

Abstract
Nipah virus (NiV) is an emerging zoonotic paramyxovirus that causes severe and often fatal disease in pigs and humans. There are currently no vaccines or treatments approved for human use. Studies in small-animal models of NiV infection suggest that antibody therapy may be a promising treatment. However, most studies have assessed treatment at times shortly after virus exposure before animals show signs of disease. We assessed the efficacy of a fully human monoclonal antibody, m102.4, at several time points after virus exposure including at the onset of clinical illness in a uniformly lethal nonhuman primate model of NiV disease. Sixteen African green monkeys (AGMs) were challenged intratracheally with a lethal dose of NiV, and 12 animals were infused twice with m102.4 (15 mg/kg) beginning at either 1, 3, or 5 days after virus challenge and again about 2 days later. The presence of viral RNA, infectious virus, and/or NiV-specific immune responses demonstrated that all subjects were infected after challenge. All 12 AGMs that received m102.4 survived infection, whereas the untreated control subjects succumbed to disease between days 8 and 10 after infection. AGMs in the day 5 treatment group exhibited clinical signs of disease, but all animals recovered by day 16. These results represent the successful therapeutic in vivo efficacy by an investigational drug against NiV in a nonhuman primate and highlight the potential impact that a monoclonal antibody can have on a highly pathogenic zoonotic human disease.
INTRODUCTION
Hendra virus (HeV) and Nipah virus (NiV) are members of the genus Henipavirus (family Paramyxoviridae) that can cause severe respiratory illness and encephalitis in horses, pigs, and humans (1, 2). HeV emerged in Australia in 1994 and has been associated with at least 40 outbreaks in horses and seven human infections with four fatalities. NiV was first identified during an outbreak of severe encephalitis in Malaysia and Singapore in 1998 to 1999 that involved 268 cases and 106 deaths, with pigs serving as the intermediate amplifying host (3). Since 1998, there have been more than a dozen recognized occurrences of human NiV infection, primarily in Bangladesh and India (4, 5), with the most recent outbreak in Bangladesh (6). In most outbreaks since 1998 to 1999, the mortality rate among humans has been higher (>75%) along with evidence of multiple rounds of person-to-person transmission (7).
Several species of fruit bats of the Pteropus genus, known as flying foxes, appear to be the principal natural reservoirs of both NiV and HeV (8). NiV has been isolated from bat urine and partially eaten fruit, and direct transmission of NiV from flying foxes to humans from contaminated food sources has been suggested (9). In contrast to all other paramyxoviruses, HeV and NiV have a broad species tropism and can infect and cause disease in a wide variety of species spanning six mammalian orders (10, 11).
There are currently no vaccines or antivirals approved for combating human HeV or NiV infections. Although there has been substantial progress in the development and advancement of experimental vaccines (12, 13), advances have been slower regarding treatments. An open-label ribavirin trial was performed in 140 patients during the initial outbreak of NiV in Malaysia in 1998; however, the results of this study are difficult to draw conclusions from because it was not a controlled clinical study (14). In addition, three of the seven recorded human HeV cases were treated with ribavirin, one of which survived (15). More recently, the utility of ribavirin as a countermeasure for HeV infection was assessed in a controlled study using African green monkeys (AGMs) (16). Although there was a small benefit in delaying death, there was no survival benefit in this model, suggesting that ribavirin is not an effective countermeasure against lethal henipavirus disease in primates.
Currently, the most promising post-exposure treatment for henipavirus infection appears to be an experimental human monoclonal antibody (mAb). The human mAb m102.4 targets the ephrin-B2 and ephrin-B3 receptor binding site of the HeV and NiV G glycoprotein. m102.4 is a potent cross-reactive neutralizing antibody in vitro (17) and has been shown to protect ferrets from lethal NiV challenge (18) and AGMs from lethal HeV challenge when administered as a post-exposure treatment (19). Here, we assessed the utility of using m102.4 as a therapeutic intervention in the AGM model of NiV infection, because this model most accurately reflects the human condition (20).
RESULTS
Human mAb m102.4 protection of AGMs after NiV challenge
An initial pilot study was conducted to assess the utility of m102.4 against NiV in AGMs when administered shortly after virus challenge. Here, three animals were given m102.4 intravenously at 10 hours and again 3 days after infection. All treated animals remained clinically healthy, whereas the control animal rapidly succumbed to disease on day 8 after infection (fig. S1 and tables S1 and S2). Subsequently, studies were geared toward extending the therapeutic window of m102.4 in NiV-infected animals. Specifically, m102.4 was administered to AGMs intravenously twice, with the first dose given 1 day after infection and the second dose 3 days after virus challenge (Fig. 1A, black arrows). To determine whether the m102.4 therapeutic window could be further extended, we also included two additional treatment groups: one group received m102.4 beginning at 3 days after infection and again at 5 days after infection (Fig. 1A, green arrows), and the third treatment group received m102.4 at 5 days after infection and again at 7 days after virus challenge (Fig. 1A, blue arrows). No adverse reactions were observed after m102.4 infusion in any of the 12 treated animals. The four control subjects, consistent with historical controls, succumbed to NiV disease between day 8 and day 10 after infection (Fig. 1B, red) and showed severe sustained behavior changes (depression and decreased activity), loss of appetite, dyspnea, thrombocytopenia, and changes in coagulation factors (Table 1). In addition, two of the four control animals had elevated temperatures (≥103.5°F), and three of the control animals had neurologic signs (muscle twitches and paresis). In contrast, subjects treated with m102.4 at day 1 after infection and again 2 days later (D1/D3) survived challenge (Fig. 1B, gray) and showed no clinical signs of disease with normal hematology, clinical chemistry, and coagulation assay results (Table 1 and tables S3 to S5). The four animals treated at day 3 after infection and again 2 days later (D3/D5) survived NiV challenge (Fig. 1B, green) and did not show any changes in behavior; three of these animals had very mild and variable clinical changes in hematology, clinical chemistry, or coagulation assays (Table 1). In the third treatment group, all four animals treated at day 5 after infection and again 2 days later (D5/D7) also survived NiV challenge (Fig. 1B, blue); however, two of the four animals had overt clinical disease that included depression, respiratory signs, loss of appetite, muscle twitches, and/or elevated temperature (≥103.5°F) (Table 1). Additionally, all four animals treated at days 5 and 7 had changes in hematology, clinical chemistry, and/or coagulation assays (Table 1). Clinical illness continued to be observed in these D5/D7 animals and resolved between days 7 and 17 depending on the animal, with all four animals completely recovering by day 17 (Table 1). Regardless of treatment group, the surviving animals all had detectable neutralizing antibody after treatment and through the end of the study unlike the control group (Table 2).
Fig. 1. (A) Diagram of the m102.4 treatment regimen and sampling days after NiV challenge in AGMs.
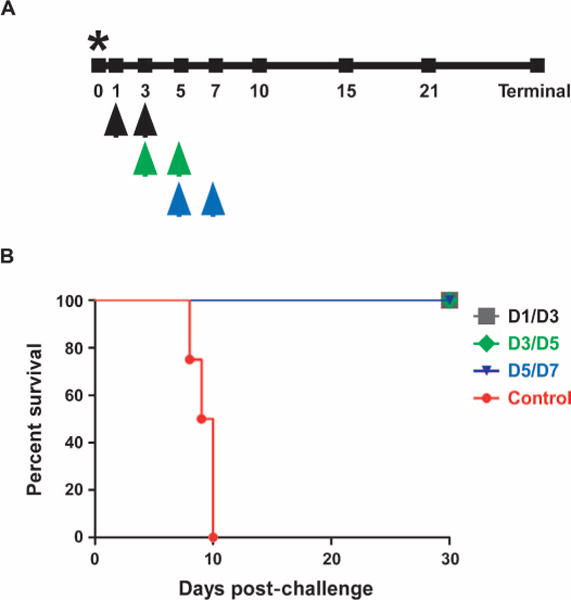
Black arrows, day 1/day 3 treatment; green arrows, day 3/day 5 treatment; blue arrows, day 5/day 7 treatment. Asterisk depicts the day of NiV challenge. (B) Kaplan-Meier survival curve for the control group (red, n = 4), D1/D3 group (gray, n = 4), D3/D5 group (green, n = 4), and D5/D7 group (blue, n = 4).
Table 1.
Clinical description and outcome of NiV-challenged AGMs
| Subject no. | Gender | Group | Clinical illness | Clinical and gross pathology |
|---|---|---|---|---|
| R272 | Female | Control | Depression (d8); lethargy (d8); loss of appetite (d8); dyspnea (d8); open-mouth breathing (d8); dehydration (d7, d8). Animal expired on d8. | Thrombocytopenia (d7–d8); lymphopenia (d7); hypoalbuminemia (d8); >10-fold ↑ in AST (d8); 5-fold ↑ in BUN (d8); >3-fold ↑ in CRE (d8); >10-fold ↑ in CRP (d7–d8); >5-fold ↑ in APTT (d8); >2-fold ↑ in fibrinogen (d8); excess blood-tinged pleural fluid; severely inflated, enlarged lungs with multifocal areas of congestion and hemorrhage; dark liver. |
| V753 | Female | Control | Depression (d8–d10); lethargy (d8–d10); loss of appetite (d8–d10); severe dyspnea (d9–d10); dehydration (d7–d10); muscle twitches (d10). Animal euthanized on d10. | Mild thrombocytopenia (d3); >3-fold ↑ in CRP (d3, d5, d7, d10); >2-fold ↑ in PT (d10); >2-fold ↑ in fibrinogen (d3, d5, d7); excess blood-tinged pleural fluid; moderately inflated, enlarged lungs with multifocal areas of congestion and hemorrhage particularly on the lower lobes. |
| R318 | Female | Control | Fever (d7); depression (d8–d9); lethargy (d8–d9); loss of appetite (d8–d9); dyspnea (d9); dehydration (d7–d9); left hindlimb paresis (d9). Animal euthanized on d9. | Thrombocytopenia (d5); lymphopenia (d3, d5, d7); 2-fold ↑ in APTT (d9); excess blood-tinged pleural fluid; moderately inflated, enlarged lungs with multifocal areas of congestion and hemorrhage particularly on the right lobes and the lower left lobe; mild splenomegaly. |
| O201216 | Male | Control | Fever (d10); depression (d10); lethargy (d10); loss of appetite (d10); dyspnea (d7–d10); open-mouth and abdominal breathing (d7–d10); muscle twitches (d10); paresis of both hindlimbs (d10). Animal euthanized on d10. | Thrombocytopenia (d7, d10); lymphopenia (d7); >3-fold ↑ in GGT (d10); >7-fold ↑ in CRP (d10); >2-fold ↑ in fibrinogen (d10); excess blood-tinged pleural fluid; inflated, enlarged lungs with multifocal areas of congestion and hemorrhage particularly of the lower right lobe; accumulation of fluid on pericardial cavity; spleen congested and friable; petechial hemorrhages on mucosal surface of urinary bladder; congestion of brain with possible meningeal hemorrhage. |
| R300 | Female | D1/D3 | None | None |
| Y083 | Female | D1/D3 | None | None |
| R429 | Female | D1/D3 | None | None |
| O7245 | Male | D1/D3 | None | None |
| R183 | Female | D3/D5 | Fever (d15) | >2-fold increase CRP (d5) |
| Y809 | Male | D3/D5 | None | Thrombocytopenia (d7) |
| R145 | Female | D3/D5 | None | >4-fold ↑ in CRP (d5); >2-fold ↑ in fibrinogen (d5) |
| O7823 | Female | D3/D5 | None | None |
| O7383 | Male | D5/D7 | Fever (d7); mild depression (d10–d13); abdominal breathing (d11–d16); muscle twitches on right ear (d11–d14) | Thrombocytopenia (d10); mild thrombocytopenia (d7); >2-fold ↑ in CRP (d7) |
| O7818 | Female | D5/D7 | Fever (d7); anorexia (d8–d12); mild dyspnea (d8–d9) | Mild thrombocytopenia (d7); >7-fold ↑ in CRP (d7); >2-fold ↑ in CRP (d10); >2-fold ↑ in fibrinogen (d10) |
| O918745 | Male | D5/D7 | None | Mild thrombocytopenia (d7, d10) |
| R198 | Female | D5/D7 | None | Thrombocytopenia (d7); >5-fold ↑ in CRP (d7); >2-fold ↑ in fibrinogen (d7) |
APTT, activated partial thromboplastin time; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CRE, creatinine; CRP, C-reactive protein; GGT, γ-glutamyltransferase; PT, partial thromboplastin time.
Table 2. NiV serum neutralization titers in control or m102.4-treated AGMs.
Reciprocal serum dilution at which 50% of virus was neutralized. NT, not tested.
| Treatment regimen | Subject no. | Day 0* | Day 3 | Day 5 | Day 7 | Day 15 | Days 30–34 |
|---|---|---|---|---|---|---|---|
| Days 1 and 3 | R300 | <20 | 5120 | 5120 | 5120 | 5120 | 2560 |
| Y083 | <20 | 5120 | 5120 | 5120 | 5120 | 2560 | |
| R429 | <20 | 5120 | 5120 | 5120 | 5120 | 1280 | |
| O7245 | <20 | 2560 | 5120 | 5120 | 2560 | 640 | |
| Days 3 and 5 | R183 | <20 | <20 | 5120 | 5120 | 5120 | 1280 |
| Y809 | <20 | <20 | 5120 | 5120 | 5120 | 2560 | |
| R145 | <20 | <20 | 5120 | 5120 | 5120 | 5120 | |
| O7823 | <20 | <20 | 5120 | 10240 | 5120 | 1280 | |
| Days 5 and 7 | O7383 | <20 | NT | NT | 5120 | 5120 | 2560 |
| O7818 | <20 | NT | NT | 5120 | 5120 | 2560 | |
| O918745 | <20 | NT | NT | 5120 | 5120 | 2560 | |
| R198 | <20 | NT | NT | 5120 | 5120 | 2560 | |
| None | R272 | <20 | NT | NT | <20 | ||
| V753 | <20 | NT | NT | <20 | |||
| R318 | <20 | NT | NT | <20 | |||
| O20121 | <20 | NT | NT | <20 |
Day after NiV challenge.
NiV load in swabs and tissues
To determine whether there was NiV replication in animals after challenge, we assessed shedding of virus using quantitative reverse transcription polymerase chain reaction (qRT-PCR) on nasal, oral, and rectal swabs (Fig. 2, A to C, respectively), with viremia also screened by qRT-PCR on whole blood samples (Fig. 2D). NiV genome equivalents (GEq) were observed in animals to varying degrees for nasal and oral swabs from the control (red), D3/D5 (green), and D5/D7 (blue) groups (Fig. 2, A and B) and also in blood samples from these three groups (Fig. 2D). However, none of the animals in the D1/D3 (gray) treatment had any detectable NiV RNA in any swab or blood sample (Fig. 2, A to D). NiV RNA was also detected systemically in the tissues of control animals R272 and O201216 (Fig. 2E, red). NiV RNA was not detected in the other two control animals, although tissues from these animals were positive for NiV antigen by immunohistochemistry (see below). There was no detectable NiV RNA in tissues from any animal in the D1/D3 group, whereas there was one animal (Y809) from the D3/D5 group with detectable NiV RNA in respiratory tissue, the spleen, and neural tissue (Fig. 2E, green). NiV RNA was detected in a tissue from each animal in the D5/D7 group, with O7383 positive in respiratory tissue and the remaining three animals having detectable RNA in spleen (Fig. 2E, blue). Overall, detectable NiV RNA in tissues of animals correlated with detection of NiV RNA in oral swabs and blood (Fig. 2, B and D). We also performed viral infectivity assays on whole blood and swab samples of all animals. Consistent with previous findings (20), infectious virus proved difficult to detect in these samples, with plaques detected in only two samples [25 plaque-forming units (PFU)/ml detected from the oral swab of control animal R272 and 475 PFU/ml detected from the rectal swab of control animal R318].
Fig. 2. Viral load from AGMs as detected by GEq/ml by qRT-PCR from nasal swabs (A), oral swabs (B), rectal swabs (C), and blood (D) and by GEq/g of tissue (E).

Gray, group D1/D3; green, group D3/D5; blue, group D5/D7; red, control group. RU, right upper; RM, right middle; RL, right lower; LU, left upper; LM, left middle; LL, left lower; Cer. Sp., cervical spinal. Error bars are SD.
Histopathological and immunohistochemical analysis of NiV-infected AGMs
Gross examination of all four control animals showed characteristic respiratory disease with enlarged lungs with multifocal areas of congestion and hemorrhage and an excess of serosanguinous fluid in the pleural cavity (Table 1). Histopathologic examination of tissue sections showed various degrees of lesions in the four control animals consistent with historical controls (12, 20). Noteworthy lesions included interstitial pneumonia with pulmonary endothelial syncytial cell formation, necrosis and hemorrhage of the splenic white pulp, necrosis and syncytial cell formation of the pancreas, and lymphocytic cystitis. The alveolar spaces contained edema fluid, polymerized fibrin, foamy alveolar macrophages, and cellular debris. Strong immunoreactivity for NiV antigen was present in the endothelium of the lungs, spleen, liver, kidney, testes, conjunctiva, nasal mucosa, and brain (frontal lobe of the cerebrum and brainstem) (Fig. 3, Lung, Spleen, and Brainstem Control panels, and fig. S2, testes control panel). In addition, moderate to strong immunoreactivity for NiV antigen was detected focally in clusters of transitional epithelium of the urinary bladder, mononuclear cells in the subcapsular and medullary sinuses of lymph nodes, medullary cells in the adrenal gland, islet cells of the pancreas, and neurons of the brainstem (Fig. 3, Brainstem Control panel). No lesions or immunoreactivity for NiV antigen was detected in tissue sections of any of the m102.4-treated AGMs (Fig. 3, D1/D3, D3/D5, and D5/D7 panels).
Fig. 3. Lack of NiV antigen in representative m102.4-treated tissues and localization of NiV antigen in representative control tissues by immunohistochemical staining.

Lung, spleen, and brainstem (representative reticular formations of the pons region) were labeled with an N protein–specific polyclonal rabbit antibody, and images were taken (lung, 40×; spleen, 40×; brainstem, 40×). Scale bars, 50 mm.
NiV F seroconversion of m102.4-treated AGMs
To assess the role of the host immune response in mediating protection from NiV disease, we measured circulating levels of antibodies directed against the NiV F (fusion) glycoprotein in sera of the NiV-infected AGMs (Fig. 4). Anti-NiV F antibodies were present in sera of all D5/D7 m102.4-treated AGMs and in one AGM (O7823) from the D3/D5 group on day 15 after infection, and these antibody levels increased by days 30 to 34 in these animals. One animal (O7245) from the D1/D3 group had anti-NiV F antibodies at day 34, whereas there was no detectable NiV RNA in any sample taken for this animal. The four control AGMs did not seroconvert. This was not surprising given the severe disease observed in these control animals, which succumbed between 8 and 10 days after exposure. Additionally, the seroconversion of the m102.4-treated animals to NiV F and the subsequent increase in titer by the study endpoint further confirm that all treated AGMs became infected with NiV, including at least one of the animals in the D1/D3 group, where no overt clinical signs of illness were observed.
Fig. 4. Detection of NiV F–specific antibodies from m102.4-treated and nontreated AGMs.

Mean fluorescence intensities (MFI) are shown on the y axis and represent binding of specific immunoglobulin (IgG and IgM) to NiV F. Error bars represent the SD of fluorescence intensity across 100 beads for each sample.
DISCUSSION
Here, we have addressed the possibility of providing an efficacious postexposure therapy to prevent NiV disease that is suitable for use in people after accidental or natural exposure and/or infection by NiV. Human henipavirus case fatality rates are about 57% for HeV and as high as 88% for NiV (4, 5). Because there have been annual emergences/spillovers of both of these viruses for the past 7 or more years, and with an increasing number of research facilities working with infectious HeV and NiV, the development of effective countermeasures against these pathogens has become a critical need. An increasing body of scientific data supports the hypothesis that antibody is sufficient to protect against lethal HeV- and NiV-mediated disease after virus exposure. Early antibody efficacy trials were done using either polyclonal hamster serum or mouse mAb (21–23), both of which are likely unsuitable for use in humans. More recent studies demonstrated that the human antibody m102.4 was able to confer complete protection to ferrets when administered shortly after NiV (18) challenge or to AGMs when given after HeV challenge (19). However, none of the previous studies have evaluated efficacy of m102.4 when administered at the time of circulating viremia as detected by qRT-PCR and during the onset of clinical signs of disease as we did in this study. In the AGM model that we used, all control animals succumbed between 8 and 10 days after NiV challenge, with infectious NiV detected in blood beginning at days 3 and 5 and initial clinical evidence of illness beginning at day 5. Although all animals in the treatment groups were protected against lethal NiV disease, the detection of NiV RNA in the neural tissue of one animal in the D3/D5 group requires further investigation considering the rare recrudescence in neural tissue of humans that have survived an initial henipavirus infection (24, 25). Unfortunately, the logistics of maintaining nonhuman primates in a Biosafety Level 4 (BSL-4) facility for long periods needed to assess recrudescence (greater than 1 year) is impractical.
The results reported here are an important demonstration of a protective outcome in a nonhuman primate model of NiV infection and pathogenesis using an antiviral therapeutic. The human mAb m102.4 used here has now demonstrated to be the only effective post-exposure therapy against NiV and, in a previous work, HeV (19), and has done so in a nonhuman primate species, considered to be an essential model for the purposes of moving anti-henipavirus therapies forward for potential licensure. Although here protection was demonstrated against the original NiV strain Malaysia, the effectiveness of m102.4 against other NiV strains should be explored. Indeed, m102.4 is an effective neutralizing mAb against all known HeV and NiV isolates, but whether it will be an effective therapy in vivo against the highly pathogenic NiV-Bangladesh strain needs to be determined.
Together, the data derived from the efficacy experiments to date indicate that the anti-henipavirus human mAb m102.4 is the only available effective antiviral that could be used to combat infection in people by NiV or HeV. Indeed, the Queensland Health Authorities announced in late 2013 that the first phase 1 clinical safety trial of m102.4 in human subjects will commence in 2014 (26). To date, six individuals exposed to either HeV or NiV have been given high-dose m102.4 therapy, and all have remained well with no adverse events.
MATERIALS AND METHODS
Study design
An initial pilot study was performed at the Rocky Mountain Laboratories (RML) to assess the potential protective efficacy of m102.4 against NiV in AGMs when administered shortly after virus challenge. Here, four AGMs were randomized with Microsoft Excel into a group of three animals and a single control animal, with the treated animals receiving m102.4 at 10 hours and again at day 3 after exposure to NiV. The single control animal was not treated. On the basis of the results of this study at RML, a larger study was performed at the Galveston National Laboratory (GNL) to determine whether the treatment window could be extended after NiV exposure including at times when animals are viremic by RT-PCR during the initial signs of clinical illness. Here, 16 AGMs were randomized with Microsoft Excel into experimental groups of four animals each and a control group of four animals. Experimental groups were treated with m102.4 at either days 1 and 3, 3 and 5, or 5 and 7 after NiV challenge. The control group was treated with saline. A number of parameters were monitored during the course of the study at the GNL including survival, clinical observations, hematology, serum biochemistry, blood coagulation, viremia and viral load in swabs and tissues by qRT-PCR and plaque assay, serum antibodies, and tissue pathology. An abbreviated analysis was done for the pilot study at RML and included survival, hematology, serum biochemistry, and RT-PCR on blood, swabs, and tissues. The overall objective of the study as a whole was to assess survival rates with all other measurements being considered secondary objectives. This study was not blinded. Because there were small numbers of animals in each group, all values are shown in the Supplementary tables. The study endpoint for surviving AGMs varied between 28 days at RML and days 30 and 34 at GNL so that complete necropsies could be performed on all animals.
Animal challenge
NiV was obtained from the Centers for Disease Control and Prevention and originated from the 1998 to 1999 outbreak in Malaysia. The NiV was obtained as a passage 2 stock and was passed one additional time on Vero cells. For the RML pilot study, four adult AGMs weighing 4 to 7 kg were inoculated by intratracheal instillation with~5 × 105 PFU of NiV (Malaysia strain). Three animals were infused with m102.4 beginning ~10 hours after challenge and again 3 days after challenge (10H/D3). The control animal was not treated. Each dose of m102.4 (~15 mg/kg) was administered intravenously. Animals were anesthetized for antibody infusion and for blood collection and collection of swabs of nasal, oral, and rectal mucosa on days 0, 3, 5, 7, 10, 14, 21, and 28 after infection (study endpoint). For studies at the GNL, 16 adult AGMs weighing 3 to 6 kg were inoculated by intratracheal instillation with ~5 × 105 PFU of NiV. Four animals were infused with m102.4 beginning 1 day after challenge and again 3 days after challenge (D1/D3); four animals were infused with m102.4 beginning 3 days after challenge and again 5 days after challenge (D3/D5); four animals were infused with m102.4 beginning 5 days after challenge and again 7 days after challenge (D5/D7). The four control animals were infused with saline. Each dose of m102.4 (~15 mg/kg) was administered intravenously. Animals were anesthetized for antibody infusion and clinical examination including temperature, blood collection, and swabs of nasal, oral, and rectal mucosa on days 0, 3, 5, 7, 10, 15, 21, and 30 to 34 after infection (study endpoint).
Measurement of serum F glycoprotein–specific antibodies
Antibodies to the F glycoprotein were measured in NiV-infected AGMs by including a recombinant soluble F (sF) glycoprotein–coupled microsphere in the assay by coupling of sF to microsphere #43 (Luminex Corp.) (19). Plasma from NiV-infected AGMs was inactivated by γ-irradiation (~5 mrads) before testing. Sera and plasma were assayed at 1:5000 and 1:10,000 dilutions. Assays were performed on a Luminex 200 IS machine equipped with Bio-Plex Manager Software (v 5.0) (Bio-Rad Laboratories). MFI and the SD of fluorescence intensity across 100 beads were determined for each sample.
Virus isolation
Virus titration was performed by plaque assay on Vero cells as previously described (20). Briefly, increasing 10-fold dilutions of the samples were adsorbed to Vero monolayers in duplicate wells (0.2 ml per well); thus, the limit for detection was 25 PFU/ml.
Hematology, serum biochemistry, and blood coagulation
Total white blood cell counts, white blood cell differentials, red blood cell counts, platelet counts, hematocrit values, total hemoglobin concentrations, mean cell volumes, mean corpuscular volumes, and mean corpuscular hemoglobin concentrations were analyzed from blood collected in tubes containing EDTA using a laser-based hematologic analyzer (Hemavet at RML and Beckman Coulter at GNL). Serum samples were tested for concentrations of albumin, amylase, alanine amino-transferase, aspartate aminotransferase, alkaline phosphatase, blood urea nitrogen, C-reactive protein, calcium, creatinine, γ-glutamyltransferase, glucose, total protein, and uric acid by using a Piccolo point-of-care analyzer and Biochemistry Panel Plus analyzer discs at GNL (Abaxis). RML studies used the General Chemistry 13 analyzer discs (Abaxis), which included total bilirubin instead of C-reactive protein. Citrated plasma samples were analyzed for coagulation parameters (partial thromboplastin time, activated partial thromboplastin time, thrombin time, and fibrinogen) on the STart4 instrument using the PTT Automate, STA Neoplastine CI Plus, STA Thrombin, and Fibri-Prest Automate kits, respectively (Diagnostica Stago).
Histopathology and immunohistochemistry
Necropsy was performed on all subjects. Tissue samples of all major organs were collected for histopathologic and immunohistochemical examination, immersion-fixed in 10% neutral buffered formalin, and processed for histopathology as previously described (20). For immuno-histochemistry, specific anti-NiV immunoreactivity was detected using an anti-NiV N protein rabbit primary antibody at a 1:5000 dilution (12). In brief, tissue sections were processed for immunohistochemistry using the Dako Autostainer. Secondary antibody used was biotinylated goat anti-rabbit IgG (Vector Laboratories) at 1:200 followed by Dako LSAB2 streptavidin–horseradish peroxidase. Slides were developed with Dako DAB chromagen and counterstained with hematoxylin. Nonimmune rabbit IgG was used as a negative control.
NiV TaqMan PCR
EDTA blood and swab sample (100 ml) was added to 600 ml of AVL viral lysis buffer (Qiagen) for RNA extraction. For tissues, about 100 mg was stored in RNAlater (Qiagen). Tissues were then homogenized in 600 μl of RLT buffer using Qiagen tissue lyser and stainless steel beads. Blood samples in AVL viral lysis buffer and tissue samples in RLT buffer were removed from the BSL-4 laboratory using approved protocols. RNA was isolated from blood and swabs using the QIAamp Viral RNA kit (Qiagen) and from tissues using the RNeasy Mini kit (Qiagen). RNA was isolated from blood or tissues and analyzed using primers/probe targeting the N gene and intergenic region between N and P of NiV for qRT-PCR as previously described (27). Threshold cycle (CT) values representing NiV genomes were analyzed with CFX Manager Software, and data are shown as GEq. To create the GEq standard, RNA from NiV challenge stocks was extracted, and the number of NiV genomes was calculated using Avogadro’s number and the molecular weight of the NiV genome.
NiV serum neutralization assays
Neutralization titers against NiV were determined by a conventional serum neutralization assay. Briefly, sera were serially diluted twofold and incubated with ~100 PFU of NiV for 1 hour at 37°C. Virus and antibodies were then added to individual wells of six-well plates of Vero cells. Plates were stained with neutral red 2 days after infection, and plaques were counted 24 hours after staining. The 50% neutralization titer was determined as the serum dilution at which there was a 50% reduction in plaque counts versus control wells.
Supplementary Material
Fig. S1. Survival and qRT-PCR results for RML study.
Fig. S2. Immunohistochemistry isotype control.
Table S1. Hematology results for RML study.
Table S2. Clinical chemistry results for RML study.
Table S3. Hematology results for GNL study.
Table S4. Clinical chemistry results for GNL study.
Table S5. Blood coagulation results for GNL study.
Acknowledgments
We thank the staff of the RML animal care staff and the University of Texas Medical Branch Animal Resources Center for animal husbandry. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the University of Texas Medical Branch, the U.S. Army, or the NIH. NiV was provided by T. Ksiazek.
Funding: This study was supported by the Department of Health and Human Services, NIH, grants AI082121 (to T.W.G.) and AI054715 and AI077995 (to C.C.B.) and in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), NIH.
Footnotes
Author contributions: T.W.G., C.E.M., K.N.B., H.F., and C.C.B. conceived and designed the experiments. K.N.B., Z.Z., D.S.D., and C.C.B. developed the m102.4 antibody used in the studies. F.F. and H.F. performed the Nipah challenge experiments in nonhuman primates at RML. T.W.G., C.E.M., and J.B.G. performed the Nipah challenge experiments at the GNL. J.B.G. and K.N.A. performed the clinical pathology assays. J.B.G. performed the NiV infectivity and neutralization assays. Y.-P.C. and K.N.B. developed and optimized the anti-NiV antibody assays, and K.N.A. performed the anti-NiV antibody assays. C.E.M. and K.N.A. performed the PCR assays. T.W.G., C.E.M., J.B.G., Y.-P.C., K.N.A., F.F., Z.Z., D.S.D., K.N.B., H.F., and C.C.B. analyzed the data. K.A.F. and D.P.S. performed histologic and immunohistochemical analysis of the data. T.W.G., C.E.M., and C.C.B. wrote the paper. All authors saw and approved the final version of the manuscript.
Competing interests: C.C.B. and D.S.D. are U.S. federal employees, and D.S.D., Z.Z., and C.C.B. are co-inventors on U.S. Patents 7,988,971 and 8,313,746: “Human monoclonal antibodies against Hendra and Nipah viruses” [assignees are The United States of America as represented by the Department of Health and Human Services (Washington, DC) and Henry M. Jackson Foundation for the Advancement of Military Medicine Inc. (Bethesda, MD)]. All other authors declare no competing interests. The opinions or assertions contained herein are the private ones of the author(s) and are not to be construed as official or reflecting the views of the Department of Defense, the Uniformed Services University of Health Sciences, and the NIAID, NIH.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/6/242/242ra82/DC1
REFERENCES AND NOTES
- 1.Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, Ksiazek TG, Zaki SR, Paul G, Lam SK, Tan CT. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet. 1999;354:1257–1259. doi: 10.1016/S0140-6736(99)04299-3. [DOI] [PubMed] [Google Scholar]
- 2.Selvey LA, Wells RM, McCormack JG, Ansford AJ, Murray K, Rogers RJ, Lavercombe PS, Selleck P, Sheridan JW. Infection of humans and horses by a newly described morbillivirus. Med J Aust. 1995;162:642–645. doi: 10.5694/j.1326-5377.1995.tb126050.x. [DOI] [PubMed] [Google Scholar]
- 3.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW. Nipah virus: A recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 4.Broder CC. Henipavirus outbreaks to antivirals: The current status of potential therapeutics. Curr Opin Virol. 2012;2:176–187. doi: 10.1016/j.coviro.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luby SP, Gurley ES. Epidemiology of henipavirus disease in humans. Curr Top Microbiol Immunol. 2012;359:25–40. doi: 10.1007/82_2012_207. [DOI] [PubMed] [Google Scholar]
- 6.International Society for Infectious Diseases. ProMED-mail. 2014 [Google Scholar]
- 7.Luby SP, Gurley ES, Hossain MJ. Transmission of human infection with Nipah virus. Clin Infect Dis. 2009;49:1743–1748. doi: 10.1086/647951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halpin K, Hyatt AD, Fogarty R, Middleton D, Bingham J, Epstein JH, Rahman SA, Hughes T, Smith C, Field HE, Daszak P. Henipavirus Ecology Research Group, Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am J Trop Med Hyg. 2011;85:946–951. doi: 10.4269/ajtmh.2011.10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luby SP, Rahman M, Hossain MJ, Blum LS, Husain MM, Gurley E, Khan R, Ahmed BN, Rahman S, Nahar N, Kenah E, Comer JA, Ksiazek TG. Foodborne transmission of Nipah virus, Bangladesh. Emerg Infect Dis. 2006;12:1888–1894. doi: 10.3201/eid1212.060732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong KT, Ong KC. Pathology of acute henipavirus infection in humans and animals. Patholog Res Int. 2011;2011:567248. doi: 10.4061/2011/567248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geisbert TW, Feldmann H, Broder CC. Animal challenge models of henipavirus infection and pathogenesis. Curr Top Microbiol Immunol. 2012;359:153–177. doi: 10.1007/82_2012_208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bossart KN, Rockx B, Feldmann F, Brining D, Scott D, LaCasse R, Geisbert JB, Feng YR, Chan YP, Hickey AC, Broder CC, Feldmann H, Geisbert TW. A Hendra virus G glycoprotein subunit vaccine protects African green monkeys from Nipah virus challenge. Sci Transl Med. 2012;4:146ra107. doi: 10.1126/scitranslmed.3004241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendez D, Büttner P, Speare R. Response of Australian veterinarians to the announcement of a Hendra virus vaccine becoming available. Aust Vet J. 2013;91:328–331. doi: 10.1111/avj.12092. [DOI] [PubMed] [Google Scholar]
- 14.Chong HT, Kamarulzaman A, Tan CT, Goh KJ, Thayaparan T, Kunjapan SR, Chew NK, Chua KB, Lam SK. Treatment of acute Nipah encephalitis with ribavirin. Ann Neurol. 2001;49:810–813. doi: 10.1002/ana.1062. [DOI] [PubMed] [Google Scholar]
- 15.Playford EG, McCall B, Smith G, Slinko V, Allen G, Smith I, Moore F, Taylor C, Kung YH, Field H. Human Hendra virus encephalitis associated with equine outbreak, Australia, 2008. Emerg Infect Dis. 2010;16:219–223. doi: 10.3201/eid1602.090552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rockx B, Bossart KN, Feldmann F, Geisbert JB, Hickey AC, Brining D, Callison J, Safronetz D, Marzi A, Kercher L, Long D, Broder CC, Feldmann H, Geisbert TW. A novel model of lethal Hendra virus infection in African green monkeys and the effectiveness of ribavirin treatment. J Virol. 2010;84:9831–9839. doi: 10.1128/JVI.01163-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Z, Bossart KN, Bishop KA, Crameri G, Dimitrov AS, McEachern JA, Feng Y, Middleton D, Wang LF, Broder CC, Dimitrov DS. Exceptionally potent cross-reactive neutralization of Nipah and Hendra viruses by a human monoclonal antibody. J Infect Dis. 2008;197:846–853. doi: 10.1086/528801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bossart KN, Zhu Z, Middleton D, Klippel J, Crameri G, Bingham J, McEachern JA, Green D, Hancock TJ, Chan YP, Hickey AC, Dimitrov DS, Wang LF, Broder CC. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute Nipah virus infection. PLOS Pathog. 2009;5:e1000642. doi: 10.1371/journal.ppat.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bossart KN, Geisbert TW, Feldmann H, Zhu Z, Feldmann F, Geisbert JB, Yan L, Feng YR, Brining D, Scott D, Wang Y, Dimitrov AS, Callison J, Chan YP, Hickey AC, Dimitrov DS, Broder CC, Rockx B. A neutralizing human monoclonal antibody protects african green monkeys from Hendra virus challenge. Sci Transl Med. 2011;3:105ra103. doi: 10.1126/scitranslmed.3002901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geisbert TW, Daddario-DiCaprio KM, Hickey AC, Smith MA, Chan YP, Wang LF, Mattapallil JJ, Geisbert JB, Bossart KN, Broder CC. Development of an acute and highly pathogenic nonhuman primate model of Nipah virus infection. PLOS One. 2010;5:e10690. doi: 10.1371/journal.pone.0010690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillaume V, Contamin H, Loth P, Georges-Courbot MC, Lefeuvre A, Marianneau P, Chua KB, Lam SK, Buckland R, Deubel V, Wild TF. Nipah virus: Vaccination and passive protection studies in a hamster model. J Virol. 2004;78:834–840. doi: 10.1128/JVI.78.2.834-840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guillaume V, Contamin H, Loth P, Grosjean I, Courbot MC, Deubel V, Buckland R, Wild TF. Antibody prophylaxis and therapy against Nipah virus infection in hamsters. J Virol. 2006;80:1972–1978. doi: 10.1128/JVI.80.4.1972-1978.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillaume V, Wong KT, Looi RY, Georges-Courbot MC, Barrot L, Buckland R, Wild TF, Horvat B. Acute Hendra virus infection: Analysis of the pathogenesis and passive antibody protection in the hamster model. Virology. 2009;387:459–465. doi: 10.1016/j.virol.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Goh KJ, Tan CT, Chew NK, Tan PS, Kamarulzaman A, Sarji SA, Wong KT, Abdullah BJ, Chua KB, Lam SK. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N Engl J Med. 2000;342:1229–1235. doi: 10.1056/NEJM200004273421701. [DOI] [PubMed] [Google Scholar]
- 25.Wong KT, Robertson T, Ong BB, Chong JW, Yaiw KC, Wang LF, Ansford AJ, Tannenberg A. Human Hendra virus infection causes acute and relapsing encephalitis. Neuropathol Appl Neurobiol. 2009;35:296–305. doi: 10.1111/j.1365-2990.2008.00991.x. [DOI] [PubMed] [Google Scholar]
- 26.Queensland Government. World-first Hendra treatment one step closer. http://statements.qld.gov.au/Statement/2013/10/31/worldfirst-hendra-treatment-one-step-closer [accessed 17 April 2014].
- 27.Mire CE, Versteeg KM, Cross RW, Agans KN, Fenton KA, Whitt MA, Geisbert TW. Single injection recombinant vesicular stomatitis virus vaccines protect ferrets against lethal Nipah virus disease. Virol J. 2013;10:353. doi: 10.1186/1743-422X-10-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Survival and qRT-PCR results for RML study.
Fig. S2. Immunohistochemistry isotype control.
Table S1. Hematology results for RML study.
Table S2. Clinical chemistry results for RML study.
Table S3. Hematology results for GNL study.
Table S4. Clinical chemistry results for GNL study.
Table S5. Blood coagulation results for GNL study.