

Abstract
Approximately 42% of patients with sepsis undergo acute kidney injury (AKI), which evidently influences patient survival. However, effective therapy strategies are lacking, thus, the present study investigated the protective effects of dexmedetomidine (DEX), a highly selective α-2 adrenoceptor agonist, in rat sepsis models. Rat sepsis models were generated through lipopolysaccharide injection (LPS; 5 mg/kg) in the tail vein. Rats were pretreated with DEX (10 µg/kg) 10 min before LPS injection to observe its protective effects. Of note, a unique α-2-adrenergic receptor antagonist, yohimbine (YOH; 1 mg/kg, intraperitoneally), was also used to antagonize the protective effects of DEX 30 min before DEX exposure. Thirty-two male Sprague Dawley rats were randomly divided into the Sham, LPS, DEX + LPS and YOH + DEX + LPS groups (n=8/group). All the rats were sacrificed 4 h later to observe the pathological changes of renal tissue, including plasma creatinine (Cr), blood urea nitrogen (BUN), kidney injury molecule-1 (KIM-1) and high mobility group protein 1 (HMGB-1) expression. Interleukin 6 (IL-6), IL-18 and tumor necrosis factor α (TNF-α) were all determined to examine the mechanisms of LPS-induced AKI relative to inflammatory reaction. The results indicated that AKI induced by LPS was serious. Renal pathological injury, plasma Cr, BUN, IL-6, IL-18 and TNF-α were all evidently increased in varying degrees. KIM-1 and HMGB-1 expression was upregulated in the LPS group (P<0.05 vs. Sham group). However, when rats were pretreated with DEX, AKI induced by LPS was decreased significantly. Renal pathological injury, plasma Cr, BUN, IL-6, IL-18, TNF-α, and KIM-1 and HMGB-1 expression were all reduced (P<0.05 vs. LPS group). In addition, exposure of the α-2-adrenergic receptor antagonist, YOH, eliminated this reduction. In conclusion, DEX protected against sepsis-induced AKI through depressing the inflammatory reaction, mechanisms of which may be associated with α-2 receptors inhibition.
Keywords: acute kidney injury, dexmedetomidine, α-2 adrenergic receptor, kidney injury molecule
Introduction
Sepsis, also called septicemia, is a serious complication of patients undergoing pathogen infection, trauma, burn, hypoxia, reperfusion injury and surgery, which could affect myocardial contractility, decreased organ oxygen uptake capacity, and can lead to septic shock and multiple organ dysfunction syndrome. This state is called systemic inflammatory response syndrome (1,2). The incidence of sepsis is extremely high in the intensive care unit (ICU), with a mortality of ≤40–80%. As reported, the kidney is one of the most susceptible target organs of sepsis (3). Approximately 50% of patients suffer from acute kidney injury (AKI) caused by sepsis. Notably, mortality of these patients is as high as 70% (4,5). However, the mechanisms of sepsis-induced AKI remain unclear and effective therapy strategies are also lacking.
Inflammatory factors, such as interleukin 6 (IL-6), IL-18 and tumor necrosis factor α (TNF-α), play an important role in the pathological and physiological process, particularly IL-18. As recently reported, IL-18 was a sensitive and specific biomarker of the AKI diagnosis and predicted mortality of patients (6). From other aspects, IL-18 induced expression of TNF-α and IL-6 and made the inflammatory reaction more serious, which contributed to organ injuries (7). Thus, modulation of the inflammatory reaction in sepsis is extremely important to protect against organ injuries, including kidneys.
As a commonly used anesthetic, dexmedetomidine (DEX) is widely used in the ICU and the surgical room. DEX is a highly selective α-2 adrenoceptor agonist and inhibits sympathetic activity effectively (8,9). In recent years, increasing basic and clinical studies have proved that DEX protected against different types of organs. Duan et al (10) reported that DEX was not neurotoxic and attenuated neuroapoptosis; other studies indicated that DEX application following cardiac surgery was associated with a lower incidence of atrial arrhythmias (11); more noteworthy, it was recently reported that DEX reduced intestinal and hepatic injury following hepatectomy and protected against ischemia/reperfusion injury in rat kidney through inhibiting apoptosis (12,13). However, mechanisms of the DEX protective effects are unclear, particularly for renal protection. Thus, the purpose of the present study was to explore the protective effects of DEX on the renal tissue of rats in lipopolysaccharide (LPS)-induced AKI models and its relative mechanisms mediated by α-2 adrenoceptor activities.
Materials and methods
Animals and treatment
All the experiments followed the National Institutes of Health criteria for the care and use of laboratory animals. The study was also approved by the Laboratory Animal Care Committee of Sun Yat-sen University (Guangzhou, China). Male Sprague Dawley rats (180–220 g) were purchased from the Laboratory Animal Center of Sun Yat-sen University and randomly divided into four groups: Sham, LPS, DEX + LPS and yohimbine (YOH) + DEX + LPS. Every group contained 8 samples. Rats were fasted for 12 hours, but not forbidden to drink. The rat sepsis models were generated through LPS injection (5 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) in the tail vein (14). Rats were pretreated with DEX (10 µg/kg; Jiangsu Hengrui Medicine Co., Ltd., Lianyungang, Jiangsu, China) 10 min before LPS injection (15). A unique α-2-adrenergic receptor antagonist, YOH (1 mg/kg; Sigma Aldrich), was injected intraperitoneally 30 min before DEX exposure (16). All the rats were sacrificed 4 h later. Blood samples were obtained from the abdominal artery to measure creatinine (Cr) and blood urea nitrogen (BUN); IL-6, IL-18 and TNF-α. The left kidney was removed, fixed in 10% formalin for tissue pathological analysis, and the right was used to detect the expression of kidney injury molecule-1 (KIM-1) and high mobility group protein 1 (HMGB-1).
Assessment of kidney damage
Cr and BUN were measured in blood samples with an automatic biochemistry analyzer (Hitachi 7600-020/7170A; Hitachi High-Technologies Corp., Tokyo, Japan). Kidney specimens were fixed in 10% buffered formalin, embedded in paraffin and processed for hematoxylin and eosin staining (Nanjing Keygen Biotech Co., Ltd., Nanjing, Jiangsu, China) for morphological examination. Each microsection was counted in five randomly selected areas/slide at magnification, x400. The renal injury quantification method was performed using a 0–3 grading system, as described by Hamar et al (17).
Measurement of plasma IL-6, IL-18 and TNF-α
Blood samples were collected in heparinized tubes and centrifuged at 2,500 × g for 15 min. Plasma was stored at −80˚C. Serum IL-6, IL-18 and TNF-α were measured by ELISA kits according to the manufacturer's instructions (IL-6, IL-18 and TNF-α assay kits; all purchased from Nanjing Keygen Biotech. Co., Ltd).
Western blot analysis of KIM-1 and HMGB-1
Western blot analysis was used to calculate the protein expression following the renal tissue protein extraction. The sample was solubilized in sodium dodecyl sulfate (SDS) loading buffer (Bio-Rad, Hercules, CA, USA) by boiling. The samples were loaded onto a 10% polyacrylamide gel (Invitrogen Life Technologies, Carlsbad, CA, USA) and SDS-PAGE (Bio-Rad) was conducted. The samples were subsequently transferred to a polyvinylidene difluoride (Bio-Rad) membrane. Membranes were further incubated overnight at 4˚C with specific antibodies against KIM-1 (cat. no. sc-47495, 1:2,000 dilution; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and HMGB-1 (cat. no. sc-26351, 1:2,000 dilution; Santa Cruz Biotechnology, Inc.). All the blots were subsequently washed and incubated with respective horseradish peroxidase-coupled secondary antibodies (cat. no. sc-2020, 1:1,500 dilution; Santa Cruz Biotechnology, Inc.) at room temperature for one hour. The protein bands were detected by an enhanced chemiluminescent detective system (Amersham Biosciences UK Ltd., Little Chalfont, UK) and were quantified using the Quantity One software package (Bio-Rad). GAPDH (1:2,000 dilution; Thermo Fisher Scientific Inc., Fremont, CA, USA) was presented as the internal control to calculate the ratio of optical density, and values were compared with those of the Sham groups.
Statistical analysis
All the data were analyzed using SPSS software, version 12.0 (SPSS, Inc., Chicago, IL, USA). The differences between groups were analyzed using one-way analysis of variance, followed by Tukey post hoc comparisons. A two-tailed P<0.05 was considered to indicate a statistically significant difference.
Results
DEX decreases BUN and Cr levels in LPS-induced AKI and YOH reverses the protective effect of DEX
In order to explore the protective effect of DEX on AKI in sepsis, rat sepsis models were established with LPS injection in the tail vein. The results in Fig. 1 showed that serum Cr and BUN levels evidently increased, nearly two-fold compared to the Sham group, which illustrated that the kidney was damaged severely. When rats were pretreated with DEX, levels of serum Cr and BUN were reduced; however, this protective effect of DEX was reversed by the α-2-adrenergic receptor antagonist, YOH. When α-2 adrenoceptor was antagonized by YOH, the protective effect of DEX was diminished.
Figure 1.
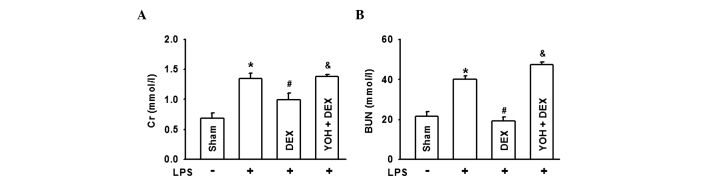
Lipopolysaccharide (LPS)-induces damage to the renal function, which was followed by treatment with dexmedetomidine (DEX) and yohimbine (YOH) in the rats. The levels of (A) creatinine (Cr) and (B) blood urea nitrogen (BUN) when rats were exposed to LPS, DEX or YOH. LPS was injected (5 mg/kg) in the tail vein to generate the rat sepsis models. DEX (10 µg/kg) was pretreated 10 min before LPS injection. YOH (1 mg/kg) was injected intraperitoneally 30 min before DEX exposure. (n=8 in each group). *P<0.05 vs. Sham group; #P<0.05 vs. LPS group; &P<0.05 vs. LPS + DEX group.
DEX ameliorates the pathological damage of kidneys in LPS-induced AKI and YOH reverses the protective effect of DEX
In order to further confirm the protective effect of DEX, the change of renal pathological morphology was analyzed in the rat sepsis models according to the Hamar score method (17). The results in Fig. 2 indicated that in the Sham group, normal kidney tissues were without lumen expansion and epithelial cell flattening in the glomerular and renal tubule; however, when rats were exposed to LPS, kidney pathological damage was clear: Renal tubular epithelial cell degeneration, renal tubular cavity expansion and tube formation and renal interstitial inflammatory cell infiltration are all serious. DEX application protected against LPS-induced renal damage, which was reversed by YOH. Rat pathological injury was coincidental with the levels of serum Cr and BUN in Fig. 1. The results in Figs. 1 and 2 indicated that the protective effect of DEX was relative with its function of activating α-2 adrenoceptor and inhibiting sympathetic activity.
Figure 2.

Lipopolysaccharide (LPS)-induces damage to the renal tissue, which was followed by treatment with dexmedetomidine (DEX) and yohimbine (YOH) in the rats. Histopathological changes in the kidney tissue (hematoxylin and eosin staining; original magnification, x200). LPS was injected (5 mg/kg) in the tail vein to generate the rat sepsis models. DEX (10 µg/kg) was pretreated 10 min before LPS injection. YOH (1 mg/kg) was injected intraperitoneally 30 min before DEX exposure. Arrow: Dilatant renal tubules and renal interstitial infiltration of the inflammatory cells.
DEX downregulates KIM-1 and HMGB-1 expression of kidneys in LPS-induced AKI and YOH reverses this type of effect of DEX
To more specifically establish the protective role of DEX in LPS-induced AKI, KIM-1 and HMGB-1 expression, as two types of more specific and sensitive biomarker for the diagnosis of early damage of kidneys, were determined in Fig. 3. When rats were pretreated with LPS, KIM-1 and HMGB-1 expression evidently increased indicating that kidney damage was significant. DEX application ameliorated KIM-1 and HMGB-1 expression increasing in kidneys of LPS-exposure rats. Similarly, YOH counteracted the protective effects of DEX.
Figure 3.
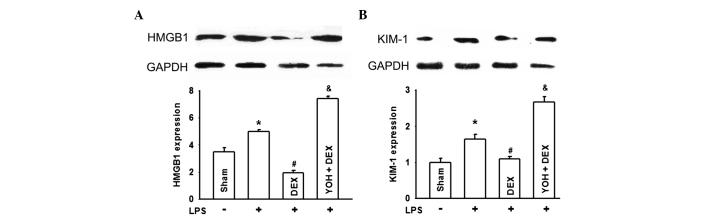
Lipopolysaccharide (LPS)-induces changes of the high mobility group protein 1 (HMGB-1) or kidney injury molecule-1 (KIM-1) expression, followed by treatment with dexmedetomidine (DEX) and yohimbine (YOH) in the rats. The changes of (A) HMGB1 and (B) KIM-1 expression when rats were exposed to LPS, DEX or YOH. LPS was injected (5 mg/kg) in the tail vein to generate the rat sepsis models. DEX (10 µg/kg) was pretreated 10 min before LPS injection. YOH (1 mg/kg) was injected intraperitoneally 30 min before DEX exposure. (n=8 in each group). *P<0.05 vs. Sham group; #P<0.05 vs. LPS group; &P<0.05 vs. LPS + DEX group.
DEX ameliorates the inflammatory reaction of kidneys in LPS-induced AKI and YOH reverses the protection effects of DEX
To the best of our knowledge, AKI caused by sepsis was always associated with inflammatory reactions (18,19). Thus, in order to explore the protective mechanisms of DEX for AKI, representative inflammatory factors, such as IL-6, IL-18 and TNF-α were detected. Fig. 4 demonstrates that IL-6, IL-18 and TNF-α were all significantly increased in the LPS group and DEX pretreatment downregulated the increase of these three types of inflammatory factors. Similarly, YOH application reversed this type of DEX effect. These results suggested that the protective function of DEX was associated with its anti-inflammatory effects through activating the α-2 adrenoceptor.
Figure 4.
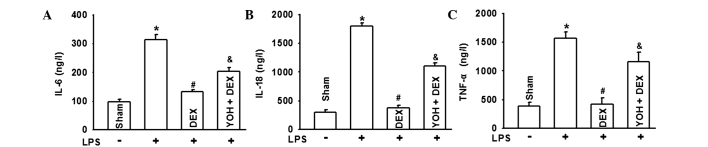
Lipopolysaccharide (LPS) induces changes of interleukin 6 (IL-6), IL-18 or tumor necrosis factor α (TNF-α), followed by treatment with dexmedetomidine (DEX) and yohimbine (YOH) in the rats. The changes of (A) IL-6, (B) IL-18 and (C) TNF-α when rats were exposed to LPS, DEX or YOH. LPS was injected (5 mg/kg) in the tail vein to generate the rat sepsis models. DEX (10 µg/kg) was pretreated 10 min before LPS injection. YOH (1 mg/kg) was injected intraperitoneally 30 min before DEX exposure. (n=8 in each group). *P<0.05 vs. Sham group; #P<0.05 vs. LPS group; &P<0.05 vs. LPS + DEX group.
Discussion
Patients with sepsis undergoing AKI is extremely common in clinical practice, particularly in ICU, which influences patient survival. Approximately 80% of acute renal failure is caused by renal tubular injury (20,21). The pathophysiological process of AKI induced by sepsis is extremely complex and severely affects patient survival. To the best of our knowledge, numerous different types of mechanisms were associated with this complication. Renal ischemia-reperfusion injury, inflammatory reaction, oxidative stress injury, nitric oxide release, cell apoptosis, endothelial dysfunction and the direct injury of endotoxin all participated in this process (19,22–26). However, mechanisms of sepsis-induced AKI remain unclear and effective therapies are lacking.
Rat sepsis models with LPS injection from tail vein were used to explore the protective effects of DEX against AKI. The analysis of the experimental results found that pretreatment with DEX ameliorated AKI, including renal function, pathological damage and two types of specific and sensitive biomarkers for the diagnosis of early kidney damage: KIM-1 and HMGB-1 expression. However, all the protective effects could be reversed by the α-2-adrenergic receptor antagonist, YOH. It is suggested that the protective effect of DEX was associated with its function of activating α-2 adrenoceptor. Of note, the inflammatory factor increase in the LPS group could be diminished by YOH application. All the results indicated that DEX ameliorated AKI through decreasing α-2-adrenergic receptor-mediated inflammatory reaction. Thus, DEX application in clinical practice may be a totally new therapy for this type of serious complication.
In the present study, KIM-1 and HMGB-1 expression, two types of specific and sensitive biomarkers for the diagnosis of early kidney damage, were determined. HMGB1 contributed to the pathogenesis of several chronic inflammatory and autoimmune diseases, including kidney disease. HMGB1 mediated the inflammatory response upon binding to mediators of inflammation, such as Toll-like receptor-2 (TLR-2) or TLR-4 and activated additional proinflammatory cytokines release, such as TNF-α or IL-6. Another type of protein is KIM-1, a type 1 transmembrane tubular protein with an immunoglobulin and mucin domain, which is undetectable in normal kidneys, but is markedly induced in experimental renal injury. KIM-1 is associated with T-cell activation and immune response (27) and is also considered as a unique marker for diagnosis of early kidney damage with high sensitivity and specificity (28–31). In the present study, HMGB1 and KIM-1 expression were coincidental with renal function and pathological damage. When AKI was significant, KIM-1 and HMGB-1 expression was also increased; as DEX application ameliorated AKI, they were also decreased. These two proteins are good markers for AKI diagnosis, but more noteworthy, changes of KIM-1 and HMGB-1 expression provided an indication that LPS-induced AKI was associated with an inflammatory reaction, as these two types of protein not only mediated the inflammatory response, but also promoted proinflammatory cytokines release. Thus, the function of the inflammatory reaction in AKI and whether the protective effect of DEX was associated with inflammatory reaction depression were explored. Fig. 4 showed that the representative inflammatory factors, IL-6, IL-18 and TNF-α, increased in the LPS-induced AKI group, which could be reversed by DEX application.
As reported, the inflammatory reaction has always played an important role in the pathogenesis of AKI (32). Lu et al (33) reported that nine polymorphism genes are closely associated with AKI, including TNF-α and IL-6 in particular. A large number of studies illustrated these inflammatory factors not only lead to AKI, but also promoted it to become worse. As reported previously, IL-18 is mainly produced from macrophages, which can effectively predict the mortality of patients. Urinary IL-18 was significantly increased with 90% sensitivity and specificity in the diagnosis of AKI; thus, it can be used as a sensitive index of patients undergoing AKI (34); and more notably, IL-18 induced the expression of TNF-α and IL-6. This made the inflammatory reaction more serious and contributed to organ injuries (7). In the present experiment, DEX application decreased these inflammatory factors and protected against LPS-induced AKI, which may be a useful therapy for AKI; however, the protective mechanism of DEX remains unclear.
DEX is a highly selective α-2-adrenergic receptor antagonist and effectively inhibited sympathetic activity widely. In recent years, investigators have found that DEX also played an important role in organ protection from numerous different aspects, such as anti-inflammation, oxidative stress inhibition or apoptosis regulation. Villela et al (35) reported that DEX protected against kidney damage through its diuretic effect. Other studies clarified that DEX decreased renal cell apoptosis through activating α-2 adrenoceptor and downregulating TLR4 expression (36). DEX also had preventive effects on acute lung injury through mediating oxidative stress (37). In the present study, DEX ameliorated LPS-induced AKI through reducing the levels of inflammatory factors, such as IL-6, IL-18 and TNF-α. Thus, DEX application was beneficial for kidney protection, particularly in sepsis.
When the unique α-2-adrenergic receptor antagonist, YOH, was used, the protective effect of DEX was counteracted and the inflammatory factors, IL-6, IL-18 and TNF-α, were reversed, suggesting that DEX protected against AKI through α-2-adrenergic receptor activation. α-2-adrenergic receptors are widely distributed in various tissues and organs, including the renal proximal tubule, collecting duct and microvascular system (38). As reported previously, α-2-adrenergic receptor activation regulated the function of the sympathetic nerve and led to the cholinergic anti-inflammatory pathway, which downregulated different types of inflammatory factors (39,40). Other studies reported that α-2-adrenergic receptor activation inhibited Janus kinase/signal transducers and activators of transcription and TLR-4/nuclear factor-κB/mitogen-activated protein kinases signal pathways to exert anti-inflammatory effects (41). These conclusions mean that DEX played an important role in anti-inflammation. Thus, DEX protected against LPS-induced AKI through α-2-adrenergic receptor activation, which decreased the inflammatory factors, IL-6, IL-18 and TNF-α.
Acknowledgements
The present study was supported by the Medical Research Foundation of Guangdong Province (grant nos. A2012565 and B2014141); and the National Natural Science Foundation of China (grant no. 81401628).
References
- 1.Puri VK. Of mice and MODS, TNF-alpha, and sepsis. Crit Care Med. 1998;26:1160. doi: 10.1097/00003246-199807000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Oda S, Hirasawa H, Sugai T, Shiga H, Nakanishi K, Kitamura N, Sadahiro T, Hirano T. Comparison of Sepsis-related Organ Failure Assessment (SOFA) score and CIS (cellular injury score) for scoring of severity for patients with multiple organ dysfunction syndrome (MODS) Intensive Care Med. 2000;26:1786–1793. doi: 10.1007/s001340000710. [DOI] [PubMed] [Google Scholar]
- 3.Bagshaw SM, George C, Bellomo R. ANZICS Database Management Committee: Early acute kidney injury and sepsis: A multicentre evaluation. Crit Care. 2008;12:R47. doi: 10.1186/cc6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 5.Rajapakse S, Rodrigo C, Rajapakse A, Kirthinanda D, Wijeratne S. Renal replacement therapy in sepsis-induced acute renal failure. Saudi J kidney Dis Transpl. 2009;20:553–559. [PubMed] [Google Scholar]
- 6.Nisula S, Yang R, Poukkanen M, et al. The FINNAKI Study Group: Predictive value of urine interleukin-18 in the evolution and outcome of acute kidney injury in critically ill adult patients. Br J Anaesth. 2014 Dec 3; doi: 10.1093/bja/aeu382. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Zhang ZX, Lian D, Haig A, Bhattacharjee RN, Jevnikar AM. IL-37 inhibits IL-18 induced tubular epithelial cell expression of pro-inflammatory cytokines and renal ischemia reperfusion injury. Kidney Int. 2015;87:396–408. doi: 10.1038/ki.2014.295. [DOI] [PubMed] [Google Scholar]
- 8.Miranda ML, Balarini MM, Bouskela E. Dexmedetomidine Attenuates the Microcirculatory Derangements Evoked by Experimental Sepsis. Anesthesiology. 2014 Oct 13; doi: 10.1097/ALN.0000000000000491. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 9.Geloen A, Chapelier K, Cividjian A, Dantony E, Rabilloud M, May CN, Quintin L. Clonidine and dexmedetomidine increase the pressor response to norepinephrine in experimental sepsis: A pilot study. Crit Care Med. 2013;41:e431–e438. doi: 10.1097/CCM.0b013e3182986248. [DOI] [PubMed] [Google Scholar]
- 10.Duan X, Li Y, Zhou C, Huang L, Dong Z. Dexmedetomidine provides neuroprotection: Impact on ketamine-induced neuroapoptosis in the developing rat brain. Acta Anaesthesiol Scand. 2014;58:1121–1126. doi: 10.1111/aas.12356. [DOI] [PubMed] [Google Scholar]
- 11.Turan A, Bashour CA, You J, Kirkova Y, Kurz A, Sessler DI, Saager L. Dexmedetomidine sedation after cardiac surgery decreases atrial arrhythmias. J Clin Anesth. 2014;26:634–642. doi: 10.1016/j.jclinane.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Wang ZX, Huang CY, Hua YP, Huang WQ, Deng LH, Liu KX. Dexmedetomidine reduces intestinal and hepatic injury after hepatectomy with inflow occlusion under general anaesthesia: A randomized controlled trial. Br J Anaesth. 2014;112:1055–1064. doi: 10.1093/bja/aeu132. [DOI] [PubMed] [Google Scholar]
- 13.Si YN, Bao HG, Xu L, Wang XL, Shen Y, Wang JS, Yang XB. Dexmedetomidine protects against ischemia/reperfusion injury in rat kidney. Eur Rev Med Pharmacol Sci. 2014;18:1843–1851. [PubMed] [Google Scholar]
- 14.Bayraktar O, Tekin N, Aydın O, Akyuz F, Musmul A, Burukoglu D. Effects of S-allyl cysteine on lung and liver tissue in a rat model of lipopolysaccharide-induced sepsis. Naunyn Schmiedebergs Arch Pharmacol. 2014 Dec 6; doi: 10.1007/s00210-014-1076-z. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 15.Restitutti F, Laitinen MR, Raekallio MR, Vainionpää M, O'Brien RT, Kuusela E, Vainio OM. Effect of MK-467 on organ blood flow parameters detected by contrast-enhanced ultrasound in dogs treated with dexmedetomidine. Vet Anaesth Analg. 2013;40:e48–e56. doi: 10.1111/vaa.12058. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Yu X, Wang F, Wang Y, Wang Y, Li H, Lv X, Lu D, Wang H. Yohimbine promotes cardiac NE release and prevents LPS-induced cardiac dysfunction via blockade of presynaptic α2A-adrenergic receptor. PLoS ONE. 2013;8:e63622. doi: 10.1371/journal.pone.0063622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamar P, Song E, Kökény G, Chen A, Ouyang N, Lieberman J. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2004;101:14883–14888. doi: 10.1073/pnas.0406421101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ratliff BB, Rabadi MM, Vasko R, Yasuda K, Goligorsky MS. Messengers without borders: Mediators of systemic inflammatory response in AKI. J Am Soc Nephrol. 2013;24:529–536. doi: 10.1681/ASN.2012060633. [DOI] [PubMed] [Google Scholar]
- 19.Westenfelder C. Programmed anti-inflammatory macrophages protect against AKI and promote repair through trophic actions. Kidney Int. 2012;81:939–941. doi: 10.1038/ki.2012.9. [DOI] [PubMed] [Google Scholar]
- 20.Singbartl K, Kellum JA. AKI in the ICU: Definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012;81:819–825. doi: 10.1038/ki.2011.339. [DOI] [PubMed] [Google Scholar]
- 21.Honore PM, Jacobs R, Joannes Boyau O, et al. Septic AKI in ICU patients. Diagnosis, pathophysiology and treatment type, dosing, and timing: A comprehensive review of recent and future developments. Ann Intensive Care. 2011;1:32. doi: 10.1186/2110-5820-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatachalam MA, Weinberg JM. The conundrum of protection from AKI by adenosine in rodent clamp ischemia models. Kidney Int. 2013;84:16–19. doi: 10.1038/ki.2013.101. [DOI] [PubMed] [Google Scholar]
- 23.Myrvang H. Acute kidney injury: Obesity is associated with AKI after surgery via oxidative stress. Nat Rev Nephrol. 2012;8:433. doi: 10.1038/nrneph.2012.121. [DOI] [PubMed] [Google Scholar]
- 24.Choe JY, Park KY, Kim SK. Oxidative stress by monosodium urate crystals promotes renal cell apoptosis through mitochondrial caspase dependent pathway in human embryonic kidney 293 cells: Mechanism for urate-induced nephropathy. Apoptosis. 2015;20:38–49. doi: 10.1007/s10495-014-1057-1. [DOI] [PubMed] [Google Scholar]
- 25.Anjaneyulu M, Chopra K. Effect of irbesartan on the antioxidant defence system and nitric oxide release in diabetic rat kidney. Am J Nephrol. 2004;24:488–496. doi: 10.1159/000080722. [DOI] [PubMed] [Google Scholar]
- 26.Yatim KM, Oberbarnscheidt MH. Endotoxin and AKI: Macrophages Protect after Preconditioning. J Am Soc Nephrol. 2014 Nov 14; doi: 10.1681/ASN.2014101042. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodríguez-Iturbe B, Johnson RJ, Herrera-Acosta J. Tubulointerstitial damage and progression of renal failure. Kidney Int Suppl. 2005;99:S82–S86. doi: 10.1111/j.1523-1755.2005.09915.x. [DOI] [PubMed] [Google Scholar]
- 28.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62:237–244. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 29.Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV. Urinary kidney injury molecule-1: A sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol. 2006;290:F517–F529. doi: 10.1152/ajprenal.00291.2005. [DOI] [PubMed] [Google Scholar]
- 30.Liangos O, Perianayagam MC, Vaidya VS, et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol. 2007;18:904–912. doi: 10.1681/ASN.2006030221. [DOI] [PubMed] [Google Scholar]
- 31.Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365:417–430. doi: 10.1016/S0140-6736(05)17831-3. [DOI] [PubMed] [Google Scholar]
- 32.Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172:2629–2635. doi: 10.4049/jimmunol.172.4.2629. [DOI] [PubMed] [Google Scholar]
- 33.Lu JCT, Coca SG, Patel UD, Cantley L, Parikh CR. Translational Research Investigating Biomarkers and Endpoints for Acute Kidney Injury (TRIBE-AKI) Consortium: Searching for genes that matter in acute kidney injury: A systematic review. Clin J Am Soc Nephrol. 2009;4:1020–1031. doi: 10.2215/CJN.05411008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall IE, Yarlagadda SG, Coca SG, Wang Z, Doshi M, Devarajan P, Han WK, Marcus RJ, Parikh CR. IL-18 and urinary NGAL predict dialysis and graft recovery after kidney transplantation. J Am Soc Nephrol. 2010;21:189–197. doi: 10.1681/ASN.2009030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villela NR, do Nascimento Júnior P, de Carvalho LR, Teixeira A. Effects of dexmedetomidine on renal system and on vasopressin plasma levels. Experimental study in dogs. Rev Bras Anestesiol. 2005;55:429–440. doi: 10.1590/s0034-70942005000400007. [DOI] [PubMed] [Google Scholar]
- 36.Hanci V, Yurdakan G, Yurtlu S, Turan IO, Sipahi EY. Protective effect of dexmedetomidine in a rat model of α-naphthylthiourea-induced acute lung injury. J Surg Res. 2012;178:424–430. doi: 10.1016/j.jss.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 37.Cekic B, Geze S, Ozkan G, et al. The effect of dexmedetomidine on oxidative stress during pneumoperitoneum. BioMed research international. 2014;2014:760323. doi: 10.1155/2014/760323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu J, Sun P, Zhao H, Watts HR, Sanders RD, Terrando N, Xia P, Maze M, Ma D. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit Care. 2011;15:R153. doi: 10.1186/cc10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Si Y, Bao H, Han L, et al. Dexmedetomidine protects against renal ischemia and reperfusion injury by inhibiting the JAK/STAT signaling activation. J Transl Med. 2013;11:141. doi: 10.1186/1479-5876-11-141. [DOI] [PMC free article] [PubMed] [Google Scholar]