | P | ||
| PRCC | 13 | 8 | 7 | 8 | 8 | 7 | |||||
| 1 type PRCC | 6 | 4 | 1 | 5 | 4 | 4 | |||||
| **2 type PRCC | 7 | 4 | 6 | 0.0291 | 3 | 4 | 3 | ||||
| *Non-PRCC | 31 | 2 | 0.0003 | 4 | 0.0131 | 2 | 0.0003 | 4 | 0 | 0.0001 | |
| **CCRCC | 10 | 0 | 0.0059 | 0 | 0.0003 | 1 | 0.0288 | 0 | 0.0059 | 0 | 0.0075 |
| **CRCC | 12 | 0 | 0.0016 | 0 | 0.0001 | 0 | 0.0016 | 0 | 0.0016 | 0 | 0.0052 |
| **Xp11.2RCC | 9 | 2 | 0.0990 | 4 | 0.1312 | 1 | 0.0306 | 4 | 0.6656 | 0 | 0.0167 |
| -2q12-32 | P | ^3p+ | P | ^^-10p12-15 | P | -11p11-15 | P | ^^-13p11-ter | P | ||
| CRCC | 12 | 6 | 4 | 7 | 5 | 7 | |||||
| CRCC C | 8 | 4 | 0 | 7 | 3 | 7 | |||||
| **CRCC S | 4 | 2 | 4 | 0.0020 | 0 | 0.0101 | 2 | 0 | 0.0101 | ||
| *Non-CRCC | 32 | 2 | 0.0036 | 0 | 0.0046 | 3 | 0.0023 | 0 | 0.0008 | 4 | 0.0062 |
| **CCRCC | 10 | 1 | 0.0743 | 0 | 0.0010 | 0 | 0.0003 | 0 | 0.0396 | 0 | 0.0003 |
| **PRCC | 13 | 0 | 0.0052 | 0 | 0.0004 | 1 | 0.0005 | 0 | 0.0372 | 1 | 0.0005 |
| **Xp11.2RCC | 9 | 1 | 0.1588 | 0 | 0.0014 | 2 | 0.0152 | 0 | 0.0451 | 3 | 0.0497 |
| +Xp11-12 | P | +12q12-24 | P | -14q13-24 | P | ||||||
| Xp11.2RCC | 9 | 6 | 6 | 4 | |||||||
| *Non-Xp11.2RCC | 35 | 0 | 0.0000 | 6 | 0.1946 | 8 | 0.3803 | ||||
| **CCRCC | 10 | 0 | 0.0030 | 1 | 0.0572 | 0 | 0.0325 | ||||
| **PRCC | 13 | 0 | 0.0011 | 5 | 0.6656 | 8 | 1.0 | ||||
| **CRCC | 12 | 0 | 0.0015 | 0 | 0.0062 | 0 | 0.0210 | ||||
Pearson χ2 test;
Fisher exact test;
+: gain; -: lose;
P value from the comparison between CRCC S group and other type RCC;
P value from the comparison between common CRCC group and other type RCC.
In CCRCC, the most frequently occurring chromosomal gains and losses were 7q (9/10), 16p (8/10), 5q (6/10), and 3p (9/10), 8p (8/10), 1p (7/10), 4p, 4q, 9p (6/10), 9q, and 14q (3/10). The gain of 16p11-13 is more frequent in CCRCC than in other types of RCC (Non-CCRCC, P = 0.001, PRCC, P = 0.0186, CRCC, P = 0.0281, Xp11.2RCC, P = 0.0108); the loss of 3p21-25 is more frequent in CCRCC than in chrRCC (P = 0.0001) and Xp11.2RCC (P = 0.0007); the loss of 15q is more frequent in CCRCC than in CRCC (P = 0.0287).
In PRCC, gains were seen in chromosome arms 7p, 7q, 12q (8/13), 16q, 20p, 20q (7/13), 8q, 16p, 17q (6/13), 12p, 17p (5/13), and 8p (4/13), and losses occurred frequently on chromosome 14q, 18q (8/13), 13q (7/13), 3p, 4p, 6q (6/13), 1p, 4q, 9p (5/13), Yq (4/13), and Xp (3/13). The gain of 8q21-24 was more apparent in type 2 PRCC than in type 1 PRCC (P = 0.0291), CCRCC (P = 0.0003), and CRCC (P = 0.0001); the loss of 18q12-ter and Xp11-q13/Y is more frequent than in other types of RCC (-18q12-ter: Non-PRCC, P = 0.003, CCRCC, P = 0.0288, CRCC, P = 0.0016, Xp11.2RCC, P = 0.0306, -Xp11-q13/Y: Non-PRCC, P = 0.001, CCRCC, P = 0.0075, CRCC, P = 0.0052, Xp11.2RCC, P = 0.0167), the gain of 7q13-22 and loss of 14q13-24 is relative more apparent in PRCC contrasted with CCRCC (+7q13-22: P = 0.0059, -14q13-24, P = 0.0059), and CRCC (+7q13-22: P = 0.0016, -14q13-24, P = 0.0016).
For CRCC, gains were seen in chromosome arms 1q (7/12), 3q (6/12), 1p (5/12), and 3p, 4q, 9q, 16p (4/12), and losses occurred frequently on chromosome 1p, 17p (8/12), 10p, 13p (7/12), 2q, 8p (6/12), 11p, 21q (5/12), and 6q, 13q (4/12). The gain of 3p, loss of 11p11-15, and 13p11-ter significantly differ between common CRCC and groups associated with sarcomatous change (P = 0.0020, P = 0.0101, P = 0.0101). The gain of 3p is more frequent in CRCC accompanied by sarcomatous change than in other types of RCC (CCRCC, P = 0.0010, PRCC, P = 0.0004, Xp11.2RCC, P = 0.0014), loss of 10p12-15, and 13p11-ter is more frequent in common CRCC than in other types of RCC (-10p12-15: CCRCC, P = 0.0003, PRCC, P = 0.0005, Xp11.2RCC, P = 0.0152; 13p11-ter: CCRCC, P = 0.0003, PRCC, P = 0.0005, Xp11.2RCC, P = 0.0497). The loss of 11p11-15 is more frequent in CRCC than in other types of RCC (CCRCC, P = 0.0396, PRCC, P = 0.0372, Xp11.2RCC, P = 0.0152), and the frequency of loss of 2q12-32 significantly differs between CRCC and PRCC (P = 0.0052).
In Xp11.2RCC, gains were seen in chromosome arms Xp (6/9), 7q, 12q (5/9), 8p, 8q, 16q, 17p, 17q, 20q (4/9), and 5q, 7p, 12p (3/9), and losses occurred frequently on chromosome 3p, 9q, 14q (4/9), and 16p (3/9). Gain of Xp11-12 is more frequent in Xp11.2RCC than in other types of RCC (CCRCC, P = 0.0030, PRCC, P = 0.0011, CRCC, P = 0.0015); loss of 14q13-24 is more frequent than in CCRCC (P = 0.0325) and CRCC (P = 0.0210); gain of 12q12-24 differs between Xp11.2RCC and CRCC (P = 0.0062).
Comparison of chromosomal changes with clinicopathological parameters
Follow-up data revealed the prognosis of RCC is associated with clinicopathologic stage (P = 0.004) and patient age (P = 0.002). The mortality risk of stage II RCC is 1.684 (0.341-8.311) vs. stage I RCC but the difference is not significant (P = 0.523). In comparison to stage I RCC, the mortality risk of stage III and IV RCC are 5.119 (95% CI: 1.052-25.679; P = 0.043), 11.187 (2.173-57.597; P = 0.004), respectively. Advanced age is associated with increased mortality risk. After controlling for clinicopathologic stage, age and gender, the multivariate Cox regression analysis showed the gain of chromosome 12p11-ter (P = 0.034, RR = 3.502, 95% CI 1.097-11.182), 12q14-ter (P = 0.002, RR = 5.115, 95% CI 1.847-14.170), 16q21-24 (P = 0.044, RR = 2.629, 95% CI 1.027-6.731), 17p12-ter (P = 0.017, RR = 3.643, 95% CI 1.262-10.512) and the loss of 18q12-23 (P = 0.049, RR = 2.911, 95% CI 1.006-8.425) is correlated with prognosis of RCC (Table 2). The type and stage distribution of the cases harboring 2p11-ter, 12q14-ter, 16q21-24, 17p12-ter, 18q12-23 aberrations were show in Figures 4 and 5, which correlated with poorer prognosis. The regions were more common in Xp11.2RCC, 2 type PRCC and CRCC associated with sarcomatous change vs. other subtype of RCC, and more frequent in stage III-IV than stage I-II.
Figure 4.
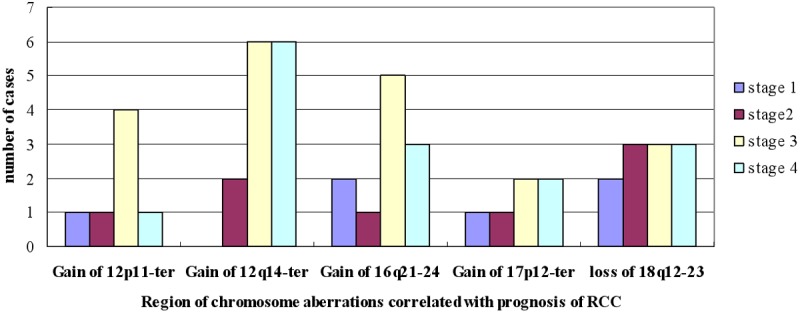
Stage distribution of the cases harboring 2p11-ter, 12q14-ter, 16q21-24, 17p12-ter, 18q12-23 aberrations.
Figure 5.
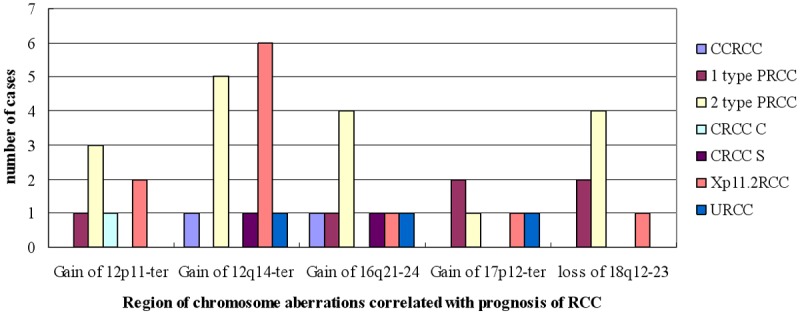
Type distribution of the cases harboring 2p11-ter, 12q14-ter, 16q21-24, 17p12-ter, 18q12-23 aberrations.
Discussion
Renal cell carcinoma is a group of malignancies arising from the epithelium of the renal tubules where histological classification of tumor subtypes is sometimes equivocal. Comparative genomic hybridization is a convenient and rapid way to screen for chromosomal changes. There have been some studies of renal cell carcinoma by CGH, but most focus on CCRCC and PRCC [12-16]. This study is the first attempt to use this method in differential diagnosis of 4 main subtypes RCC, and advances our understanding of the molecular basis of renal cell carcinoma, which may provide clues to new genes involved RCC tumorigenesis.
In this preliminary study, we performed genome-wide screening to detect genetic changes associated with clinical parameters in primary renal cell carcinoma. We detected DNA gains and losses in all 46 cases investigated; losses were more common than gains. Losses (in order of frequency) were detected at chromosomes 1, 3, 13, 14, and 8. For CCRCC, there were consistent losses of whole or partial arms of several chromosomes, notably chromosomes 3, 8, 1, 4, 9, and 14. The chromosomes with consistent losses in PRCC were 14, 18, 13, 3, 4, 6, 1, 9, and Y. In CRCC, chromosomes 1, 17, 10, 13, 2, 8, 11, 21, and 6 consistently exhibited losses. Chromosomes 3, 9, 14, and 16 sustained consistent losses in Xp11.2RCC. For unRCC, the chromosomes sustaining chromosomal losses were 1, 2, 3, 5, 6, 11, 16, 17, and 20. Gains were infrequent in the 5 subtypes. These results are consistent with previous reports [15,17-19].
Our study shows the CGH assay is a useful complementary method for differential diagnosis of the 4 main subtypes of RCC. The comparisons of chromosome aberrations in CCRCC, PRCC, CRCC, and Xp11.2 RCC revealed characteristic differences. These results are mainly consistent with previous reports [14,15,20-22], but we also identified new regions that are helpful in differential diagnosis of RCC. For CCRCC, in addition to loss of 3p21-25, the loss of 15q and gain of 16p11-13 are also relatively frequent in comparison to other subtypes of RCC. In PRCC, gain of 7p13-22 and 8q21-24 and loss of 18q12-ter, 14q13-24, and Xp11-q13/Y are more frequent than in other types of RCC, and gain of 8q21-24 is more characteristic of type 2 than type 1 PRCC. For CRCC, the loss of 2q12-32, 10p12-15, 11p11-15, and 13p is helpful in differential diagnosis with other types of RCC, and the gain of 3p and loss of 11p11-15 and 13p significantly differ between common CRCC and CRCC accompanied by sarcomatous change. Gain of Xp11-12 is more frequent in Xp11.2RCC than in other types of RCC.
Based on Cox regression analysis, 5 chromosome region aberrations were poor prognostic indicators in RCC, including the gain of chromosome 12p12-ter, 12q14-ter, 16q21-24, and 17p12-ter and loss of 18q12-23, which may provide clues to new genes involved in RCC tumorigenesis.
The first region 12p11-ter was associated with RCC in Cox analysis (P = 0.034, RR = 3.502, 95% CI 1.097-11.182). This region, which contains the P27 gene, occurs frequently and is a strong predictor of poor survival in RCC [23-26]. Recent research has shown that p27 is phosphorylated at T157 of the NLS, causing inhibition of phosphatidylinositol 3-kinase (PI3K), reducing AKT activity and T157 phosphorylation and inducing nuclear relocalization of p27. Clinical testing of these findings may provide a rational method for use of PI3K/AKT pathway inhibitors in patients with RCC [25].
Our finding on 12q14-ter is notable because the small locus on 12q24.31 for rs4765623, which maps to SR-B1, the scavenger receptor class B, member 1 gene, has recently been implicated in RCC [27]. The SCARB1 gene encodes a cell-surface receptor that binds to high-density lipoprotein cholesterol (HDL-C) and mediates HDL-C uptake. Polymorphisms in the SR-BI gene (SCARB1) are associated with variations in plasma lipoprotein profile and other risk factors for cardiovascular disease [28-31]. Its role in cancer biology is not well established, as the proportion of all cases of RCC attributable to excess weight and obesity has been estimated to be about 40% in the United States and up to about 30% in European countries [5,32,33]. The mechanisms by which obesity influences renal carcinogenesis are not clear. Further investigation is required to study its association with RCC risk.
The third region 16q21-24 may harbor a tumor suppressor gene that controls cell proliferation and loss of function leads to a growth advantage and transformation of low-grade to high-grade tumors. The identity of this gene or genes remains unknown. One interesting note is that E-cadherin is located on 16q21-24, which modulates cell adhesion and cell polarity. The repression of E-cadherin in renal tubular cells may participate in the events of epithelial to mesenchymal transition (EMT) that plays an important role in progressive kidney disease [34], and the E-cadherin repressor Snail is associated with cancer invasion and prognosis [35].
The genes on 17p12-ter remain unclear, but one interesting gene is TP53, a well-known tumor suppressor gene. There is some data on the relationship between TP53 and RCC, although the results are controversial. Some researchers believe RCC patients with tumors expressing increased p53 and MDM2 may have the poorest overall survival [36]. TP53, Profilin 1 may be a key element in the pathological processes of RCC; it has the potential to serve as a diagnostic or progression biomarker and therapeutic target in RCC [37]. Other results suggest p53 expression may not play a role in prognosis of RCC [38].
The loss of 18q12-23 is significant because the small locus on 18q21.3 has been implicated in RCC [39], and recent research suggests inactivation of genes at 18q12-23, including SMAD 2/4/7, Smad-ubiquitination regulatory factor 2 (Smurf2), TGFBI, TCF-4, receptor activator of NF-kappaB ligand (RANKL) gene, may be involved in the tumorigenesis of RCC. For example, immunoreactivity to nuclear phosphorylated Smad2 was significantly lower in RCC than in normal renal tissues [40]. The level of Smurf2 was greater in RCC tissues of patients with advanced clinical stages vs. normal tissues [40]. TGFBI can promote metastasis of RCC cells depending on inactivation of the VHL tumor suppressor; TGFBI could be a therapeutic target against RCC in the future [41]. The imbalance between TCF-4 gene splicing isoforms with long and short reading frames is associated with RCC progression through inhibition of the apoptotic pathway [42]. RANKL and its receptor, receptor activator of NF-kappaB (RANK) was observed in metastatic RCC in the bone and other organs, suggesting they play a role in metastasis to the bone and other organs. Multivariate Cox analysis revealed that the RANKL-RANK-OPG system is involved not only in bone metastasis of RCCs but also in metastasis to other organs through the stimulation of cancer cell migration [43].
In summary, comparative genomic hybridization analysis revealed novel genomic imbalances in primary renal cell carcinoma. The results of this study suggest comparative genomic hybridization is a useful complementary method for differential diagnosis of RCC and for detecting alterations in large, critical chromosomal regions in renal cell carcinoma. Five chromosome regions with aberrations achieved bad prognosis significance of RCC, including the gain of chromosome 12p11-ter, 12q14-ter, 16q21-24, and 17p12-ter and the loss of 18q12-23, which may provide clues to new genes involved in RCC tumorigenesis. Further analysis to map genes to specific regions is underway to determine the contributions of these genes to the development of renal cell carcinoma.
Acknowledgements
Supported by grants from the National Natural Science Foundation of China (NSFC, No. 81060209) and from the International S&T Cooperation Program of China (2010DFB34100).
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA. 1999;281:1628–1631. doi: 10.1001/jama.281.17.1628. [DOI] [PubMed] [Google Scholar]
- 3.Hock LM, Lynch J, Balaji KC. Increasing incidence of all stages of kidney cancer in the last 2 decades in the United States: an analysis of surveillance, epidemiology and end results program data. J Urol. 2002;167:57–60. [PubMed] [Google Scholar]
- 4.Lam JS, Klatte T, Kim HL, Patard JJ, Breda A, Zisman A, Pantuck AJ, Figlin RA. Prognostic factors and selection for clinical studies of patients with kidney cancer. Crit Rev Oncol Hematol. 2008;65:235–262. doi: 10.1016/j.critrevonc.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Cho E, Adami HO, Lindblad P. Epidemiology of renal cell cancer. Hematol Oncol clin North Am. 2011;25:651–665. doi: 10.1016/j.hoc.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Ljungberg B, Campbell SC, Choi HY, Jacqmin D, Lee JE, Weikert S, Kiemeney LA. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615–621. doi: 10.1016/j.eururo.2011.06.049. [DOI] [PubMed] [Google Scholar]
- 7.Tang PA, Vickers MM, Heng DY. Clinical and molecular prognostic factors in renal cell carcinoma: what we know so far. Hematol Oncol clin North Am. 2011;25:871–891. doi: 10.1016/j.hoc.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Sun M, Shariat SF, Cheng C, Ficarra V, Murai M, Oudard S, Pantuck AJ, Zigeuner R, Karakiewicz PI. Prognostic factors and predictive models in renal cell carcinoma: a contemporary review. Eur Urol. 2011;60:644–661. doi: 10.1016/j.eururo.2011.06.041. [DOI] [PubMed] [Google Scholar]
- 9.Zou H, Pang LJ, Hu WH, Li F, Li HA, Jiang JF, Liang WH, Sun ZZ, Wang C, Lang JY. [Study on clinicopathologic features and immunophenotype of 114 cases of renal cell carcinoma] . Zhonghua Bing Li Xue Za Zhi. 2008;37:726–731. [PubMed] [Google Scholar]
- 10.Eggener S. TNM Staging for Renal Cell Carcinoma: time for a new method. Eur Urol. 2010;58:517–519. doi: 10.1016/j.eururo.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Erratum: Validation of the 2009 TNM Classification for Renal Cell Carcinoma: Comparison with the 2002 TNM Classification by Concordance Index. Korean J Urol. 2011;52:654. doi: 10.4111/kju.2011.52.8.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuura K, Nakada C, Mashio M, Narimatsu T, Yoshimoto T, Tanigawa M, Tsukamoto Y, Hijiya N, Takeuchi I, Nomura T, Sato F, Mimata H, Seto M, Moriyama M. Downregulation of SAV1 plays a role in pathogenesis of high-grade clear cell renal cell carcinoma. BMC Cancer. 2011;11:523. doi: 10.1186/1471-2407-11-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vieira J, Henrique R, Ribeiro FR, Barros-Silva JD, Peixoto A, Santos C, Pinheiro M, Costa VL, Soares MJ, Oliveira J, Jeronimo C, Teixeira MR. Feasibility of differential diagnosis of kidney tumors by comparative genomic hybridization of fine needle aspiration biopsies. Genes Chromosomes Cancer. 2010;49:935–947. doi: 10.1002/gcc.20805. [DOI] [PubMed] [Google Scholar]
- 14.Szponar A, Zubakov D, Pawlak J, Jauch A, Kovacs G. Three genetic developmental stages of papillary renal cell tumors: duplication of chromosome 1q marks fatal progression. Int J Cancer. 2009;124:2071–2076. doi: 10.1002/ijc.24180. [DOI] [PubMed] [Google Scholar]
- 15.Hagenkord JM, Gatalica Z, Jonasch E, Monzon FA. Clinical genomics of renal epithelial tumors. Cancer Genet. 2011;204:285–297. doi: 10.1016/j.cancergen.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Monzon FA, Hagenkord JM, Lyons-Weiler MA, Balani JP, Parwani AV, Sciulli CM, Li J, Chandran UR, Bastacky SI, Dhir R. Whole genome SNP arrays as a potential diagnostic tool for the detection of characteristic chromosomal aberrations in renal epithelial tumors. Mod Pathol. 2008;21:599–608. doi: 10.1038/modpathol.2008.20. [DOI] [PubMed] [Google Scholar]
- 17.Wilhelm M, Veltman JA, Olshen AB, Jain AN, Moore DH, Presti JC Jr, Kovacs G, Waldman FM. Array-based comparative genomic hybridization for the differential diagnosis of renal cell cancer. Cancer Res. 2002;62:957–960. [PubMed] [Google Scholar]
- 18.Kallio JP, Mahlamaki EH, Helin H, Karhu R, Kellokumpu-Lehtinen P, Tammela TL. Chromosomal gains and losses detected by comparative genomic hybridization and proliferation activity in renal cell carcinoma. Scand J Urol Nephrol. 2004;38:225–230. doi: 10.1080/00365590310025399. [DOI] [PubMed] [Google Scholar]
- 19.Yoshimoto T, Matsuura K, Karnan S, Tagawa H, Nakada C, Tanigawa M, Tsukamoto Y, Uchida T, Kashima K, Akizuki S, Takeuchi I, Sato F, Mimata H, Seto M, Moriyama M. High-resolution analysis of DNA copy number alterations and gene expression in renal clear cell carcinoma. J Pathol. 2007;213:392–401. doi: 10.1002/path.2239. [DOI] [PubMed] [Google Scholar]
- 20.Algaba F, Akaza H, Lopez-Beltran A, Martignoni G, Moch H, Montironi R, Reuter V. Current pathology keys of renal cell carcinoma. Eur Urol. 2011;60:634–643. doi: 10.1016/j.eururo.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 21.Kuroda N, Toi M, Hiroi M, Enzan H. Review of papillary renal cell carcinoma with focus on clinical and pathobiological aspects. Histol Histopathol. 2003;18:487–494. doi: 10.14670/HH-18.487. [DOI] [PubMed] [Google Scholar]
- 22.Chen M, Ye Y, Yang H, Tamboli P, Matin S, Tannir NM, Wood CG, Gu J, Wu X. Genome-wide profiling of chromosomal alterations in renal cell carcinoma using high-density single nucleotide polymorphism arrays. Int J Cancer. 2009;125:2342–2348. doi: 10.1002/ijc.24642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruck S, Merseburger AS, Hennenlotter J, Scharpf M, Eyrich C, Amend B, Sievert KD, Stenzl A, Bedke J. High cytoplasmic expression of p27(Kip1) is associated with a worse cancer-specific survival in clear cell renal cell carcinoma. BJU Int. 2012;109:1565–70. doi: 10.1111/j.1464-410X.2011.10649.x. [DOI] [PubMed] [Google Scholar]
- 24.Sgambato A, Camerini A, Genovese G, De Luca F, Viacava P, Migaldi M, Boninsegna A, Cecchi M, Sepich CA, Rossi G, Arena V, Cittadini A, Amoroso D. Loss of nuclear p27(kip1) and alpha- dystroglycan is a frequent event and is a strong predictor of poor outcome in renal cell carcinoma. Cancer Sci. 2010;101:2080–2086. doi: 10.1111/j.1349-7006.2010.01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Jonasch E, Alexander A, Short JD, Cai S, Wen S, Tsavachidou D, Tamboli P, Czerniak BA, Do KA, Wu KJ, Marlow LA, Wood CG, Copland JA, Walker CL. Cytoplasmic sequestration of p27 via AKT phosphorylation in renal cell carcinoma. Clin Cancer Res. 2009;15:81–90. doi: 10.1158/1078-0432.CCR-08-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Fu Q, Lv J, Wang F, Ding K. Prognostic implication of p27Kip1, Skp2 and Cks1 expression in renal cell carcinoma: a tissue microarray study. J Exp Clin Cancer Res. 2008;27:51. doi: 10.1186/1756-9966-27-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purdue MP, Johansson M, Zelenika D, Toro JR, Scelo G, Moore LE, Prokhortchouk E, Wu X, Kiemeney LA, Gaborieau V, Jacobs KB, Chow WH, Zaridze D, Matveev V, Lubinski J, Trubicka J, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Bucur A, Bencko V, Foretova L, Janout V, Boffetta P, Colt JS, Davis FG, Schwartz KL, Banks RE, Selby PJ, Harnden P, Berg CD, Hsing AW, Grubb RL 3rd, Boeing H, Vineis P, Clavel-Chapelon F, Palli D, Tumino R, Krogh V, Panico S, Duell EJ, Quiros JR, Sanchez MJ, Navarro C, Ardanaz E, Dorronsoro M, Khaw KT, Allen NE, Bueno-de-Mesquita HB, Peeters PH, Trichopoulos D, Linseisen J, Ljungberg B, Overvad K, Tjonneland A, Romieu I, Riboli E, Mukeria A, Shangina O, Stevens VL, Thun MJ, Diver WR, Gapstur SM, Pharoah PD, Easton DF, Albanes D, Weinstein SJ, Virtamo J, Vatten L, Hveem K, Njolstad I, Tell GS, Stoltenberg C, Kumar R, Koppova K, Cussenot O, Benhamou S, Oosterwijk E, Vermeulen SH, Aben KK, van der Marel SL, Ye Y, Wood CG, Pu X, Mazur AM, Boulygina ES, Chekanov NN, Foglio M, Lechner D, Gut I, Heath S, Blanche H, Hutchinson A, Thomas G, Wang Z, Yeager M, Fraumeni JF Jr, Skryabin KG, McKay JD, Rothman N, Chanock SJ, Lathrop M, Brennan P. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 2011;43:60–65. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundvold H, Helgeland H, Baranski M, Omholt SW, Vage DI. Characterisation of a novel paralog of scavenger receptor class B member I (SCARB1) in Atlantic salmon (Salmo salar) BMC Genet. 2011;12:52. doi: 10.1186/1471-2156-12-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerda A, Genvigir FD, Rodrigues AC, Willrich MA, Dorea EL, Bernik MM, Arazi SS, Oliveira R, Hirata MH, Hirata RD. Influence of polymorphisms and cholesterol-lowering treatment on SCARB1 mRNA expression. J Atheroscler Thromb. 2011;18:640–651. doi: 10.5551/jat.6544. [DOI] [PubMed] [Google Scholar]
- 30.Cerda A, Genvigir FD, Arazi SS, Hirata MH, Dorea EL, Bernik MM, Bertolami MC, Faludi AA, Hirata RD. Influence of SCARB1 polymorphisms on serum lipids of hypercholesterolemic individuals treated with atorvastatin. Clin Chim Acta. 2010;411:631–637. doi: 10.1016/j.cca.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Ordovas JM, Gao G, Province M, Straka RJ, Tsai MY, Lai CQ, Zhang K, Borecki I, Hixson JE, Allison DB, Arnett DK. The SCARB1 gene is associated with lipid response to dietary and pharmacological interventions. J Hum Genet. 2008;53:709–717. doi: 10.1007/s10038-008-0302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nature reviews. Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 33.Bergstrom A, Hsieh CC, Lindblad P, Lu CM, Cook NR, Wolk A. Obesity and renal cell cancer--a quantitative review. Br J Cancer. 2001;85:984–990. doi: 10.1054/bjoc.2001.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burns WC, Thomas MC. The molecular mediators of type 2 epithelial to mesenchymal transition (EMT) and their role in renal pathophysiology. Expert Rev Mol Med. 2010;12:e17. doi: 10.1017/S1462399410001481. [DOI] [PubMed] [Google Scholar]
- 35.Mikami S, Katsube K, Oya M, Ishida M, Kosaka T, Mizuno R, Mukai M, Okada Y. Expression of Snail and Slug in renal cell carcinoma: E-cadherin repressor Snail is associated with cancer invasion and prognosis. Lab Invest. 2011;91:1443–1458. doi: 10.1038/labinvest.2011.111. [DOI] [PubMed] [Google Scholar]
- 36.Noon AP, Vlatkovic N, Polanski R, Maguire M, Shawki H, Parsons K, Boyd MT. p53 and MDM2 in renal cell carcinoma biomarkers for disease progression and future therapeutic targets? Cancer. 2010;116:780–790. doi: 10.1002/cncr.24841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minamida S, Iwamura M, Kodera Y, Kawashima Y, Ikeda M, Okusa H, Fujita T, Maeda T, Baba S. Profilin 1 overexpression in renal cell carcinoma. Int J Urol. 2011;18:63–71. doi: 10.1111/j.1442-2042.2010.02670.x. [DOI] [PubMed] [Google Scholar]
- 38.Baytekin F, Tuna B, Mungan U, Aslan G, Yorukoglu K. Significance of P-glycoprotein, p53, and survivin expression in renal cell carcinoma. Urol Oncol. 2011;29:502–507. doi: 10.1016/j.urolonc.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Hirata H, Matsuyama H, Matsumoto H, Korenaga Y, Ohmi C, Sakano S, Yoshihiro S, Naito K. Deletion mapping of 18q in conventional renal cell carcinoma. Cancer Genet Cytogenet. 2005;163:101–105. doi: 10.1016/j.cancergencyto.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Fukasawa H, Yamamoto T, Fujigaki Y, Misaki T, Ohashi N, Takayama T, Suzuki S, Mugiya S, Oda T, Uchida C, Kitagawa K, Hattori T, Hayashi H, Ozono S, Kitagawa M, Hishida A. Reduction of transforming growth factor-beta type II receptor is caused by the enhanced ubiquitin-dependent degradation in human renal cell carcinoma. Int J Cancer. 2010;127:1517–1525. doi: 10.1002/ijc.25164. [DOI] [PubMed] [Google Scholar]
- 41.Shang D, Liu Y, Yang P, Chen Y, Tian Y. TGFBI-promoted adhesion, migration and invasion of human renal cell carcinoma depends on inactivation of von Hippel-Lindau tumor suppressor. Urology. 2012;79:966, e1–7. doi: 10.1016/j.urology.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Shiina H, Igawa M, Breault J, Ribeiro-Filho L, Pookot D, Urakami S, Terashima M, Deguchi M, Yamanaka M, Shirai M, Kaneuchi M, Kane CJ, Dahiya R. The human T-cell factor-4 gene splicing isoforms, Wnt signal pathway, and apoptosis in renal cell carcinoma. Clin Cancer Res. 2003;9:2121–2132. [PubMed] [Google Scholar]
- 43.Mikami S, Katsube K, Oya M, Ishida M, Kosaka T, Mizuno R, Mochizuki S, Ikeda T, Mukai M, Okada Y. Increased RANKL expression is related to tumour migration and metastasis of renal cell carcinomas. J Pathol. 2009;218:530–539. doi: 10.1002/path.2567. [DOI] [PubMed] [Google Scholar]