

Abstract
Apremilast is a novel agent for the treatment of inflammatory based autoimmune disorders. The objective of this study was to assess the pharmacokinetic effects of co administration of apremilast and methotrexate on both agents. This was an open-label, multi-center, 3-treatment period, sequential study conducted in otherwise healthy subjects with psoriatic arthritis or rheumatoid arthritis who were receiving a stable oral dose of methotrexate between 7.5 to 20 mg once weekly. Subjects received their dose of methotrexate on Days 1 and 8 of the study in addition to Apremilast 30 mg oral every 12 hours on Days 3–8. Pharmacokinectic profiles of methotrexate and 7-OH methotrexate were characterized after methotrexate alone (Day 1) and after co-administration of methotrexate and Apremilast (on Day 8). The pharmacokinetic profile of Apremilast was characterized after Apremilast alone (on Day 7) and after co-administration of methotrexate and Apremilast (on Day 8). The 90% confidence interval of the ratio of the geometric means for the Cmax and AUC parameters for methotrexate, 7-OH methotrexate, and Apremilast alone and after co-adminstration are all within the FDA acceptance range for equivalency (80–125%). This study showed that methotrexate and apremilast can be co-administered without any effect on the pharmacokinetic exposure of either agent.
Keywords: phosphodiesterase 4 inhibitor, inflammatory autoimmune disorders, drug interaction
Introduction
Apremilast is an oral phosphodiesterase 4 inhibitor for the treatment of inflammatory based autoimmune disorders, such as psoriatic arthritis, at a dose of 20 and 30 mg given twice daily.1,2 Apremilast is primarily eliminated as metabolites with less than 3% excreted unchanged in urine.3,4 In vitro apremilast is a substrate and a weak inhibitor of P-glycoprotein (P-gp; IC50 > 50 µM), however, good oral bioavailability of ∼73% in humans suggest P-gp in the gastrointestinal tract does not limit the oral absorption of apremilast.4 In addition P-gp does not play an important role in elimination of intact apremilast, since less than 3% of orally administered apremilast is excreted in unchanged form. Apremilast is not a substrate for other transporters such as breast cancer resistance protein (BCRP), organic anion transporter (OAT)1,OAT3, organic cation transporter (OCT)2, organic anion transporting polypeptide (OATP)1B1, or OATP1B3. It does not inhibit OAT3, BCRP, multidrug resistance protein (MRP)1, MRP2, MRP4, OATP1B3, and OCT2, and is a weak inhibitor of, OAT1, OATP1B1, and MRP3 (IC50 > 10 µM).
Methotrexate is a disease modifying antirheumatic drug (DMARD) where patients are to start at 5–10 mg of methotrexate administered per week and can titrate up to doses of 30 mg per week to achieve optimal clinical response.5 Methotrexate is eliminated almost entirely in an unchanged form in urine, which involves glomerular filtration and active tubular secretion.6 A small fraction (∼10%) of methotrexate is metabolized to 7-hydroxy methotrexate which is primarily excreted by the kidney. Seven-hydroxymethotrexate is believed to be equipotent with respect to toxicological properties and therapeutic benefits.
Based on the distinct dispositional profiles of apremilast and methotraxate, it is hypothesized that once weekly methotrexate and multiple dose apremilast could be coadministered to patients with rheumatoid arthritis or psoriasis without any effect on the pharmacokinetics of either agent. Therefore this study was undertaken to test this hypothesis by assessing the effects apremilast on methotrexate pharmacokinetics and the effect of methotrexate on apremilast pharmacokinetics.
Materials and Methods
The current study is an open-label, multi-center, 3-treatment period, sequential study conducted in otherwise healthy subjects with psoriasis, psoriatic arthritis, or rheumatoid arthritis who were being treated with methotrexate. It included male and female subjects in good health, other than the underlying diseases for which they were receiving a stable dose of methotrexate. The subjects were determined to be in good health by assessment of past medical history, physical examination, vital signs, 12-lead electrocardiogram, and clinical laboratory tests completed during screening and at baseline. Subjects received once weekly oral methotrexate therapy at a stable dose (between 7.5 and 20 mg/week) for at least 3 months prior to the start of the study.
The clinical portion of the study was conducted at 2 centers: Altoona Center for Clinical Research, Duncansville, PA and Metroplex Clinical Research Center, Dallas, TX. Subjects were prohibited from taking the following medications during the course of the study:DMARD and/or other immunosuppressive medications (aside from methotrexate); any investigational medication; herbal products containing St. John's Wort or garlic (alimentary garlic excluded); drugs excluded as per the US Food and Drug Administration (FDA) regulatory approved package insert for methotrexate, or grapefruit juice, cephalosporin, and the penicillin group of antibiotics.
Subjects who successfully completed screening, entered the clinic on Day −1 for a baseline evaluation and then received a single dose of methotrexate (7.5–20 mg) on the morning on Day 1. They received their first dose of Apremilast 30 mg on the morning of Day 3 while still in the clinic and then they continued to take Apremilast 30 mg every 12 hours on an outpatient basis from the evening of Day 3 through the morning dose on Day 6. Subjects returned to the clinic on Day 6 and continued with the Apremilast regimen in the clinic through the evening on Day 8 (for a total of 12 Apremilast doses). On Day 8 a single dose of methotrexate was administered with the morning apremilast dose. Serial pharmacokinetic samples were collected after both doses of methotrexate and after Day 7 and Day 8 dosing of apremilast.
Apremilast was supplied by Celgene Corporation as 30-mg tablets, (lot# B080093). Methotrexate was sourced by the investigators. However both clinic sites obtained the methotrexate from the same manufacturer (Boehringer Ingelheim Roxane Laboratories, lot number 857861B).
The clinical study protocol, informed consent document, and appropriate study-related documents were reviewed and approved by independent IRBs. The study was conducted in compliance with the Declaration of Helsinki, International Conference on Harmonization Guideline for Good Clinical Practice, and applicable regulatory requirements. All study subjects provided written informed consent prior to the start of any study-specific procedures.
Pharmacokinetic Sampling
Blood samples were collected for determination of plasma methotrexate and 7-hydroxy methotrexate concentrations immediately prior to and at the following times after the administration of methotrexate (on Day 1 and 8): 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 24, 36, and 48 hours. Blood samples were collected for determination of plasma apremilast concentrations just prior to the apremilast dose on the evening of Day 6, and serially on Day 7 and Day 8. Serial blood samples were collected prior to and at 0.5, 1, 1.5, 2, 3, 4, 6, 9, and 12 hours following the morning dose on Day 7 and 8 (12-hour sample was collected prior to administration of the evening dose). Subjects received the study treatment in a fasted state on the morning of pharmacokinetic sampling days (i.e., Day's 1, 7, and 8).
Blood Collection and Analytical Methodology
Blood samples were drawn through an indwelling venous cannula or by venipuncture from each subject at each timepoint and immediately placed in ice water. Blood was separated by centrifugation (approximately 1,500g × 10 minutes at 4°C) and the plasma harvested, within 30 minutes of collection. Citrated plasma samples for analysis of apremilast and heparanized plasma samples for analysis of methotrexate/7-hydroxy methotrexate were then stored at −70°C until assayed. Assay of Apremilast, methotrexate, and 7-hydroxy methotrexate in plasma samples were performed by QPS, LLC (Newark, DE). Plasma concentrations of apremilast, methotrexate, and 7-hydroxy methotrexate were determined by means of validated high performance liquid chromatography/tandem mass spectrometric (LC–MS/MS) methods. For the apremilast analytical method, CC-10004 and its internal standard (IS) CC-16305 were quantitatively extracted from 100 µL of human plasma sample containing 0.1 M citric acid using a liquid-liquid extraction method with methyl tert-butyl ether and reconstituted with 200 µL of H2O:methanol:formic acid/80:20:0.1 (v:v:v). The sample extract was loaded onto a Synergy Hydro-RP 30 mm × 2 mm, 4 µm, Phenomenex, Inc. (Torrance, CA) for separation. The mobile phase was composed of mobile phase: both H2O:Formic Acid/100:0.1 (v:v) as well as MeOH:Formic Acid/100:0.1 (v:v). The HPLC effluent was introduced into an API-4000 Tandem Mass Spectrometer equipped with an ESI source for CC-10004. Positive ions were detected in the multiple reaction monitoring (MRM) mode with precursor → product ion pairs of 461.16 m/z → 257.05 m/z for CC-10004, 465.16 m/z → 261.05 m/z for CC-16305. The apremilast assay ranged of 1–1,000 ng/mL with a intra-day coefficient of variation (CV) of ≤11.2 and relative error of −0.4 to 9.5% and a inter-day CV of ≤7.7% and a relative error 4.4–8.3%. For the methotrexate and 7-OH methotrexate method, methotrexate, and 7-hydroxy methotrexate and their IS methotrexatemethyl-d3, and [methyl-13C,U-2H3]-7-hydroxymethotrexate were quantitatively extracted from 300 µL of human plasma sample using a solid-phase extraction with OasisTM 30 µm (30 mg) cartridges and reconstituted with 200 µL of methanol:water/10:90 (v:v). The sample extract was loaded onto a Prodigy 5 µ Phenyl-3 (PH-3) 50 mm × 2.0 mm, Phenomenex for separation. The mobile phase was composed of H2O:Formic Acid/100:0.1 (v:v) and Acetonitrile:Formic Acid/100:0.1 (v:v). The HPLC effluent was introduced into an API-4000 Tandem Mass Spectrometer equipped with an ESI source for methotrexate and 7-hydroxy methotrexate. Positive ions were detected in the MRM mode with precursor → product ion pairs of 455.3 m/z → 308.2 m/z for methotrexate, 458.3 m/z → 311.2 m/z for methotrexate-d3, 471.2 m/z → 190.9 m/z for 7-hydroxy methotrexate, and 475.2 m/z → 190.9 m/z for [methyl-13C,U-2H3]-7-hydroxymethotrexate.The methotrexate assay ranged from 1 to 500 ng/mL with a intra-day CV of ≤5.4 and relative error of −4.4 to 1.1% and a inter-day CV of ≤ 3.9% and a relative error −3.3 to −1.3%. The 7-hydroxy methotrexate assay ranged from 1 to 500 ng/mL with a intra-day CV of ≤7.2 and relative error of −8.0 to −0.1% and a inter-day CV of ≤4.7% and a relative error −4.7 to 3.1%.
Pharmacokinetic and Statistical Analysis
In order to assess the steady-state pharmacokinetics of Apremilast with and without co-administration of methotrexate, the following pharmacokinetic parameters were calculated for Apremilast using noncompartmental analysis: Area under the plasma concentration-time curve during a dosing interval at steady-state (AUC0–tau), Maximum observed plasma concentration (Cmax), Time to maximum plasma concentration (tmax), Steady-state apparent total plasma clearance (CLss/F), and Minimum observed plasma concentration (Cmin).
In order to assess the single dose pharmacokinetics of methotrexate administered with and without co-administration of Apremilast the following PK parameters were calculated for methotrexate and 7-hydroxy methotrexate using noncompartmental analysis: area under the plasma concentration-time curve from time 0 to the last quantifiable concentration (AUC0–t), area under the plasma concentration-time curve from time 0 extrapolated to infinity (AUC0–∞), maximum observed plasma concentration (Cmax), time to maximum plasma concentration (tmax), and estimate of the terminal elimination half-life in plasma 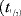 . The following parameters were calculated for methotrexate only: apparent total plasma clearance (CL/F), apparent volume of distribution (Vz/F).
. The following parameters were calculated for methotrexate only: apparent total plasma clearance (CL/F), apparent volume of distribution (Vz/F).
Pharmacokinetic parameters were derived using noncompartmental methods with WinNonlin® Professional Version 5.2 or higher, (Pharsight Corp., Mountain View, CA). The effect of multiple dose apremilast on the pharmacokinetics of methotrexate was evaluated statistically using an analyses of variance (ANOVA) on the natural log transformed AUC0–t, AUC0–∞, and Cmax of methotrexate and 7-hydroxy methotrexate to estimate the mean ratio of methotrexate administered with and without apremilast. The model included treatment as fixed effect and subject as random effect. The ratio of geometric means (“methotrexate + apremilast”/“methotrexate”) and its 90% CI were provided. For tmax, Hodges–Lehmann estimate and its 90% CI were calculated for the median difference between the 2 treatments (methotrexate administered with and without apremilast).
Subjects received individualized methotrexate dose ranging from 5 to 20 mg, titrated to optimize clinical outcome. However the pharmacokinetic data were analyzed without dose-normalization, as it has been previously established that the oral bioavailability of methotrexate declines at higher doses (15 mg or greater). The uptake of methotrexate by the gastrointestinal tract is primarily mediated by a saturable transporter and this decline is thought to occur because of saturation of this transporter.7
To assess the effect of methotrexate on the steady-state pharmacokinetics of apremilast, a similar ANOVA analysis was performed on the natural log transformed AUCtau and Cmax of apremilast. The model included treatment as fixed effect and subject as random effect. The ratio of geometric means (“methotrexate + apremilast”/“apremilast”) and its 90% CI were provided. For tmax Hodges–Lehmann estimate and its 90% CI were calculated for the median difference between the 2 treatments (apremilast administered with and without methotrexate). Healthy volunteer studies indicate that steady-state is achieved by 36 hours with Apremilast administered BID. An analysis was performed to verify that steady-state had been reached prior to the serial apremilast pharmacokinetic sampling. For this analysis the natural log transformed apremilast trough concentrations were analyzed using a repeated measure ANOVA model with the Helmert contrasts. The model included time point as fixed effect and subject as random effect.
Safety assessments were performed throughout the course of the study (i.e., upon signature of informed consent through a follow up visit scheduled on 9–10 days after the last dose). This includes collection of adverse events throughout the study, and assessment of physical exam, clinical laboratory tests, vital signs, and 12-lead electrocardiogram collected periodically.
Results
Fifteen subjects were successfully enrolled, received all planned doses of study drug and completed all study procedures as per protocol. Among the 15 subjects, a majority of the subjects (12/15) had rheumatoid arthritis and the remaining 3 subjects had psoriatic arthritis. Four subjects (27%) were male and the remaining 11 (73%) were female, which was expected for this disease population. These 15 subjects had a mean age of 52 ± 10 years (range of 35–64 years), a mean weight of 80 kg (range of 69–96 kg), a mean height of 167 cm (range of 155–183 cm); and a mean BMI of 29 kg/m2 (range of 24–32 kg/m2). Thirteen (87%) subjects were white and 2 (13%) subjects were black. A majority of the study subjects received either 15 mg (n = 6) or 20 mg (n = 6) of once weekly methotrexate. The remaining subjects received 10 mg (n = 1), 12.5 mg (n = 1), or 17.5 mg (n = 1) of once weekly methotrexate.
Pharmacokinetic Results
The semi log means (standard deviation) plasma methotrexate concentration-time curves for the subjects who received 20 mg of methotrexate with and without Apremilast are displayed in Figure 1. The methotrexate pharmacokinetic parameters were calculated for each subject and treatment group (i.e., methotrexate alone and methotrexate with Apremilast) the arithmetic mean for each parameter is listed in Table1. The 90% CI for Cmax, AUC0–t, and AUC0–∞ are listed in Table1 and were all within the acceptance range for equivalency (FDA acceptance range of 80–125%). The 90% CI of the median difference between the 2 treatments included 0 for tmax (P = 0.2646), indicating that there was no statistically significant difference for tmax (Table1).
Figure 1.
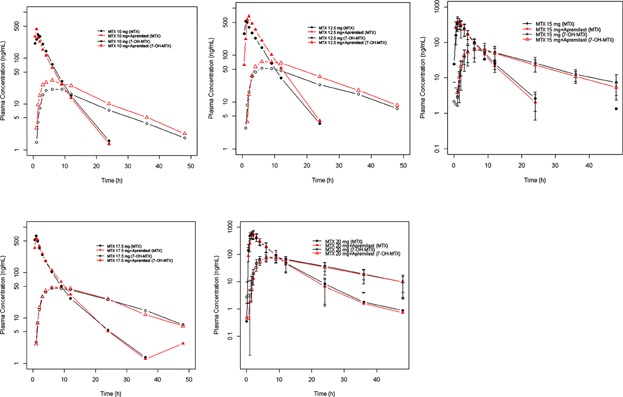
Methotrexate (MTX) and 7-OH methotrexate (7-OH MTX) plasma concentration-time profile following administration of 10 mg (N = 1), 12.5 mg(N = 1), 15 mg(N = 6), 17.5 mg (N = 1), 20 mg (N = 6) MTX alone or in combination with apremilast (mean ± SD presented for N > 1).
Table 1.
Summary of Methotrexate (MTX) Plasma Pharmacokinetic Parameters after Administration of Methotrexate Along (i.e., Day 1) and after Co-Administration of Methotrexate and Apremilast (i.e., Day 8)
| Treatment | Arithmetic Mean (Standard Deviation)a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| tmaxb (hr) | Cmax (ng/mL) | AUC0–t (hr ng/mL) | AUC0–∞ (hr ng/mL) | t1/2 (hr) | CL/F (mL/min) | ||||||
| MTX | MTX + Apremilast | MTX | MTX + Apremilast | MTX | MTX + Apremilast | MTX | MTX + Apremilast | MTX | MTX + Apremilast | MTX | |
| MTX 10 mg (N = 1) | 1.45 | 0.98 | 323 | 427 | 1,390 | 1,300 | 1,380 | 1,300 | 3.5 | 3.5 | 120 |
| N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | |
| MTX 12.5 mg (N = 1) | 1.00 | 2.07 | 528 | 669 | 2,360 | 3,020 | 2,360 | 3,010 | 3.6 | 3.2 | 88 |
| N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | |
| MTX 15 mg (N = 6) | 1.27 (0.98–1.52) | 1.25 (0.50–1.52) | 423 (110) | 412 (127) | 1,740 (563) | 1,730 (446) | 1,600c (520) | 1,720 (451) | 3.5c (0.55) | 3.3 (14.4) | 177c (81.1) |
| N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 5 | N = 6 | N = 5 | N = 6 | N = 5 | |
| MTX 17.5 mg (N = 1) | 1.00 | 1.00 | 646 | 565 | 2,250 | 2,270 | 2,250 | NC | 5.6 | NC | 129 |
| N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | |
| MTX 20 mg (N = 6) | 1.50 (1.00–2.07) | 2.07 (1.00–2.08) | 607 (144) | 586 (162) | 2,950 (957) | 2,820 (971) | 2,960 (971) | 2,830 (986) | 5.6 (2.8) | 4.5 (1.4) | 122 (33.7) |
| N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | |
| Ratio of geometric means | 0.235d | 99.49 | 99.90 | 100.39 | NC | NC | |||||
| 90% CI of Ratio of geometric means | (−0.22, 0.5)e | (92.67, 106.81) | (93.35, 106.90) | (92.89, 108.50) | NC | NC | |||||
| Intra-subject CV% | 0.2646f | 11.07 | 10.57 | 11.19 | NC | NC | |||||
| N = 15 | N = 15 | N = 15 | N = 14 | NC | NC | ||||||
N, number of subjects; CV, coefficient of variation; Cmax, maximum observed plasma concentration; tmax, time to maximum plasma concentration; t1/2, estimate of the terminal elimination half-life in plasma; AUC0–t, area under the plasma concentration-time curve from time 0 to the last quantifiable concentration; AUC0–∞, area under the plasma concentration-time curve from time 0 extrapolated to infinity; CL/F, apparent total plasma clearance when dosed orally; Vz/F, apparent total volume of distribution when dosed orally; NA, not applicable.
Arithmetic mean when appropriate (i.e., N > 1).
tmax is summarized by median and range (minimum to maximum), when appropriate.
Mean excludes data with R2 < 0.9.
Median difference (“MTX + Apremilast” − “MTX”).
90% CI of the median difference are from Hodges–Lehmann Estimate.
P-value is from Wilcoxon signed-rank test.
The semi log mean (and standard deviation) plasma concentration-time curves for 7-hydroxy methotrexate concentrations for subjects treated with 20 mg of methotrexate with and without co-administration of Apremilast are displayed in Figure 2. The individual and arithmetic mean (for groups with n > 1) pharmacokinetic parameters and statical analyses of each methotrexate dose group for 7-hydroxy methotrexate are listed in Table2. The 90% CI for Cmax, AUC0–t, and AUC0–∞ were within the acceptance range for equivalency (FDA acceptance range of 80–125%). The 90% CI of the median difference between the 2 treatments included 0 for tmax (P = 0.9902), indicating that there was no statistically significant difference for tmax.
Figure 2.
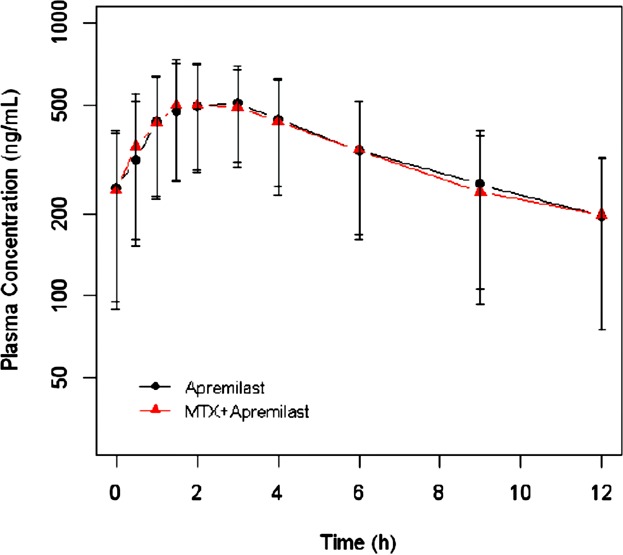
Mean (±SD) steady-state CC-10004 plasma concentrations by time and treatment following apremilast administration of 30 mg BID (Q12h) alone or in combination with methotrexate (N = 15).
Table 2.
Summary of 7-Hydroxy Methotrexate (7-OH MTX) Plasma Pharmacokinetic Parameters of after administration of methotrexate alone (i.e. Day 1) and methotrexate after co-administration with Apremilast (i.e., Day 8)
| Treatment | Arithmetic Mean (Standard Deviation)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| tmaxb (hr) | Cmax (ng/mL) | AUC0–t (hr ng/mL) | AUC0–∞ (hr ng/mL) | t1/2 (hr) | ||||||
| 7-OH MTX | 7-OH MTX + Apremilast | 7-OH MTX | 7-OH MTX + Apremilast | 7-OH MTX | 7-OH MTX + Apremilast | 7-OH MTX | 7-OH MTX + Apremilast | 7-OH MTX | 7-OH MTX + Apremilast | |
| MTX 10 mg (N = 1) | 5.98 | 6.00 | 21.0 | 32.9 | 430 | 620 | 460 | 650 | 11.9 | 10.9 |
| N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | |
| MTX 12.5 mg (N = 1) | 6.00 | 6.00 | 52.8 | 72.8 | 1,220 | 1,670 | 1,370 | 1,820 | 14.2 | 12.0 |
| N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | |
| MTX 15 mg (N = 6) | 7.52 (6.00–12.00) | 8.99 (6.00–9.07) | 64.7 (34.4) | 66.1 (29.0) | 1,330 (605.3) | 1,280 (490.1) | 1,470 (701.5) | 1,370 (506.4) | 12.0 (1.76) | 11.4 (1.86) |
| N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | |
| MTX 17.5 mg (N = 1) | 6.00 | 9.00 | 45.4 | 48.9 | 1,170 | 1,170 | 1,300 | 1,290 | 13.0 | 12.2 |
| N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | N = 1 | |
| MTX 20 mg (N = 6) | 7.54 (5.95–9.00) | 6.09 (5.97–9.00) | 83.3 (20.0) | 76.5 (22.4) | 1,800 (461.6) | 1,650 (630.5) | 1,980 (580.1) | 1,870 (834.5) | 12.0 (2.45) | 13.1 (4.08) |
| N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | |
| Ratio of geometric means | 0.005c | 104.01 | 98.59 | 98.24 | NC | |||||
| 90% CI of ratio of geometric means | (−1.405, 1.45)d | (92.82, 116.55) | (86.86, 111.91) | (86.85, 111.13) | NC | |||||
| Intra-subject CV% | 0.9902e | 17.84 | 19.89 | 19.34 | NC | |||||
| N = 15 | N = 15 | N = 15 | N = 15 | NC | ||||||
N, number of subjects; CV, coefficient of variation; Cmax, maximum observed plasma concentration; tmax, time to maximum plasma concentration; t1/2, estimate of the terminal elimination half-life in plasma; AUC0–t, area under the plasma concentration-time curve from time 0 to the last quantifiable concentration; AUC0–∞, area under the plasma concentration-time curve from time 0 extrapolated to infinity; CL/F, apparent total plasma clearance when dosed orally; Vz/F, apparent total volume of distribution when dosed orally; NA, not applicable.
Arithmetic mean when appropriate (i.e., N > 1).
tmax is summarized by median and range (minimum to maximum), when appropriate.
median difference (“MTX + Apremilast” − “MTX”).
90% CI of the median difference are from Hodges–Lehmann Estimate.
P-value is from Wilcoxon signed-rank test.
Blood samples for the determination of Apremilast (i.e., CC-10004) concentrations were collected pre-dose and 0.5–12 hours post-dose on Day 7 and 8. The semi log mean (standard deviation) plasma concentration-time curves for Apremilast concentration when apremilast was administered with and without methotrexate are displayed in Figure 2. The statistical analysis of Day 6, 7, and 8 predose apremilast concentrations to evaluate steady-state (using Helmert contrasts) showed that apremilast concentrations reached steady-state by the morning of Day 7 (P > 0.05). For this analysis one sample collected on Day 6 pre-pm dose concentration was below the limit of quantification, and was set to the limit of quantification (1 ng/mL) in the analysis. Day 6 pre-pm dose concentrations for 7 subjects were not used in the analysis because samples were collected outside the acceptable time deviation window (by more than 2 hours). The apremilast pharmacokinetic parameters and statistical analysis is listed in Table3. The 90% CI for Cmax and AUCtau were within the acceptance range for equivalency (FDA acceptance range of 80–125%). The 90% CI of the median difference between the 2 treatments included 0 for tmax (P = 1.000), indicating that there was no statistically significant difference for tmax.
Table 3.
Summary of CC-10004 (APR) Plasma Pharmacokinetic Parameters of after Administration of Apremilast Alone (i.e., Day 7) and Apremilast after Co-Administration with Methotrexate (MTX) (i.e., Day 8)
| Treatment | Arithmetic Mean (Standard Deviation) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| tmaxa (hr) | Cmax (ng/mL) | Cmin (ng/mL) | AUC0–τ (hr ng/mL) | CLss/F (mL/min) | ||||||
| APR | APR + MTX | APR | APR + MTX | APR | APR + MTX | APR | APR + MTX | APR | APR + MTX | |
| Apremilast 30 mg Q12H (N = 15) | 2.00 (0.95–4.00) | 2.00 (0.98–4.08) | 584 (198) | 564 (204) | 248 (153) | 242 (153) | 4,110 (1981) | 4,120b (1991) | 153 (78.6) | 156b (86.3) |
| N = 15 | N = 15 | N = 15 | N = 15 | N = 15 | N = 15 | N = 15 | N = 13 | N = 15 | N = 13 | |
| Ratio of geometric means | 0c | 95.00 | NC | 99.30 | NC | |||||
| 90% CI of ratio of geometric means | (−0.75, 0.74)d | (87.93, 102.65) | NC | (92.84, 106.20) | NC | |||||
| Intra-subject CV% | 1.000e | 12.07 | NC | 9.66 | NC | |||||
| N = 15 | N = 15 | NC | N = 15 | N = 13 | NC | |||||
N, number of subjects; CV, coefficient of variation; Cmax, maximum observed plasma concentration; tmax, time to maximum plasma concentration; t1/2, estimate of the terminal elimination half-life in plasma; AUC0–t, area under the plasma concentration-time curve from time 0 to the last quantifiable concentration; AUC0–∞, area under the plasma concentration-time curve from time 0 extrapolated to infinity; CL/F, apparent total plasma clearance when dosed orally; Vz/F, apparent total volume of distribution when dosed orally; NA, not applicable.
tmax is summarized by median and range (minimum to maximum).
Mean excludes data with R2 < 0.9.
Median difference (“APR + MTX” − “APR”).
90% CI of the median difference are from Hodges–Lehmann Estimate.
P-value is from Wilcoxon signed-rank test.
Safety
There were no deaths, serious adverse events (SAEs), or treatment emergent adverse events (TEAEs) that led to study discontinuation. Overall, 10 of 15 subjects (67%) enrolled in the study reported 53 TEAEs. Forty-one of the TEAEs reported by 6 out of 15 subjects (40%) were suspected to be related to study drug. The most frequently reported TEAEs were headache (reported by 6/15 subjects [40%]); nausea (reported by 8/15 subjects [53%]); dyspepsia and diarrhea (reported by 3/15 subjects [20%] each); and abdominal pain, dysgeusia, pain, and anxiety (reported by 2/15 subjects [13%] each). TEAEs resolved without intervention by the end of the study, with the exception of 1 TEAE of subcutaneous abscess. The incidence of TEAEs reported was higher in during the Period where apremilast was administered alone (i.e., Day 3–7), (10/15 subjects [67%] reported 45 events); than during the period where methotrexate was administered alone (i.e., Day 1–3), (2/15 subjects [13%] reported 2 events), or during the period where both treatments were administered together (i.e., Day 8–10)) (5/15 subjects [33%] reported 6 events). Overall, no trends or clinically meaningful changes were observed in clinical laboratory analytes, vital signs, ECG intervals, or physical examination findings throughout the study.
Discussion
Apremilast is a novel PDE4 inhibitor with anti-inflammatory activity. This compound is currently in clinical development for the treatment of inflammatory conditions such as psoriasis, psoriatic arthritis, and ankylosing spondylitis.8–11 Methotrexate is an antifolate drug widely used at high dosages in the treatment of malignancies, whereas it is used at low dosages in the same inflammatory conditions. This study assessed and confirmed that methotrexate and apremilast can be co-administered to patients with rheumatoid arthritis or psoriasis without any effect on the pharmacokinetic exposure of either agent.
This study established that Apremilast administered at steady-state would not affect the pharmacokinetics of methotrexate. Lack of effect was seen over a range of once weekly methotrexate doses (5 to 20 mg) where most subjects were receiving doses at the higher end of the range (i.e., 15 and 20 mg). The major circulating metabolite of Apremilast is a pharmacologically inactive glucuronide conjugate of O-demethylated Apremilast, designated as M12. Prior studies in healthy subjects have shown that when Apremilast was administered 30 mg BID, M12 was the most abundant metabolite of apremilast with AUC and Cmax being approximately 1.7-fold and 1.25-fold the concentration of Apremilast.12 While M-12 was not quantified in this study, the lack of change in methotrexate pharmacokinetics following Apremilast administration and subsequent metabolism leading to plasma exposure of M-12 also established that the M12 major metabolite of Apremilast did not affect the pharmacokinetics of methotrexate.
Methotrexate absorption is thought to be mediated by multiple transporters including MRP2, MRP3, BCRP, and proton coupled folate transport (PCFT).13 It is proposed that proton pump inhibitors reduce the function of PCFT.14 There are reports of interactions between methotrexate and proton pump inhibitors leading to an increased toxicity associated with high dose methotrexate.15 When studied at lower oral doses in rheumatoid arthritis subjects, no interaction was seen between methotrexate and proton pump inhibitors.16 MRP 2, MRP3, and BCRP are thought to have a larger impact on elimination of methotrexate.17 The drug–drug interactions that have been reported between methotrexate and NSAIDS, phenylbutazone, and probenecid are considered due to the inhibition of the uptake mediated by OAT3 as well as OAT1 and OAT4. The glucuronide conjugates of these compounds are also implicated in drug–drug interactions with methotrexate which involves inhibition of apical ATP-binding cassette transporters (MRP2 and MRP4).18–20 Transporters have been implicated in a majority of the interactions with methotrexate. In vitro studies showed that apremilast is not an inhibitor of hOAT or the ATP-binding cassette transporters (i.e., MRP2, MRP3, and BCP) which is involved in the absorption and elimination of MTX and 7-OH MTX, indicating Apremilast is unlikely to have PK drug–drug interaction with MTX. The present clinical study confirms the results from this in vitro studies. It is also important to note that the dose range of methotrexate being studied is limited to the low doses used in treating inflammatory conditions; the results of this study may not be extrapolated to higher doses of methotrexate used in malignancy.
Methotrexate did not affect the steady-state pharmacokinectics of Apremilast in this study. This is consistent with the fact that disposition of methotrexate is mainly via renal excretion and does not involve hepatic metabolism. Since Apremilast is primarily cleared via metabolism methotrexate was not expected to inhibit/induce metabolism of Apremilast. This study clearly showed that methotrexate did not affect the pharmacokinetics of Apremilast. Prior studies have shown that the apparent clearance of apremilast is 8.7 L/hr in patients compared to ∼10 L/hr in healthy subjects, the CL/F for the doses evaluated in the current study ranged from 5.28 to 10.62 L/hr which is consistent with these results.
The safety profile of apremilast in this study is consistent with previously conducted clinical pharmacology studies in healthy subjects. There were no deaths or SAEs reported during study conduct. No subjects were withdrawn due to TEAEs. The incidence of TEAEs and TEAEs assessed as having a “suspected” relationship to study drug (treatment related) was higher following apremilast treatment given alone BID than following MTX treatment alone or MTX (single administration) with apremilast administered together. However, this difference in the incidence of the TEAE's that occurred during the study is not unexpected. Subjects had been stabilized on methotrexate for at least 3 months prior to the start of the study and this is likely the first time subjects are receiving Apremilast, this would affect the incidence of TEAE's.
Thus apremilast, a new novel oral anti inflammatory agent, can be given with methotrexate, an established first line agent for many rheumatic diseases such as psoriasis, psoriatic arthritis, and rheumatoid arthritis, without any concern for drug–drug interaction of an increase/decrease in exposure of either agent and/or altered safety profile.
Acknowledgments
The authors thank Prachi Wickremasingha, Pharm.D. for assistance with manuscript preparation. The authors also thank colleagues Xiaomin Wang, Ph.D. and Liangang Liu, Ph.D. for their technical contributions to bioanalysis, and biostatistical analysis.
Declaration of Conflicting Interests
The authors declare no conflicts of interest.
References
- 1.Shutty B, West C, Pellerin M, Feldman S. Apremilast as a treatment for psoriasis. Expert Opin Pharmacother. 2012;13:1761–1770. doi: 10.1517/14656566.2012.699959. [DOI] [PubMed] [Google Scholar]
- 2.Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583–1590. doi: 10.1016/j.bcp.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann M, Kumar G, Schafer P, et al. Disposition, metabolism and mass balance of [14C]apremilast following oral administration. Xenobiotica. 2011;41:1063–1075. doi: 10.3109/00498254.2011.604745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu A, Wan Y, Laille E, et al. Washington, DC: AAPS Annual Meeting and Exposition; 2011. Absolute bioavailability of apremilast using a [14C] labeled microtracer IV solution concomitantly with an oral dose [abstract] October 23-27, 2011; [Google Scholar]
- 5.Weinblatt ME. Methotrexate in rheumatoid arthritis: a quarter century of development. Trans Am Clin Climatol Assoc. 2013;124:16–25. [PMC free article] [PubMed] [Google Scholar]
- 6.Seideman P, Beck O, Eksborg S, Wennberg M. The pharmacokinetics of MTX and its 7-hydroxy metabolite in patients with rheumatoid arthritis. Br J Clin Pharmacol. 1993;35:409–412. doi: 10.1111/j.1365-2125.1993.tb04158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian H, Cronstein BN. Understanding the mechanisms of action of methotrexate—Implications for the treatment of rheumatoid arthritis. Bull NYU Hosp Jt Dis. 2007;65:168–173. [PubMed] [Google Scholar]
- 8.Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis: results of a phase 3, randomized, controlled trial [abstract L13] Arthritis Rheum. 2012;64:4172–4173. [Google Scholar]
- 9.Papp K, Cather J, Rosoph L, et al. The efficacy of apremilast, a phosphodiesterase-4 inhibitor, in the treatment of moderate to severe psoriasis: results of a phase 2 randomised study. Lancet. 2012;380:738–746. doi: 10.1016/S0140-6736(12)60642-4. [DOI] [PubMed] [Google Scholar]
- 10.Reich K, Papp K, Leonardi C, et al. Miami, FL: Annual Meeting of the American Academy of Dermatology; 2013. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate to severe psoriasis: 16-week results of a phase 3, randomized, controlled trial (ESTEEM 1) [oral presentation] pp. 1–5. March. [DOI] [PubMed] [Google Scholar]
- 11.Schett G, Wollenhaupt J, Papp K, et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2012;64:3156–3167. doi: 10.1002/art.34627. [DOI] [PubMed] [Google Scholar]
- 12.Wu A, Dvorchik B, Cahell C, et al. Double-blind, randomized, multiple-dose, crossover, fourtreatment, four-period, four sequence study to investigate the effects of apremilast on the QT interval in healthy male subjects abstract #152. J Clin Pharmacol. 2010;50:1058–1095. [Google Scholar]
- 13.Murakami T, Yokooji T, Mori N. Study on absorption sites of quinidine and methotrexate in rat intestine. Pharmazie. 2010;65:440–447. [PubMed] [Google Scholar]
- 14.Urquhart BL, Gregor JC, Chande N, Knauer MJ, Tirona RG, Kim RB. The human proton-coupled folate transporter (hPCFT): modulation of intestinal expression and function by drugs. Am J Physiol Gastrointest Liver Physiol. 2010;298:G248–G254. doi: 10.1152/ajpgi.00224.2009. [DOI] [PubMed] [Google Scholar]
- 15.Bezabeh S, Mackey AC, Kluetz P, Jappar D, Korvick J. Accumulating evidence for a drug–drug interaction between methotrexate and proton pump inhibitors. Oncologist. 2012;17:550–554. doi: 10.1634/theoncologist.2011-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vakily M, Amer F, Kukulka MJ, Andhivarothai N. Coadministration of lansoprazole and naproxen does not affect the pharmacokinetic profile of methotrexate in adult patients with rheumatoid arthritis. J Clin Pharmacol. 2005;45:1179–1186. doi: 10.1177/0091270005280100. [DOI] [PubMed] [Google Scholar]
- 17.Vlaming ML, van Esch A, van de Steeg E, et al. Impact of Abcc2 [multidrug resistance-associated protein (Mrp) 2], Abcc3 (Mrp3), and Abcg2 (breast cancer resistance protein) on the oral pharmacokinetics of methotrexate and its main metabolite 7-hydroxymethotrexate. DMD. 2011;39:1338–1344. doi: 10.1124/dmd.111.038794. [DOI] [PubMed] [Google Scholar]
- 18.Nozaki Y, Kusuhara H, Kondo T, et al. Species difference in the inhibitory effect of nonsteroidal anti-inflammatory drugs on the uptake of methotrexate by human kidney slices. JPET. 2007;322:1162–1170. doi: 10.1124/jpet.107.121491. [DOI] [PubMed] [Google Scholar]
- 19.Takeda M, Khamdang S. Narikawa S, et al. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J Pharmacol Exp Ther. 2002;2:666–671. doi: 10.1124/jpet.102.034330. [DOI] [PubMed] [Google Scholar]
- 20.Breedveld P, Zelcer N, Pluim D, et al. Mechanism of the pharmacokinetic interaction between MTX and benzimidazoles: potential role of breast cancer resistance protein in clinical drug-drug interactions. Cancer Res. 2004;64:5804–5811. doi: 10.1158/0008-5472.CAN-03-4062. [DOI] [PubMed] [Google Scholar]