

Abstract
FUSE-binding protein (FBP)-interacting repressor (FIR) is a c-myc transcriptional suppressor. A splice variant of FIR that lacks exon 2 in the transcriptional repressor domain (FIRΔexon2) upregulates c-myc transcription by inactivating wild-type FIR. The ratio of FIRΔexon2/FIR mRNA was increased in human colorectal cancer and hepatocellular carcinoma tissues. Because FIRΔexon2 is considered to be a dominant negative regulator of FIR, FIR heterozygous knockout (FIR+/−) C57BL6 mice were generated. FIR complete knockout (FIR−/−) was embryonic lethal before E9.5; therefore, it is essential for embryogenesis. This strongly suggests that insufficiency of FIR is crucial for carcinogenesis. FIR+/− mice exhibited prominent c-myc mRNA upregulation, particularly in the peripheral blood (PB), without any significant pathogenic phenotype. Furthermore, elevated FIRΔexon2/FIR mRNA expression was detected in human leukemia samples and cell lines. Because the single knockout of TP53 generates thymic lymphoma, FIR+/−TP53−/− generated T-cell type acute lymphocytic/lymphoblastic leukemia (T-ALL) with increased organ or bone marrow invasion with poor prognosis. RNA-sequencing analysis of sorted thymic lymphoma cells revealed that the Notch signaling pathway was activated significantly in FIR+/−TP53−/− compared with that in FIR+/+TP53−/− mice. Notch1 mRNA expression in sorted thymic lymphoma cells was confirmed using qRT-PCR. In addition, flow cytometry revealed that c-myc mRNA was negatively correlated with FIR but positively correlated with Notch1 in sorted T-ALL/thymic lymphoma cells. Moreover, the knockdown of TP53 or c-myc using siRNA decreased Notch1 expression in cancer cells. In addition, an adenovirus vector encoding FIRΔexon2 cDNA increased bleomycin-induced DNA damage. Taken together, these data suggest that the altered expression of FIRΔexon2 increased Notch1 at least partially by activating c-Myc via a TP53-independent pathway. In conclusion, the alternative splicing of FIR, which generates FIRΔexon2, may contribute to both colorectal carcinogenesis and leukemogenesis.
Keywords: FBP interacting repressor (FIR), splicing variant, haplo-insufficiency, leukemia, P53, T-ALL
INTRODUCTION
DNA damage affects carcinogenesis, transcription, alternative splicing, and cell cycle control; however, the precise mechanism behind these affects remains largely unexplored. FUSE-binding protein (FBP) is a transcription factor that stimulates c-myc expression [1-3]. FBP-interacting repressor (FIR) is a c-myc transcriptional repressor that functions by suppressing the TFIIH/P89/XPB helicase (P89) [4-7]; hence, enhanced FIR showed antitumor effect in mouse xenografted model by suppressing c-myc [8-10]. Markedly, a splice variant of FIR that lacks exon 2 in the transcriptional repression domain (FIRΔexon2) elevates c-Myc protein expression in vitro [11]. FIRΔexon2 mRNA is frequently upregulated in human colorectal cancers [12] as well as hepatocellular carcinoma [13], where it stimulates tumor growth by preventing FIR from suppressing c-myc [13]. FIRΔexon2 functions as a dominant negative regulator of FIR; therefore it reduces FIR function. Recent studies suggested that DNA damage induces alternative splicing of several genes including FIR [14,15]. Specifically, FIR/FIRΔexon2 monitors the DNA damage response by potentially interacting with DNA-PKcs or Ku-86 [14]. Therefore, DNA damage may induce persistent c-myc upregulation via FIRΔexon2 in cancer cells, whereas it induces TP53 in normal cells
FIR is a splice variant of PUF60, reported as a splicing factor that lacks the exon 5 consists of 17 amino acids [16]. SAP155, a subunit of the SF3b spliceosome complex, interacts directly with PUF60 in vitro [17] and could be co-immunoprecipitated with FIR (or FIRΔexon2)-FLAG beads in vivo [18]. Furthermore, SAP155 is required for proper FIR pre-mRNA splicing; therefore, SAP155-FIR complex formation inhibits the well-established functions of both SAP155 and FIR, disturbing splicing and the transcriptional suppression of c-myc [18,19]. Accordingly, the FIR/FIRΔexon2/SAP155 interaction, which affects FIR and p21Kip1 splicing, links the DNA damage response to c-myc regulation [19]. In fact SAP155 mutations, which potentially affect FIR/FIRΔexon2/SAP155 formation, were reported not only in myeloid lineage tumors but also lymphoid lineage tumors [20-23]. Consequently, an aberrant FIR/FIRΔexon2/SAP155 interaction is responsible for cancer development and differentiation and is a potent target for cancer screening and treatment [13, 19].
The upregulation of c-Myc and Notch1 with TP53 loss-of-function is critical for T-ALL pathogenesis [24]. This mechanism involves the loss of F-box WD repeat-containing protein 7 (FBW7/FBWX7), which was reported to induce sustained c-Myc and Notch1 expression via a post-transcriptional mechanism, resulting in TP53-deficient T-ALL [25, 26]. FBW7 is required for the polyubiquitination-mediated proteasomal degradation of c-Myc. Accordingly, FBW7 modulates leukemia-initiating cell (LIC) activity by regulating c-Myc stability [25], and thereby plays a role in the pathogenesis [26]. However, the mechanism of c-Myc upregulation in T-ALL in the absence of TP53, FBW7, or Notch1 mutations is unclear. In this study, the significance of disturbed FIR expression was examined by generating FIR+/− mice to assess the dominant negative effect of FIRΔexon2. This study indicated that the alternative splicing of FIR links the DNA damage response to c-myc regulation and revealed how the alteration of FIR affects c-Myc, Notch1, or TP53 during the pathogenesis of T-ALL in a FIR+/−TP53−/− mouse model.
RESULTS
FIR−/− mice were embryonic lethal at E9.5 or earlier
The design of the FIR-targeting vector (Figure 1A), as well as the wild-type, targeted, and deleted FIR alleles used to prepare the FIR+/− mice are shown in Figure S1. The FIR homozygous knockout mouse FIR−/− was prepared by the cross-fertilization of FIR+/− mice (Figure 1B). A total of 86 mice were analyzed after the genetically confirmed mating of FIR+/− mice. FIR+/− and FIR+/+ were recovered at close to the expected Mendelian ratio of 48:26 (~2:1; Table 1). There were 12 dead embryos (hypothetically FIR−/−): six between E13.5 and E14.5 and six on E9.5 (Table 1). No live FIR−/− mice were observed at birth, E13.5–E14.5, or E9.5 (Figure 1B, Table 1). FIR−/− mice exhibited early developmental defects and die by E4.5 or earlier (Dr. David Levens, NCI, USA). FIR total knockout, FIR−/−, mouse is embryonic lethal before E9.5, suggesting that FIR is essential for embryogenesis. Proteins expressed during embryogenesis disappear during development but are re-expressed in cancers [27, 28], suggesting that FIR is crucial for carcinogenesis as well.
Figure 1. c-Myc mRNA was activated in the peripheral blood cells of inducible FIR heterozygous knockout mouse and FIR/IRΔexon2 mRNA expression in human clinical leukemia/malignant lymphoma samples.

(A) Genetic construction for inducible FIR+/− mouse. Primers locations for detecting ES genome, LoxP insertion, neomycin cassette and probes for Southern blot analysis were indicated. Expected DNA sizes for Southern blot analysis are also shown. (B) Family tree to obtain FIR homo knockout mouse FIR−/−, by cross-fertilization between FIR hetero knockout mouse: FIR+/−. FIR−/− mouse was revealed to be embryonic lethal at least before E9.5 by cross-fertilized between FIR hetero knockout mouse. The relative expression of c-myc and FIR mRNAs from lung, intestinal mucosae, heart muscle, kidneys, livers and peripheral blood (PB) were examined. (C) c-myc mRNA of PB of FIR hetero knockout mice was three-times higher than those of wild mouse. (D) FIR mRNA expression level of FIR hetero knockout mice was exactly half of those of wild mouse. (E) qRT-PCR of PUF60, FIR PUF60Δexon2, FIRΔexon2 mRNA were indicated by RT-PCR. Samples: leukemia cells from peripheral blood (PB) and bone marrow (BM) of adult patients (listed in table 2). (F) The ratio of FIR/FIRΔexon2 mRNA level of leukemia cells was significantly higher than those of non-leukemia or control samples (Student's t-test). (G) mRNA extracted from HeLa (human cervical squamous carcinoma cells), RKO (human colon adenocarcinoma cells), and Jurkat (human immortalized T lymphocyte) cells was examined for their FIR splicing variants expression.
Table 1. The number of FIR hetero and homo knockout mice during the time of observation.
| FIR+/+ | FIR+/− | FIR−/− | supposed FIR−/− | ||
|---|---|---|---|---|---|
| Time at obervation | Wild mice | FIR hetero knockout mice | FIR homo knockout mice | Dead embryo | Total |
| At birth | 12 | 17 | 0 | 0 | 29 |
| E13.5 to E14.5 | 9 | 20 | 0 | 6 | 35 |
| E9.5 | 5 | 11 | 0 | 6 | 22 |
| Total | 26 | 48 | 0 | 12 | 86 |
FIR+/− mice exhibited increased c-myc mRNA expression but had no significant deleterious phenotype
The relative expression of c-myc (Figure 1C and FIR (Figure 1D) mRNA in the lungs, intestines, heart, kidney, liver, and peripheral blood (PB) of FIR+/− mouse was approximately half of that detected in wild-type mice. However, FIR+/− mouse had no apparent pathogenic phenotype. Recently, five individuals were reported with de novo interstitial 8q24.3 deletions ranging from 65 kb to 1 Mb on the chromosome that includes FIR (PUF60). These deletions had a clinical phenotype that was associated with multiple systemic phenotypes but no hematological malignancy or lymphoma [29, 30]. This suggests that the haploinsufficiency of PUF60 (FIR) with c-myc mRNA elevation alone is not sufficient to drive the pathogenesis of T-ALL.
FIR is alternatively spliced in human leukemia
To explore how c-myc is activated in T-ALL/lymphoma, the alternative splicing of FIR, the ratio of FIR/FIRΔexon2, and c-myc mRNA expression were examined in human leukemia samples (Figure 1E, Table S1). qRT-PCR for the cDNA of full-length FIR variants was performed in bone marrow or peripheral blood samples using primers to amplify the amino terminal region. At least four variants (FIR, PUF60, FIRΔexon2, and PUF60Δexon2) were expected from the alternative use of the two potential exons [12]. The ratio of FIR/FIRΔexon2 mRNA (Figure 1F) was significantly higher in leukemia cells compared with that in non-leukemia or control samples from adults (Figures 1G) and children (data not shown). This suggests that the alternative splicing of FIR and the ratio of FIR/FIRΔexon2 may contribute to c-myc upregulation in T-ALL. Notably, c-Myc upregulation alone by FIR haploinsufficiency did not generate leukemia. Thus, FIR splicing variants rather than FIR haploinsufficiency significantly contributes toward promoting the progression of T-ALL/lymphoma via a c-Myc-independent pathway.
FIR+/−TP53−/− promotes the bone marrow invasion of T-cell malignant lymphoma
Because the FIR+/− mice suggested significant c-myc upregulation in the PB without a significant pathogenic phenotype, we generated FIR+/−TP53+/− double compound heterozygous knockout mice and cross-fertilized or mated female TP53+/− with male FIR+/−TP53+/− mice because of the low fertility of FIR+/−TP53+/− mice (Figure 2A). The genotypes of the FIR+/−TP53−/− and FIR+/+TP53−/− mice were confirmed by PCR analysis of genomic DNA (Figure 2B). FIR+/−TP53−/− and FIR+/+TP53−/− mice were sacrificed when a loss of 10%–15% bodyweight during growth or a systemic disorder such as dyspnea with a loss of movement was observed. A demonstrative macroscopic view of the organs of FIR+/−TP53−/−, FIR+/+TP53−/−, and wild-type mice are shown in Figure 2C. Atypical cells in FIR+/−TP53−/− mice that were observed in the PB, bone marrow (BM), liver, spleen, and thymus are shown in Figures 3A and B. Next, flow cytometry analysis of the PB, spleen, thymus, and BM of thymic lymphoma of FIR+/+TP53−/− mice was performed (Figure 3C). These analyses revealed that both FIR+/+TP53−/− and FIR+/−TP53−/− mice exhibited T-ALL/T-cell-type thymic lymphoma. Therefore, the single knockout of TP53−/− was sufficient to cause T-ALL/T-cell-type thymic lymphoma. Flow cytometry also revealed that the size of the thymic lymphoma cells was apparently larger in FIR+/−TP53−/− mice compared with that in control mice (Figure 3D, gated area).
Figure 2. Preparation of FIR+/−P53−/− and FIR+/+P53−/− mouse.

(A) FIR+/− and P53−/− were obtained by cross-fertilization between FIR+/−P53−/− and FIR+/+P53−/− mice. FIR+/−P53+/− double compound hetero knockout mouse was prepared and mated each other, or female P53+/− was mated with male FIR+/−P53+/− mouse to obtain FIR+/−P53−/− because FIR+/−P53+/− showed low fertility. (B) Genotyping of FIR+/+P53−/−, FIR+/−P53+/+ and FIR+/−P53−/− and wild mice were confirmed by PCR. (C) Thymic lymphoma was observed in FIR+/+p53−/− mice.
Figure 3. Histologic features and flow cytometry analysis of FIR+/−P53−/−.

(A) Atypical cells were indicated by Giemsa stain in bone marrow and peripheral blood of FIR+/−P53−/− mouse (C610). (B) Histologic features of bone marrow, liver, spleen and thymus in FIR+/−P53−/− (F338) and wild mouse by Hematoxylin-Eosin stain. (C) Flow cytometry analysis of lymphocytic cells with CD4 and CD8 as indicated markers. Mac1 and Gr1 were used for myelocytic markers. Flow cytometry analysis revealed that lymphocytic atypical cells (left) were CD4low+CD8+ phenotype (gated area) but no significant findings in myeloid cells (right) in FIR+/−P53−/− mouse (C610), and diagnosed as T-cell type acute lymphocytic/lymphoblastic leukemia (T-ALL)/lymphoma. (D) Cell size of gated area was measured by flow cytometry analysis (FSC: Forward Scatter).
Haploinsufficiency of FIR developed rapid T-ALL progression with bone marrow invasion
The weight of the thymus was significantly heavier in FIR+/−TP53−/− and FIR+/+TP53−/− mice compared with that in wild-type or FIR+/−TP53+/+ mice. In addition, the weight of the spleen was significantly heavier in FIR+/+TP53−/− mice than that in wild-type or FIR+/−TP53+/+ mice. The WBC count was increased significantly in FIR+/−TP53−/− and FIR+/+TP53−/− mice compared with that in wild-type. Conversely, the RBC count was significantly lower in FIR+/+TP53−/− compared with that in wild-type mice. The platelet count was also significantly lower in FIR+/−TP53−/− and FIR+/+TP53−/− mice compared with that in wild-type or FIR+/−TP53+/+ mice (Figures 4A, B). FIR+/−TP53−/− mice with T-ALL/lymphoma exhibited a significantly lower bodyweight than did FIR+/+ (Figure 4C). One-hundred percent of wild-type and FIR+/−TP53−/− mice survived during the study period, and the overall survival rate (Kaplan–Meier) of FIR+/+TP53−/− mice was better than that of FIR+/−TP53−/− (Figure 4D). There were no significant differences in the nose-to-anus length and bodyweight of FIR+/− compared with that of wild-type mice (FIR+/+) (Figure S2). These results suggest that FIR haploinsufficiency promoted the progression of T-ALL/lymphoma, reduced bodyweight, and was associated with a poorer prognosis. The incidence of T-ALL with > 10% bone marrow infiltration of blast cells was higher in FIR+/−TP53−/− mice (5 of 23; 21.7%, including three live mice before analysis) compared with that in FIR+/+TP53−/− (1 of 19; 5.3%, including two live mice before analysis) (Figures 4E, F). These results demonstrated that high levels of c-Myc promoted an increased occurrence of T-ALL and bone marrow infiltration in this mouse model. Of the FIR+/−TP53−/− mice (N = 20), 10 had T-ALL (50.0%), 15 had thymic lymphoma (75%), and five experienced bone marrow invasion (25.0%). In contrast, of the 17 FIR+/+P53−/− mice, seven had T-ALL (41.2%), 10 had thymic lymphoma (58.8%), and one exhibited bone marrow invasion (5.9%) (Figure 4F).
Figure 4. Summary of organs' weight and blood tests, body weight and overall survival of curves of FIR−/−P53+/+, FIR+/−P53+/+, FIR+/+P53−/−, and FIR+/−P53−/− mice.
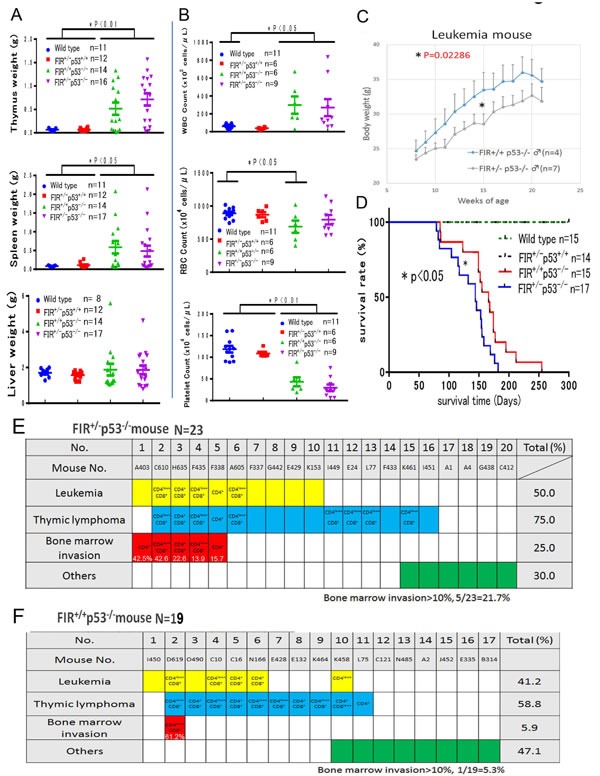
(A) The weight of thymus of FIR+/−P53−/− and FIR+/+P53−/− was significantly heavier than that of wild or FIR+/−P53+/+ mouse (P<0.05). The weight of spleen of FIR+/+P53−/− -were significantly heavier than that of wild or FIR+/−P53+/+ mouse (P<0.05). The weight of liver of wild mouse, FIR+/−P53+/+, FIR+/+P53−/−, and FIR+/−P53−/− was no significant difference. (B) WBC count of FIR+/−P53−/− and FIR+/+P53−/− was significantly increased than that of wild mouse. RBC count of FIR+/+P53−/− was significantly less than that of wild mouse (P<0.05). Platelet count of FIR+/−P53−/− and FIR+/+P53−/− was significantly less than that of wild or FIR+/−P53+/+ mouse (P<0.05). (C) Body weight of FIR+/−P53−/− mice was significantly lighter than that of FIR+/+P53−/−. Statistical signifincace was calculated by Student's t-test. (D) The overall survival curves of four genetically different group: wild, FIR+/−p53+/+, FIR+/−p53−/−, and FIR+/+p53−/− mice. FIR+/−p53+/+ and FIR+/+p53+/+ were survived 100% up to 25 weeks after birth without obvious tumor formation, body weight loss or other physical disabilities. On the contrary, the overall survival curves (Kaplan-Meier method) of FIR+/−p53−/− and FIR+/+p53−/− mice were declined around 70 days after birth. Overall survival curves of four genetically different group: wild, FIR+/−p53+/+, FIR+/−p53−/−, and FIR+/+p53−/− mice were compared by log-rank test. (E) T-ALL with more than 10 % bone marrow infiltration of blast cells in FIR+/−P53−/− mice was 5 out of 23 (21.7%) including three pre-analytical alive mouse in FIR+/+P53−/− (1 out of 19=5.3%) including two pre-analytical alive mouse. (F) In FIR+/−P53−/− mice (N=23), T-ALL: 10 (50.0%), thymic lymphoma: 15 (75%), bone marrow invasion: 5 (25.0%). Whereas in FIR+/+P53−/− mice (N=17) T-ALL: 7 (41.2%), thymic lymphoma: 10 (58.8%), bone marrow invasion: 1 (5.9%). Blank in colored column indicated undetermined or not tested for cell surface marker.
Comparison of RNA-sequencing analysis of sorted thymic lymphoma cells from FIR+/−TP53−/− or FIR+/+TP53−/− mice
RNA-sequencing was used to compare the gene expression profiles of sorted thymic lymphoma cells from FIR+/−TP53−/− and FIR+/+TP53−/− mice. The top 100 activated genes were analyzed (Figure S3A), and data revealed that the Notch signaling pathway was activated more in CD4+CD8+ thymic lymphoma cells from FIR+/−TP53−/− mice compared with those from FIR+/+TP53−/−mice(Table S3A–C). RNA-sequencing analysis of sorted CD4+CD8+ thymic lymphoma cells revealed that the Notch signaling (Figure 5) and tight junction pathways (Figure S4A) were activated more significantly in FIR+/−TP53−/− (H635) than FIR+/+TP53−/− (N166) mice. In contrast, analysis of CD4low+CD8+ thymic lymphoma cells demonstrated that the focal adhesion pathway (Figure S4B) was activated more significantly in FIR+/−TP53−/− (A605) than in FIR+/+TP53−/− (D619) mice. The upregulation of c-myc and notch1 mRNA was confirmed by qRT-PCR (Table S4, Figure S3C–E). Notch1 mRNA was more activated in both CD4+CD8+ and CD4low+CD8+ thymic lymphoma cells from FIR+/−TP53−/− mice compared with those from FIR+/+TP53−/− mice (Figure S3C, D). In contrast, c-myc mRNA was activated in whole peripheral blood cells in two FIR+/−TP53+/+ mice examined in this study (Figure S3E).
Figure 5. Signaling pathway activated in sorted thymic lymphoma cells in FIR+/−TP53−/− mice.
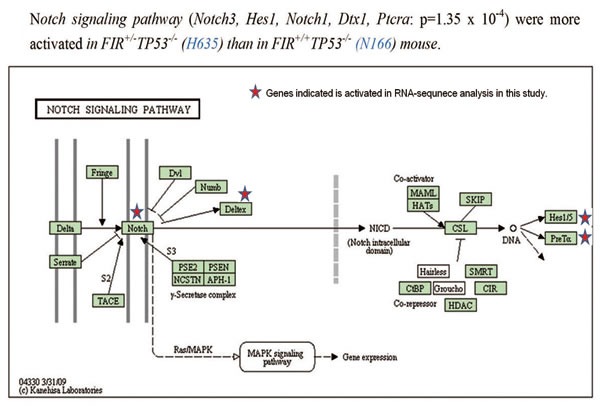
(A) KEGG pathway analysis showed that Notch Signaling pathway was more activated in FIR+/−TP53−/− mice compared with FIR+/−TP53−/− and FIR+/+TP53−/− mice, with the Notch3, Hes1, Notch1, Dtx1 and Ptcra genes upregulated (P=1.4×10−4). Other activated pathways were also shown in Figure S6.
c-Myc and FIR proteins were negatively correlated and c-myc and notch1 mRNAs were positively correlated in sorted thymic lymphoma/T-ALL cells
Because FIR is believed to be a c-myc transcriptional repressor, c-Myc expression was examined in atypical or lymphoma cells from FIR+/−P53−/− mice. As expected, c-Myc expression was higher in atypical/lymphoma cells compared with that in non-atypical cells or normal lymphocytes (Figure 6A). Furthermore, there was a significant negative correlation between c-myc and FIR mRNA expression in CD4+CD8+ and CD4low+CD8+ thymic lymphoma cells (Figure 6B. These results strongly suggest that FIR suppresses c-myc expression both in vitro and in vivo. Notably, atypical/lymphoma cells from FIR+/+TP53−/− mice also expressed high levels of c-Myc. These results also suggest that activated c-myc mRNA, which does not directly reflect an increase in c-Myc protein, is inadequate for the pathogenesis of T-ALL. Therefore, the pathogenesis of T-ALL/lymphoma observed in FIR+/+P53−/− mice was at least partly Notch1 upregulation because c-myc and Notch1 mRNAs were positively correlated (Figures 6C, D). Together, the current FIR haploinsufficiency mouse model revealed that T-ALL/lymphoma was generated via a p53-dependent pathway, but that its progression was potentially due to a c-Myc-independent mechanism because the TP53 single knockout alone exhibited sustained c-Myc expression.
Figure 6. c-Myc protein is enhanced and showed inverse correlation with FIR in thymic lymphoma cells and promotes bone marrow invasion.

(A) Flow cytometry of thymic lymphoma cells in FIR+/+P53−/− or FIR+/−P53−/− mice are indicated. CD4+ cells of FIR+/+P53−/− (upper column 1) has two populations indicated c-Myc-high (1a) and c-Myc–low intensity (1b). Notably, c-Myc-high (1a) population, presumably lymphoma cells, has c-Myc-low peak, indicating non-tumor cells cluster. CD4+CD8+ cells (2c) also indicated c-Myc-high intensity population. CD4low+CD8+ (3d) showed c-Myc-high intensity population indicated by FACS (bottom). Red line: Thymus cells of wild type FIR+/+P53+/+ mouse. Thin blue line: non-tumor cells of FIR+/+P53−/− or FIR+/−P53−/− mice. Orange line: Tumor cells of FIR+/+P53−/− or FIR+/−P53−/− mice. (B) Inverse correlation with significance between c-myc and FIR mRNA expression in CD4low+CD8+ or CD4+CD8+thymus lymphocytes obtained from FIR+/+P53−/− or FIR+/−P53−/− mice. Relative c-myc (or FIR)/HPRT mRNA expression of CD4low+CD8+ thymic lymphoma/leukemia cells of FIR+/+p53−/− (light green) was 8.6 (0.81), thymic lymphoma cells of FIR+/−p53−/− (yellow) was 10.7 (0.58), and thymic lymphoma/leukemia cells of FIR+/−p53−/− (blue) was 18.0 (0.37) times as compare to thymic cells of wild mouse (dark orange), respectively (right). The relationship between c-myc/HPRT mRNA (x-axis) and FIR/HPRT mRNA (y-axis) was y=-0.4557x+0.093 (R2=0.9503). Relative c-myc (or FIR)/HPRT mRNA expression of CD4+CD8+ thymic lymphoma cells of FIR+/+p53−/− (light green) was 3.3 (0.56), thymic lymphoma/leukemia cells of FIR+/+p53−/− (yellow) was 6.1 (0.48), and thymic lymphoma/leukemia cells of FIR+/+p53−/− (blue) was 5.2 (0.56) times as compare to thymic cells of wild mouse (dark orange), respectively. The relationship between c-myc/HPRT mRNA (x-axis) and FIR/HPRT mRNA (y-axis) was y=-1.1161x+0.086 (R2=0.8131). (C) (D) c-myc mRNA and Notch1 mRNA expression was positively correlated each other in sorted thymic lymphoma cells extracted from mice of different genetic backgrounds.
Knocking down TP53 and c-myc using siRNA suppressed Notch1 expression
Bleomycin (BLM)-induced DNA damage induces FIR splicing [14]. The alternative splicing of FIR contributes to the transcriptional regulation of c-myc, which is critical for cell cycle control. Because Notch1 expression is pivotal for the pathogenesis of T-ALL, TP53 or c-myc were knocked down using siRNA whereas BLM was treated as DNA damaging agent to examine the relationship among DNA damage, the alternative splicing of FIR, and cell cycle control. Knocking down both TP53 and c-myc using siRNA significantly suppressed Notch1 expression without disturbing FIR expression (Figures 7A-C, arrows). TP53 expression was not affected significantly by the enforced expression by FIR//FIRΔexon2 using an adenovirus vector (Figure 7D). DNA damage affects the alternative splicing of FIR, which contributes to the transcriptional activation of c-myc via a dominant negative effect on endogenous FIR [14]. Activated c-Myc accelerates the cell cycle by suppressing p27Kip1 expression, which leads to the accumulation of DNA damage [14, 15]. These results suggest that disturbed FIR expression or the altered splicing of FIR may contribute to the pathogenesis of T-ALL via upregulating c-Myc-Notch1 axis independent on TP53 (Figure 7E). Therefore, FIR splicing is a novel mechanism that links DNA damage to c-myc regulation (Figure 7E).
Figure 7. Alternative splicing of FIR connects DNA damage response, c-myc activation and cell cycle control.
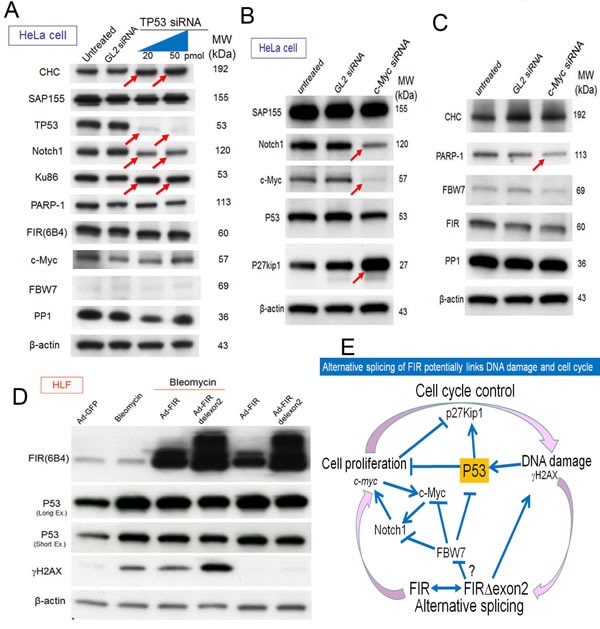
(A) 20 and 50pmol of TP53 siRNA were transfected into HeLa cells. GL2 siRNA was transfected as the negative control. After 72h of incubation, whole-cell extracts were analyzed by western blotting for relevant protein expressions. (B, C) 20pmol c-Myc siRNA was transfected into HeLa cells. GL2 siRNA is for negative control. After 48h of incubation, whole-cell extracts were analyzed by western blotting for relevant protein expressions. (D) 3.76 × 108 VP/ml (10 MOI) of Ad-FIR or Ad-FIRΔexon2 adenovirus vectors and DNA damaging agent bleomycin (30 μg/ml) were either co-treated or single treated into HLF cell. 3.76 × 108 VP/ml (10 MOI) of GFP adenovirus (Ad-GFP) was treated as negative control. After 48h of incubation, whole-cell extracts were analyzed by western blotting. Severity of the DNA damage caused by bleomycin treatment is indicated by γH2AX expression. (E) Schematic view of haploinsufficiency, as a dominant negative-alternative splicing model of FIR in T-ALL pathogenesis. DNA damage affects alternative splicing of FIR that contributes to c-myc transcriptional activations. Activated c-Myc accelerates cell cycle by suppressing P27Kip1 and in turn accumulates DNA damage. The altered expression of FIRΔexon2 increased Notch1 at least partially by activating c-Myc via a TP53-independent pathway.
DISCUSSION
The conditional FIR+/− mouse exhibited prominent c-myc upregulation, particularly in the PB among other organs but without a significant pathogenic phenotype (Figure 1). The increased mRNA expression of FIR/FIRΔexon2 was detected in human leukemia cell lines and clinical samples (Figure 1E–G). FIR−/− was embryonic lethal in mice before E9.5 (Table 1). FIR+/−TP53−/− mice developed T-ALL (Figure 2) and exhibited an increased incidence of organ or bone marrow invasion (Figure 3). The bodyweight of FIR+/−TP53−/− mice was less than that of FIR+/+TP53−/− mice, and overall survival was reduced in FIR+/−TP53−/− compared with that in FIR+/+TP53−/− mice, presumably due to the rapid progression of T-ALL (Figure 4). The Notch signaling pathway was more activated in sorted CD4+CD8+ thymic lymphoma cells isolated from FIR+/−TP53−/− (H635) compared with those from FIR+/+TP53−/− (N166) mice, as revealed by RNA-sequencing analysis (Figure 5). Quantitative RT-PCR confirmed that c-myc mRNA expression was negatively correlated with FIR mRNA expression but positively correlated with Notch1 mRNA in sorted T-ALL/thymic lymphoma cells (Figure 6). Furthermore, knocking down TP53 or c-myc using siRNA suppressed Notch1 expression; however, TP53 expression was not affected significantly by the enforced expression of FIR/FIRΔexon2 using an adenovirus vector (Figures 7A–D). Thus, alternative splicing of FIR expression increased Notch1 through c-Myc upregulation independent on TP53-pathway (Figure 7E).
DNA damage affects alternative splicing by modulating the elongation activity and/or the phosphorylation status of RNA polymerase II [15,31]. In addition, BLM-induced DNA damage alters FIR splicing, which contributes to the transcriptional upregulation of c-myc via dominant negative effect on endogenous FIR [12, 13]. c-Myc accelerates the cell cycle by suppressing p27Kip1 expression, eventually leading to the accumulation of DNA damage. Therefore, FIR splicing is a novel mechanism that connects DNA damage to the regulation of c-myc. The sustained c-Myc and Notch1 protein expression level also needs to be regulated by their stability and degradation process. So, how does FIR splicing contribute to these processes that are pivotal for T-ALL pathogenesis? One possibility is that FIR/FIRΔexon2 interferes with FBW7 (F-box WD repeat-containing protein 7). FBW7 is a polyubiquitin ligase that acts on Cdc4 phospho-degron (CPD) consensus sequence (−-TP--S/E--)-containing proteins, including c-Myc and Notch1 [32-36]. Therefore, leukemic cells from FBW7-deficient mice exhibited the marked accumulation of Notch-1 and c-Myc proteins, which led to the development of T-ALL [25]. The WD-like motifs (W425 and D399) in the CPD-binding propeller pocket of FBW7 are positioned close to each other in the 3D structure after protein folding, and they potentially interact with FIR-UHM (LNGRWFAGRKVVA) (unpublished data). Purified FIRΔexon2 exhibited a higher binding affinity for Skp1-FWB7 than did FIR in a HiTrap Ni affinity column [37], suggesting that FIRΔexon2 may interact with FBW7 (data not shown). Therefore, FIRΔexon2 may sustain c-Myc protein levels post-transcriptionally by inhibiting the FBW7-mediated pathway. This situation is further complicated because the dimerization site of FIR (PUF60), the RRM2 domain, is not affected by the splicing of exon 2 or 5 in terms of FBP/FIR/FIRΔexon2/PUF60/SAP155 interaction [38, 39]. The difference of the FBP/FIR/FIRΔexon2/PUF60/SAP155 interaction or regulation among organs also needs to be revealed in carcinogenesis in further study.
The KRAS G12D and Notch1 mutations contribute cooperatively to the leukemogenic transformation of normal T-cells in mouse models [40]. Notch1 mutations, which activate c-myc transcription, were identified in > 50% of T-ALL cases [41-43], suggesting that upregulation of Notch1 or c-Myc phosphorylation occurred via the EGFR/KRAS/MEK/ERK pathway [37]. How does the splicing of FIR affect Notch1 or c-Myc phosphorylation? Phosphorylated-ERK (p-ERK) is a substrate of protein phosphatase 1 (PP1). Both Ad-FIR and Ad-FIRΔexon2 activated p-ERK but did not affect PP1 in Jurkat and HeLa cells (data not shown). Ad-FIRΔexon2, but not Ad-FIR, elevated pSer62-c-Myc expression much more than expected by c-myc mRNA level in HeLa cells [16]. Therefore, FIRΔexon2 sustains c-Myc activation via both transcriptional and post-transcriptional mechanisms. Notably, FLAG-tagged FIR or FIRΔexon2 were co-immunoprecipitated with TOPOII-alpha, SAP155, TRRAP, and filamine-A in pull-down assays [14, 18, 19]; these proteins contain the GKKRVRWADLE sequence, which is specific for interactions with PP1 [44]. Because PP1 inhibits the RAS/BRAF/MEK/ERK pathway [45] and also regulates c-Myc phosphorylation at Ser-62 (pSer62-c-Myc) [46], FIRΔexon2 may interfere with the function of PP1. However, further studies are required to reveal the detailed mechanism behind this potential effect.
Finally, five individuals had de novo interstitial 8q24.3 (chr8: 144,868,670–144,933,911; USCS Genome Browser hg19, http://genome.ucsc.edu.) deletions ranging from 65 kb to 1 Mb on the chromosome that encodes Scrib [scribbled homologue (MIM607733)], FIR (PUF60), and NRBP2 (nuclear receptor binding protein 2) [29]. Patients with 8q24.3 deletions showed clinical phenotypes associated with coloboma, congenital heart defects, limb abnormalities, psychomotor retardation, and convulsions; however, no hematologic malignancies or lymphoma have been described [30]. Detecting FIR/FIRΔexon2 for diagnosis, or the use of specific antibodies against FIRΔexon2 or chemicals that inhibit the FIR/FIRΔexon2/SAP155 interaction may have clinical applications in T-ALL.
Together, these data suggest that the alternative splicing of FIR may link DNA damage to c-myc regulation. Haploinsufficiency of the c-myc transcriptional repressor FIR and the FIR+/−TP53−/− genotype in mice potently promoted the progression of T-ALL/lymphoma, at least in part by activating the Notch signaling pathway with c-myc/c-Myc upergulation. The alternative splicing of FIR contributes to not only colorectal carcinogenesis but also leukemogenesis.
MATERIALS AND METHODS
Human leukemia samples
Human leukemia, control or adult healthy volunteer samples were obtained from Chiba University Hospital (adult patients) with written informed consent (Table S1).
Generation of animals and ethical approval
C57BL/6NCrSlc mice were obtained from Japan SLC. Mice were bred and maintained in the animal research facility of the Graduate School of Medicine, Chiba University (Chiba, Japan) in accordance with institutional guidelines. This study was approved by the institutional review committees of Chiba University. All experiments using mice received approval from the Chiba University Administrative Panel for Animal Care. Littermates were used as controls in all experiments.
Constructing the FIR-targeting vector
Construct of FIR targeting vector was indicated (Figures 1 and S1).
Homologous recombination of the FIR-targeting vector in ES cells
First, FIR targeting vector was injected by electroporation for homologous recombination ES cells into (C57BL/6) to prepare genetically modified ES cells (C57BL/6). 23 (clone nos. 26, 29, 31, 45, 84, 105, 112, 114, 117, 125, 145, 146, 172, 176, 178, 179, 185, 188, 191, 238, 244, 245, and 265) of 279 clones were identified in which the FIR-targeting vector had integrated in the chromosomes of ES cells using PCR (Figure S5A). The LoxP site of the integrated FIR-targeting vector was confirmed using PCR with suitable primers (Figure S5B). The integration of the FIR-targeted allele into the genome was confirmed using Southern blotting with 5′ and 3′ probes (Figure S5C). The FIR genomic sequence located between the loxP sites was excised using Cre-recombinase (Figure S5D). After verifying that the ES cells had integrated the FIR-targeting allele successfully using PCR and Southern blotting, the ES cells were microinjected into blastocysts from BALB/c mice. The resulting blastocysts were inoculated into the uterus of ICR mice (Table S2). Male FIRfl/+ chimeric mice (chimera mouse) were cross-fertilized with C57BL/6 female mice to obtain F1 FIR heterozygous mice.
Generating inducible FIR heterozygous knockout mice, FIR+/−
ES cells were purchased from DS Pharma Biomedical Co., Ltd. (Osaka, Japan). The FIR-targeting vector was prepared from Bac clone using PCR, and the FIR heterozygous knockout mice were prepared using the Cre-loxP system in C57BL6 mice [47] (Unitech Co., Ltd, Chiba, Japan) (Figure S1B). The primers and probes used to prepare FIR heterozygous knockout mice are shown in Figure 1A and Table S2. To assess the function of FIR in hematopoiesis, FIR was deleted conditionally by crossing FIRfl/+ mice with CAG-Cre transgenic mice, which express Cre ubiquitously (FIR+/−) (Figure S3). The efficient deletion of FIR in fetal cells from FIR+/− mice was confirmed using genomic PCR (Figure 1, primers loxP_F1, forward; and loxP_R2; reverse; Figure S6). The number of chimeric mice, born from clone number 26, 105 and 145 were indicated (Table S2). Genetically modified ES cells (C57BL/6) were microinjected into the blastocysts of BALB/c mice. Among the 23 positive clones that contained the FIR-targeting vector, clones 26, 105, and 145 (Figure S6C) were microinjected into the blastocyst cavity of BALB/c mice and were then transplanted into the uterus of pseudo-pregnant ICR mice. Seven chimeric mice were obtained successfully (Figure S6A and Table S2). Cross-fertilization was performed between male FIRfl/+ (C57BL/6) and female C57BL/6 mouse to obtain the sperm carrying the FIRfl/+ genome (Figure S6B). Finally, FIRfl/+ mice were cross-fertilized with CAG-Cre transgenic mice to obtain FIRfl/+/Cre(+), which were conditionally inducible FIR heterozygous knockout (FIR+/−) mice (Figure S6C). FIR heterozygous knockout mice were confirmed using the Cre-LoxP system (Figure S6D).
Southern blotting of genomic DNA
Three probes were prepared and used for Southern blotting. The 5′-probe (new probe) was located upstream of the short arm in the neomycin gene, and the 3′-probe was located downstream of the long arm (Figure S1). The primers and qRT-PCR conditions used for Southern blotting are shown in Table S5A.
Registration of FIR heterozygous knockout mice [FIRfl/+/Cre(+)]
FIR heterozygous knockout mice were established, registered, and made available at the National Institute of Biomedical Innovation (http://animal.nibio.go.jp/j_FIR.html) and the experimental animal division of the RIKEN Bioresource Center, Japan (RBRC No. RBRC05542; http://www2.brc.riken.jp/lab/animal/search.php). Briefly, two loxP sites were inserted upstream of FIR exon 3 and downstream of exon 5, respectively, and a PGK-neo cassette and a loxP site was inserted downstream of exon 5. FIR-deficient mice could be generated by crossing with tissue-specific Cre mice to give Mus musculus C57BL/6-FIR<tm1>/CU.
TP53-null mice; TP53−/−
The p53-null mice (TP53−/−) were purchased from RIKEN BRC (Bio-Resource Center, Tsukuba, Japan; BRC_No 01361, strain name C57BL-p53+/−).
CAG-Cre-transgenic mice
These mice were a gift from Dr T. Miki [47].
Measuring the bodyweight and survival curves of mice
The bodyweight of all mice was measured twice a week after the age of 7 weeks. The nose-to-anus length was also measured (Figure S2).
Immunocytochemistry
Cancer cells were prepared for immunocytochemistry as described previously [8].
Flow cytometry and cell sorting
The antibodies used for immunostaining and flow cytometry are listed in Table S6. Flow cytometry and cell sorting were performed as described previously [48, 49]. Briefly, linage surface marker antibodies for thymocytes, splenocytes, peripheral blood cells, and bone marrow were Gr-1 (Ly-G6, bone-marrow derived cells), Mac-1 (CD11b, granulocyte, macrophage, etc), B220, CD4 (helper/induced T cell marker), CD8α (cytotoxic T cell marker), CD45.2 (Leukocyte common antigen). Dead cells were eliminated by staining with propidium iodide (1 μg/mL; Sigma-Aldrich). After cell surface staining, intracellular staining was performed using a FITC-conjugated anti-c-Myc antibody. IntraprepTM (Beckman Coulter) was used for fixation and permeabilization. All flow cytometric analyses and cell sorting were performed on FACSAria II or FACSCanto II (BD Biosciences) and analyzed using FACSDiva software (BD Biosciences) and Flowjo (Tree Star, Ashland, OR, USA).
siRNA
c-myc and TP53 siRNA duplexes were purchased from Sigma-Aldrich (Tokyo, Japan). The transient transfection of siRNAs was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfected cells were cultured for 48–72h at 37°C in a CO2 incubator. The target sequences for the siRNAs are listed in Supplementary Table 4.
Bleomycin treatment
The DNA-damaging agent bleomycin was purchased from Sigma-Aldrich (sulfate powder from Streptomyces verticillus; Tokyo, Japan; Lot no. BCBG6499V; PCode, 101203713), dissolved in distilled H2O at a concentration of 5 mg/mL, and stored at −20°C. HLF cells were seeded in 6-well plates and incubated at 37°C/5% CO2 until confluent (approximately 24 h). Immediately before drug treatment, the media were removed and replaced with fresh culture media. Cells were treated with 30 μg/mL bleomycin alone or co-treated with adenovirus vectors.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using TRIzol LS solution (Invitrogen) and reverse-transcribed using the ThermoScript RT-PCR system (Invitrogen) with oligo-dT primers. qRT-PCR was performed using an ABI Prism 7300 Thermal Cycler (Applied Biosystems) with FastStart Universal Probe Master (Roche) and Universal Probe Library (Roche). Primers and probes for mouse were listed (Table S5B).
FIR and FIRΔexon2 adenovirus vectors
FIR and FIRΔexon2 adenovirus vectors were prepared as described previously [18].
Protein extraction and western blotting
Culture media were removed and the cells were washed twice with cold (4°C) PBS, lysed with 1:20 β-mercaptoethanol and 2x sample buffer, and incubated at 100°C for 5 min. Whole cell lysates were assayed for protein content (Bio-Rad, Hercules, CA, USA), and 10 μg protein samples were separated using SDS-PAGE on 7.5% and 10%–20% XV PANTERA gels. They were then transferred to polyvinylidene fluoride membranes using a tank transfer apparatus, and the membranes were blocked with 0.5% skimmed milk in PBS overnight at 4°C. Membranes were incubated with primary antibodies for 1 h at room temperature, followed by three 10-min washes with PBS/0.01% Tween 20. Membranes were then incubated with secondary antibodies, followed by three 15-min washes with PBS/0.01% Tween 20. Details of the antibodies used in this study are listed in Table S6. Antigens on the membranes were detected using enhanced chemiluminescence detection reagents (GE Healthcare UK Ltd., Buckinghamshire, UK).
RNA-sequencing
Total RNA was extracted form sorted CD4+CD8+ and CD4low+CD8+ thymic lymphoma cells from FIR+/−TP53−/− and FIR+/+TP53−/− mice using TRIzol. The RNA quality was analyzed using a 2100 Bioanalyzer system (Agilent, Santa Clara, CA) to confirm that their RINs (RNA integrity numbers) were > 7.0. RNA-seq was performed to analyze genome-wide gene expression levels. Specifically, RNA-seq libraries were prepared using a TruSeqStranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA) followed by sequencing using a HiSeq1500 genome sequencer (Illumina), according to the manufacturer's protocol. The gene expression levels in FIR+/−TP53−/− mice were compared with those in FIR+/+TP53−/− mice, and the top 100 upregulated genes (Table S2C) were analyzed using KEGG (Kyoto Encyclopedia of Genes and Genomes) software (http://www.genome.jp/kegg/). Signaling pathways with a FDR (false discovery rate) < 1.0 were selected as significantly activated pathways.
Statistical analysis
The expression of SAP155 and FIR was compared in the lungs, intestine, heart, kidney, and liver of FIR heterozygous knockout adult mice and 14-day-old fetal mice (E14) using Student's t-tests and the Wilcoxon test. The WBC, RBC, and platelet counts, organs' weight curve, and the ratio of FIRΔexon2/FIR mRNA were analyzed statistically using Student's t-tests. Overall survival curves were generated using the Kaplan–Meier method and analyzed statistically using log-rank tests. Statistical analyses were performed using GraphPad Prism version 6.0 for Windows (GraphPad Software, San Diego, CA, USA).
Accession number and genetic information of FIR genome
Ensemble NM_014281.
SUPPLEMENTARY MATERIAL, FIGURES AND TABLES
Acknowledgments
The authors thank Unitech CO., LTD (Chiba, Japan) for preparing the FIR-targeting vector. The authors also thank Dr Toshiko Ishitsuka (Kajiwara) and Ms Kaori Ono for technical assistance.
Footnotes
Conflict of interest
We have no potential conflicts of interest to disclose.
Grant Support
This study was supported in part by Grant-in-Aid 18591453 for priority areas in cancer research, 21st COE (Center Of Excellence) program from the Ministry of Education, Science, Sports and Culture of Japan and the “Seed Finding Programs”, “Mini-Feasibility Study Project” of the JST (Japan Science and Technology) Agency and “Futaba Electronics Memorial Foundation” in Japan to K.M.
REFERENCES
- 1.Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, Levens D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994;8:465–80. doi: 10.1101/gad.8.4.465. [DOI] [PubMed] [Google Scholar]
- 2.Bazar L, Meighen D, Harris V, Duncan R, Levens D, Avigan M. Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J Biol Chem. 1995;270:8241–8. doi: 10.1074/jbc.270.14.8241. [DOI] [PubMed] [Google Scholar]
- 3.Avigan MI, Strober B, Levens D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem. 1990;265:18538–45. [PubMed] [Google Scholar]
- 4.Liu J, He L, Collins I, Ge H, Libutti D, Li J, Egly JM, Levens D. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell. 2000;5:331–41. doi: 10.1016/s1097-2765(00)80428-1. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Akoulitchev S, Weber A, Ge H, Chuikov S, Libutti D, Wang XW, Conaway JW, Harris CC, Conaway RC, Reinberg D, Levens D. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell. 2001;104:353–63. doi: 10.1016/s0092-8674(01)00223-9. [DOI] [PubMed] [Google Scholar]
- 6.Kouzine F, Wojtowicz D, Yamane A, Resch W, Kieffer-Kwon KR, Bandle R, Nelson S, Nakahashi H, Awasthi P, Feigenbaum L, Menoni H, Hoeijmakers J, Vermeulen W, Ge H, Przytycka TM, Levens D, Casellas R. Global regulation of promoter melting in naive lymphocytes. Cell. 2013;153:988–99. doi: 10.1016/j.cell.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kouzine F, Gupta A, Baranello L, Wojtowicz D, Ben-Aissa K, Liu J, Przytycka TM, Levens D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat Struct Mol Biol. 2013;20:396–403. doi: 10.1038/nsmb.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamura A, Matsushita K, Takiguchi Y, Shimada Hideaki, Tomonaga T, Matsubara H, Inoue M, Hasegawa M, Sato Y, Levens D, Tatsumi K, Nomura F. Synergistic effect of non-transmissible Sendai virus vector encoding the c-myc suppressor FUSE-binding protein-interacting repressor plus cisplatin in treatment of malignant pleural mesothelioma. Cancer Science. 7(102):1366–73. doi: 10.1111/j.1349-7006.2011.01931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsushita K, Shimada H, Ueda Y, Inoue M, Hasegawa M, Tomonaga T, Matsubara H, Nomura F. Non-transmissible Sendai virus vector encoding c-myc suppressor FBP-interacting repressor for cancer therapy. World J of Gastroenterology. 2014;20(15):4316–28. doi: 10.3748/wjg.v20.i15.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka N, Araki K, Mizokami D, Miyagawa Y, Yamashita T, Tomifuji M, Ueda Y, Inoue M, Hasegawa M, Matsushita K, Nomura F, Shimada H, Shiotani A. Sendai virus-mediated c-myc suppressor far-upstream element binding protein interacting repressor gene transfer suppresses head and neck squamous cell carcinoma. Gene Therapy. 2014 doi: 10.1038/gt.2014.123. In press. [DOI] [PubMed] [Google Scholar]
- 11.Matsushita K, Tomonaga T, Shimada H, Shioya A, Higashi M, Matsubara H, Harigaya K, Nomura F, Libutti D, Levens D, Ochiai T. An essential role of alternative splicing of c-myc suppressor FIR in carcinogenesis. Cancer Res. 2006;66:1409–17. doi: 10.1158/0008-5472.CAN-04-4459. [DOI] [PubMed] [Google Scholar]
- 12.Kajiwara T, Matsushita K, Itoga S, Tamura M, Tanaka N, Tomonaga T, Matsubara H, Shimada H, Habara Y, Matsuo M, Nomura F. SAP155-mediated c-myc suppressor far-upstream element-binding protein-interacting repressor splicing variants are activated in colon cancer tissues. Cancer Sci. 2013;104:149–56. doi: 10.1111/cas.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malz M, Bovet M, Samarin J, Rabenhorst U, Sticht C, Bissinger M, Roessler S, Bermejo JL, Renner M, Calvisi DF, Singer S, Ganzinger M, Weber A, Gretz N, Zörnig M, Schirmacher P, Breuhahn K. Overexpression of far upstream element (FUSE) binding protein (FBP)-interacting repressor (FIR) supports growth of hepatocellular carcinoma. Hepatology. 2014;60(4):1241–50. doi: 10.1002/hep.27218. [DOI] [PubMed] [Google Scholar]
- 14.Rahmutulla B, Matsushita K, Satoh M, Seimiya M, Tsuchida S, Kubo S, Shimada H, Ohtsuka M, Miyazaki M, Nomura F. Alternative splicing of FBP-interacting repressor coordinates c-Myc, P27Kip1/cyclinE and Ku86/XRCC5 expression as a molecular sensor for bleomycin-induced DNA damage pathway. Oncotarget. 2014;5:2404–17. doi: 10.18632/oncotarget.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahmutulla B, Matsushita K, Nomura F. Alternative splicing of DNA damage response genes and gastrointestinal cancers. World J Gastroenterol. 2014;20(46):17305–13. doi: 10.3748/wjg.v20.i46.17305. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Page-McCaw PS, Amonlirdviman K, Sharp PA. PUF60: a novel U2AF65-related splicing activity. RNA. 1999;5:1548–60. doi: 10.1017/s1355838299991938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corsini L, Hothorn M, Stier G, Rybin V, Scheffzek K, Gibson TJ, Sattler M. Dimerization and protein binding specificity of the U2AF homology motif of the splicing factor Puf60. J Biol Chem. 2009;284:630–39. doi: 10.1074/jbc.M805395200. [DOI] [PubMed] [Google Scholar]
- 18.Matsushita K, Kajiwara T, Tamura M, Satoh M, Tanaka N, Tomonaga T, Matsubara H, Shimada H, Yoshimoto R, Ito A, Kubo S, Natsume T, Levens D, Yoshida M, Nomura F. SAP155-mediated splicing of FUSE-binding protein-interacting repressor (FIR) serves as a molecular switch for c-myc gene expression. Mol Cancer Res. 2012;10:787–99. doi: 10.1158/1541-7786.MCR-11-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushita K, Tamura M, Tanaka N, Tomonaga T, Matsubara H, Shimada H, Matsubara H, Shimada H, Levens D, He L, Liu J, Yoshida M, Nomura F. Interactions between SAP155 and FUSE-binding protein-interacting repressor bridges c-myc and P27Kip1 expression. Mol Cancer Res. 2013;11:689–98. doi: 10.1158/1541-7786.MCR-12-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 21.Cazzola M, Rossi M, Malcovati L. Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood. 2013;121:260–9. doi: 10.1182/blood-2012-09-399725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Je EM, Yoo NJ, Kim YJ, Kim MS, Lee SH. Mutational analysis of splicing machinery genes SF3B1, U2AF1 and SRSF2 in myelodysplasia and other common tumors. Int J Cancer. 2013;133:260–5. doi: 10.1002/ijc.28011. [DOI] [PubMed] [Google Scholar]
- 23.Te Raa GD, Derks IA, Navrkalova V, Skowronska A, Moerland PD, van Laar J, Oldreive C, Monsuur H, Trbusek M, Malcikova J, Lodén M, Geisler CH, Hüllein J, Jethwa A, Zenz T, Pospisilova S, Stankovic T, van Oers MH, Kater AP, Eldering E. The impact of SF3B1 mutations in CLL on the DNA damage response. Leukemia. 2014 doi: 10.1038/leu.2014.318. Nov 5. doi: 10.1038/leu.2014.318. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, Wei W. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 2012;586:1409–18. doi: 10.1016/j.febslet.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A, Shi J, Vakoc C, Sandy P, Shen SS, Ferrando A, Aifantis I. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552–66. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levens D, Aplan PD. Notching up MYC gives a LIC. Cell Stem Cell. 2013;13:8–9. doi: 10.1016/j.stem.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fanali C, Lucchetti D, Farina M, Corbi M, Cufino V, Cittadini A, Sgambato A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: controversies and perspectives. World J Gastroenterol. 2014;20(4):923–42. doi: 10.3748/wjg.v20.i4.923. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langan CR, Mullinax JE, Raiji MT, Upham T, Summers T, Stojadinovic A, Avital I. Colorectal Cancer Biomarkers and the Potential Role of Cancer Stem Cells. J Cancer. 2013;4(3):241–250. doi: 10.7150/jca.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dauber A, Golzio C, Guenot C, Jodelka FM, Kibaek M, Kjaergaard S, Leheup B, Martinet D, Nowaczyk MJ, Rosenfeld JA, Zeesman S, Zunich J, Beckmann JS, Hirschhorn JN, Hastings ML, Jacquemont S, Katsanis N. SCRIB and PUF60 are primary drivers of the multisystemic phenotypes of the 8q24. 3 copy-number variant. Am J Hum Genet. 2013;93:798–811. doi: 10.1016/j.ajhg.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verheij JB, de Munnik SA, Dijkhuizen T, de Leeuw N, Olde Weghuis D, van den Hoek GJ, Rijlaarsdam RS, Thomasse YE, Dikkers FG, Marcelis CL, van Ravenswaaij-Arts CM. An 8. 35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur J Med Genet. 2009;52:353–357. doi: 10.1016/j.ejmg.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Dutertre M, Sanchez G, Barbier J, Corcos L, Auboeuf D. The Emerging role of pre-messenger RNA splicing in stress responses: sending alternative messages and silent messengers. RNA Biol. 2011;8(5):740–7. doi: 10.4161/rna.8.5.16016. [DOI] [PubMed] [Google Scholar]
- 32.Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF (Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–73. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau AW, Fukushima H, Wei W. The Fbw7 and beta-TRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci. (Landmark Ed.) 2012;17:2197–212. doi: 10.2741/4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Y, Li G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev. 2012;1:75–87. doi: 10.1007/s10555-011-9330-z. [DOI] [PubMed] [Google Scholar]
- 35.Tu K, Zheng X, Zhou Z, Li C, Zhang J, Gao J, Yao Y, Liu Q. Recombinant human adenovirus-p53 injection induced apoptosis in hepatocellular carcinoma cell lines mediated by p53-Fbxw7 pathway, which controls c-Myc and cyclin E. PLoS One. 2013;8:e68574. doi: 10.1371/journal.pone.0068574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 37.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–43. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 38.Hsiao HH, Nath A, Lin CY, Folta-Stogniew EJ, Rhoades E, Braddock DT. Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry. 2010;49:4620–34. doi: 10.1021/bi9021445. [DOI] [PubMed] [Google Scholar]
- 39.Cukier CD, Hollingworth D, Martin SR, Kelly G, Diaz-Moreno I, Ramos A. Molecular basis of FIR-mediated c-myc transcriptional control. Nat Struct Mol Biol. 2010;17:1058–64. doi: 10.1038/nsmb.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong G, Du J, Liu Y, Meline B, Chang YI, Ranheim EA, Wang J, Zhang J. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J Biol Chem. 2013;288:18219–27. doi: 10.1074/jbc.M113.475376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res. 2008;14:5314–7. doi: 10.1158/1078-0432.CCR-07-4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rakowski LA, Garagiola DD, Li CM, Decker M, Caruso S, Jones M, Kuick R, Cierpicki T, Maillard I, Chiang MY. Convergence of the ZMIZ1 and NOTCH1 pathways at C-MYC in acute T lymphoblastic leukemias. Cancer Res. 2013;73:930–41. doi: 10.1158/0008-5472.CAN-12-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal. 2014;26:149–61. doi: 10.1016/j.cellsig.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 44.Moorhead GB, Trinkle-Mulcahy L, Nimick M, De Wever V, Campbell DG, Gourlay R, Lam YW, Lamond AI. Displacement affinity chromatography of protein phosphatase one (PP1) complexes. BMC Biochem. 2008;9:28–35. doi: 10.1186/1471-2091-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manfroid I, Martial JA, Muller M. Inhibition of protein phosphatase PP1 in GH3B6, but not in GH3 cells, activates the MEK/ERK/c-fos pathway and the human prolactin promoter, involving the coactivator CPB/p300. Mol Endocrinol. 2001;15:625–37. doi: 10.1210/mend.15.4.0615. [DOI] [PubMed] [Google Scholar]
- 46.Lee T, Yao G, Nevins J, You L. Sensing and integration of Erk and PI3K signals by Myc. PLoS Comput Biol. 2008;4:e1000013. doi: 10.1371/journal.pcbi.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakai K, Miyazaki Ji. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–24. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 48.Mochizuki-Kashio M, Mishima Y, Miyagi S, Negishi M, Saraya A, Konuma T, Shinga J, Koseki H, Iwama A. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood. 2011;118(25):6553–61. doi: 10.1182/blood-2011-03-340554. [DOI] [PubMed] [Google Scholar]
- 49.Oguro H, Yuan J, Ichikawa H, Ikawa T, Yamazaki S, Kawamoto H, Nakauchi H, Iwama A. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell. 2010;6(3):279–86. doi: 10.1016/j.stem.2010.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.