

Abstract
Objective
Activation of inflammatory pathways plays a critical role in the development of abdominal aortic aneurysms (AAA). Notch1 signaling is a significant regulator of the inflammatory response; however, its role in AAA is unknown.
Methods and Results
In an angiotensin II (AngII)-induced mouse model of AAA, activation of Notch1 signaling was observed in the aortic aneurysmal tissue of Apoe−/− mice and a similar activation of Notch1 was observed in aneurysms of humans undergoing AAA repair. Notch1 haploinsufficiency significantly reduced the incidence of AAA in Apoe−/− mice in response to AngII. Reconstitution of bone marrow-derived cells from Notch1+/−; Apoe−/− mice (donor) in lethally irradiated Apoe−/− mice (recipient) decreased occurrence of aneurysm. Flow cytometry and immunohistochemistry demonstrated that Notch1 haploinsufficiency prevented the influx of inflammatory macrophages at the aneurysmal site by causing defects in macrophage migration and proliferation. Additionally, there was an overall reduction in the inflammatory burden in the aorta of the Notch1+/−;Apoe−/− mice as compared to the Apoe−/− mice. Lastly, pharmacologic inhibition of Notch1 signaling also prevented AAA formation and progression in Apoe−/− mice.
Conclusion
Our data suggest that decreased levels of Notch1 protect against the formation of AAA by preventing macrophage recruitment and attenuating the inflammatory response in the aorta.
Keywords: aneurysm, Notch1 signaling, inflammation, macrophages
Abdominal aortic aneurysm (AAA) is characterized by extensive remodeling of the aortic wall, which results in a weakened and dilated aorta that is prone to rupture.1–4 The incidence of AAA is associated with the clinical risk factors of smoking, hypercholesterolemia and hypertension, and it affects ~8% males over 65 years of age.1 Rupture of an AAA carries a risk of death up to 90% and is among the leading causes of death in the United States.5 Currently, there are no available non-surgical therapies to treat AAA.6, 7
A hallmark of AAA in both diseased humans and animal models is the presence of extensive inflammatory macrophages and lymphocytes by histologic examination.8 These infiltrating cells exacerbate tissue injury by releasing cytokines, chemokines and adhesion molecules.9, 10 Human aneurysmal aortas along with mouse models of AAA, demonstrate upregulation of IL-6 and MCP-1 in the aortic wall.8, 11, 12 Inflammatory macrophages cause activation of matrix metalloproteinases (MMPs)resulting in both the degradation of collagen and its associated collagenous matrix along with elastin fragmentation and smooth muscle cell apoptosis.13 These pathways act in synergy to progressively weaken the aortic wall, predisposing it to rupture.2, 3, 14
The Notch signaling pathway is important in a wide spectrum of developmental processes, but recently it has been implicated in the molecular pathogenesis of cancer and cardiovascular disease.15, 16 The Notch signaling pathway consists of a family of four Notch receptors (1–4) that interact with the Jagged and Delta family of ligands. In the canonical signaling pathway, Notch1 is activated after receptor-ligand binding at the cell surface resulting in the release and translocation of the Notch1 intracellular domain (NICD) to the nucleus where it functions as a transcriptional activator.17 Mice lacking Notch1 suffer embryonic lethality with profound cardiovascular defects, whereas heterozygous deletion of Notch1 is associated with mild aortic valve calcification.18, 19 Notch1 signaling has also been shown to be critical for the development and activation of lymphocytes and macrophages in various cell culture studies.15, 20, 21 The role of Notch1 signaling in inflammatory aneurysmal disease, however, has not been addressed.
To obtain insight into the role of Notch1 signaling in aortic aneurysm development, we generated Notch1+/−;Apoe−/− mice and conducted our studies in the AngII-induced mouse model of AAA. Although AngII-infusion studies have reported striking differences as compared to AAA pathophysiology in human patients, it resembles the human AAA in male propensity, localized dilation of aorta and histological features of the aortic injury.22 Here, we show that the Notch1 signaling pathway is activated in the aneurysmal aorta of the AngII-treated Apoe−/− mice, and that haploinsufficiency or pharmacologic inhibition of Notch1 significantly reduces the incidence of AAA by a macrophage-mediated mechanism.
Materials and Methods
A detailed description of the Methods is available in the online-only Supplement.
Generation of Notch1+/−;Apoe−/− Mice
Apoe−/− and Notch1+/−;Apoe−/− littermates were generated as described in the Supplemental Methods. Animal experiments were approved by Institutional Animal Care and Use Committee at the Research Institute at Nationwide Children’s Hospital.
Human Aortic Tissue Samples
Tissue specimens were collected from the infrarenal abdominal aorta of patients undergoing AAA repair (n=3). Non-aneurysmal infrarenal aortic tissue samples (n=3) were collected at the time of autopsy of individuals with no evidence of AAA. The collection of the human tissues was approved by the Institutional Review Board of Wayne State University, Detroit, Michigan, USA.
Angiotensin-II Infusion and DAPT Treatment
Mini-osmotic pumps containing AngII (1000ng/min/kg) or saline were implanted subcutaneously in anesthetized male mice (8–10 weeks old) following standard protocol. A group of Apoe−/− mice (n=10) were injected with a Notch inhibitor (DAPT).23, 24
Transabdominal Ultrasound Imaging
For in vivo imaging of the abdominal aorta, two dimensional (B-mode) transabdominal ultrasound images were obtained at weekly intervals after the implantation of osmotic pumps using a VisualSonics Vevo2100 imaging system (Ontario, Canada) with a mechanical transducer (MS400).
Histology and Immunostaining
Experimental animals were euthanized and the aortas were dissected, fixed in 10% formalin, and maximum aortic diameters measured. Serial sections were stained with hematoxylin and eosin (HE), elastin and immunohistochemistry (IHC) with antibodies to NICD, mouse monocyte-macrophage marker (Moma2), and monocyte chemotactic protein-1 (mcp1), as described.25, 26 Specificity of NICD, Moma2 and Mcp1 staining was determined using non-specific IgG against the source of host species.
Bone Marrow Transplantation Studies and Quantification of Leukocytes
Recipient mice were irradiated with single dose of 1000 Rads from a Cesium source. Bone marrow-derived cells were obtained from the tibia and femur of donor mice and were injected into the tail vein of 7–8 week old irradiated recipient mice (1 × 107 cells/ml).
Cell Isolation and Flow Cytometry
After 7 days of AngII infusion, macrophages were isolated from the abdominal aorta and lymphocytes were isolated from the abdominal aorta, spleen and peripheral blood. Cells were sorted with fluorescence-activated cell sorting (FACS) after staining with macrophage (Cd11b and Cd14) and lymphocyte markers (Cd3, Cd4, Cd8).
Macrophage Migration and Proliferation Studies
Mice were injected intraperitoneally with sterile thioglycollate broth to elicit in vivo macrophage proliferation. For the scratch assay, cells were grown as a monolayer and then scraped to create an injury. To assess proliferation, immunostaining was performed on these fixed cells with Ki67. For chemotactic-induced migration, cells were grown in 8 μM polycarbonate filter transwell membrane plates (upper chamber) and lower chamber media was supplemented with 100 nM n-formyl-met-leu-phe, (FMLP)27, 28. Bone marrow-derived macrophages (BMDM) were isolated from femurs and tibias of Apoe−/− and Notch1+/−;Apoe−/− mice. BMDM were stimulated for 24 hours with 100 ng/ml LPS and Ifn-γ (20 ng/ml) for M1 polarization or Il-4 (20 ng/ml) for M2 polarization.
RNA Isolation and qRT-PCR
The suprarenal aorta of approximately 5 mm in length was frozen in RNAlater, and mRNA extracted using TRIzol reagent after homogenizing tissue with TissueLyser II. Macrophages were washed and RNA extracted using RNAeasy kit (Qiagen). cDNA was synthesized and subjected to real-time quantitative RT-PCR (qRT-PCR).
Statistical Analysis
Statistical comparisons were performed using either Student’s t-test or one-way ANOVA followed by the Bonferroni’s Multiple Comparison Test. GraphPad PRISM V5.0 (San Diego, CA) was used for these comparisons and a P<0.05 was considered significant. For the statistical analysis of actual incidence, Fisher’s exact test was employed using the SAS software (Cary, NC). To analyze the BMT data for the two group comparisons at each time point, two sample t or Wilcoxon Sum Rank test and Log rank test was used depending on data distribution.
Results
Notch1 is Activated in Response to AngII in Apoe−/− Mice
To determine if Notch1 signaling is altered in the formation and progression of AAA, we examined the expression of NICD, the active form of Notch1, in the abdominal aorta of AngII-induced mouse model of AAA. NICD immunoreactivity was increased in the aneurysmal aorta of AngII-treated Apoe−/− miceas compared to Apoe−/− mice treated with saline at day 7 (Figure 1A–F) and day 28 (Figure 1G–L). At day 7, increased NICD signaling was restricted to the vicinity of vascular injury and was localized with inflammatory macrophages as determined by double immunofluorescence (IF) using the monocyte and macrophage marker (Moma2) (Supplemental Figure IA–F in the online-only data). At day 28, expression of NICD protein was extended to both the adventitial and medial layers of the aneurysmal abdominal aorta (Figure 1J–L and Supplemental Figure IG–L). Consistent with immunostaining, qRT-PCR data demonstrated a significant increase in mRNA expression of Notch1 and its downstream target, Hey1, in the abdominal aorta of Apoe−/− mice treated with AngII as compared to saline treated controls at day 7 and day 28 (Supplemental Figure II A–D). Consistent with our observations in mice, increased NICD signaling was also observed in the infrarenal abdominal aorta from patients undergoing AAA repair (Figure 1P–R) as compared to age-matched controls (Figure 1M–O) and appeared to be localized to inflammatory cells. Overall, activation of the Notch1 signaling pathway is observed in both mouse and human models of AAA.
Figure 1. Increased expression of Notch1 intracellular domain in aneurysmal aorta of Apoe−/− mice and human AAA.
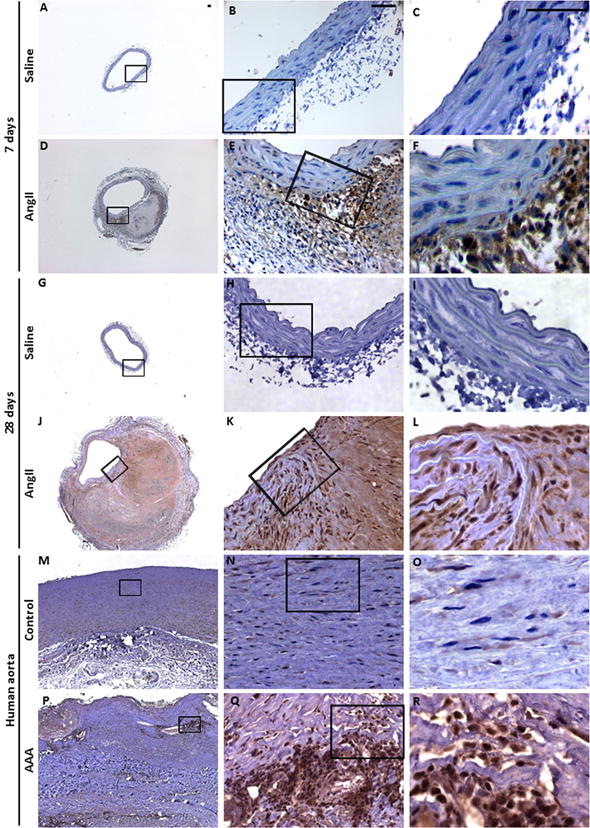
At day 7 of AngII infusion, transverse sections of the aneurysmal abdominal aorta from Apoe−/− mice (n=5) demonstrate increased expression of Notch1 intracellular domain (NICD) by immunohistochemistry treated with AngII (D–F) as compared to saline treated controls (A–C). At day 28, NICD immunoreactivity extended to the medial layer (J–L) as compared to controls (G–I). Increased NICD signaling was also observed in the infrarenal abdominal aorta from patients with AAA (P–R) as compared to age-matched controls (M–O). Scale bar, 50 μm.
Notch1 Haploinsufficiency Reduces the Occurrence of AAA in AngII-Induced Mouse Model
Next we examined if Notch1 haploinsufficiency affects the incidence of AAA in the AngII-induced mouse model of AAA. Notch1+/−, Apoe−/− and Notch1+/−;Apoe−/− mice were treated with AngII or saline for 28 days using published protocols.2, 14 Gross examination and transabdominal ultrasound imaging at 28 days of AngII demonstrated luminal expansion in suprarenal region of the abdominal aorta of Apoe−/− mice (Figure 2E, K), while no such luminal expansion in this region was observed in the Notch1+/−;Apoe−/− mice (Figure 2F, L). As expected, Notch1+/− and wild type mice treated with AngII and all mice treated with saline did not develop luminal expansion (Figure 2 and Supplemental Figure III). A similar rate of mortality was observed between Apoe−/− mice and Notch1+/−;Apoe−/− mice in response to AngII infusion (Supplemental Figure IV). No significant blood pressure (BP) differences were observed between Apoe−/− and Notch1+/−;Apoe−/− mice with AngII infusion suggesting that the protective effects of Notch1 haploinsufficiency were not related to BP (data not shown).
Figure 2. Notch1 haploinsufficiency reduces the incidence of AAA in Apoe−/− mice after 28 days of AngII infusion.
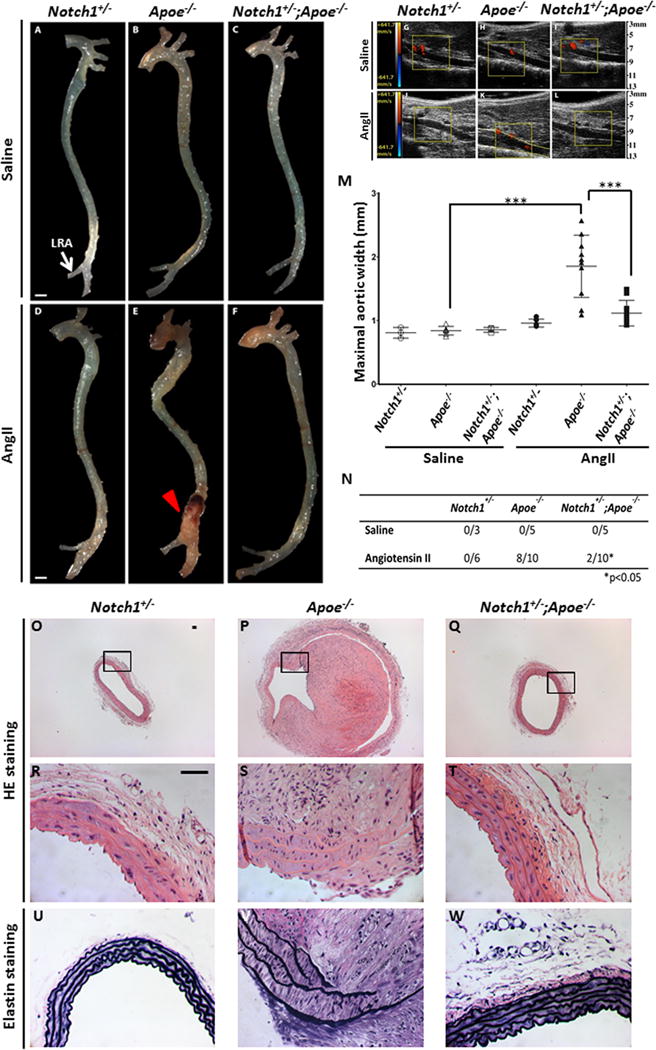
Representative aortas from mutant mice treated with saline (A–C) and AngII (D–F) are shown. LRA, left renal artery. (E) Aneurysm (red arrow) is observed in the suprarenal aorta of Apoe−/− mice treated with AngII but is absent Notch1+/− mice (D)and reduced in Notch1+/−;Apoe−/− mice treated with AngII (F). No aneurysms were seen in saline treated groups (A–C). Transabdominal ultrasound images (G–L) show that Notch1+/−;Apoe−/− mice (L) have decreased abdominal aortic luminal diameter as compared to Apoe−/− mice (K) when infused with AngII. Dashed yellow lines outline the expansion of lumen in (K). (M) Quantitative measurement of maximal aortic width (mm) in all experimental mice. Mean and standard deviations are shown. Each individual animal is represented by a symbol. ***P<0.001 when comparing Apoe−/− with AngII versus Apoe−/− with saline and Apoe−/− with AngII versus Notch1+/−;Apoe−/− treated with AngII. (N) Table showing incidence of AAA in Notch1+/−, Apoe−/− and Notch1+/−;Apoe−/− mice treated with saline or AngII. *P<0.05 when comparing Apoe−/− with AngII versus Notch1+/−;Apoe−/− treated with AngII. (O–T) Transverse sections of abdominal aorta were stained with hematoxylin and eosin (n=6 for each genotype). Adventitial remodeling along with inflammation, elastin degradation and thrombus were found in the aorta of Apoe−/− mice infused with AngII (P, S). No changes except mild adventitial thickening was observed in Notch1+/− (O, R) and Notch1+/−;Apoe−/− mice (Q, T) infused with AngII. (U–W) Elastin staining demonstrated elastin fragmentation in Apoe−/− mice (V) that was not seen in Notch1+/− (U) and Notch1+/−;Apoe−/− mice (W). Scale bar, 1 mm (A–F) and 50 μm (O–W).
Macroscopic measurement of the external diameter of suprarenal aorta demonstrated a significant increase in maximal aortic width of Apoe−/− mice treated with AngII versus saline treated Apoe−/− mice (Figure 2 E, M). In comparison, the maximal aortic width of suprarenal aorta was significantly reduced in the Notch1+/−;Apoe−/− mice treated with AngII as compared to Apoe−/− mice on similar treatment (Figure 2M). In fact, the aortic width of Notch1+/−;Apoe−/− mice in response to AngII was not significantly different from Notch1+/− mice treated with AngII or saline-treated controls (Figure 2M). In total, 80% (8/10) of Apoe−/− mice treated with AngII developed AAA, defined as a 50% increase in maximal aortic width, compared to 20% (2/10) of Notch1+/−;Apoe−/− mice treated with AngII (P<0.05; Figure 2N). Notably, except marginal adventitial thickening, no other characteristic features of AAA were observed in the Notch1+/−;Apoe−/− mice (Figure 2Q, T, W).
Histologically, Apoe−/− mice treated with AngII demonstrated cellular and architectural changes of typical AAA including thrombus formation, adventitial remodeling, inflammatory cell infiltration and elastin degradation (Figure 2P, S, V). The aortae of Notch1+/−;Apoe−/− mice infused with AngII displayed a well-defined lumen, with no elastin degradation and minimal infiltration of inflammatory cells (Figure 2Q, T, W). Histological examination of Notch1+/− mice treated with AngII or saline-treated controls also did not show any evidence of AAA (Figure 2O, R, U and Supplemental Figure III). Active caspase-3 immunostaining was prominent in the aortic smooth muscle cell (aSMC) enriched medial layer of aorta in the Apoe−/− mice infused with AngII as compared to Notch1+/−;Apoe−/− mice (Supplemental Figure V, which was not surprising as apoptosis of aSMCs is a characteristic feature of AAA).29 These data demonstrate that Notch1 haploinsufficiency significantly decreased the occurrence of pathological sequela associated with AAA in an established mouse model.
Notch1 Haploinsufficiency in Bone Marrow-Derived Cells Prevents AngII-Induced AAA Formation
The influx of inflammatory cells, consisting of macrophages and lymphocytes, to the site of aneurysm formation is critical for the development of AAA.2, 30, 31 We performed bone marrow transplantation (BMT) experiments to address the question if Notch1 haploinsufficiency in bone marrow-derived cells alters aneurysmal development in Apoe−/− mice (Figure 3; Supplemental Figure VI). After optimizing the irradiation and validating the repopulation procedure, the Apoe−/− (Group I, n=12) and Notch1+/−;Apoe−/− (Group II, n=12) were irradiated and reconstituted with bone marrow-derived cells harvested from Notch1+/−;Apoe−/− and Apoe−/− mice, respectively. Two additional groups of mice were also studied to serve as control groups: irradiated Apoe−/− mice were repopulated with bone marrow-derived cells harvested from Apoe−/− mice (Group III, n=8) and Apoe−/− mice without BMT (Group IV, n=6). Transabdominal ultrasound measurements and gross examination demonstrate that infusion of AngII into Apoe−/− mice with Notch1+/−;Apoe−/− bone marrow-derived cells (Group I) significantly decreased the progression of luminal expansion as compared to Group II (Figure 3; Supplemental Figure VI). Notch1+/−;Apoe−/− mice repopulated with bone marrow-derived cells from Apoe−/− mice (Group II) not only augmented progression of luminal expansion but also increased aortic rupture associated mortality during 28 days of AngII infusion (P<0.001, Figure 3L). Consistent with other studies, BMT in irradiated Apoe−/− mice (Group III, n=8) led to lower incidence of AngII-induced AAA compared with non-irradiated Apoe−/− mice (Group IV; data not shown).32 Notch1 haploinsufficiency did not affect total leukocyte count, monocyte or lymphocyte count in the peripheral blood (data not shown). Overall, the BMT studies suggest that protective effects of Notch1 haploinsufficiency are preserved after transplantation of bone marrow-derived cells from Notch1+/−;Apoe−/− mice into Apoe−/− mice.
Figure 3. Notch1 haploinsufficiency in bone marrow-derived cells decreased AAA incidence and mortality rate in Apoe−/− mice.
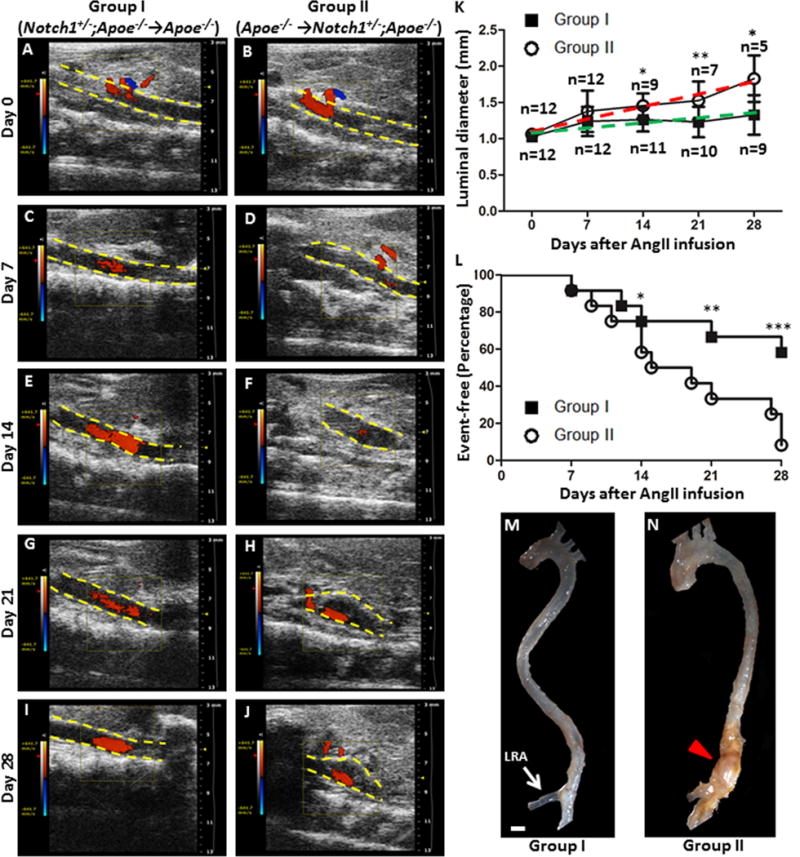
Bone marrow-derived cells from Apoe−/− mice increased AAA disease in Notch1+/−;Apoe−/− (Group II; Apoe−/− →Notch1+/−; Apoe−/−, n=12) mice as compared Apoe−/− mice which received bone marrow derived cells from Notch1+/−;Apoe−/− mice (Group I; Notch1+/−;Apoe−/−→Apoe−/−, n=12). The representative transabdominal ultrasound images at day 0 (A, B), day 7 (C, D), day 14 (E, F), day21 (G, H) and day 28 (I, J) demonstrating an increase in the suprarenal luminal diameter in Group II as compared to Group I. (K) Significant increase in the rate of luminal expansion in Group II (red) as compared to Group I (green) after 2nd week of AngII infusion (L). Significantly increased number of disease events, defined by either 50% increase in aortic diameter or sudden death. Representative images of the whole aorta from Group I and Group II mice are shown in M and N. Scale bar, 1 mm. ***P<0.001; **P<0.01; *P<0.05.
Notch1 Haploinsufficiency Reduces Macrophage Infiltration at Site of AAA
Monocyte recruitment and macrophage infiltration occur during early and later stages of AAA development; therefore we examined the suprarenal abdominal aorta at day 7 and 28 of AngII infusion for the expression of Moma2, a marker of activated macrophages. Similar to AngII exposure for 28 days (Figure 2), the aorta of Apoe−/− mice infused with AngII for 7 days showed signs of luminal expansion and elastin degradation, which was absent in Notch1+/−;Apoe−/− mice (Supplemental Figure VII). At both day 7 and day 28 of AngII infusion, the expression of Moma2 in the abdominal aorta of Notch1+/−;Apoe−/− mice was decreased as compared to Apoe−/− mice as determined by IHC (Figures 4A–H). Mcp1 plays a critical role in the recruitment and infiltration of macrophages in response to external stimuli. IHC of the aneurysmal region revealed a significantly increased expression of Mcp1 in the inflammatory cells of Apoe−/− mice, which was abrogated in the aorta of Notch1+/−; Apoe−/− mice at both 7 and 28 days of AngII treatment (Figures 4I–P, Supplemental Figure VIII). Of note, although marginal Mcp1 expression was observed in the SMC-enriched medial layer of Notch1+/−;Apoe−/− mice, the adventitial thickening observed in the abdominal aortic region of Notch1+/−;Apoe−/− mice was devoid of MCP-1 positive inflammatory cells (arrowheads in Figure 4F, H, N, P).
Figure 4. Minimal macrophage recruitment and Mcp1 expression in the aorta of Notch1+/−;Apoe−/− mice after 7 and 28 days of AngII infusion.

Immunohistochemistry showing decreased expression of Moma2, a macrophage marker, in the aneurysmal tissue at day 7 and day 28 of AngII-treated Notch1+/−ApoE−/− mice (B, F, and D, H) as compared to ApoE−/− mice (A, E and C, G). Decreased Mcp1 expression in the abdominal aorta of Notch1+/−;ApoE−/− mice (J, N and L, P) in response to AngII as compared to ApoE−/− mice (I, M and K, O). Moma2 and Mcp1 expression was specifically decreased in adventitial layer (F, H, N, P; arrowheads). (Q) qRT-PCR demonstrates reduced mRNA levels of Mcp1, Il6, Cxcl10, Vegf, iNOS, Icam1 and Vcam1 in the abdominal aorta of Notch1+/−;Apoe−/− mice as compared to Apoe−/− mice treated with AngII for 28 days (n=3, in triplicate). (R) qRT-PCR demonstrates reduced expression of a similar panel of inflammatory mediators in LPS-stimulated primary macrophages isolated from the peritoneal cavity of Notch1+/−;Apoe−/− mice as compared to Apoe−/− mice. Macrophages from 5 mice were pooled for each experiment; data shown represents 3 experiments performed in triplicate. ***P<0.001; **P<0.01; *P<0.05. Means and standard deviations are shown. Scale bar, 50 μm.
AngII is postulated to play a central role in initiating inflammation in the aorta by increasing the expression of chemokines, adhesion molecules and cytokines in macrophages.2, 30, 33 To determine if Notch1 haploinsufficiency reduces the inflammatory response in the AngII model of AAA, we examined the mRNA expression levels of a panel of cytokines, chemokines and pro-inflammatory mediators by qRT-PCR in the aortas of Apoe−/− and Notch1+/−; Apoe−/− mice treated with AngII for 28 days. Significant reduction of the expression of chemokines and cytokines (Mcp1, Il6, Cxcl10) and the pro-inflammatory mediators (Vegf, iNOS, Icam1, Vcam1) was found in the abdominal aorta of Notch1+/−;Apoe−/− mice as compared to Apoe−/− mice (Figure 4Q).
To determine if a similar decreased inflammatory response occurs ex vivo, we isolated primary macrophages from the peritoneal cavity of Apoe−/− and Notch1+/−; Apoe−/− mice and subjected them to LPS stimulation (100 ng/ml) for 3 hours. LPS, a potent activator of macrophages, is known to up-regulate expression of Notch1 and its downstream targets and mimic inflammatory response of AAA in as similar fashion.15, 34, 35 The expression of Mcp1, Il6, Tnf-α and iNOS in macrophages from Notch1+/−;Apoe−/− mice was ~60% less than in macrophages from Apoe−/− mice in response to LPS treatment (Figure 4R). As expected, the expression of Notch1 was also reduced by about 60% in the macrophages from Notch1+/−; Apoe−/− mice (data not shown). In summary, Notch1 haploinsufficiency decreases macrophage infiltration and resultant inflammatory response, as measured by cytokine and chemokine expression, in response to external stress.
Notch1 Haploinsufficiency Causes Defects in Macrophage Migration and Proliferation by Differentially Regulating M1/M2 Polarization
Given the limitations of IHC in determining the specificity and difficulty in quantification, we assessed direct infiltration of active macrophages and lymphocytes at the site lesion by FACS analysis. Consistent with our IHC observations, the suprarenal abdominal aorta of Notch1+/−;Apoe−/− mice contained a decreased number of inflammatory macrophages with Cd11b and Cd14 staining as compared to Apoe−/− mice after 7 days of AngII infusion (Figure 5A–C). Notch1 haploinsufficiency did not alter the differentiation of Cd4+ or Cd8+ lymphocytes at the site of injury, although total number of Cd3+ T lymphocytes was significantly decreased in the suprarenal aorta of Notch1+/−; Apoe−/− mice as compared to Apoe−/− mice after 7 days of AngII infusion as determined by FACS analysis and immunostaining (Supplemental Figure IX). Interestingly, no significant difference was observed in the absolute number of Cd3+ T cells in the spleen (Supplemental Figure IX) or peripheral blood (data not shown) of these experimental mice.
Figure 5. Notch1 haploinsufficiency decreases macrophage recruitment, migration and M1 polarization.
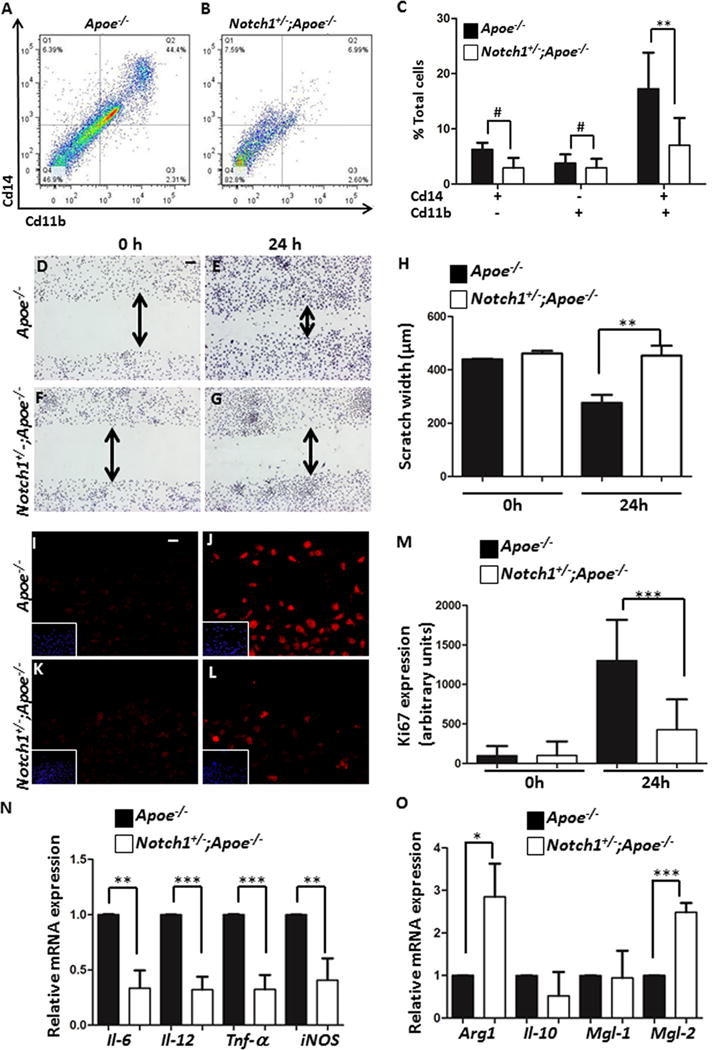
(A–C) Flow cytometry demonstrates a decreased number of Cd11b+Cd14+ macrophages in the abdominal aorta of Notch1+/−;Apoe−/− mice (B) as compared to Apoe−/− mice (A). (C) Quantification of the FACS data (n=6 for each genotype). (D–G) Scratch assay showing decreased migration of Notch1+/−;Apoe−/− macrophages to the site of injury after 24 h with quantification shown in (H). (I–L) Decreased proliferation of Notch1+/−;Apoe−/− macrophages as detected by Ki-67 immunostaining. DAPI staining (blue) is shown in inserts and quantification is in (M). (N) qRT-PCR demonstrates reduced expression of M1-polarization associated genes with LPS (100 ng/ml)/Ifn-γ (20 ng/ml) stimulation of BMDM of Notch1+/−;Apoe−/− mice as compared to Apoe−/− mice. (O) Notch1 haploinsufficiency increased M2-polarization associated genes in BMDM in response to Il4 (20 ng/ml) as compared to Apoe−/− mice (n=3). Scale bar, 50μm. ***P<0.001; **P<0.01; *P<0.05.
Since it has been shown that total lymphocyte deficiency had no detectable effect on the development of AAA in the AngII mouse model,36 we focused on the effects of Notch1 haploinsufficiency on macrophage functions. We first examined if Notch1 signaling has direct effects on macrophage migration or proliferation. In a classical scratch assay, Notch1 haploinsufficiency significantly decreased the migration of peritoneal-derived primary macrophages, towards the injury site after 24h (Figure 5D–H). After 24h, decreased proliferation was observed in Notch1+/−; Apoe−/− macrophages as compared to Apoe−/− macrophages as determined by staining with proliferation marker (Ki67; Figure 5I–M). Increased Ki67 staining localized with macrophages was also observed in the abdominal aorta of Apoe−/− mice, whereas no such co-expression was observed in the aorta of Notch1+/−;Apoe−/− mice (Supplemental Figure XA–F). In a transwell culture system, FMLP (a potent and specific chemotactic agent)-induced migration of primary macrophages was significantly reduced with Notch1 haploinsufficiency (Supplemental Figure XG). To determine, if Notch1 haploinsufficiency affects proliferation of primary macrophages in vivo, thioglycollate injection was used to elicit recruitment of macrophages to the peritoneal cavity. Notch1 haploinsufficiency resulted in the diminished influx of macrophages by almost 50% at day 4 of the thioglycollate infusion (Supplemental Figure XH). Recent studies have suggested integral role for Nocth1 to regulate M1 polarization of naïve macrophages mediated by synthesis of interferon regulatory factor 8 (Irf8) protein.37 As expected, expression of Irf8 was upregulated in the abdominal aorta of Apoe−/− mice in response to AngII at day 7 whereas Notch1 haploinsufficiency prevented upregulation of Irf8 (Supplemental Figure X I, J). To determine whether Notch1 regulates polarization of macrophages, we evaluated markers for inflammatory M1-associated genes in response to LPS/Ifn-γ stimulation and M2-associated genes in response to Il4 stimulation in the bone-marrow derived macrophages (BMDM). Increased expression of Il6, Il12, Tnf-α and iNOS genes was observed in the BMDM of Apoe−/− mice in response to LPS/Ifn-γ whereas no change was seen with Notch1 haploinsufficiency (Figure 5N). More importantly, in response to Il4, the expression of arginase1 (Arg1) and macrophage galactose N-acetyl-galactosamine specific lectin 2 (Mgl2) was higher in BMDM from Notch1+/−;Apoe−/− mice as compared to Apoe−/− mice suggesting that Notch1 deficient macrophages are polarized towards the M2 fate (Figure 5O). The M1 polarization of macrophages in response to LPS/Ifn-γ was further confirmed by FACS analysis (Supplemental Figure X; M, N). M1 polarization of macrophages in Apoe−/− mice was associated with increased Irf8 staining, which was reduced by Notch1 haploinsufficiency (Supplemental Figure X; K, L). Taken together, the results demonstrate that Notch1 haploinsufficiency causes defects in the migration and proliferation functions of macrophages by differentially regulating M1/M2 polarization of macrophages thus preventing them from infiltrating the site of aneurysm formation in the AngII mouse model.
Pharmacologic Inhibition of Notch Signaling Attenuates Aneurysm Development in an AngII-Induced Mouse Model of AAA
While the BMT studies and flow cytometry data demonstrate a specific role for Notch1 signaling in macrophages, Notch1 is known to be necessary for the proper development of the vasculature.18 To determine if protective effects of Notch1 haploinsufficiency on AAA formation are not secondary to the embryonic functions of Notch1 and to investigate therapeutic potential of Notch1 inhibition, we examined whether pharmacologic inhibition of Notch1 protects against the formation of AAA in adult Apoe−/− mice infused with AngII. Mice were treated with DAPT (10 mg/kg) three times a week, starting one week prior to initiating the AngII infusion or 3 days after the AngII infusion and continuing for 28 days.23, 24 These time periods were chosen to determine if DAPT mimics the effects of Notch1 haploinsufficiency on AAA formation and to determine its therapeutic potential to prevent the progression of AAA after a small aortic dilation is established (Supplemental Figure XI). Treatment with DAPT resulted in a significant reduction in the aortic diameter and incidence of AAA at both time intervals as compared to untreated Apoe−/− mice and was similar to Notch1+/−; Apoe−/− mice (Figure 6A–D). Histologic analysis of the aorta of the Apoe−/− mice treated with DAPT 7 days prior to AngII infusion Figure 6F, I) demonstrated normal aortic wall architecture without infiltration of inflammatory cells as compared to untreated mice (Figure 6E, H). Although marginal adventitial thickening was observed in Apoe−/− mice treated with DAPT treatment 3 days after AngII infusion, no visible inflammation was observed (Figure 6G, J). Further, pharmacologic inhibition of Notch (DAPT) had similar defects in the migration and proliferation of macrophages in a murine macrophage cell line (RAW 264.7 cells; data not shown). DAPT also down regulated the inflammatory response of these macrophages in response to LPS as compared to non-DAPT treated cells (data not shown). Significantly decreased gene expression of Notch1 and its downstream Hey1 expression in the abdominal aorta of these mice was observed with DAPT treatment as compared to vehicle-treated Apoe−/− mice (Supplemental Figure XII A–B). These studies demonstrate that the reduced incidence of AAA seen with genetic deficiency of Notch1 are not the result of developmental differences in the aortic wall and also demonstrate a potential therapeutic strategy for treatment of AAA.
Figure 6. Pharmacologic inhibition of Notch signaling attenuates aneurysm development in an AngII-induced mouse model of AAA.
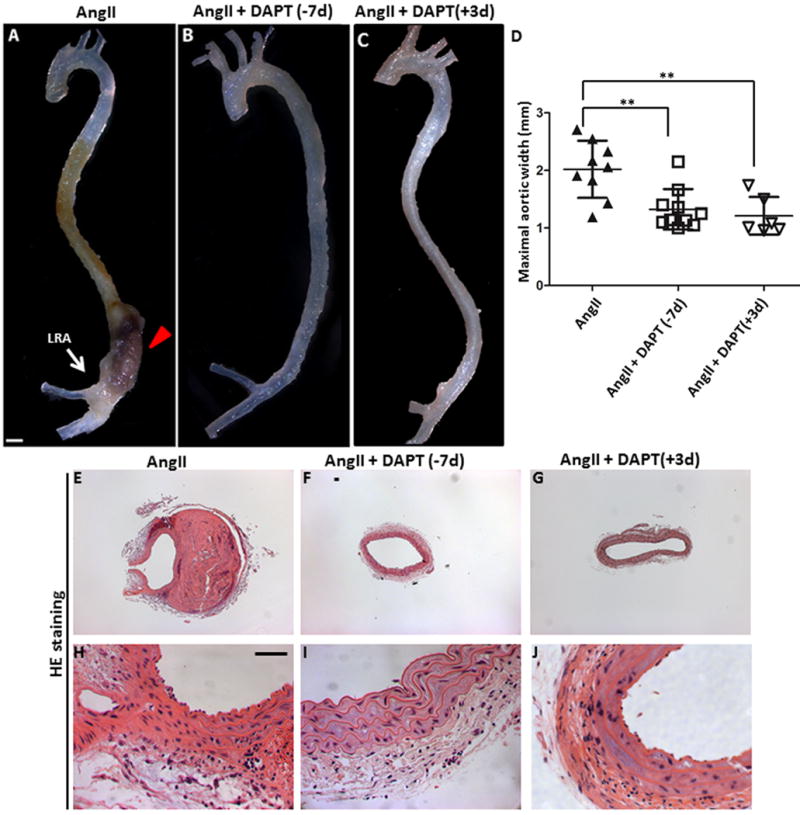
Representative aortas from (A) Apoe−/− (n=9), (B) Apoe−/− mice treated with 10 mg/kg of DAPT 7 days prior to AngII infusion (n=10) and (C) Apoe−/− mice treated with 10 mg/kg of DAPT 3 days after AngII infusion. All mice received AngII infusion for 28 days. Scale bar, 1mm. (D) Quantitative measurement of aortic width (mm) in these mice. Each symbol represents an individual animal. (E–J) Histologic sections show no evidence of aneurysm in Apoe−/− mice that received DAPT (F, I, G, J). Scale bar, 50 μm. Means and standard deviations are shown in D.
Discussion
The Notch1 signaling pathway has been implicated in numerous developmental processes and disease states.15, 16, 18 Here, we provide the first evidence that Notch1 signaling is activated in the abdominal aorta from the AngII-induced mouse model of AAA and in patients with AAA. We also demonstrate that Notch1 haploinsufficiency significantly reduces the incidence of AAA in Apoe−/− mice infused with AngII. The protective effects of Notch1 haploinsufficiency on AAA are mediated by decreased recruitment of inflammatory macrophages at the site of aneurysm. Bone marrow studies demonstrate that Notch1 haploinsufficiency in inflammatory cells is sufficient to reduce the development of AAA. Gene expression studies demonstrate that Notch1 haploinsufficiency results in decreased expression of Il6, Mcp1 and adhesion molecules, which play critical roles in macrophage recruitment at the site of injury in response to stress. Flow cytometry confirmed the selective recruitment of inflammatory macrophages in the suprarenal aorta in response to AngII. Notch1 inhibition reduces M1 polarization of macrophages and promotes their M2 fate thus causing defects in macrophage migration and proliferation. Consistent with this, pharmacologic inhibition of Notch signaling by DAPT attenuated dilation of the abdominal aorta in the AngII-induced model of AAA. These findings suggest that Notch1 actively participates in the process of inflammation by directly regulating cytokines and chemokines critical for macrophage recruitment in the pathogenesis of aneurysm (Supplemental Figure XIII). Deficiency of Notch1, by preventing this inflammatory cascade, preserves the anti-inflammatory environment and normal aortic wall architecture thereby attenuating dilation of the suprarenal abdominal aorta.
While the role of the Notch signaling pathway as a critical regulator of cell fate during development is well established,16, 38 there is emerging evidence that Notch signaling is also critical in the pathogenesis of a variety of inflammatory diseases. Although NOTCH1 mutations have been associated with bicuspid valve, aortic valve calcification and thoracic aortic aneurysm (TAA), no study has exclusively examined the role of Notch1 in the pathogenesis of AAA.39–42 A recent study showed strong correlation of bicuspid valve with TAA, but interestingly none of the patients in their study had AAA, suggesting different pathophysiology for TAA and AAA.43 There is growing evidence that activation of the Notch1 signaling pathway regulates the expression of an inflammatory cascade that includes Il6, Mcp1 along with a variety of key inflammatory genes such as iNOS, Cxcl10, Icam1 and Vcam.15 A recent study suggests that Notch1 signaling alone is sufficient to switch on Il6 mRNA transcription in macrophages.12 Although, deficiency of Il6 and Mcp1 has been shown to prevent AAA formation, role of Cxcl10 receptor in AAA is conflicting. While increased expression of Cxcl10 is reported in AngII-exposed Apoe−/− mice, deficiency of Cxcl10 has also been shown to augment AAA formation.44 Overall, our work provides the first evidence suggesting a role for Notch1 signaling in AAA and further studies are required to decipher the specific role of these inflammatory factors in AAA.
Although remarkable progress has been made in the recent years in understanding the role of Notch1 signaling in immune system, the complexity of Notch signaling because of its multiple receptors and ligands is not completely understood. Data obtained from our chemotactic studies and immunostaining combined with BMT studies support the hypothesis that Notch1 haploinsufficiency primarily interferes with the migration, proliferation and activation of macrophages. Consistent with our findings, a recent study reported that myeloid specific reduction in Notch1 decreases macrophage recruitment and localization during angiogenesis.45 However, we do not exclude the possibility that Notch1 regulates expression of pro-inflammatory cytokines in other cells including aortic SMCs and ECs in a similar manner as in macrophages. It is worthwhile to mention here that a modest levels of Mcp1 expression observed in the intimal endothelial layer and medial smooth muscle layer of the aorta in Notch1+/−;Apoe−/− mice seem to be insufficient for the recruitment of macrophages (Figure 4 and Supplemental Figure VIII). Since monocyte infiltration appears to precede aSMC apoptosis in developing AAA, factors released by monocyte and macrophages may play a primary role in the overall perpetuation of inflammation by regulating the function of aSMCs and ECs in the setting of AAA.46, 47 Further studies are needed to decipher the crosstalk between macrophages and aSMCs in the setting of AAA.
In our study, Notch1 haploinsufficiency reduced infiltration of Cd3+ T lymphocytes at the site of aneurysm formation. Consistent with other studies, differential effects of Notch1 haploinsufficiency on cytotoxic Cd4+ or Cd8+ T lymphocyte differentiation in the aneurysmal aorta or spleen were not detectable in response to AngII suggesting that Notch1 pathway activation is a not a feature of all T cells involved in vascular inflammation and is disease specific.21, 48 Moreover, total lymphocyte deficiency, achieved by developing Apoe−/− mice that lacked the recombination activating gene-1 (Rag1) gene, fails to affect the development of AAA in AngII mouse model suggesting that lymphocytes are dispensable for AAA.36
The clinical management of AAA is based on the control of primary risk factors such as tobacco use, dyslipidemia, hypertension, atherosclerosis and infection.6, 7 To prevent the progression or stimulate the regression of established AAA, there is a critical need to develop pharmacologic interventions that can selectively target one or more features of AAA. A number of strategies have been proposed to achieve the objective of impeding AAA progression, but available options fall short of this goal. Our data demonstrate that Notch1 is an important player in the inflammatory process in the setting of AAA and suggest that treatment with Notch1-specific inhibitors may be a potentially promising strategy for slowing aneurysm development. Notch1 inhibitors are s potential therapeutic agents for cancer and leukemia.49, 50 Based upon recent findings that Notch inhibition decreases macrophage infiltration45 and directly regulates macrophage polarization37, it is plausible that targeting Notch1 signaling may hold a promising target-based therapy for developing AAA. Beneficial effects of pharmacologic inhibition of γ-secretase have been reported to blunt the inflammatory response in a mouse model of atherosclerosis and reduce plaque formation51 and also to prevent vascular inflammation in an experimental model of giant cell arthritis.48 Further investigations in other animal models of AAA (elastase and CaCl2 infusion) are needed to determine the clinical utility of Notch1 inhibition as a pharmacologic therapy.11, 14, 52
Supplementary Material
Acknowledgments
The authors wish to thank Dave Dunaway in the Flow Cytometry Core, members of the Morphology Core at the Research Institute at Nationwide Children’s Hospital for technical support, Yongjie Miao for the statistical analysis of the data and Drs. P.A. Lucchesi and B. Lilly for helpful comments on the manuscript.
Sources of Funding
This work was supported by an American Heart Association-National Center Scientific Development Grant to C.P.H. and funding from the Research Institute at Nationwide Children’s Hospital and NIH/NHLBI to V.G.
Footnotes
Disclosures
Chetan P. Hans and Vidu Garg have applied for a patent related to this work.
References
- 1.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. doi: 10.1038/nrcardio.2010.180. [DOI] [PubMed] [Google Scholar]
- 2.Daugherty A, Cassis LA. Mechanisms of abdominal aortic aneurysm formation. Curr Atheroscler Rep. 2002;4:222–227. doi: 10.1007/s11883-002-0023-5. [DOI] [PubMed] [Google Scholar]
- 3.Daugherty A, Manning MW, Cassis LA. Angiotensin ii promotes atherosclerotic lesions and aneurysms in apolipoprotein e–deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boddy AM, Lenk GM, Lillvis JH, Nischan J, Kyo Y, Kuivaniemi H. Basic research studies to understand aneurysm disease. Drug News Perspect. 2008;21:142–148. [PubMed] [Google Scholar]
- 5.Klink A, Hyafil F, Rudd J, Faries P, Fuster V, Mallat Z, Meilhac O, Mulder WJM, Michel J-B, Ramirez F, Storm G, Thompson R, Turnbull IC, Egido J, Martin-Ventura JL, Zaragoza C, Letourneur D, Fayad ZA. Diagnostic and therapeutic strategies for small abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:338–347. doi: 10.1038/nrcardio.2011.1. [DOI] [PubMed] [Google Scholar]
- 6.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurks R, Hoefer IE, Vink A, Pasterkamp G, Schoneveld A, Kerver M, de Vries JPPM, Tangelder MJ, Moll FL. Different effects of commonly prescribed statins on abdominal aortic aneurysm wall biology. Eur J of Vasc and Endovasc. 2010;39:569–576. doi: 10.1016/j.ejvs.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 8.Parry DJ, Al-Barjas HS, Chappell L, Rashid ST, Ariëns RAS, Scott DJA. Markers of inflammation in men with small abdominal aortic aneurysm. J Vasc Surg. 2010;52:145–151. doi: 10.1016/j.jvs.2010.02.279. [DOI] [PubMed] [Google Scholar]
- 9.Rush C, Nyara M, Moxon J, Trollope A, Cullen B, Golledge J. Whole genome expression analysis within the angiotensin ii-apolipoprotein e deficient mouse model of abdominal aortic aneurysm. BMC Genomics. 2009;10:298. doi: 10.1186/1471-2164-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenk G, Tromp G, Weinsheimer S, Gatalica Z, Berguer R, Kuivaniemi H. Whole genome expression profiling reveals a significant role for immune function in human abdominal aortic aneurysms. BMC Genomics. 2007;8:237. doi: 10.1186/1471-2164-8-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colonnello JS, Hance KA, Shames ML, Wyble CW, Ziporin SJ, Leidenfrost JE, Ennis TL, Upchurch GR, Jr, Thompson RW. Transient exposure to elastase induces mouse aortic wall smooth muscle cell production of mcp-1 and rantes during development of experimental aortic aneurysm. J Vasc Surg. 2003;38:138–146. doi: 10.1016/s0741-5214(03)00125-3. [DOI] [PubMed] [Google Scholar]
- 12.Wongchana W, Palaga T. Direct regulation of interleukin-6 expression by notch signaling in macrophages. Cell Mol Immunol. 2012;9:155–162. doi: 10.1038/cmi.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keeling WB, Armstrong PA, Stone PA, Bandyk DF, Shames ML. An overview of matrix metalloproteinases in the pathogenesis and treatment of abdominal aortic aneurysms. Vasc Endovasc Surg. 2005;39:457–464. doi: 10.1177/153857440503900601. [DOI] [PubMed] [Google Scholar]
- 14.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb and Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 15.Monsalve E, Perez MA, Rubio A, Ruiz-Hidalgo MJ, Baladron V, Garcia-Ramirez JJ, Gomez JC, Laborda J, Diaz-Guerra MJ. Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol. 2006;176:5362–5373. doi: 10.4049/jimmunol.176.9.5362. [DOI] [PubMed] [Google Scholar]
- 16.de la Pompa José L, Epstein Jonathan A. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Developmental Cell. 2012;22:244–254. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bray SJ. Notch signalling: A simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 18.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 19.Nus M, MacGrogan D, Martínez-Poveda B, Benito Y, Casanova JC, Fernández-Avilés F, Bermejo J, de la Pompa JL. Diet-induced aortic valve disease in mice haploinsufficient for the notch pathway effector rbpjk/csl. Arterioscler Thromb and Vasc Biol. 2011;31:1580–1588. doi: 10.1161/ATVBAHA.111.227561. [DOI] [PubMed] [Google Scholar]
- 20.Radtke F, Wilson A, Mancini SJ, MacDonald HR. Notch regulation of lymphocyte development and function. Nat Immunol. 2004;5:247–253. doi: 10.1038/ni1045. [DOI] [PubMed] [Google Scholar]
- 21.Tanigaki K, Honjo T. Regulation of lymphocyte development by notch signaling. Nat Immunol. 2007;8:451–456. doi: 10.1038/ni1453. [DOI] [PubMed] [Google Scholar]
- 22.Bruemmer D, Daugherty A, Lu H, Rateri DL. Relevance of angiotensin ii-induced aortic pathologies in mice to human aortic aneurysms. Ann NY Acad Sci. 2011;1245:7–10. doi: 10.1111/j.1749-6632.2011.06332.x. [DOI] [PubMed] [Google Scholar]
- 23.Lanz TA, Himes CS, Pallante G, Adams L, Yamazaki S, Amore B, Merchant KM. The γ-secretase inhibitor n-[n-(3,5-difluorophenacetyl)-l-alanyl]-s-phenylglycine t-butyl ester reduces aβ levels in vivo in plasma and cerebrospinal fluid in young (plaque-free) and aged (plaque-bearing) tg2576 mice. J Pharmacol Exp Ther. 2003;305:864–871. doi: 10.1124/jpet.102.048280. [DOI] [PubMed] [Google Scholar]
- 24.Loane DJ, Pocivavsek A, Moussa CEH, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature Medicine. 2009;15:377–379. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hans CP, Zerfaoui M, Naura AS, Catling A, Boulares AH. Differential effects of parp inhibition on vascular cell survival and acat-1 expression favouring atherosclerotic plaque stability. Cardiovasc Res. 2008;78:429–439. doi: 10.1093/cvr/cvn018. [DOI] [PubMed] [Google Scholar]
- 26.Oumouna-Benachour K, Hans CP, Suzuki Y, Naura A, Datta R, Belmadani S, Fallon K, Woods C, Boulares AH. Poly(adp-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein e-deficient mice: Effects on macrophage recruitment, nuclear factor-kappab nuclear translocation, and foam cell death. Circulation. 2007;115:2442–2450. doi: 10.1161/CIRCULATIONAHA.106.668756. [DOI] [PubMed] [Google Scholar]
- 27.Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J. Chemoattraction of macrophages, t lymphocytes, and mast cells is evolutionarily conserved within the human α-defensin family. J Immunol. 2007;179:3958–3965. doi: 10.4049/jimmunol.179.6.3958. [DOI] [PubMed] [Google Scholar]
- 28.Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, Jones KL, Kawamori R, Cassis LA, Tschöp MH, Bruemmer D. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. 2007;117:2877–2888. doi: 10.1172/JCI31986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. 1997;8:623–631. doi: 10.1097/00019501-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Golledge J, Tsao PS, Dalman RL, Norman PE. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation. 2008;118:2382–2392. doi: 10.1161/CIRCULATIONAHA.108.802074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial il-6/mcp1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchida HA, Poduri A, Subramanian V, Cassis LA, Daugherty A. Urokinase-type plasminogen activator deficiency in bone marrow–derived cells augments rupture of angiotensin ii–induced abdominal aortic aneurysms. Arterioscler Thromb and Vasc Biol. 2011;31:2845–2852. doi: 10.1161/ATVBAHA.111.234997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tieu BC, Ju X, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Brasier AR, Tilton RG. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J Vasc Res. 2011;48:261–272. doi: 10.1159/000320358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Outtz HH, Wu JK, Wang X, Kitajewski J. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol. 2010;185:4363–4373. doi: 10.4049/jimmunol.1000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vikatmaa P, Lajunen T, Ikonen TS, Pussinen PJ, Lepäntalo M, Leinonen M, Saikku P. Chlamydial lipopolysaccharide (clps) is present in atherosclerotic and aneurysmal arterial wall—clps levels depend on disease manifestation. Cardiovas Pathol. 2010;19:48–54. doi: 10.1016/j.carpath.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 36.Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, Daugherty A. Total lymphocyte deficiency attenuates angii-induced atherosclerosis in males but not abdominal aortic aneurysms in apoe deficient mice. Atherosclerosis. 2010;211:399–403. doi: 10.1016/j.atherosclerosis.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, Outtz H, Kitajewski J, Shi C, Weber S, Saftig P, Li Y, Ozato K, Blobel CP, Ivashkiv LB, Hu X. Notch-rbp-j signaling regulates the transcription factor irf8 to promote inflammatory macrophage polarization. Nat Immunol. 2012;13:642–650. doi: 10.1038/ni.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gridley T. Notch signaling during vascular development. Proc Natl Acad Sci USA. 2001;98:5377–5378. doi: 10.1073/pnas.101138098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in notch1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 40.Guo D-C, Papke CL, He R, Milewicz DM. Pathogenesis of thoracic and abdominal aortic aneurysms. Ann NY Acad Sci. 2006;1085:339–352. doi: 10.1196/annals.1383.013. [DOI] [PubMed] [Google Scholar]
- 41.McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM., 3rd Novel notch1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;134:290–296. doi: 10.1016/j.jtcvs.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 42.Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, Merrill WH, Hinton RB, Garg V. Inhibitory role of notch1 in calcific aortic valve disease. PLoS One. 2011;6:e27743. doi: 10.1371/journal.pone.0027743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahu A, Garg V, Boettner B, Cook S. Absence of abdominal aneurysm in adults with coarctation and/or bicuspid aortic valve – is this really an “aortopathy”? J Am Coll Cardiol. 2010;55:A40.E382–A340.E382. (Abstract) [Google Scholar]
- 44.King VL, Lin AY, Kristo F, Anderson TJT, Ahluwalia N, Hardy GJ, Owens AP, Howatt DA, Shen D, Tager AM, Luster AD, Daugherty A, Gerszten RE. Interferon-γ and the interferon-inducible chemokine cxcl10 protect against aneurysm formation and rupture. Circulation. 2009;119:426–435. doi: 10.1161/CIRCULATIONAHA.108.785949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Outtz HH, Tattersall IW, Kofler NM, Steinbach N, Kitajewski J. Notch1 controls macrophage recruitment and notch signaling is activated at sites of endothelial cell anastomosis during retinal angiogenesis in mice. Blood. 2011;118:3436–3439. doi: 10.1182/blood-2010-12-327015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong W, Zhao Y, Prall A, Greiner TC, Baxter BT. Key roles of cd4+ t cells and ifn-γ in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172:2607–2612. doi: 10.4049/jimmunol.172.4.2607. [DOI] [PubMed] [Google Scholar]
- 47.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin ii-infused, apolipoprotein e-deficient mice. Arterioscler Thromb and Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 48.Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, Goronzy JJ, Weyand CM. Blocking the notch pathway inhibits vascular inflammation in large-vessel vasculitis /clinical perspective. Circulation. 2011;123:309–318. doi: 10.1161/CIRCULATIONAHA.110.936203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bin Hafeez B, Adhami VM, Asim M, Siddiqui IA, Bhat KM, Zhong W, Saleem M, Din M, Setaluri V, Mukhtar H. Targeted knockdown of notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clinical Cancer Research. 2009;15:452–459. doi: 10.1158/1078-0432.CCR-08-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, Randolph S, Smeal T. Evaluation of selective γ-secretase inhibitor pf-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Molecular Cancer Therapeutics. 2010;9:1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 51.Aoyama T, Takeshita K, Kikuchi R, Yamamoto K, Cheng XW, Liao JK, Murohara T. Γ-secretase inhibitor reduces diet-induced atherosclerosis in apolipoprotein e-deficient mice. Biochem Bioph Res Co. 2009;383:216–221. doi: 10.1016/j.bbrc.2009.03.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaer RA, DeRubertis BG, Hynecek R, Kent KC, Faries PL. Models of abdominal aortic aneurysm: Characterization and clinical applications. Vascular. 2006;14:343–352. doi: 10.2310/6670.2006.00059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.