

Abstract
Increasing evidences suggest that p120 catenin (p120ctn) exerts important functions in the regulation of pro-inflammatory molecules. However, the relationship among p120ctn, inflammatory responses and blood-brain barrier (BBB) dysfunction as they are the initiator of sepsis is not unknown. In this study, we found that p120ctn expression was correlated with an increase in the permeability of BBB and a decrease in the expression of tight-junction proteins in human brain microvascular endothelial cells (HBMECs) after LPS challenge. Transfection with p120ctn small interfering RNA (siRNA) induced disruption of BBB integrity, monocyte migration across BBB and inflammatory responses at basal level and after LPS treatment. Conversely, over-expression of p120ctn with adenovirus significantly ameliorated BBB disruption and inflammatory responses in LPS-treated cells. Mechanistically, up-regulation of p120ctn inhibited LPS-induced NF-κB activation by suppressing IKKβ and IκBα phosphorylation, IκBα degradation. Therefore, we conclude that p120ctn improves the BBB dysfunction and inflammatory responses through the inhibition of NF-κB activation, suggesting that forced p120ctn expression may provide a novel therapeutic strategy to attenuate LPS-induced BBB compromise and sepsis.
Keywords: p120ctn, LPS, BBB, HBMECs, inflammatory response
Introduction
Brain microvascular endothelial cells form a blood-brain barrier (BBB) between the blood and the surrounding brain tissue and facilitate the exchange of nutrients and proteins between these compartments [1-3]. In normal condition, BBB plays a dominant role in the regulation of paracellular flux and permeability, which are primarily mediated by the tight junction proteins, such as occludin and claudin-5 [1,4]. Once stimulated by a variety of environmental, toxic, degenerative, and microbial insults, the tight junctions become loosened and BBB is breakdown [5,6]. Of note, such abnormal changes have been observed in brain microvascular endothelial cells, animal models and clinical studies during the development of sepsis [7-9]. These findings suggest that elucidation of the underlying mechanisms of BBB dysfunction may help to better understand sepsis and to develop a novel therapeutic agent.
Sepsis, an uncontrolled systemic inflammatory response, can induce BBB dysfunction, maintaining a leading cause of morbidity and mortality [7,10]. It has been documented that high levels of lipopolysaccharide (LPS) in the blood indicates infection or sepsis [7]. LPS, the major component of the cell wall of Gram-negative bacteria, induces inflammatory gene expression through nuclear factor-κB (NF-κB) activation, which in turn, results in neuro-inflammation [11-13]. Although NF-κB signaling pathway has been suggested to function as an important regulator in inflammatory disease, possible regulators are not completely understood.
P120 catenin (p120ctn) was first found as an Armadillo repeat protein, which functions as a substrate for Scr- and receptor-tyrosine kinases [14]. Accumulating evidence indicates that p120ctn is a critical adherent junction protein in the regulation of barrier function [15,16]. Interestingly, recent work in human endothelial cells has documented that several pro-inflammatory adhesion molecules, including ICAM-1, VCAM-1, E-selectin and P-selectin, could be regulated by p120ctn at transcriptional level [16]. Moreover, p120ctn has been found to be closely associated with NF-κB activation, because the increased NF-κB activity was observed in p120ctn null epidermal cells and in the skin of p120ctn conditional knock-out mice [17,18]. These results demonstrate the critical role of p120ctn in inflammation response. However, the relationship between p120ctn and brain endothelial cell inflammation and the mechanism by which p120ctn regulates NF-κB activation are unknown. Therefore, in this study, we aimed to investigate the role of p120ctn in LPS-induced brain endothelial inflammatory response and the underlying molecular mechanisms. Our work suggests p120ctn may be a new strategy to improve BBB dysfunction.
Methods and materials
Materials and reagents
EGM-2 medium, fetal bovine serum (FBS), penicillin and streptomycin were obtained from Gibco (Carlsbad, CA). FITC-labeled Dextran, lipopolysaccharide (LPS), phosphate buffered saline (PBS), Hank’s Balanced Salt Solution (HBSS) and calcein-AM were purchased from Sigma Chemical Co. (St. Louis, MO). Hiperfect Transfection Reagent, RNeasy Micro Kit and PCR primers were from Qiagen (Valencia, CA). p65, p50, p-IκBα, IκBα, p-IKKβ, IKKβ and all secondary peroxidase-conjugated antibodies were obtained from Cell Signaling Technology (Danvers, MA). Antibodies targeting against occludin, claudin-5 and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). P120ctn antibody and collagen-coated FluoroBlok-tinted tissue culture inserts were from BD Biosciences (Franklin Lakes, NJ).
Cell culture
Human brain microvascular endothelial cells (HBMECs) were obtained from Sciencell (ScienCell Research Laboratories, Carlsbad, CA) and maintained in EGM-2 medium supplemented with 5% FBS, and antibiotics (100 U/mL penicillin and 100 U/mL streptomycin) in a 37°C incubator with 5% CO2.
Permeability of the BBB in vitro
Permeability for FITC-labeled dextran through the BBB was assayed as previously described with minor modifications [7]. HBMECs (2×104) were seeded on collagen-coated FluoroBlok-tinted tissue culture inserts (pore size 3 μm) and incubated in mixed medium (endothelial cell growth medium: astrocyte-conditioned medium, 1:1). After treatment of LPS (1 μg/mL) and FITC-dextran (150 kDa; 100 μg/ml) for the indicated times, samples were collected from the acceptor chambers and FITC-dextran amount in the samples were measured with a fluorescence spectrophotometer at an excitation of 488 nm and an emission of 520 nm using a microplate reader (Infinite F500 mMulti Reader, TECAN, Mannedorf, Swittzerland). The FITC-dextran concentration in the samples was calculated from the standard curves generated by FITC-dextran in PBS. To investigate the effect of p120ctn on permeability of the BBB, HBMECs were transfected with p120ctn siRNA or adenovirus for 48 h first, followed by incubation with LPS for another 24 h.
Western blot analysis
HBMECs were washed twice with cold PBS and lysed in RIPA buffer (Beyotime, Jiangsu, China). Cell lysates were harvested by centrifuging at 12,000 g for 12 min at 4°C, and the protein concentration was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA). Equal proteins were fractionated in an 8% to 12% gradient gel and transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA). The membranes were incubated with the following primary antibodies: p120ctn, occluding, claudin-5, p65, p50, p-IκBα, IκBα and p-IKKβ (1:1000); β-actin (1:4000). After washing and incubation with the appropriate secondary peroxidase-conjugated antibodies (1:1000) for 1 hour, signals were detected by an enhanced chemiluminescence reagent (Thermo Scientific, Pittsburgh, PA) and quantified by densitometry with Gel-Pro3.0 image software (Media Cybernetics, Silver spring, MD).
Q-PCR (Quantitative real-time PCR) analysis
Total RNA was isolated from cultured HBMECs using RNeasy Micro Kit according to the manufacturer’s instructions. The cDNA was synthesized from 1 μg of RNA using the superScript III First-Strand Synthesis system (Invitrogen, Grand Island, NY). Real-time PCR was carried out with a Fast RT-PCR system (ABI 7300, Applied Biosystems, Grand Island, NY) using a Fast SYBR® Green Master Mix Kit (Applied Biosystems). The PCR conditions were as follow: 37°C for 4 mins, followed by 32 cycles at 95°C for 15 s, 60°C for 1 min, 72°C for 30 s. The level of mRNA expression for GAPDH was measured and used an internal control. The specific primers (Table S1) used for PCR were developed using Primer Express 2.0 software (Applied Biosystems).
Overexpression of p120ctn in HBMECs
Human p120ctn cDNA was amplified and cloned into pCMV-Tag2 (Invitrogen) between BamH1 and Xho1 restriction sites. Then the recombinant shuttle plasmid containing p120ctn gene was linearized and was transfected into 293A cells with Hiperfect Transfection Reagent to generate recombinant adenovirus. LacZ was purchased from Clontech (Mountain View, CA). The titers of virus were assayed using by p24 ELISA kit (Cell Biolabs, San Diego, CA). For overexpression of p120ctn in HBMECs, the cells were cultured in serum-free medium containing appropriate multiplicity of infection (MOI) of p120ctn adenovirus (Ad-p120ctn). After 6 h, HBMECs were transferred into complete medium and cultured for 48 h. In this study, the cells were infected with 100 MOI Lacz or Ad-p120ctn for 48 h, followed by LPS stimulation.
Transfection of small interfering RNA (siRNA)
The siRNA oligonucleotide duplexes against human p120ctn gene (GenBank Accession No. NM_001331; 5’-GGACCUUACUGAAGUUAUUUU-3’) and a negative siRNA (negative) were obtained from Qiagen. The siRNA were transfected with Hiperfect Transfection Reagent according to the manufacturer’s instructions. In this study, HBMECs were transiently transfected with 20 nM negative or p120ctn siRNA for 48 h prior to LPS incubation.
Monocytes isolation and migration
Fresh blood from healthy human was obtained from the Blood Center of the Affiliated People’s Hospital of Jiangsu University and layered in lymphocyte separation medium (LSM, BioWhitaker Inc., Walkersville, MD). After centrifugation, the monocytes (leukocytes)-enriched pellet was re-suspended and the red blood cells were lysed in a hypotonic buffer (water: HBSS, 1:1). This procedure was repeated twice, and then the monocytes were harvested and counted. For migration assay, after transfection with p120ctn siRNA or adenovirus for 48 h, HBMECs (2×104) were digested and seeded on collagen-coated FluoroBlok-tinted tissue culture inserts. LPS-treated or -nontreated monocytes were labeled with 5 μM calcein-AM for 30 min. Then the labeled monocytes (2×105) were added to the upper chamber and placed on HBMECs, and then allow to migrate for 2 h. The fluorescence of the migrated monocytes was measured with an Infinite F500 mMulti Reader with emission and excitation wavelength of 485 nm and 535 nm.
Transient transfection and luciferase reporter gene assay
HBMECs in 6-well plates were transfected with NFκB promoter-luciferase (Clontech, CA, USA) and β-galactosidase plasmid, or co-transfected the above plasmids with Lacz or Ad-p120ctn. After 48 h, cells were stimulated with LPS for 2 h. Cell lysates were assayed for luciferase activity and β-galactosidase activity using a Secrete-PairTMDual Luminescence Assay kit (Gene Copoeia, MD) as measured with a luminometer (Infinite F500 mMulti Reader). The luminometer reported relative light units, which were normalized by β-galactosidase activity.
Nuclear translocation of NFκB
Nuclear proteins were extracted with a nuclear/cytosol Fractionation Kit according to the manufacturer’s instructions (Bio Vision, Milpitas, CA), and were analyzed by western blot as mentioned above.
Statistical analysis
All data were expressed as mean ± SEM. The regression analysis was determined by the Pearson correlation test. 1-way ANOVA followed by the Bonferroni multiple comparison post hoc test with a 95% confidence interval was employed in SPSS 16.0 system (SPSS Inc., Chicago, IL). P value of less than 0.05 was considered to be statistically significant.
Results
LPS decreased p120ctn expression and disrupted the BBB integrity
Previous studies have suggested that p120ctn is an important regulator in the transcription of pro-inflammatory molecules [16]. To investigate whether p120ctn is involved in inflammatory response in endothelial cells, we treated HBMECs with LPS (1 μg/mL) for the indicated times and examined the expression of p120ctn. As shown in Figure 1A, LPS decreased the expression of p120ctn in a time-dependent manner. Compared with control (0 h), at 3, 6, 12, 24 h, p120ctn protein expression was reduced to 87.75 ± 7.92%, 69.43 ± 7.64%, 52.87 ± 6.38% and 39.85 ± 4.37%, respectively. Moreover, we found LPS dramatically increased the BBB permeability (Figure 1B). The concentration of FITC-dextran reached the maximal level at 24 h, which was negatively correlated with the expression of p120ctn (Figure 1C). Tight junctions have been documented to play an important role in regulating the BBB integrity (Liu et al., 2012b). To unravel whether LPS-induced abnormality of tight junctions contributes to the increased BBB permeability, we determined the expression of occludin and claudin-5 in HBMECs exposed to LPS. After treatment of LPS, occludin and claudin-5 expression were both significantly decreased in a time-dependent manner (Figure 1D). Interestingly, the expression of p120ctn was positively correlated with the expression of occludin and claudin-5 (Figure 1E and 1F). These data suggest that the decrease of p120ctn expression may be involved in LPS-induced the disruption of BBB integrity.
Figure 1.
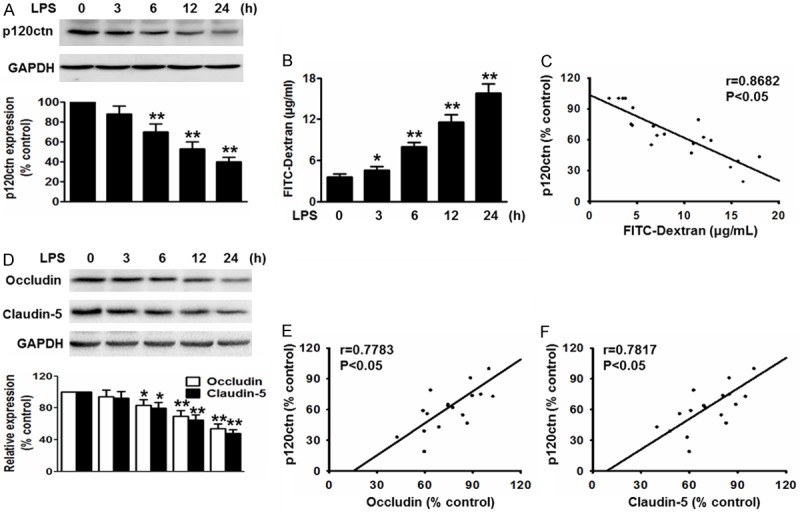
P120ctn protein expression paralleled with LPS-induced the decrease of blood-brain barrier (BBB) integrity and the expression of tight-junction proteins in HBMECs. Cells were stimulated with LPS (1 μg/mL) over a range of indicated time points. A. The expression of p120ctn was examined by western blotting using GAPDH as an internal control. B. The concentration of FITC-dextran in LPS was measured as described in methods section. C. Correlation between p120ctn expression and the FITC-dextran concentration. D. The expression of occludin and claudin-5 were analyzed by western blotting. E and F. P120ctn expression was positively correlated with the expression of occludin and claudin-5. Group (0 h) was considered as control group. All data were expressed as mean ± SEM. *P<0.05, **P<0.01 vs. control, n=6.
Lack of p120ctn disrupted the BBB integrity through impairment of tight junctions
To examine whether p120ctn is involved in LPS-induced impairment of the BBB integrity, we first determined the effect of p120ctn knockdown on the leakage of FITC-dextran. Silencing efficiency of siRNA was detected by western blot and RT-PCR. Compared to control, siRNA targeting against p120ctn at 20 nM significantly decreased p120ctn protein expression by 77.24% and mRNA expression by 72.18%, respectively, while negative did not change the expression of p120ctn (Figure S1A, S1B). Blockade of p120ctn increased the leakage of FITC-dextran and accelerated LPS-induced increase of FITC-dextran (Figure 2A). Furthermore, deficiency of p120ctn remarkably decreased the expression of occludin and claudin-5 in basal level and after LPS stimulation (Figure 2B), indicating that lack of p120ctn-induced the BBB leakage was due to the loss of tight-junction proteins. On the contrary, we determined the effect of p120ctn over-expression on the BBB integrity. The efficiency of infection was evaluated by western blot and RT-PCR (Figure S1C, S1D). Over-expression of p120ctn obviously decreased the leakage of FITC-dextran and increased the expression of occludin and claudin-5 in basal level and after LPS stimulation (Figure 2C, 2D), suggesting p120ctn is critical for regulating the BBB permeability after LPS induced damage.
Figure 2.
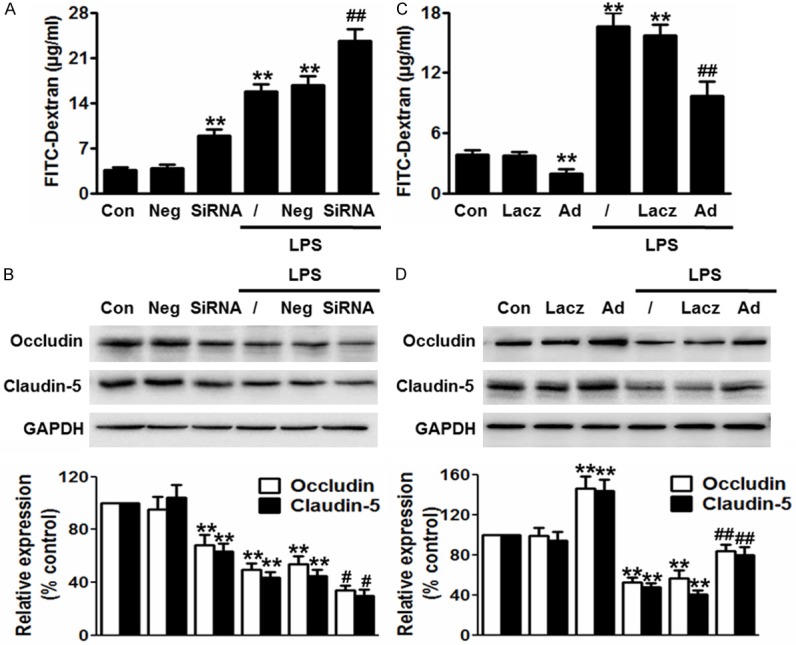
Effect of p120ctn on LPS-induced the disruption of BBB integrity. (A-D) P120ctn expression was knockdown by siRNA or over-expressed by adenovirus for 48 h, and then was incubated with LPS for another 24 h. The concentration of FITC-dextran (A and C) and the expression of occludin and claudin-5 (B and D) were measured as described in methods section. All data were expressed as mean ± SEM. **P<0.01 vs. control, #P<0.05, ##P<0.01 vs. LPS alone, n=6.
P120ctn modulated monocytes transendothelial migration in vitro BBB models
To further explore the biologic significance of LPS-decreased p120ctn expression, we examined the effect of p120ctn on monocytes migration. Compared with control group, blockade of p120ctn increased transenthelial migration of untreated monocytes by 1.4-fold and increased migration of LPS-treated monocytes by 7.2-fold (Figure 3A). However, over-expression of p120ctn inhibited monocytes transmigration at basal level and significantly diminished LPS-induced monocytes migration in vitro BBB model (Figure 3B).
Figure 3.
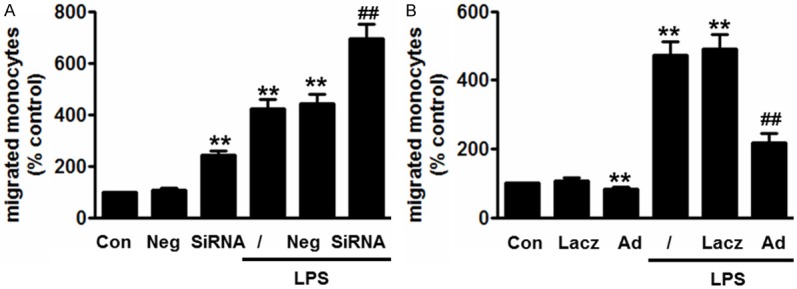
Effect of p120ctn on monocyte migration across in vitro BBB models. (A and B) HBMECs were treated with siRNA (A) or Ad-p120ctn (B) for 48 h first, followed by digestion on culture inserts. LPS-treated or -nontreated monocytes were labeled with 5 μM calcein-AM for 30 min. Then the labeled monocytes were added to the HBMECs and then migrate for 2 h. The fluorescence of the migrated monocytes was measured with a fluorescence plate reader. **P<0.01 vs. control, #P<0.05, ##P<0.01 vs. LPS alone, n=6.
Blockade of p120ctn promoted LPS-induced inflammatory response in HBMECS
To investigate whether the alteration of inflammatory response contributes to the enhanced permeability to LPS-induced BBB dysfunction in p120ctn knockdown cells, we determined the expression of the inflammatory mediators of ICAM-1, VCAM-1, endothelial activation marker-E-selectin, MCP-1, IL-1β and CXCL1 by RT-PCR. In HBMECs transfected with p120ctn siRNA showed increased mRNA expression of ICAM-1, VCAM-1, E-selectin, MCP-1, IL-1β and CXCL1 at basal level that persisted after treatment with LPS (Figure 4A). However, inverse results were obtained in Ad-p120ctn-infected cells (Figure 4B), indicating a protective role of p120ctn in LPS-induced inflammation in HBMECs.
Figure 4.
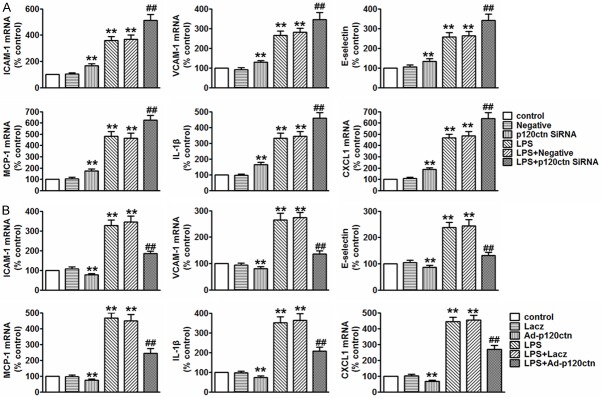
P120ctn inhibited LPS-induced secretion of inflammatory mediators in endothelial cells. (A and B). The levels of ICAM-1, VCAM-1, E-selectin, MCP-1, IL-1β and CXCL1 in HBMECs transfected with negative or p120ctn siRNA for 48 h prior to LPS treatment for 24 h (A, upper panel), or in HBMECs infected with Lacz or Ad-p120ctn for 48 h prior to LPS incubation (B, lower panel). All data are presented as mean ± SEM. **P<0.01 vs. control, ##P<0.01 vs. LPS alone, n=6.
P120ctn inhibited LPS-induced NF-κB activation by suppressing IKKβ/IκBα signaling
NF-κB is a major transcription factor involved in the inflammatory response [19]. To examine whether p120ctn mediates the activation of NF-κB, the luciferase reporter assay was performed. We found that over-expression of p120ctn could inactivate NF-κB promoter luciferase activity. LPS remarkably increased the luciferase activity of NF-κB, which was obviously ameliorated after up-regulation of p120ctn (Figure 5A). Additionally, NF-κB nuclear accumulation was analyzed by western blotting using p65 and p50 antibodies. HBMECs were co-transfected with Lacz or Ad-p120ctn for 48 h, followed by incubation of LPS over a range of indicated time points. As shown in Figures, translocation of p65 was triggered within 40 min and reached a maximal level at 80 min and declined thereafter in Lacz-treated cells. Although the nuclear accumulation of p65 showed peak within 40 min after up-regulation of p120ctn and was not different from those in Lacz-treated cells at 40 min, it was declined rapidly over the time points tested (Figure 5B). As expected, similar tendency was observed in the nuclear accumulation of p50 (Figure 5C), suggesting p120ctn regulates negatively NF-κB activation mainly through the inhibition of p65 and p50 nuclear accumulation.
Figure 5.
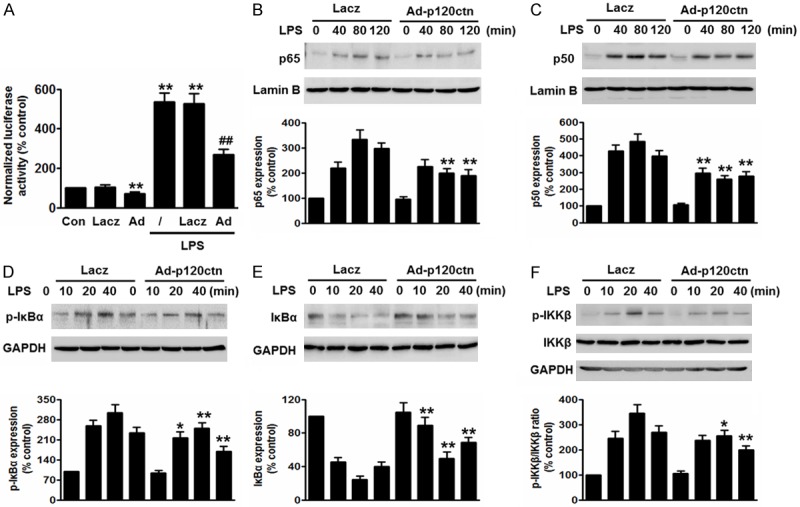
Over-expression of p120ctn inhibited LPS-induced NF-κB activation by suppressing IKKβ/IκBα signaling. (A) HBMECs were treated as mentioned in methods section. NF-κB luciferase activity was measured using the β-galactosidase activity as an internal control. (B-F) HBMECs were infected with Lacz or Ad-p120ctn for 48 h, and then incubated with LPS for different times as indicated. (B and C) Nuclear fractions were isolated and detected by western blot using p65 (B) and p50 (C) antibodies. Cell lysates were subjected to western blot analysis using p-IκBα (D), IκBα (E), and p-IKKβ and IKKβ (F) antibodies. All data are expressed as mean ± SEM. *P<0.05, **P<0.01 vs. control or similarity treated control, ##P<0.01 vs. LPS alone, n=6.
To answer whether NF-κB upstream signaling pathway, IKKβ/IκBα, is involved in p120ctn-inhibited NF-κB activation, IKKβ and IkBα phosphorylation, IkBα degradation was measured. We found LPS induced phosphorylation of IkBα at 10 min and reached maximal level at 20 min in Lacz-treated cells. However, the phosphorylation of IkBα was significantly inhibited after up-regulation of p120ctn, as compared to corresponding time points (Figure 5D). In addition, IkBα expression was decreased after LPS treatment and was peaked at 20 min. Nevertheless, over-expression of p120ctn dramatically antagonized against LPS-induced the degradation of IkBα (Figure 5E). Notably, no difference was observed in the onset of IKKβ phosphorylation. However, up-regulation of p120ctn obviously inhibited IKKβ phosphorylation after 10 min, as compared with those from Lacz-treated cells (Figure 5F). These data indicate that inhibition of IkBα degradation and IkBα and IKKβ phosphorylation underlies, at least in part, the inactivation of NF-κB after over-expression of p120ctn.
Discussion
In the present study, we observed p120ctn expression was significantly inhibited after LPS challenge in HBMECs. This finding was consistent with a previous study in human bronchial epithelial cells, which exhibited similar patterns in p120ctn expression following LPS treatment [15]. Moreover, we first demonstrated that LPS-decreased p120ctn expression was accompanied by an increase in BBB permeability and a decrease in tight junctions in the same time-dependent manner. These results together suggest that p120ctn is involved in the regulation of BBB function. In support, using knockdown and over-expression of p120ctn, we evidenced that deficiency of p120ctn increased BBB permeability, decreased tight junction proteins and enhanced monocyte migration across BBB model in vitro at basal level and after LPS treatment. Inverse results were obtained when the cells were over-expressed with p120ctn, demonstrating a protective role of p120ctn in BBB disruption.
During sepsis, BBB dysfunction impairs its ability to protect the brain from potentially neurotoxic substances, which triggers inflammatory response [7,9]. Therefore, elucidating the mechanisms linking inflammation and BBB dysfunction is important to understand sepsis pathogenesis and may provide a novel therapeutic strategy. Studies in p120ctn null cells and knockout mice have suggested a critical role of p120ctn in inflammation [17,18]. Nevertheless, it is uncertain whether p120ctn is involved in brain endothelial inflammatory response induced by LPS. Here, several representative adhesion molecules and chemokines including IVCAM-1, VCAM-1, E-selectin, MCP-1, IL-1β and CXCL1, were analyzed. We found that modulation of p120ctn expression itself could obviously affect the expression of these pro-inflammatory cytokines. Our results are consistent with a previous study, which showed an increase in pro-inflammatory cytokines expression in p120ctn null epidermal cells without stimulation [17]. However, the precise mechanism of p120ctn-mediated inhibition of inflammatory response in HBMECs has not been elucidated.
In most unstimulated cells, NF-κB, containing p65 (RelA) and p50 (NF-κB1), resides in the cytoplasm in an inactive complex with IκBs [20]. IκBα is the most important isoform of IκBs [15]. Upon on stimulation, IKK (inhibitor of NF-κB) β is phosphorylated, leading to trigger IκBα phosphorylation, ubiquitination and degradation [21]. Activated NF-κB allows p65 and p50 to translocate from the cytoplasm to the nucleus, which in turn, promotes gene transcription [22]. In this study, we found up-regulation of p120ctn inhibited LPS-induced IKKβ and IκBα phosphorylation, blocked LPS-induced IκBα degradation, and suppressed NF-κB translocation. Of note, luciferase reporter gene assay exhibited a notable change in NF-κB activity after only up-regulation of p120ctn expression without LPS treatment. This is different from a study which showed NF-κB activity remained unchanged in basal level in bronchial epithelial cell line 16HBE 14o-cell [15]. The varied results may be due to different cell-type-specific response.
In summary, our data confirm the protective role of p120ctn during BBB injury in vitro, and demonstrate that p120ctn inhibited LPS-induced inflammatory response by NF-κB inactivation, suggesting modulation of p120ctn expression may be a novel strategy to prevent and treat sepsis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 2.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 3.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 4.Matter K, Balda MS. Signalling to and from tight junctions. Nat Rev Mol Cell Biol. 2003;4:225–236. doi: 10.1038/nrm1055. [DOI] [PubMed] [Google Scholar]
- 5.Banks WA, Ercal N, Price TO. The blood-brain barrier in neuroAIDS. Curr HIV Res. 2006;4:259–266. doi: 10.2174/157016206777709447. [DOI] [PubMed] [Google Scholar]
- 6.Toborek M, Lee YW, Flora G, Pu H, Andras IE, Wylegala E, Hennig B, Nath A. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol. 2005;25:181–199. doi: 10.1007/s10571-004-1383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Z, Hu J, Gao X, Liang H, Liu Z. Activation of AMPK attenuates lipopolysaccharide-impaired integrity and function of blood-brain barrier in human brain microvascular endothelial cells. Exp Mol Pathol. 2014;97:386–392. doi: 10.1016/j.yexmp.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Mina F, Comim CM, Dominguini D, Cassol-Jr OJ, Dall Igna DM, Ferreira GK, Silva MC, Galant LS, Streck EL, Quevedo J, Dal-Pizzol F. Il1-beta involvement in cognitive impairment after sepsis. Mol Neurobiol. 2014;49:1069–1076. doi: 10.1007/s12035-013-8581-9. [DOI] [PubMed] [Google Scholar]
- 9.Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014;13:630–636. doi: 10.1016/S1474-4422(14)70017-1. [DOI] [PubMed] [Google Scholar]
- 10.Akrout N, Sharshar T, Annane D. Mechanisms of brain signaling during sepsis. Curr Neuropharmacol. 2009;7:296–301. doi: 10.2174/157015909790031175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dokka S, Shi X, Leonard S, Wang L, Castranova V, Rojanasakul Y. Interleukin-10-mediated inhibition of free radical generation in macrophages. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1196–1202. doi: 10.1152/ajplung.2001.280.6.L1196. [DOI] [PubMed] [Google Scholar]
- 12.Wright G, Singh IS, Hasday JD, Farrance IK, Hall G, Cross AS, Rogers TB. Endotoxin stress-response in cardiomyocytes: NF-kappaB activation and tumor necrosis factor-alpha expression. Am J Physiol Heart Circ Physiol. 2002;282:H872–879. doi: 10.1152/ajpheart.00256.2001. [DOI] [PubMed] [Google Scholar]
- 13.Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Hasko J, Krizbai IA. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds AB, Herbert L, Cleveland JL, Berg ST, Gaut JR. p120, a novel substrate of protein tyrosine kinase receptors and of p60v-src, is related to cadherin-binding factors beta-catenin, plakoglobin and armadillo. Oncogene. 1992;7:2439–2445. [PubMed] [Google Scholar]
- 15.Wang M, Li N, Li J, Ma Y, Li D, Qin L, Wang X, Wu R. Involvement of p120 in LPS-induced NF-kappaB activation and IL-8 production in human bronchial epithelial cells. Toxicol Lett. 2010;195:75–81. doi: 10.1016/j.toxlet.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 16.O’Donnell JJ 3rd, Zhuge Y, Holian O, Cheng F, Thomas LL, Forsyth CB, Lum H. Loss of p120 catenin upregulates transcription of pro-inflammatory adhesion molecules in human endothelial cells. Microvasc Res. 2011;82:105–112. doi: 10.1016/j.mvr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Moreno M, Song W, Pasolli HA, Williams SE, Fuchs E. Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci U S A. 2008;105:15399–15404. doi: 10.1073/pnas.0807301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prigent M, Barlat I, Langen H, Dargemont C. IkappaBalpha and IkappaBalpha /NF-kappa B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3-binding protein 2. J Biol Chem. 2000;275:36441–36449. doi: 10.1074/jbc.M004751200. [DOI] [PubMed] [Google Scholar]
- 21.Song W, Yang Z, He B. Bestrophin 3 ameliorates TNFalpha-induced inflammation by inhibiting NF-kappaB activation in endothelial cells. PLoS One. 2014;9:e111093. doi: 10.1371/journal.pone.0111093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nam J, Aguda BD, Rath B, Agarwal S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: experiments and modeling. PLoS One. 2009;4:e5262. doi: 10.1371/journal.pone.0005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.