

Abstract
Severe aplastic anemia (SAA) is a bone marrow failure disease induced by hyperfunctional autoimmunic Th1 lymphocytes. Memory T cells (TM) are a component of the adaptive immune system. They ensure the host of more aggressive and faster immune response to efficiently eliminate the specific antigens after re-exposure and thus play a key role in T-cell functions. In this study we investigate the quantities and functions of memory T cells in SAA patients before and after immunosuppressive therapy (IST) to further clarify the mechanism of SAA apoptosis of bone marrow hematopoietic cells. Results showed that the percentage of CD4+ effector T cells in peripheral blood and bone marrow lymphocytes was decreased in SAA patients. The ratio of CD4+ memory T lymphocytes to CD8+ memory T subsets (CD4+/CD8+TM) in SAA patients was also lower. The percentage of CD8+ effector T cells in peripheral blood and CD8+ central memory T cells in the bone marrow lymphocytes was significantly higher in newly diagnosed patients. Furthermore, the median expressions of perforin and granzyme B on memory T cells were higher in SAA patients compared to those in normal controls. After IST, the quantities and functions of memory T cells return to normal level. Therefore, we concluded that the abnormal immunomodulatory ability on memory T cells may contribute to the imbalance of Th1/Th2 subsets and thus lead to over-function of T lymphocytes and hematopoiesis failure in SAA.
Keywords: Severe aplastic anemia, memory T cells, perforin, ganzyme B
Introduction
Severe aplastic anemia (SAA) is a kind of bone marrow failure mainly characterized by severe peripheral pancytopenia. Recently SAA has been recognized an immune-mediated destruction of hematopoietic cells caused by abnormally activated T lymphocytes (especially CD8+ cells), leading to hyperfunction of downstream Th1 cells and cytotoxic T lymphocytes [1,2]. Anti-lymphocyte globulin/anti-thymocyte globulin-based intensive immunosuppressive treatment (IIST) has achieved increased clinical efficacy in recent years [3,4].
Memory T cells play an important role in host defense against viruses, malignancies and allogeneic cells. According to the ability to express chemokine receptor CCR7, CD4+ and CD8+ memory T cells can be divided into central memory T cells (TCM, CD3+CD45RA-CCR7+), and effector memory T cells (TEM, CD3+ CD45RA-CCR7-). TEM circulate through nonlymphoid peripheral tissues with effector phenotypes and have distinct migratory and functional characteristics [5], and produce IFN-γ, perforin, ganzyme B, and IL-4 but little IL-2. TCM circulate through secondary lymphoid tissues with strong proliferative capacity and might contribute to the maintenance of TEM pool, produce IL-2 but little IFN-γ and no perforin. However, the origin of these memory subsets and whether they are interrelated are not completely understood. Recently study have found that CD45RA+ cells in autoimmune diseases are often reduced, particularly in the active or relapse stage of the disease, representing the transformation of quiescent cells to activated memory lymphocytes [6]. In systemic lupus erythematosus (SLE), multiple sclerosis (MS) and other autoimmune diseases, it has been shown that the quantitative abnormality of memory T cells correlates with the inflammatory process of the disease. Moreover, these patients have more memory T cells with impaired function. Therefore, immune memory plays a significant role in the pathogenesis of autoimmune disease. Nevertheless little is known about any potential role for them in SAA. The memory T cells that damage bone marrow hematopoiesis in SAA patients show heterogeneity. The abnormalities of quantities and functions of memory T cells and its subsets with the potent effector capacity may relate to the abnormal immune status in the pathogenesis of SAA. This study aimed to investigate the quantities and functions of memory T cells in SAA patients to further clarify SAA apoptosis of bone marrow hematopoietic cells.
Materials and methods
Study subjects
A total of 45 patients with SAA including 26 men and 19 women with median age 30 (age range 8-56) were enrolled in the present study, including 11 newly diagnosed cases and 34 cases in remission after immunosuppressive therapy (IST). All patients were newly diagnosed in the Hematology Department of General Hospital Tianjin Medical University from November 2013 to November 2014, according to international AA Study Group Criteria (Marsh et al., 2009). The disease was considered SAA if pancytopenia with at least two of the following parameters were met: a neutrophil count less than 0.5×109/L, a platelet count less than 20×109/L, and a reticulocyte count less than 20×109/L with hypocellular bone marrow. VSAA was diagnosed in the cases SAA with the neutrophil count <0.2×109/L [16,17]. Patients were excluded if they had congenital AA or other autoimmune diseases. All patients were screened for paroxysmal nocturnal hemoglobinuria (PNH) by flow cytometry using anti-CD55 and anti-CD59 antibodies, and no PHN clones had been found. Patients’ features are listed in Table 1. Patients in remission were those who improved after immunosuppressive therapy (antithymocyte globulin, cyclosporine, glucocorticoid) and hematopoietic stimulating factors (granulocyte colony-stimulating factor, recombinant human erythropoietin, recombinant human thrombopoietin, and/or IL-11 in combination). All of the patients in remission achieved bone marrow hematopoietic recovery and became transfusion independent, while some had normal peripheral blood cell counts but still required drug therapy (Table 2).
Table 1.
Characteristics of untreated SAA patients
| Case | Age/sex | Granulocyte (×109/L) | Hemoglobin (g/L) | Platelet (×109/L) | Abnormal chromosome | Therapy |
|---|---|---|---|---|---|---|
| 1 | 13/F | 0.21 | 50 | 14 | Absence | None |
| 2 | 43/M | 0.34 | 72 | 12 | Absence | None |
| 3 | 22/M | 0.06 | 36 | 5 | Absence | None |
| 4 | 18/M | 0.41 | 74 | 10 | Absence | None |
| 5 | 8/F | 0.29 | 68 | 9 | Absence | None |
| 6 | 29/M | 0.16 | 56 | 7 | Absence | None |
| 7 | 27/F | 0.23 | 61 | 13 | Absence | None |
| 8 | 16/F | 0.25 | 53 | 9 | Absence | None |
| 9 | 20/M | 0.38 | 70 | 11 | Absence | None |
| 10 | 23/F | 0.34 | 64 | 10 | Absence | None |
| 11 | 15/F | 0.42 | 72 | 14 | Absence | None |
Table 2.
Characteristics of remission SAA patients
| Case | Age/sex | Granulocyte (×109/L) | Hemoglobin (g/L) | Platelet (×109/L) | Abnormal chromosome | Therapy |
|---|---|---|---|---|---|---|
| 1 | 19/F | 5.34 | 99 | 121 | Absence | ATG+ CsA |
| 2 | 56/M | 6.89 | 101 | 98 | Absence | ATG+ CsA |
| 3 | 37/M | 7.59 | 132 | 103 | Absence | ATG+ CsA |
| 4 | 32/F | 6.21 | 108 | 94 | Absence | ATG+ CsA |
| 5 | 16/F | 10.38 | 97 | 135 | Absence | ATG+ CsA |
| 6 | 10/F | 7.45 | 93 | 126 | Absence | ATG+ CsA |
| 7 | 54/M | 9.76 | 112 | 89 | Absence | ATG+ CsA |
| 8 | 34/F | 8.39 | 128 | 94 | Absence | ATG+ CsA |
| 9 | 29/F | 4.66 | 94 | 141 | Absence | ATG+ CsA |
| 10 | 41/F | 8.47 | 96 | 113 | Absence | ATG+ CsA |
| 11 | 30/F | 5.74 | 105 | 76 | Absence | ATG+ CsA |
| 12 | 46/M | 9.21 | 109 | 99 | Absence | ATG+ CsA |
| 13 | 33/F | 9.55 | 99 | 117 | Absence | ATG+ CsA |
| 14 | 26/M | 6.36 | 110 | 132 | Absence | ATG+ CsA |
| 15 | 44/F | 10.75 | 102 | 122 | Absence | ATG+ CsA |
| 16 | 48/M | 8.04 | 111 | 98 | Absence | ATG+ CsA |
| 17 | 39/M | 7.94 | 103 | 85 | Absence | ATG+ CsA |
| 18 | 41/F | 7.09 | 98 | 91 | Absence | ATG+ CsA |
| 19 | 39/F | 10.86 | 92 | 86 | Absence | ATG+ CsA |
| 20 | 40/M | 8.93 | 121 | 145 | Absence | ATG+ CsA |
| 21 | 53/F | 4.68 | 98 | 66 | Absence | ATG+ CsA |
| 22 | 28/F | 8.37 | 114 | 92 | Absence | ATG+ CsA |
| 23 | 16/F | 5.82 | 119 | 104 | Absence | ATG+ CsA |
| 24 | 33/F | 9.67 | 106 | 108 | Absence | ATG+ CsA |
| 25 | 37/M | 7.15 | 100 | 93 | Absence | ATG+ CsA |
| 26 | 46/M | 5.46 | 112 | 122 | Absence | ATG+ CsA |
| 27 | 37/F | 6.28 | 96 | 73 | Absence | ATG+ CsA |
| 28 | 43/M | 8.54 | 98 | 79 | Absence | ATG+ CsA |
| 29 | 51/F | 4.38 | 123 | 95 | Absence | ATG+ CsA |
| 30 | 28/F | 6.17 | 93 | 78 | Absence | ATG+ CsA |
| 31 | 23/M | 6.39 | 97 | 81 | Absence | ATG+ CsA |
| 32 | 20/M | 7.66 | 104 | 86 | Absence | ATG+ CsA |
| 33 | 19/F | 10.40 | 115 | 110 | Absence | ATG+ CsA |
| 34 | 24/M | 3.63 | 107 | 105 | Absence | ATG+ CsA |
25 normal healthy individuals were selected as controls (12 men and 13 women, median age 29, age range 22-56, Table 3). The study was approved by the Ethics Committee of Tianjin Medical University General Hospital. Informed written consents were obtained from all patients and normal controls or their parents in accordance with the Declaration of Helsinki.
Table 3.
Characteristics of normal controls
| Case | Age/sex | Granulocyte (×109/L) | Hemoglobin (g/L) | Platelet (×109/L) | Abnormal chromosome |
|---|---|---|---|---|---|
| 1 | 23/M | 8.13 | 124 | 151 | Absence |
| 2 | 24/M | 7.39 | 145 | 286 | Absence |
| 3 | 24/M | 6.98 | 160 | 213 | Absence |
| 4 | 24/F | 9.56 | 139 | 137 | Absence |
| 5 | 25/M | 8.26 | 127 | 265 | Absence |
| 6 | 24/F | 9.14 | 122 | 201 | Absence |
| 7 | 24/F | 7.30 | 158 | 186 | Absence |
| 8 | 26/M | 7.74 | 132 | 194 | Absence |
| 9 | 42/F | 8.91 | 137 | 241 | Absence |
| 10 | 33/F | 5.88 | 146 | 190 | Absence |
| 11 | 34/F | 6.23 | 141 | 266 | Absence |
| 12 | 29/M | 9.04 | 125 | 148 | Absence |
| 13 | 31/M | 5.78 | 159 | 169 | Absence |
| 14 | 28/F | 5.68 | 154 | 253 | Absence |
| 15 | 29/M | 6.03 | 128 | 210 | Absence |
| 16 | 29/F | 8.52 | 134 | 155 | Absence |
| 17 | 37/M | 9.11 | 147 | 172 | Absence |
| 18 | 56/M | 4.23 | 121 | 134 | Absence |
| 19 | 35/F | 5.16 | 126 | 209 | Absence |
| 20 | 49/F | 5.87 | 144 | 214 | Absence |
| 21 | 30/F | 5.47 | 137 | 233 | Absence |
| 22 | 22/M | 7.22 | 143 | 127 | Absence |
| 23 | 36/M | 6.39 | 119 | 145 | Absence |
| 24 | 41/F | 4.41 | 151 | 138 | Absence |
| 25 | 43/F | 5.23 | 128 | 195 | Absence |
Monoclonal antibodies
Anti-CCR7-PE, anti-CD3-APC, anti-CD45RA-FITC, anti- CD4-PerCP, anti-CD8-PerCP and the mouse isotype controls were purchased from Becton Dickinson (Franklin Lakes, NJ, USA).
Measurement of quantities of memory T cells and its subsets from peripheral blood and bone marrow
Blood and bone marrow samples were collected in heparin-anticoagulant tubes from the patients and normal individuals. Effector memory T cells (TEM, CD3+CD45RA-CCR7-) and central memory T cells (TCM, CD3+ CD45RA-CCR7+) were identified with a single platform four-color flow cytometric analysis. 50 µl whole blood or bone marrow was immunostained in TruCount tubes (BD), followed by lysis in 1.0 milliliter FACS RBC lysing solution (BD). Data acquisition and analysis were carried out by using FACS-Caliburflow cytometer (BD Biosciences, US), with the Cell Quest software (Becton Dickinson, version 3.1) (Figure 1).
Figure 1.
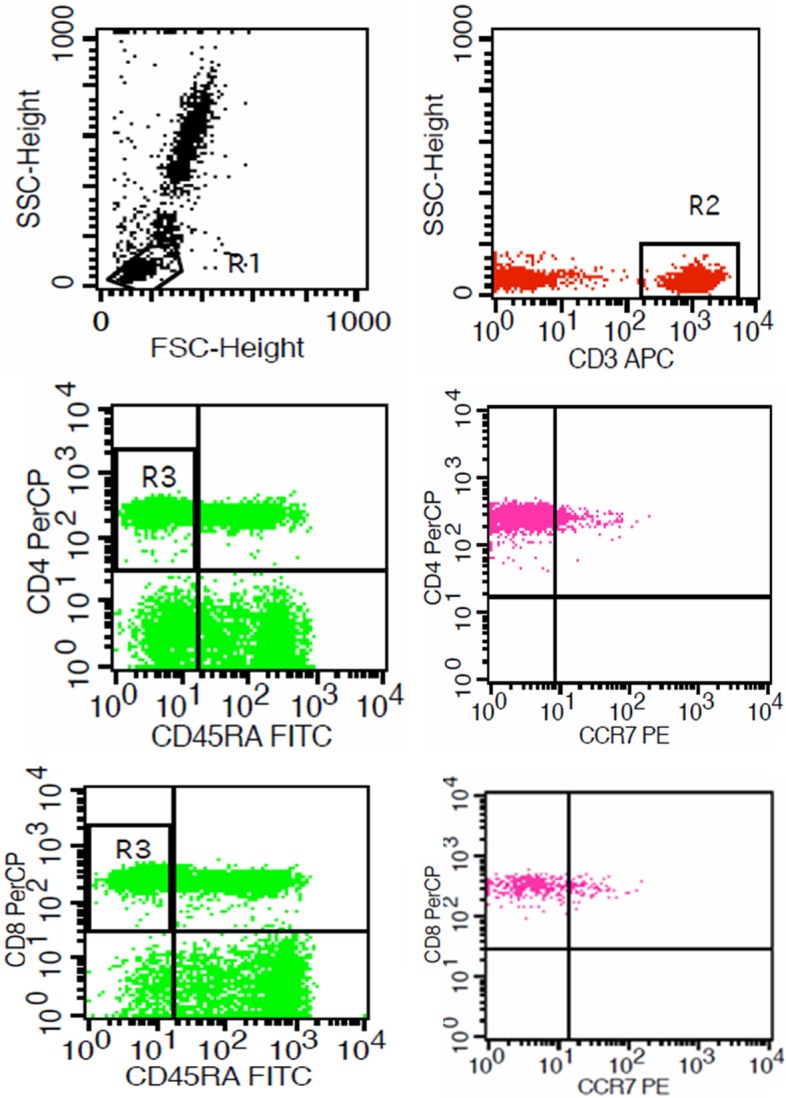
Flow cytometry (FCM) tests. Autoantibodies were detected on CCR7-PE, CD3-APC, CD45RA-FITC, CD4-PerCP (or CD8-PerCP).
Isolation and purification of CD8+TEM lymphocytes
Ten milliliters fresh human peripheral blood or bone marrow was obtained from SAA patients and normal controls. Peripheral blood mononuclear cells (PMMNCs) were isolated by density gradient centrifugation using Ficoll-Paque Plus solution (Amersham Bioscience, Uppsala, Sweden), followed by staining with fluorophore-conjugated monoclonal antibodies as described before (as mentioned above). CD8+ effector memory T cells were sorted and collected using a FacsAria flow cytometer, and the purity of these sorted cells was also measured. After detected by the multiparameter flow cytometry (BD Biosciences) and analyzed using the Cell Quest software program (Version 3.1, Becton Dickinson), the purity of CD8+ effector memory T cells was more than 90% (Figure 2).
Figure 2.
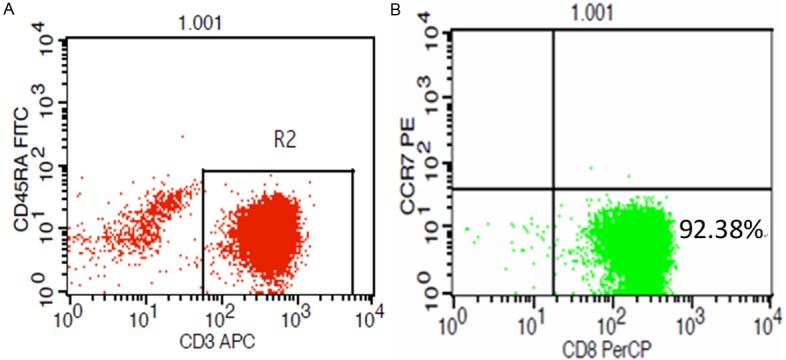
A and B. The purity of isolated CD8+ effector memory lymphocytes. After being isolated using a Facs Aria flow cytometer, we measured using multiparameter flow cytometry. The purity of CD8+ effector memory lymphocytes (CD3+CD8+CD45RA-CCR7-) was more than 90%.
RNA isolation and quantitative real-time PCR
PBMCs in human peripheral memory T cells were lysed in TRIzol reagent (Invitrogen, USA). Total RNA was dissolved in RNase-free water and quantified using a UV spectrophotometer (NanoDrop ND-1000, Thermo Scientific, USA). And 1 µg of RNA was reversed transcripted using TIANScript RT Kit (Invitrogen, USA). The sequences of primers specific for granzyme B, perforin and β-actin used have been listed below (Table 4), which were designed and synthesized by Sangon Biotech, Shanghai, China.
Table 4.
Primers Used for quantitative real-time PCR detection
| Target gene | Primer sequences | Product |
|---|---|---|
| Perforin | F:5’-GAGGAGAAGAAGAAGAAGCACAA-3’ | 200 bp |
| R: 5’-AGGGGTTCCAGGGTGTAGTC-3’ | ||
| Ganzyme B | F: 5’-CCAGCAGTTTATCCCTGTGAA-3’ | 235 bp |
| R:5’-CACCTCTTGTAGTGTGTGTGAGTG-3’ | ||
| β-actin | F: 5’-TTGCCGACAGGATGCAGAA-3’ | 100 bp |
| R: 5’-GCCGATCCACACGGAGTACT-3’ |
Real-time PCR was performed with 3 µL of each cDNA working solution in a final volume of 25 µL containing 12.5 µL 2x SYBR Green Real-time PCR Master Mix (TianGen Biotech) and 400 nM of each sense and antisense primer. The real-time PCR was carried out in a BIO-RAD iQ5 Real-Time System (BIO-RAD, USA), using the following thermal cycling profile for all genes of interest: 95°C for 5 min, followed by 40 cycles of amplification (95°C for 15 s, indicated annealing temperature for 15 s, 72°C for 45 s). After normalization of the data according to the expression of b-actin mRNA, the levels of perforin and granzyme B were calculated by the 2-ΔΔCt method ((Ct, target gene-Ct, β-actin)sample-(Ct, target gene-Ct, β-actin)).
Statistical analysis
All statistical calculations were performed using SPSS Statistics 18.0. Statistical analysis was performed with the parametric unpaired t-test for normally distribution data, and the nonparametric test for skewed data. Statistical significance was accepted if P<0.05.
Results
Proportion of TEM subsets in untreated SAA patients
The percentage of CD4+ TEM cells in peripheral blood lymphocytes was (22.21±6.97)% in untreated SAA patients, and (28.18±8.18)% in normal controls. The percentage of CD4+ TEM cells in untreated SAA patients was lower than that in normal controls (P<0.05). The percentage of CD8+ TEM cells in peripheral blood lymphocytes was (18.24±7.12)% in untreated SAA patients, and (11.83±5.52)% in normal controls. The percentage of CD8+ TEM cells in untreated SAA patients was higher than normal controls (P<0.05). The ratio of CD4+ memory T lymphocytes to CD8+ memory T subsets in untreated SAA patients was also lower than remission group and normal controls (P<0.05). The percentage of CD4+ TEM cells in the bone marrow lymphocytes was (17.27±1.07)% in untreated SAA patients, and (27.30±4.29)% in normal controls. The percentage of CD4+ TEM cells in untreated SAA patients was lower than that in normal controls (P<0.05). There was no statistical difference between newly diagnosed SAA patients and normal controls in CD8+ TEM subsets in the bone marrow (P>0.05) (Figure 3).
Figure 3.
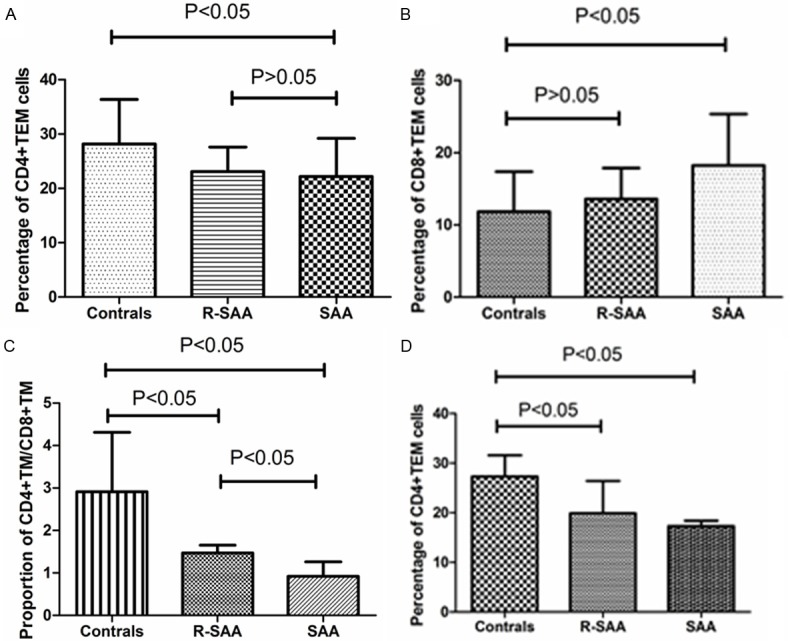
The changes of TEM cells and its subsets after intensive immune suppressive therapy in severe aplastic anemia patients. A. The percentage of CD4+ TEM cells in SAA, R-SAA and healthy controls in peripheral blood; B. The percentage of CD8+ TEM cells in SAA, R-SAA and healthy controls in peripheral blood; C. The ratio of CD4+ memory T lymphocytes to CD8+ memory T subsets in SAA, R-SAA and healthy controls in peripheral blood; D. The percentage of CD4+ TEM cells in SAA, R-SAA and healthy controls in the bone marrow.
Proportion of TCM subsets in untreated SAA patients
The percentage of CD8+ TCM cells in the bone marrow lymphocytes was (0.56±0.35)% in untreated SAA patients, and (0.28±0.10)% in normal controls. The percentage of CD8+ TCM cells in untreated SAA patients was higher than that in normal controls (P<0.05). No significant differences in the proportion of TEM cell subsets between groups were detected in the bone marrow (Figure 4).
Figure 4.
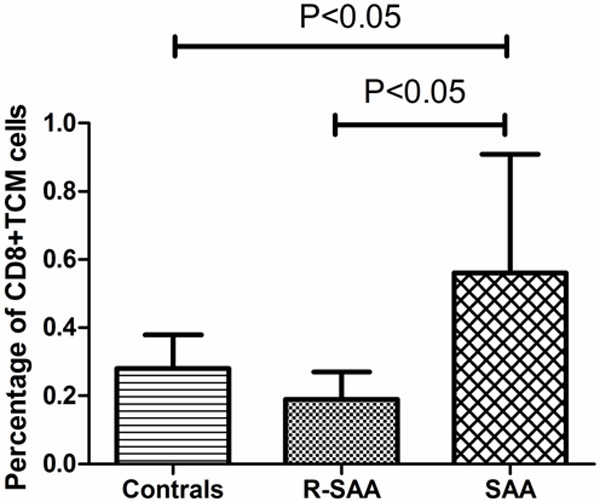
Percentage of CD8+TCM cells in SAA, R-SAA and healthy controls in the bone marrow.
mRNA expression abnormalities of perforin and granzyme B in PWBCs from SAA patients
To determine the function of CD8+ memory T cells in SAA, the perforin and granzyme B mRNA expression on peripheral blood CD8+ TEM cells from untreated SAA patients, recovery patients and normal controls were evaluated. The levels of mRNA of perforin and granzyme B genes were dramatically increased CD8+ TEM cells from our patient samples compared with normal controls in the peripheral blood.
We measured the levels of mRNA of these genes in AA patient samples and identified significantly higher expression of perforin in the SAA untreated group (5.46, range: 4.31-6.25) relative to the remission group (3.71, range: 1.44-5.83) and the control group (0.82, range: 0.39-1.22; P<0.05). Meanwhile, the relative Granzyme B mRNA levels evidently increased in the peripheral blood CD8+ TEM cells from untreated SAA patients (4.54, range: 2.71-5.56 ) compared to remission group (3.59, range: 1.86-5.48) and normal controls (0.97, range: 0.21-1.50; P<0.05; Figure 5).
Figure 5.
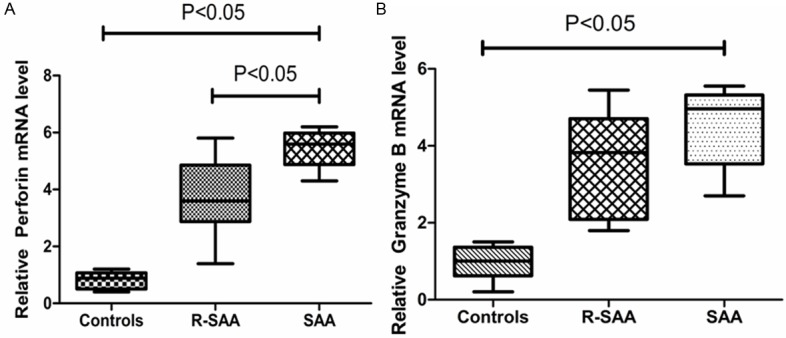
Perforin and granzyme B mRNA expression in different groups.
Discussion
Severe aplastic anemia is a potentially life-threatening disease whose high mortality is usually due to complications such as infection, hemorrhage and severe anemia. It is known that our immune system is usually in a state of autoimmune tolerance in order to maintain the relatively stable internal environment of body. But stable dysfunction of the immune system can produce its reactive antibodies and/or its reactive T/B lymphocytes to attack its own cells or molecules, leading to autoimmune disease. In the past decades, studies have shown that SAA was an autoimmune disease with hematopoietic stem/progenitor cells impaired by hyperfunctional autoimmunic T lymphocytes (especially CD8+ cells) which activated abnormally by Th1 cells. The increased expression of TNF-α, IFN-γ, and IL-2 from SAA patients indicates that hematopoietic stem/progenitor cells are destroyed through Th1 cell response [7]. Liu et al. reported about increased expression of Fas antigen on bone marrow CD34+ cells of patients with SAA. Fas antigen, a receptor molecule that has contributed great progress to human programmed cell death and apoptosis, is enhanced by INF-γ and TNF-α. The recognition of the Fas expression antigen by the FasL expression CTL might give rise to excessive apoptosis of bone marrow hematopoietic cells in patients with SAA. Furthermore, CD34+ cells are likely the main targets of SAA immune injury [8]. Patients with SAA have a significant increase in CD8+ suppressor T lymphocytes [9,10]. Moreover, the expression of perforin, granular enzyme and TNF-β were elevated obviously in these effector T cells as well as many other hematopoietic negative regulatory factors in SAA patients, suggesting that hematopoietic stem/progenitor cells might destroyed through lymphokine induced apoptosis [11-13]. Previous studies have demonstrated that activated DC1 cells increased in the bone marrow of SAA patients, promoting Th0 cells to polarize to Th1 cells and even increases in the quantity and function of CD8+ T cells, thus cause the hyperfunction of T lymphocytes and hematopoiesis failure in SAA [14,15].
The present study was aimed to elucidate the effect of memory T cells and its subsets from both in peripheral and bone marrow on SAA patients and obtain more evidence for the bone marrow failure in SAA. Here we have studied memory T cells in peripheral blood from patients with SAA and normal controls. Our results have demonstrated that there was a significant decrease in the proportions of CD4+ /CD8+ TEM cells in the peripheral blood from individuals with severe aplastic anemia, and the proportion of TM subsets (CD4+ TM/CD8+ TM) restored to normal levels in these patients after IIST. Furthermore, the ratio of CD8+ TCM cells in the bone marrow of SAA patients increases obviously. Based on these results, we suggested that abnormal immunomodulatory ability on memory T cells is responsible for T-cell proliferation, imbalance of Th1/Th2 subsets and even in the processes of bone marrow failure of SAA.
Memory T cells are the components of the adaptive immune system. They circulate between peripheral tissues, lymphoid organs and blood to ensure the host of more aggressive and faster immune response to efficiently eliminate the specific antigens after re-exposure [16]. According to the ability to express chemokine receptor CCR7, CD4+ and CD8+ memory T cells can be divided into central memory T cells (TCM, CD3+CD45RA-CCR7+), and effector memory T cells. Different surface molecular markers on memory T cells affect its tissue localization. TEM is mainly distributed in the blood, lymph nodes and peripheral non-lymphoid tissues and express high levels of integrin β1 and β2, cutaneous lymphocyte antigen (CLA) and CCR1, CCR3, CCR5 chemokine receptors. Those TEM cells who homing to the skin express CCR4 and CCR10 molecules. Other TEM cells who homing to the intestinal tract express integrin α4β7. Activated TEM cells migrate to peripheral sites of inflammation, and rapidly secrete both Th1 and Th2 cytokines [17]. TCM express CD62L, CCR7, CCR4, CCR6, CXCR3, CCR1 and CCR2 molecules, most of them exist in secondary lymphoid tissues and organs such as spleen, blood and lymph nodes. Additionally, memory T cells are critically dependent on instructive signals from specific cytokines, such as IL-7, to remain in a functionally competent state [18,19]. IL-15 is the central driver of CD8+ memory T cell [20]. Such memory cells can rapidly secrete IFN-γ and IL-2, with increased sensitivity to TCR signaling and a reduced need for co-stimulation [21]. Following a second exposure to the same antigen, the CD8+ memory T cells develop into secondary effectors and eventually differentiate into secondary memory T cells. While the CD4+ T memory cells have not been extensively studied and complicated by the existence of multiple Th subsets [22].
In addition to play a role in virus, bacteria and other pathogens, TM cells also related to the occurrence of many autoimmune diseases. In type 1 diabetes (T1D), memory T cells protect from the progress of the disease through secretion of immunoregulatory cytokines [23]. In systemic lupus erythematosus (SLE) [24], it has been shown that the quantitative abnormality of memory T cells correlates with the inflammatory process of the disease. Moreover, patients with multiple sclerosis (MS) have more memory T cells (especially CD8+ memory T cells) with impaired function, and this may be related to maintain continuous process after the occurrence of immune response in multiple sclerosis patients [25]. These studies above strongly suggest that the memory T cells are involved in the autoimmune disease. Memory T cells show enhanced effector function which is more likely to be activated and have a rapid production of effector molecules. This process is controlled by transcription factors, such as Eomesodermin (EOMES) and T-box transcription factor 21 [26,27]. T-bet is a kind of T-box transcription factor, which is the key regulator of Th1 development and function. There is evidence that T-bet and the cognate transcription factors EOMES closely related to the differentiation of effect and memory T cells [28]. The expression of T-bet in TM will be reduced while the expression of EOMES will be elevated. Moreover, those cells with high expression of T-bet will differentiate into effector cells, while the cells with low expression of T-bet will differentiate into memory cells [29]. Past studies have shown that EOMES directly regulates expression of perforin, granzyme B, and TNF-α, and promotes effector function in CD8+ T cells. Granzymes, a cell death-inducing serine proteases released from cytotoxic T lymphocytes and natural killer cells, considered to be the responsible molecule for target cell cytosol. Perforin, a Ca (2+)-dependent pore-forming protein, is released in the presence of Ca2+ and aggregate to form pores on the target cell membranes. These pores can cause disruption of the balance of osmotic pressure inside and outside of the cell and finally lead to the death of the target cell [30]. A critical cofactor for the granule exocytosis pathway is granzyme B, which enters the target cell through the pores formed by perforin where they cleave specific substrates that initiate DNA fragmentation and apoptosis [31,32]. Studies have found that CD8+ TEM can rapidly exert effector functions when re-exposure to antigen, including the release of perforin and granzyme B [33]. Patients with SLE were characterized by higher proportions of perforin and/or granzyme B-positive lymphocytes, and the increase in circulating perforin or granzyme B-positive CD8+ T cells thoroughly reflected the activity of the disease [34]. The results obtained in the present study allowed us to better delineate a still-unknown role for CD8+ TEM lymphocytes in SLE pathogenesis. No studies showing the specific mechanism for memory T cells in SAA patients have yet been reported.
Our previous study has demonstrated that the quantity of CD8+ TEM cells in peripheral blood was significantly increased in SAA patients compared with normal controls, then next we use the method of qPCR to verify whether there is a hyperfunctional state of TEM in peripheral blood. The results indicate that the median expressions of perforin and granzyme B on CD8+ TEM cells were higher in SAA patients compared to those in normal controls. Based on these results, we consider that hyperfunction of memory T cells possibly caused by genetic mutation or antigenic stimulus could enhance the cytotoxicity of CD8+ T cells in SAA. The activated TM cells proliferate extremely quickly upon stimulating repeatedly by unknown antigen, causing the hyperfunction of antigen-specific lymphocytes attack against hematopoietic tissue, and thus ultimately led to the apoptosis of hematopoietic cells. It is noteworthy that the body’s memory T cells return to a dormant state after the majority of the specific antigen is removed, and Th1 cells decreased gradually with hematopoietic function recovery. Additionally, because of the increase expression of CCR7 and CD62L, a mass of TCM cells can infiltrate to the bone marrow of SAA patients to attack hematopoietic stem/progenitor cells, which might also contribute to the immunopathogenesis of SAA. The activation of CD4+ memory T cells is associated with the deterioration of SAA, and the activation of CD8+ memory T cells may reflect the systemic immune dysfunction in SAA patients. Especially the abnormal changes of TCM cells subgroup, which may be related to sustain continuous process of the SAA after the occurrence of immune response.
In conclusion, the abnormal quantity and function of memory T cells in humans with severe aplastic anemia may be one of the reasons that cause the over-function of T lymphocytes and thus lead to hematopoiesis failure in SAA. Our research might allow the development of new therapeutic strategies based on memory T cells for the treatment of SAA. Nevertheless, the biological characteristics of memory T cells and its subsets need further study to explore the real target.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81170472, 81370607, and 81400085), Tianjin Municipal Natural Science Foundation (12JCZDJC21500), Health Industry Research and Special Projects (201202017), Tianjin Science and Technology support key project plan (20140109), Health Industry Research and Special Projects (201202017), Tianjin Cancer Research of Major Projects (12ZCDZSY17900 and 12ZCDZSY18000), and the Tianjin Health Industry Key Projects (11KG135).
Disclosure of conflict of interest
None.
References
- 1.Bacigalupo A. Aplastic anemia: pathogenesis and treatment. Hematology Am Soc Hematol Educ Program. 2007;2007:23–28. doi: 10.1182/asheducation-2007.1.23. [DOI] [PubMed] [Google Scholar]
- 2.Marsh JC, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, Keidan J, Laurie A, Martin A, Mercieca J, Killick SB, Stewart R, Yin JA British Committee for Standards in Haematology. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147:43–70. doi: 10.1111/j.1365-2141.2009.07842.x. [DOI] [PubMed] [Google Scholar]
- 3.Shao ZH. Standardized diagnosis and treatment of aplastic anemia. Chin J Intern Med. 2010;30:311–313. [Google Scholar]
- 4.HBCMA. Aplastic anemia diagnosis and treatment expert consensus. Hematology Branch of the Chinese Medical Association Red Blood Cell Disease (Anemia) Study Group. Chin J Hematol. 2010;31:790–792. [Google Scholar]
- 5.Okada R, Kondo T, Matsuki F, Takata H, Makiguchi M. Phenotypic classification of human CD4+ T cell subsets and their differentiation. Int Immunol. 2008;20:1189–99. doi: 10.1093/intimm/dxn075. [DOI] [PubMed] [Google Scholar]
- 6.Gattornol M, prigione l, Morandi F, Gregorio A, Chiesa S, Ferlito F, Favre A, Uccelli A, Gambini C, Martini A, Pistoia V. Phenotypic and functional characterisation of CCR7+ and CCR7-CD4+ memory T cells homing to the joints in juvenile idiopathic arthritis. Arthritis Res Ther. 2005;7:R256–67. doi: 10.1186/ar1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, Bluestone JA, Abbas AK, Dooms Hb. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci U S A. 2012;109:12668–73. doi: 10.1073/pnas.1203692109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu CY, Fu R, Wang HQ, Li LJ, Liu H, Guan J, Wang T, Qi WW, Ruan EB, Qu W, Wang GJ, Liu H, Wu YH, Song J, Xing LM, Shao ZH. Fas/FasL in the immune pathogenesis of severe aplastic anemia. Genet Mol Res. 2014;13:4083–8. doi: 10.4238/2014.May.30.3. [DOI] [PubMed] [Google Scholar]
- 9.Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119–S125. doi: 10.1016/j.bbmt.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xing L, Liu C, Fu R, Wang H, Wang J, Liu X, Feng L, Li L, Liu H, Wang H, Zhang T, Shao Z. CD8+ HLA-DR+ T cells are increased in patients with severe aplastic anemia. Mol Med Rep. 2014;10:1252–8. doi: 10.3892/mmr.2014.2344. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Fu R, Wang J, Li LJ, Song J, Qu W, Wang HQ, Xing LM, Liu H, Wu YH, Guan J, Wang GJ, Wang XM, Liang Y, Ruan EB, Liu H, Shao ZH. Perforin gene mutations in patients with acquired severe aplastic anemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2011;19:431–4. [PubMed] [Google Scholar]
- 12.Liu CY, Fu R, Wang J, Liang Y, Qu W, Ruan EB, Wang HQ, Li LJ, Liu H, Wang GJ, Liu H, Wu YH, Wang XM, Song J, Xing LM, Guan J, Wang HL, Zhang T, Liu X, Shao ZH. Expression of lymphokines in CD8 (+) HLA-DR (+) T lymphocytes of patients with severe aplastic anemia. Zhonghua Yi Xue Za Zhi. 2012;92:1240–3. [PubMed] [Google Scholar]
- 13.Liu CY, Fu R, Wang HQ, Li LJ, Liu H, Guan J, Wang T, Qi WW, Ruan EB, Qu W, Wang GJ, Liu H, Wu YH, Song J, Xing LM, Shao ZH. Fas/FasL in the immune pathogenesis of severe aplastic anemia. Genet Mol Res. 2014;13:4083–8. doi: 10.4238/2014.May.30.3. [DOI] [PubMed] [Google Scholar]
- 14.Zonghong S. The research of aplastic anemia. Basic Clin Med. 2007;27:233–7. [Google Scholar]
- 15.Zonghong S, Meifeng T, Huaquan W, Limin X, Jun W, Rong F, Hong L, Yuhong W. Circulating myeloid dendritic cells are increased in individuals with severe aplastic anemia. Int J Hematol. 2011;93:156–62. doi: 10.1007/s12185-010-0761-z. [DOI] [PubMed] [Google Scholar]
- 16.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 17.Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One. 2011;6:e16245. doi: 10.1371/journal.pone.0016245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyman O, Letourneau S, Krieg C, Sprent J. Homeostatic proliferation and survival of naive and memory T cells. Eur J Immunol. 2009;39:2088e94. doi: 10.1002/eji.200939444. [DOI] [PubMed] [Google Scholar]
- 19.Zaunders JJ, Levy Y, Seddiki N. Exploiting differential expression of the IL-7 receptor on memory T cells to modulate immune responses. Cytokine Growth Factor Rev. 2014;25:391–401. doi: 10.1016/j.cytogfr.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, Bluestone JA, Abbas AK, Dooms H. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci U S A. 2012;109:12668–73. doi: 10.1073/pnas.1203692109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanzavecchia A, Sallusto F. Understanding the generation and function of memory T cell subsets. Curr Opin Immunol. 2005;17:326–32. doi: 10.1016/j.coi.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oling V, Reijonen H, Simell O, Knip M, Ilonen J. Autoantigen-specific memory CD4+ T cells are prevalent early in progression to Type 1 diabetes. Cell Immunol. 2012;273:133–139. doi: 10.1016/j.cellimm.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Fritsch RD, Shen X, Illei GG, Yarboro CH, Prussin C, Hathcock KS, Hodes RJ, Lipsky PE. Abnormal differentiation of memory T cells in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2184–2197. doi: 10.1002/art.21943. [DOI] [PubMed] [Google Scholar]
- 25.Beynon V, Quintana FJ, Weiner HL. Actiated human CD4+CD45RO+ memory T-cells indirectly inhibit NLRP3 inflammasome activation through downreguktion of P2X7R signaling. PLoS One. 2012;7:1112–1117. doi: 10.1371/journal.pone.0039576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 27.Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B) J Immunol. 2008;180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki H, Shi Z, Okuno Y, Isobe K. Are CD8+CD122+ cells regulatory T cells or memory T cells? Hum Immunol. 2008;69:751–754. doi: 10.1016/j.humimm.2008.08.285. [DOI] [PubMed] [Google Scholar]
- 29.Cui W, Kaech SM. Generation of effector CD8+ T cells andtheir conversion to memory T cells. Immunol Rev. 2010;236:151–166. doi: 10.1111/j.1600-065X.2010.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kastrukoff LF, Lau A, Wee R, Zecchini D, White R, Paty DW. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J Neuroimmunol. 2003;145:103–14. doi: 10.1016/j.jneuroim.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Pardo J, Aguilo JI, Anel A, Martin P, Joeckel L, Borner C, Wallich R, Müllbacher A, Froelich CJ, Simon MM. The biology of cytotoxic cell granule exocytosis pathway: granzymes have evolved to induce cell death and inflammation. Microbes Infect. 2009;11:452–9. doi: 10.1016/j.micinf.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. 2007;19:301–8. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molicular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175:5895–5903. doi: 10.4049/jimmunol.175.9.5895. [DOI] [PubMed] [Google Scholar]
- 34.Shah D, Kiran R, Wanchu A, Bhatnagar A. Soluble granzyme B and cytotoxic T lymphocyte activity in the pathogenesis of systemic lupus erythematosus. Cell Immunol. 2011;269:16–21. doi: 10.1016/j.cellimm.2011.03.004. [DOI] [PubMed] [Google Scholar]