

Abstract
Objective
Icosapent ethyl (IPE) is a prescription form of eicosapentaenoic acid (EPA) ethyl ester. This randomized, open-label study characterized EPA pharmacokinetics.
Methods
Four healthy subject groups received IPE for 28 days: three received 2 g/day (1 × 1,000 mg BID, 2 × 1,000 mg QD, or 2 × 500 mg BID); one received 4 g/day (2 × 1,000 mg BID) administered with meals. Blood sampling was before the morning dose on days 1, 14, 26, 28, and at specified intervals during an 18-day pharmacokinetic period. EPA was measured in plasma (total and unesterified) and red blood cells (RBCs) by liquid chromatography/tandem mass spectrometry.
Results
Mean plasma total EPA increased from 19 µg/mL to a peak (Cmax) of 366 µg/mL at 5 hours postdosing 4 g/day IPE on Day 28. Mean RBC EPA Cmax after 4 g/day was 89 µg/mL (baseline, 12 µg/mL). Mean steady state (SD) for half-life, clearance, and volume of distribution of total EPA were 79 (47) hours, 757 (283) mL/h, and 82 (56) L, respectively. Steady state for total and unesterified plasma EPA was reached by Day 28, whereas RBC levels were still increasing.
Conclusions
EPA pharmacokinetic profile demonstrated a slowly cleared, extensively distributed molecule with dose linearity and comparable exposures with BID and QD regimens.
Keywords: eicosapentaenoic acid, eicosapentaenoic acid ethyl ester, hypertriglyceridemia, fish oil, icosapent ethyl, omega-3 fatty acids, triglycerides, pharmacokinetics
Omega-3 fatty acids have been shown to have a variety of cardioprotective effects,1 and dietary intake of 1 g/day of omega-3 fatty acids has been recommended for the secondary prevention of cardiovascular disease by the American Heart Association.2 Hypertriglyceridemia is directly associated with increased risk of atherosclerotic coronary heart disease,3 and consumption of the marine omega-3 fatty acids, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) has been shown to have potent triglyceride (TG)-lowering effects.4,5
Icosapent ethyl (IPE; Vascepa® [formerly AMR101]; Amarin Pharma, Inc., Bedminster, NJ) is a high-purity prescription form of EPA ethyl ester approved by the United States Food and Drug Administration as an adjunct to diet to reduce TG levels in adult patients with severe (≥500 mg/dL) hypertriglyceridemia. The MARINE and ANCHOR pivotal studies collectively demonstrated that IPE significantly reduced TG levels in patients with very high (≥500 mg/dL and ≤2,000 mg/dL) or high (≥200 mg/dL and <500 mg/dL) TG levels in the absence and presence of statin therapy.6,7 TG-lowering occurred without raising low-density lipoprotein cholesterol (LDL-C) levels in the MARINE study,6 and in the ANCHOR study, LDL-C levels were significantly reduced.7 While the safety and efficacy of IPE have been described,6,7 the pharmacokinetics have not yet been reported.
EPA is a component of the normal diet. During absorption, EPA is distributed and (re)incorporated into circulating phospholipids, triacylglycerol, and cholesteryl esters and in the phospholipid components of cell membranes.8–11 Only a small fraction of the total circulating EPA concentration is unesterified (not incorporated in triacylglycerol, phospholipids, cholesteryl esters, and red blood cells [RBCs]). Incorporation of EPA into RBC membranes reflects membrane EPA content in other tissues12 and, because RBCs are convenient for sampling, assessment of RBC EPA in pharmacokinetic studies offers insight into the tissue content of EPA.
Several studies in humans have examined the pharmacokinetics of various formulations of EPA ethyl esters, some of which also contained DHA ethyl esters. These studies have demonstrated the absorption and appearance of EPA in plasma as free fatty acid, phospholipids, and other lipid forms, as well as the incorporation into RBC membranes following oral administration of the ethyl ester of EPA.8,13–19 Interestingly, the ethyl ester of EPA is not readily detectable in the blood following oral intake14,15,19; it is evidently completely hydrolyzed during digestion to become free fatty acid.20 This may have an effect upon the relative bioavailability of omega-3 fatty acid ethyl esters as compared to the free fatty acid forms as documented in overweight patients on low-fat diets.21
The purpose of this study was to characterize the pharmacokinetics of EPA in plasma and RBCs after multiple-dose oral administration of IPE at doses used in the MARINE6 and ANCHOR7 studies and to explore dosing regimens in healthy males and females.
Methods
Study Design
This was a phase 1, open-label, randomized, multidose study in healthy, nonsmoking men and women aged >18 and ≤55 years with a body mass index (BMI) >18 and ≤30 kg/m2. The study protocol was reviewed and approved by an institutional review board (IntegReview Ethics Review Board, Austin, TX). Written informed consent was obtained from each subject prior to performing any procedures or collecting any information. Exclusion criteria included use within 6 weeks prior to randomization through study end of any lipid-altering medications or supplements, including statins, niacin >200 mg/day, fibrates, ezetimibe, bile acid sequestrants, omega-3 fatty acid medications, and supplements or foods enriched with omega-3 fatty acids (no more than two servings of fish per week were permitted).
Following a 14-day screening period, subjects entered a 28-day treatment period and were randomized to one of four IPE dose regimen treatment groups. Group 1 received IPE 2 g daily (1,000-mg capsules; twice-daily [BID] regimen); Group 2 received IPE 4 g daily (1,000-mg capsules; BID regimen); Group 3 received IPE 2 g daily (1,000-mg capsules; once-daily [QD] regimen); and Group 4 received IPE 2 g daily (500-mg capsules; BID regimen). Treatment was administered orally with a meal in the morning and evening for BID regimens or in the morning only for the QD regimen. The content of the meals during the 28-day outpatient treatment period was not protocol-specified other than that noted above in the exclusion criteria. During the scheduled study site visits, all subjects were provided the same meals, although the content of the meals was different on different study days. On Day 28, the last dose of study drug was administered with a breakfast containing approximately 460 kcal and 25% fat. All meals at the study site were consumed within 30 minutes. The average daily dose of IPE was calculated based on the unused capsule counts across all treatment days prior to the last day of dosing. Percent compliance was calculated as actual daily dose/planned daily dose × 100. Subjects then entered a post-treatment pharmacokinetic sampling period lasting 18 days to estimate the expected long half-life of EPA. Safety assessments were evaluated in the intent-to-treat population and consisted of adverse event (AE) monitoring, clinical laboratory measurements (chemistry, hematology, and urinalysis), 12-lead electrocardiographic measurements, vital signs (systolic and diastolic blood pressure, heart rate, respiratory rate, and oral body temperature), and physical examination findings. The intent-to-treat population was defined as all randomized subjects who received at least one dose of study drug. The per-protocol population was defined as all subjects who completed the 28-day treatment period without any major protocol deviations, and who provided blood samples for EPA analyses.
Fasting EPA concentrations were measured in plasma and RBCs prior to the morning dose (Days 1, 14, 26, 28) and at serial time points (1, 3, 5, 6, 8, 10, 12, 24, 48, 72, 120, 192, 312, and 432 hours) after the morning dose on Day 28. Total plasma EPA included all EPA forms (unesterified EPA and that incorporated in phospholipids, triacylglycerols, and cholesteryl esters). In packed RBCs, EPA was measured in the cell membrane, where EPA is mainly incorporated in phospholipids.
Bioanalytical Methods
For total EPA in plasma and RBCs, lipids were isolated by acid/methanol/chloroform extraction followed by centrifugation and purified by isohexane and solid-phase extraction after confirmed complete lipid hydrolysis and transmethylation (acid/methanol, 50°C overnight). Quantitation of EPA in plasma and RBCs is based on the EPA methyl ester formed during the transmethylation. For unesterified EPA in plasma, 25 μL of an inhibitor solution (0.5 g sodium fluoride, 1.0 g l-ascorbic acid, and 0.25 g 5-methylisoxazole-3-carboxylic acid per 10 mL water) was included per 1 mL plasma sample to prevent degradation, and lipids were isolated by centrifugation after methanol/chloroform extraction (without hydrolysis or methylation) and further purified by protein precipitation and solid-phase extraction.
EPA concentrations were measured using a validated liquid chromatography/tandem mass spectrometry procedure (Charles River Laboratories Ltd, Elphinstone Research Center, Tranent, Edinburgh, Scotland, UK). Separation of the analytes was obtained with a Series 200 Perkin Elmer liquid chromatography system (Perkin Elmer, Beaconsfield, Cheshire, UK), utilizing an Ascentis® Express C18 column 30 × 2.1 mm, 2.7 μm (Sigma–Aldrich Co. Ltd, Poole, UK) with gradient elution from a 60%/40% mobile phase A/B to 100% mobile phase A in 2.5 minutes and back, with a flow rate of 1 mL/min at 60°C. Mobile phase A was acetonitrile/acetic acid (100/0.5, v/v) and mobile phase B was water/acetic acid (100/0.5, v/v). Quantitation utilized linolenic acid 13C18 as the internal standard for unesterified EPA and EPA-d5 (five deuterium atoms; two at position 19 and three at position 20) for EPA methyl ester with EPA as the reference standard for both assays. The API400™ mass spectrometer with TurboIonSpray® electrospray ion source (AB Sciex, Cheshire, UK) settings were: negative ionization mode (positive for EPA methyl esters); 750°C; −4,500 V (5,500 V for EPA methyl ester), with standard API4000 gases (nitrogen and air). Ions were monitored for EPA at m/z 301.4–257.0, linolenic acid 13C18 (internal standard) at m/z 295.3–295.2, total plasma and RBC EPA methyl ester at m/z 317.2–285.4, and EPA-d5 methyl ester at m/z 322.2–290.4.
For total plasma EPA, RBC EPA, and unesterified EPA, the lower/upper limits of quantitation were 10–1,000 µg/mL, 5–500 µg/mL, and 50–5,000 ng/mL, respectively. The assay accuracy (mean determined concentration/nominal concentration) ranged from 94.3% to 113.5% (intra-batch) and from 100.3% to 106.5% (inter-batch). The assay precision (coefficient of variation of the mean determined concentration) ranged from 1.6% to 9.3% (intra-batch) and from 3.0% to 9.6% (inter-batch).
Pharmacokinetic Analyses
Pharmacokinetic parameters were calculated with standard methods and included area under the plasma concentration versus time curve from time zero to 24 hours (AUC0–24 hours), calculated using the linear trapezoidal rule; maximum observed concentration (Cmax); minimum observed plasma concentration (Cmin, pre-dose, trough concentration); apparent terminal elimination half-life (T½) calculated from (loge2)/λz, where λz is the apparent terminal rate constant obtained from the slope of the line, fitted by linear least-squares regression, through the terminal points of the natural log of the concentration versus time plots of these points; apparent total plasma clearance (CL/F) after an oral dose, calculated from Dose/AUC0–τ, where F is the oral bioavailability; time of observed Cmax (Tmax); and apparent volume of distribution after an oral dose (Vz/F), calculated from Dose/AUC0–τ × λz, where F is the oral bioavailability. Unless otherwise specified, all reported parameters are based on baseline-subtracted concentrations (baseline is due to EPA derived from dietary sources). Evaluation of steady state was by visual inspection of concentration data and not by statistical analysis.
Results
Subjects
A total of 48 subjects were randomized to the four treatment groups, with six male and six female subjects in each group. There were six subjects who discontinued the study due to withdrawal of consent (n = 5) and an AE (n = 1; cholecystitis due to previously undiagnosed, longstanding cholelithiasis unrelated to study drug); 42 subjects completed the study (87.5%). In the intent-to-treat population, mean age (SD) was 38.8 (11.9) years, subjects were primarily white (n = 37; 77.1%), and half were male (n = 24; 50.0%). Mean (SD) weight and body mass index were 74 (12) kg and 26 (3) kg/m2, respectively.
Baseline total EPA plasma concentrations were variable between subjects, with a study-wide mean (SD) of 15.3 (15.2) µg/mL. Fourteen subjects had baseline total EPA values lower than the lower limit of quantitation (10 µg/mL). The study-wide mean (SD) baseline unesterified EPA plasma concentration was 0.099 (0.095) µg/mL, which is less than 1% of the total EPA plasma concentration. In RBCs, the study-wide mean (SD) baseline EPA concentration was 8.01 ± 9.89 µg/mL.
Mean daily IPE dose was 2 g for all groups except group 2, which had a mean daily IPE dose of 3.9 g. Compliance rates were 99.4%, 97.1%, 99.1%, and 101.3% for groups 1 through 4, respectively.
Pharmacokinetic Results
Following IPE dosing, maximum concentration of total EPA was reached in approximately 5–6 hours in plasma (Table 1 and Figure 1) and approximately 8–24 hours in RBCs (Table 1). The mean terminal half-life of total EPA in plasma was long, ranging between 70 and 89 hours for the four treatment groups. The mean oral clearance and volume of distribution of total EPA in plasma ranged between 684 and 868 mL/h and 79 and 88 L, respectively (Table 1). Steady state was reached by Day 14 in plasma in all treatment groups (Figure 2). However, RBC concentrations slowly increased over the duration of treatment, and steady state was not reached by Day 28 (data not shown). Study-wide steady-state means (SD) for half-life (mainly from plasma lipids), total plasma clearance, and volume of distribution of total EPA were 79 (47) hours, 757 (283) mL/h, and 82 (56) L, respectively.
Table 1.
Pharmacokinetic Parameters by Treatment at Day 28 (Per-Protocol Population)
| Analyte | PK parametera | Group 1: IPE 2 g/day 1 × 1,000 mg BID (n = 10) | Group 2: IPE 4 g/day 2 × 1,000 mg BID (n = 9) | Group 3: IPE 2 g/day 2 × 1,000 mg QD (n = 12) | Group 4: IPE 2 g/day 2 × 500 mg BID (n = 12) |
|---|---|---|---|---|---|
| Total EPA in plasma | Baseline (µg/mL) | 7.9 (7.0) | 19.3 (16.1) | 19.2 (17.5) | 14.7 (16.6) |
| Cmax (µg/mL) | 154.9 (49.4) | 347.2 (112.5) | 232.8 (127.6) | 210.5 (93.1) | |
| AUC0–24 hours (µg•h/mL) | 2,907 (1,160) | 6,519 (1,963) | 2,65.9 (1,136) | 3,233 (1,104) | |
| Cmin (µg/mL) | 101.0 (50.4) | 211.9 (68.2) | 75.1 (40.5) | 104.2 (42.4) | |
| Tmax (hour) | 5 (5, 8) | 5 (3, 8) | 5 (3, 12) | 6 (5, 8) | |
| T1/2 (hour) | 75.1 (46.5) | 89.3 (42.0) | 69.7 (60.9) | 83.5 (38.8) | |
| CL/F (mL/h) | 776.4 (256.9) | 683.7 (280.6) | 867.7 (328.8) | 684.5 (248.3) | |
| Vz/F (L) | 79.8 (62.6) | 88.4 (55.2) | 79.3 (71.5) | 79.3 (35.3) | |
| Total EPA in RBCb | Baseline (µg/mL) | 5.7 (4.3) | 12.1 (15.7) | 8.2 (7.1) | 6.7 (10.4) |
| Cmax (µg/mL) | 42.3 (14.0) | 76.7 (25.2) | 31.0 (11.1) | 37.6 (15.7) | |
| AUC0–24 hours (µg•h/mL) | 801.5 (268.5) | 1,472 (469.5) | 557.6 (239.7) | 573.9 (336.4) | |
| Cmin (µg/mL) | 31.2 (12.2) | 64.1 (21.8) | 20.7 (8.6) | 26.3 (17.7) | |
| Tmax (hour) | 12 (1, 72) | 8 (1, 12) | 10 (6, 48) | 24 (0, 120) | |
| T1/2 (hour) | 683.8 (NC) | 371.4 (311.5) | 303.2 (NC) | 314.4 (194.5) | |
| Unesterified EPA in plasma | Baseline (µg/mL) | 0.13 (0.12) | 0.08 (0.05) | 0.09 (0.06) | 0.10 (0.13) |
| Cmax (µg/mL) | 0.66 (0.34) | 1.4 (0.41) | 1.4 (0.49) | 0.70 (0.24) | |
| AUC0–24 hours (µg•h/mL) | 6.9 (3.1) | 18.4 (4.6) | 9.0 (2.5) | 7.5 (2.6) | |
| Cmin (µg/mL) | 0.41 (0.28) | 1.06 (0.56) | 0.36 (0.22) | 0.44 (0.24) | |
| Tmax (hour) | 5 (0, 48) | 5 (0, 24) | 5 (3, 8) | 3 (0, 24) | |
| T1/2 (hour) | 80.8 (37.5) | 97.2 (36.5) | 136.3 (80.8) | NC | |
| CL/F (mL/h) | 364,513 (214,003) | 234,329 (81,639) | 241,855 (82,203) | 326,622 (207,574) | |
| Vz/F (L) | 33,164 (8,068) | 28,200 (10,929) | 40,873 (19,580) | NC | |
AUC0–24 hours, area under the plasma concentration versus time curve from time zero to 24 hours; BID, twice daily; CL/F, apparent total plasma clearance after an oral dose; Cmax, maximum observed concentration; Cmin, minimum observed plasma concentration; EPA, eicosapentaenoic acid; IPE, icosapent ethyl; NC, not calculated due to large variability and therefore small number of subjects for whom values could be calculated; PK, pharmacokinetic; QD, once daily; RBCs, red blood cells; SD, standard deviation; T½, elimination half-life; Tmax, time of observed Cmax; Vz/F, apparent volume of distribution after an oral dose.
Mean (SD) displayed for all PK parameters except Tmax, which is displayed as median (min, max); data based on Day 28 values except for baseline.
Packed RBCs.
Figure 1.
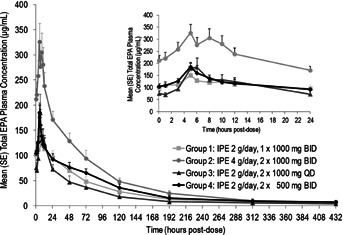
Total plasma EPA concentration versus time on Day 28 from time zero to 432 hours post-dose (inset, 0 to 24 hours post-dose). BID, twice daily; EPA, eicosapentaenoic acid; IPE, icosapent ethyl; QD, once daily; SE, standard error.
Figure 2.
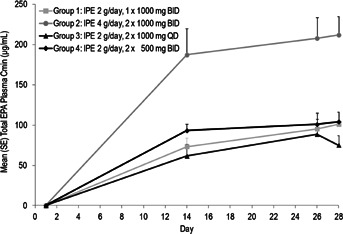
Total plasma EPA trough concentration (Cmin) during the treatment period. BID, twice daily; Cmin, minimum observed plasma concentration; EPA, eicosapentaenoic acid; IPE, icosapent ethyl; QD, once daily; SE, standard error.
A small fraction (≤0.5%) of the total EPA was determined to be unesterified. Based on AUC0–24 hours in plasma, 0.3% of the total EPA was unesterified in group 1 (2 g/day) and group 2 (4 g/day); based on maximum concentration in plasma, 0.5% and 0.4% of the total EPA was unesterified EPA in group 1 (2 g/day) and group 2 (4 g/day), respectively.
Comparisons of exposure (AUC0–24 hours and Cmax) revealed similarity between QD and BID regimens and between 1 × 1,000-mg and 2 × 500-mg formulations (Figure 3; group 1 versus group 3, AUC0–24 hours for total EPA in plasma, unesterified EPA and total EPA in RBCs, with observed differences in maximum concentration expected based on dosing regimen; group 1 versus group 4, AUC0–24 hours and maximum concentration for total plasma and unesterified EPA). Dose linearity was observed between IPE 2 g/day and 4 g/day (group 1 versus group 2; dose-normalized comparison of AUC and maximum concentration for total EPA in plasma, unesterified EPA and total EPA in RBCs). Based on graphical examination of the AUC0–24 hours and maximum concentration of total and unesterified plasma EPA concentrations, no effect of age was observed. Minimum concentration appeared to be slightly lower for males compared with females in some treatment groups, but overall, no consistent gender effect was observed based on all remaining exposure parameters.
Figure 3.
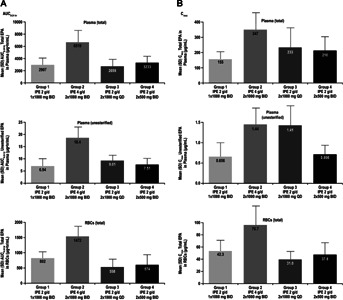
Mean EPA AUC0-24 hours (A) and Cmax (B) on Day 28. AUC0–24 hours, area under the plasma concentration versus time curve from time zero to 24 hours; BID, twice daily; Cmax, maximum observed concentration; EPA, eicosapentaenoic acid; IPE, icosapent ethyl; RBC, red blood cell; QD, once daily; SD, standard deviation.
Safety and Tolerability
Fourteen (29.2%) subjects reported at least one AE during the study (3 [25.0%] in group 1, 4 [33.3%] in group 2, 3 [25.0%] in group 3, and 4 [33.3%] in group 4). All AEs were treatment emergent and mild or moderate in intensity. The most common AEs were upper respiratory tract infection (two subjects in group 2) and headache (two subjects in group 3). One subject in group 2 discontinued study treatment as noted earlier due to cholecystitis, which was considered unrelated to study drug. There were no clinically meaningful changes in laboratory, electrocardiographic, or physical examination findings.
Discussion
The present study describes the plasma and RBC pharmacokinetics of IPE, a prescription form of EPA ethyl ester. Once-daily versus twice-daily regimens, 500-mg versus 1,000-mg formulations, and 2 g/day versus 4 g/day doses were compared, and the effects of age and gender were also evaluated.
Following oral dosing with IPE 4 g/day and 2 g/day, the study-wide mean (SD) elimination half-life of total EPA at steady state was 79 (47) hours. The study-wide mean apparent total plasma clearance of total EPA at steady state was 757 (283) mL/h and the apparent volume of distribution of total EPA at steady state was 82 (56) L. Maximum concentrations of total EPA were attained approximately 5–6 hours in plasma after dosing. Maximum EPA concentrations occurred later in RBCs compared with plasma (∼8–24 hours after dosing). Steady-state concentrations of total plasma EPA were observed 14 days after continuous dosing, whereas steady state was not reached by Day 28 in RBCs.
The slow increase in RBC concentrations observed over the duration of treatment in the present study was consistent with the slow washout of EPA after cessation of dosing (long elimination half-life) in RBCs17,22 and likely due to the known slower process of incorporation of EPA into RBC membranes.22 The maximum plasma concentration of 5–6 hours post dosing observed in this study is in agreement with that of a prescription formulation of ethyl EPA available in Japan (see approved product label). The study-wide mean elimination half-life of total plasma EPA was consistent with previously reported mean elimination half-lives ranging from approximately 1–3 days for plasma EPA in phospholipids, cholesteryl esters, and triacylglycerol.9 The half-life of EPA in plasma was 79 hours in this setting where dosing continued for 1 month; previously, where dosing was continued for longer periods of time, longer half-lives have been reported.22
Recently, the bioavailability of a novel omega-3 fatty acid formulation containing the free fatty acids forms of EPA plus DHA was investigated in comparison to prescription omega-3-acid ethyl esters (EPA plus DHA) at the same dose of 4 g/day in the Epanova® Compared to Lovaza® in a Pharmacokinetic Single-Dose Evaluation (ECLIPSE) study.21 While the study may be useful for understanding specific pharmacokinetic comparisons for the two formulations that were investigated head to head, comparisons to the present study should be approached with caution. Subjects in the ECLIPSE bioavailability study received single doses of EPA + DHA formulations and were overweight (BMI 25–35 kg/m2) whereas the present study was a multiple-dose pharmacokinetic study of an EPA-only formulation in subjects wherein BMI was specified as >18 and ≤30 kg/m2. Furthermore, the ECLIPSE study was investigated under “low-fat” and “high-fat” dosing periods. The “low-fat” periods may perhaps be better described as fasting conditions because subjects fasted 12 hours prior to dosing and did not eat again until 4 hours post dose. The “high-fat” periods may perhaps be better described as drug administration with a low-fat breakfast because the study specified that the breakfast consisted of 600 kcal total with 20 g fat, while FDA recommendations for “high-fat” meals in bioavailability studies consist of 800–1,000 kcal total with approximately 500–600 kcal of calories derived from fat (56–67 g fat).23 In contrast, dietary fat content was not specified in the present study during the 28-day outpatient treatment period; on Day 28, the last dose of IPE was administered with a breakfast containing approximately 460 kcal and 25% fat.
Based on dose-normalized maximum observed concentration and AUC of total and unesterified EPA in plasma and total EPA in RBCs, the mean exposure to EPA appears to be dose-proportional between IPE 2 and 4 g/day. This pharmacokinetic dose linearity was supported by the statistical comparisons of dose-normalized pharmacokinetic values, indicating that IPE has predictable pharmacokinetics. Administration of prescription omega-3-acid ethyl esters (EPA plus DHA) has also been shown to result in dose-dependent increases in serum EPA, but increases in serum DHA were found to be less pronounced and not dose-dependent (see approved product label).
In general, the once-daily and twice-daily dosing regimens for the same daily dose of IPE resulted in similar total AUC for total EPA in plasma. As expected, the maximum observed concentration was higher and the trough concentration was lower in the once-daily regimen compared with the twice-daily regimen. As is typical for drugs with long half-lives, the major determinant of steady-state exposure to EPA in plasma, and even more so in RBCs, was the total daily dose of IPE, irrespective of how the dose was divided over the course of the day. The relatively long half-life of EPA in plasma permits a dosing schedule with intervals of ≥12 hours, and day-to-day variability in the dosing interval is not expected to influence the average plasma or tissue exposure to EPA. In comparing the 500-mg and 1,000-mg formulations of IPE at the same daily dose, similar total plasma EPA maximum observed concentrations and AUCs were observed, indicating that, as expected, the 500-mg and 1,000-mg formulations provide comparable exposures at comparable doses.
In this investigation, age and gender had minimal effect on IPE pharmacokinetics. A limitation of this study is the relatively small number of subjects; the effects of gender and age should therefore be interpreted with caution.
Conclusions
The pharmacokinetic profile of EPA was that of a slowly cleared, extensively distributed molecule with dose linearity. With regard to daily exposure, comparable results for once- and twice-daily IPE regimens and 500- and 1,000-mg formulations at comparable doses were observed with no effects of age, and minimal effects of gender.
Declaration of Conflicting Interests
Drs. Braeckman and Soni are former employees of Amarin Pharma Inc. and were employed by Amarin Pharma Inc. at the time of this analysis and manuscript preparation. Dr. Stirtan is an employee of Amarin Pharma Inc.
Funding
This study was designed and sponsored by Amarin Pharma, Inc., Bedminster, NJ, USA. Editorial assistance was provided by Peloton Advantage, LLC, Parsippany, NJ, USA, funded by Amarin Pharma, Inc.
References
- 1.Calder PC. n-3 Fatty acids and cardiovascular disease: evidence explained and mechanisms explored. Clin Sci (Lond) 2004;107:1–11. doi: 10.1042/CS20040119. [DOI] [PubMed] [Google Scholar]
- 2.Smith SC, Jr, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. Circulation. 2011;124:2458–2473. doi: 10.1161/CIR.0b013e318235eb4d. [DOI] [PubMed] [Google Scholar]
- 3.Ginsberg HN. Hypertriglyceridemia: new insights and new approaches to pharmacologic therapy. Am J Cardiol. 2001;87:1174–1180. doi: 10.1016/s0002-9149(01)01489-8. [DOI] [PubMed] [Google Scholar]
- 4.Wei MY, Jacobson TA. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: a systematic review and meta-analysis. Curr Atheroscler Rep. 2011;13:474–483. doi: 10.1007/s11883-011-0210-3. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson TA, Glickstein SB, Rowe JD, Soni PN. Effects of eicosapentaenoic acid and docosahexaenoic acid on low-density lipoprotein cholesterol and other lipids: a review. J Clin Lipidol. 2012;6:5–18. doi: 10.1016/j.jacl.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Bays HE, Ballantyne CM, Kastelein JJ, Isaacsohn JL, Braeckman RA, Soni PN. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the Multi-center, plAcebo-controlled, Randomized, double-blINd, 12-week study with an open-label Extension [MARINE] trial) Am J Cardiol. 2011;108:682–690. doi: 10.1016/j.amjcard.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Ballantyne CM, Bays HE, Kastelein JJ, et al. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study) Am J Cardiol. 2012;110:984–992. doi: 10.1016/j.amjcard.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 8.Nordoy A, Barstad L, Connor WE, Hatcher L. Absorption of the n-3 eicosapentaenoic and docosahexaenoic acids as ethyl esters and triglycerides by humans. Am J Clin Nutr. 1991;53:1185–1190. doi: 10.1093/ajcn/53.5.1185. [DOI] [PubMed] [Google Scholar]
- 9.Zuijdgeest-van Leeuwen SD, Dagnelie PC, Rietveld T, van den Berg JW, Wilson JH. Incorporation and washout of orally administered n-3 fatty acid ethyl esters in different plasma lipid fractions. Br J Nutr. 1999;82:481–488. doi: 10.1017/s0007114599001737. [DOI] [PubMed] [Google Scholar]
- 10.Subbaiah PV, Kaufman D, Bagdade JD. Incorporation of dietary n-3 fatty acids into molecular species of phosphatidyl choline and cholesteryl ester in normal human plasma. Am J Clin Nutr. 1993;58:360–368. doi: 10.1093/ajcn/58.3.360. [DOI] [PubMed] [Google Scholar]
- 11.Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J Clin Nutr. 2006;83:1467S–1476S. doi: 10.1093/ajcn/83.6.1467S. [DOI] [PubMed] [Google Scholar]
- 12.Harris WS, Sands SA, Windsor SL, et al. Omega-3 fatty acids in cardiac biopsies from heart transplantation patients: correlation with erythrocytes and response to supplementation. Circulation. 2004;110:1645–1649. doi: 10.1161/01.CIR.0000142292.10048.B2. [DOI] [PubMed] [Google Scholar]
- 13.von Schacky C, Weber PC. Metabolism and effects on platelet function of the purified eicosapentaenoic and docosahexaenoic acids in humans. J Clin Invest. 1985;76:2446–2450. doi: 10.1172/JCI112261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawson LD, Hughes BG. Human absorption of fish oil fatty acids as triacylglycerols, free acids, or ethyl esters. Biochem Biophys Res Commun. 1988;152:328–335. doi: 10.1016/s0006-291x(88)80718-6. [DOI] [PubMed] [Google Scholar]
- 15.Lawson LD, Hughes BG. Absorption of eicosapentaenoic acid and docosahexaenoic acid from fish oil triacylglycerols or fish oil ethyl esters co-ingested with a high-fat meal. Biochem Biophys Res Commun. 1988;156:960–963. doi: 10.1016/s0006-291x(88)80937-9. [DOI] [PubMed] [Google Scholar]
- 16.el Boustani S, Colette C, Monnier L, Descomps B, Crastes de PA, Mendy F. Enteral absorption in man of eicosapentaenoic acid in different chemical forms. Lipids. 1987;22:711–714. doi: 10.1007/BF02533970. [DOI] [PubMed] [Google Scholar]
- 17.Brown AJ, Pang E, Roberts DC. Persistent changes in the fatty acid composition of erythrocyte membranes after moderate intake of n-3 polyunsaturated fatty acids: study design implications. Am J Clin Nutr. 1991;54:668–673. doi: 10.1093/ajcn/54.4.668. [DOI] [PubMed] [Google Scholar]
- 18.Hansen JB, Grimsgaard S, Nilsen H, Nordoy A, Bonaa KH. Effects of highly purified eicosapentaenoic acid and docosahexaenoic acid on fatty acid absorption, incorporation into serum phospholipids and postprandial triglyceridemia. Lipids. 1998;33:131–138. doi: 10.1007/s11745-998-0188-8. [DOI] [PubMed] [Google Scholar]
- 19.Krokan HE, Bjerve KS, Mork E. The enteral bioavailability of eicosapentaenoic acid and docosahexaenoic acid is as good from ethyl esters as from glyceryl esters in spite of lower hydrolytic rates by pancreatic lipase in vitro. Biochim Biophys Acta. 1993;1168:59–67. doi: 10.1016/0005-2760(93)90266-c. [DOI] [PubMed] [Google Scholar]
- 20.Ackman RG. The absorption of fish oils and concentrates. Lipids. 1992;27:858–862. doi: 10.1007/BF02535864. [DOI] [PubMed] [Google Scholar]
- 21.Davidson MH, Johnson J, Rooney MW, Kyle ML, Kling DF. A novel omega-3 free fatty acid formulation has dramatically improved bioavailability during a low-fat diet compared with omega-3-acid ethyl esters: the ECLIPSE (Epanova compared to Lovaza in a pharmacokinetic single-dose evaluation) study. J Clin Lipidol. 2012;6:573–584. doi: 10.1016/j.jacl.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Katan MB, Deslypere JP, van Birgelen AP, Penders M, Zegwaard M. Kinetics of the incorporation of dietary fatty acids into serum cholesteryl esters, erythrocyte membranes, and adipose tissue: an 18-month controlled study. J Lipid Res. 1997;38:2012–2022. [PubMed] [Google Scholar]
- 23. Guidance for industry. Food-effect bioavailability and fed bioequivalence studies. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research. Available at: http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf. Accessed July 19, 2013.