

Abstract
Oxaliplatin (OX) has been widely used in adjuvant and palliative treatments of advanced colon cancer; however, cancer cells ultimately become resistant in the majority of cases. Therefore, the development of a novel strategy to overcome this resistance is important for the effective treatment of colon cancer. Cell autophagy reduces the sensitivity of cancer cells to therapeutic reagents in various types of human cancer; therefore, the present study used murine CT26 colon carcinoma cells to explore whether inhibition of autophagy by 3-methyladenine (3-MA) is able to enhance OX-induced apoptosis in vitro and OX-suppressed tumor growth in vivo. CT26 cells were treated with 3-MA, OX, or 3-MA plus OX, and the autophagy, apoptosis and proliferation of the CT26 cells was investigated. Additionally, the therapeutic efficiency of the combination of 3-MA and OX treatment was evaluated in vivo by determining the survival time of the tumor-bearing mice and, thus, tumor growth rate. The treatment of CT26 cells in vitro with OX alone increased autophagy as well as apoptosis, whereas treatment with 3-MA plus OX markedly inhibited OX-induced autophagy, but increased OX-induced cell apoptosis. Furthermore, the combination of OX and 3-MA treatment significantly suppressed tumor growth in vivo and prolonged mouse survival time when compared with OX treatment alone. Similarly, 3-MA increased OX-induced cell apoptosis and decreased autophagy in xenograft tumor tissues. Thus, the administration of 3-MA may increase tumor cell sensitivity to OX by reducing its autophagic effects and enhancing its apoptotic effects. Data obtained in the present study indicates that the clinical combination of an autophagy inhibitor with OX may increase the therapeutic effect of OX and improve the clinical outcome of patients with colon cancer.
Keywords: colon cancer, autophagy, apoptosis, chemotherapy, mouse, combination
Introduction
Colon cancer was the third most common type of cancer diagnosed and the third most common cause of cancer-related mortality in the United States in 2015 (1). Colorectal cancer was the third most commonly diagnosed cancer in males and the second most common in females worldwide in 2012 according to Global Cancer Statistics (2). The five-year survival rate for colon cancer is only 10% in patients with metastases (3), potentially due to the development of drug resistance during therapy. Oxaliplatin (OX), a third generation platinum-based antineoplastic agent, is a commonly used chemotherapeutic agent for the clinical treatment of advanced colon cancer (4). Although the clinical application of OX has resulted in a significant improvement in response rate and progression-free survival in advanced colon cancer (5), ~40% of cases of colon cancer continue to develop resistance (6). Therefore, the development of novel therapeutic strategies is required to overcome the current OX resistance in advanced colon cancer.
Autophagy is a dynamic cellular protective process of cells that occurs in response to stress and an abnormal microenvironment (7). Recently, increasing evidence has indicated the importance of autophagy in human cancer, with autophagy exhibiting a dual function in cancer development. In healthy cells, autophagy acts as a tumor suppressor by clearing damaged proteins and organelle accumulation to prevent tumorigenesis (8). However, autophagy in cancer cells may promote cell survival by sustaining the cellular metabolism necessary for survival under hypoxic conditions and drug treatment, resulting in tumor growth and therapeutic resistance (8–10). In colon cancer, autophagy appears to favor cancer cell survival by protecting from tumor cell death caused by chemotherapy and radiotherapy (11–15). For example, it has been demonstrated that autophagy is responsible for the resistance of colon cancer cells to 5-fluorouracil (FU) treatment, as inhibition of autophagy enhances 5-FU-induced tumor cell apoptosis in vitro (11,12,15). In addition, OX can activate cell autophagy via endoplasmic reticulum (ER) stress and the reactive oxygen species (ROS) pathway in human colon cancer cells to induce their resistance to OX treatment (13). Although the inhibition of autophagy can increase the sensitivity of colon cancer cells to OX treatment in vitro, the in vivo therapeutic effects of autophagy inhibition on tumor growth has yet to be investigated in animal models treated with OX.
Therefore, the aim of the present study was to observe the effect of the autophagy inhibitor 3-methyladenine (3-MA) combined with OX on murine CT26 cell growth in vitro and in vivo. In addition, the potential mechanism of the combined effect induced by this combination therapy was investigated.
Materials and methods
Cells, reagents and antibodies
The CT26 mouse colon carcinoma cell line (CRL-2638; American Type Culture Collection, Manassas, VA, USA) was maintained in RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum and 100 µg/ml kanamycin sulfate at 37°C in a 5% CO2 humidified incubator. The following reagents were used in present study: OX (Eloxatin®; Sanofi, Paris, France), 3-MA (M9281; Sigma-Aldrich, St. Louis, MO, USA) and ProLong® Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Cell Signaling Technology, Inc., Danvers, MA, USA). In addition, the antibodies used in current study included, Beclin1 (Abcam, Cambridge, UK; cat no. ab62557), microtubule-associated protein light chain 3 (LC3; Abcam; cat no. ab58610), autophagy related 5 (ATG5; Abcam; cat no. ab108327), Bcl-2-associated X protein (Bax; Abcam; cat no. ab32503), caspase-3 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA; cat no. sc-7148) and p53 (Abcam; cat no. ab1431).
Immunofluorescence
A total of 2×104 CT26 cells were seeded into an 8-well chamber slide and treated with phosphate-buffered saline (PBS), OX (4 mM), 3-MA (5 nM) or OX plus 3-MA for 48 h. Cells were then washed with PBS and fixed using 4% formaldehyde. After washing, the cells were treated with 0.1% Triton X-100 containing 1% bovine serum albumin for 1 h at room temperature, followed by incubation with the LC3 antibody (dilution, 1:200) for 24 h at 4°C in a humidified chamber. Cells were washed with PBS and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at room temperature. After rinsing with PBS, the cells were placed on a coverslip by ProLong® Gold antifade reagent with DAPI and examined using an IX70 inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Acridine orange (AO) and monodansylcadaverine (MDC) staining
A total of 5×104 CT26 cells were seeded into a 4-well chamber slide and treated with PBS, OX (4 mM), 3-MA (5 nM) or OX plus 3-MA for 48 h. The treated cells were washed with PBS, stained with medium containing 1 µg/ml AO (Polysciences, Inc., Warrington, PA, USA) or 10 mM MDC (Sigma-Aldrich) in PBS for 15 min, washed with PBS and immediately examined under a fluorescence microscope.
Western blot analysis
Proteins were extracted from the treated CT26 cells using RIPA buffer (50 mM Tris-base, 1.0 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate and 1 mM phenylmethylsulfonyl fluoride) and the protein concentration was determined using a DC™ protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of the protein samples were loaded and separated on a 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (GE Healthcare Life Sciences, Chalfont, UK). After blocking with 5% non-fat milk in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h, the membranes were incubated with the indicated primary antibodies overnight at 4°C. After washing with TBST, the blots were incubated with HRP-conjugated secondary antibody for 1 h at room temperature and the target proteins were detected using enhanced chemiluminescence reagents (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, USA).
Quantitative assessment of apoptosis using flow cytometry
After trypsinization and washing with PBS, 1×106 CT26 cells were stained in 0.5 ml hypotonic fluorochrome solution (50 mg/ml propidium iodide in 0.1% sodium citrate containing 0.1% Triton X-100) overnight at 4°C. Subsequently, flow cytometric analysis was performed using a flow cytometer (EPICS Elite ESP; Beckman Coulter, Brea, CA, USA) to assess the number of apoptotic cells with hypodiploid DNA content.
Cell viability assays
CT26 cells were seeded at a concentration of 5×103 cells per well in a 96-well plate. After culturing overnight, the cells were incubated with fresh medium containing PBS, OX (4 mM), 3-MA (5 nM) or OX plus 3-MA for the indicated time periods. The number of viable cells was then estimated using an MTT assay by measuring absorbance at a wavelength of 570 nm using a microplate reader (Multiskan MK3; Bio-Rad Laboratories). The data are expressed as the mean ± standard deviation (SD) from a minimum of three independent experiments.
In vivo tumor growth
In total, 40 female BALB/c mice (age, 6–7 weeks) were purchased from the Animal Center at Sichuan University (Chengdu, China). CT26 cells (5×105 cells in 100 µl PBS) were subcutaneously injected into the BALB/c mice at the dorsal flank. After injection, the mice were randomly separated into four groups of 10 mice (control group; 3-MA group; OX group;3-MA plus OX groups). Each mouse received an intraperitional (i.p.) injection of PBS (1 ml; control and OX groups) or 3-MA (1 ml; 24 mg/kg; 3-MA and 3-MA plus OX groups) and an intravenous (i.v.) injection of PBS (100 µl; control and 3-MA groups) or OX (100 µl; 10 mg/kg; OX and 3-MA plus OX groups). The i.p. injections were performed on days 5, 10, 15, 20 and 25 after tumor cell inoculation, and the i.v. injections were administered on days 10 and 24 after tumor cell inoculation. Tumor growth was monitored by measuring the tumor diameter using a vernier caliper every two days until day 27, when the mice were sacrificed, and the tumor volume was calculated using the following equation: Length × width2 × 0.52 (16).
To determine the survival time of the mice, 100 mice received a subcutaneous inoculation of CT26 cells (5×105 cells/mouse) prior to being randomly separated into four groups of 25 mice per group. The four groups received treatment with PBS, OX, 3-MA or OX plus 3-MA, as described above, and the survival times of the mice were recorded for seven weeks after cell inoculation, at which point the experiment was terminated. All mouse experiments were reviewed and approved by the Animal Care and Use Committee of the Animal Center at Sichuan University.
Electron microscopy
Tumor tissues were obtained from three randomly-selected mice from each of the four groups (control, 3-MA, OX and OX + 3-MA) (n=12) that were sacrificed on day 27 after tumor cell inoculation. Tumor tissues were sliced into 1 mm3 sections and fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (SCB) for 1 h. After washing with SCB, the tissues were incubated with 1% osmium tetroxide in 0.1 M SCB for 1 h at room temperature, followed by washing three times with SCB. Subsequently, the tissues were dehydrated with alcohol in a graded ethanol series and then embedded in paraffin. A Reichert EM UC7 Ultramicrotome (Leica Microsystems GmbH, Wetzlar, Germany) was used to cut ultrathin sections of the tissue block. After counterstaining with 0.3% lead citrate, the ultrathin sections were examined on a Philips EM420 transmission electron microscope (Philips, Eindhoven, The Netherlands). Autophagic cells were defined as cells that had five or more autophagic vacuoles. Furthermore, the areas occupied by autophagic vacuoles within the cytoplasm were calculated using Image-Pro Plus software (version 3.0; Media Cybernetics, Inc., Silver Spring, MD, USA) and used for indexing the cytoplasmic area occupied by autophagic vacuoles.
Terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) assay
Apoptotic cells within the tumor sections were detected by performing a TUNEL assay using the DeadEnd™ Fluorometric TUNEL system (Promega Corporation, Madison, WI, USA), according to the manufacturer's instructions. The mean percentage of apoptotic cells in tumor tissues was determined by counting the number of apoptotic cells in a field and dividing this number by the total number of cells counted in the same field, selecting five high power fields for each slide.
Statistical analysis
All data are presented as the mean ± standard deviation (SD) and all statistical analyses were performed using a Student's t-test, unless stated otherwise. P<0.05 was considered to indicate a statistically significant difference.
Results
OX treatment increases autophagy in murine CT26 cells
The in vitro treatment of CT26 cells with OX markedly increased cell autophagy, as indicated by the increased LC3 staining (green color) observed in Fig. 1A. Furthermore, the autophagy inhibitor 3-MA appeared to block this OX-induced CT26 cell autophagy. OX-induced cell autophagy was also indicated by the increased presence of acidic vacuoles/autophagosomes identified by AO (red color; Fig. 1B) and MDC (blue color; Fig. 1C) staining in the CT26 cells. Consistently, OX-treated CT26 cells markedly increased the expression levels of autophagy-related proteins, such as LC3-II, Beclin1 and ATG5; however, the increase in the expression levels of all three proteins was quenched by the administration of 3-MA in combination with OX (Fig. 2). Taken together, these data indicate that OX increases cell autophagy and that the autophagic stimulation effect of OX can be blocked by the autophagy inhibitor, 3-MA.
Figure 1.

OX induces autophagy in CT26 cells. (A) Immunofluorescence staining shows the expression of microtubule-associated protein light chain 3 protein (green) in CT26 cells treated with phosphate-buffered saline (control), 3-MA, OX, and 3-MA plus OX. (B) Acridine orange staining indicates the presence of acidic vesicular organelles (red) in CT26 cells administered with different treatment regimes. (C) Monodansylcadaverine staining indicates the presence of autophagic vacuoles (blue) in CT26 cells administered with different treatment regimes. 3-MA, 3-methyladenine; OX, oxaliplatin.
Figure 2.
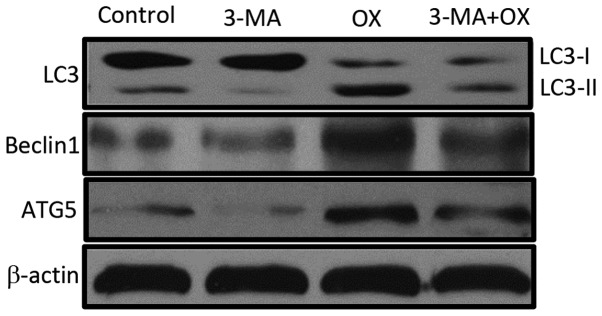
OX induces the expression of autophagy proteins in CT26 cells. Equal quantities of cell lysate from CT26 cells administered with different treatment regimes was used for western blot analysis to detect the expression levels of LC-3, Beclin1, ATG5 and β-actin (loading control). 3-MA, 3-methyladenine; OX, oxaliplatin; LC3, microtubule-associated protein light chain 3; ATG5, autophagy related 5.
OX increases the apoptosis rate of CT26 cells
Investigation of the effects of different treatment regimens on the apoptosis of CT26 cells demonstrated that, although 3-MA alone appears to exhibit minor effects on cell apoptosis (10.27% of cells exhibited sub-G1 DNA content), 3-MA significantly enhanced OX-induced apoptosis of CT26 cells when compared with OX treatment alone (86.81 vs. 39.48% cells with sub-G1 DNA content; P=0.0034; Fig. 3A). Consistently, CT26 cells treated with 3-MA and OX showed the lowest cell viability (27.5%), followed by cells treated with OX alone (53.2%) and 3-MA alone (84.86%; Fig. 3B). Furthermore, 3-MA administration increased the expression of a number of apoptosis-related proteins, including Bax, p53, and active caspase-3, which were initially induced by OX treatment (Fig. 3C). These results indicate that blocking the autophagic effect of OX may enhance its apoptotic effect on colon cancer cells.
Figure 3.

3-MA enhances OX-induced apoptosis in CT26 cells in vitro. (A) Apoptosis analysis of CT26 cells by flow cytometry. The sub-G1 cell population percentage in OX treated cells was 39.48%; however, it was increased to 86.81% in 3-MA plus OX cotreated cells. (B) The viability of CT26 cells administered with different treatment regimens was measured by performing an MTT assay. *P<0.05; **P<0.01. (C) The expression of apoptotic proteins, such as Bax, caspase-3 and p53, was detected by performing a western blot analysis in CT26 cells administered with different treatment regimens. β-actin was used as the loading control. 3-MA, 3-methyladenine; OX, oxaliplatin; Bax, Bcl-2-associated X protein.
In vivo synergistic anticancer effects of OX combined with 3-MA
To investigate the synergistic cytotoxic effects of OX combined with 3-MA in vivo, a mouse model in which colon cancer-bearing mice received different treatment regimens was established. Tumor growth was significantly slower in the mice that received OX treatment alone compared with the mice that received PBS or 3-MA alone (P<0.05). Furthermore, cotreatment of the tumor-bearing mice with OX and 3-MA suppressed tumor growth to a greater extent than OX treatment alone (Fig. 4A). Consistently, mice cotreated with OX and 3-MA exhibited significantly longer survival times compared with the mice treated with OX alone (P<0.01; Fig. 4B).
Figure 4.

Combination treatment of OX plus 3-MA enhances OX-induced tumor suppression and prolongs mouse survival. Mice subcutaneously injected with CT26 cells received different treatment strategies, as indicated. (A) Tumor growth was monitored by measuring tumor volume every two days until day 27 post cell injection. *P<0.05. (B) A Kaplan-Meier curve of mouse survival was plotted using survival data collected over a period of seven weeks after cell injection. *P<0.01 vs. OX group; log-rank test. 3-MA, 3-methyladenine; OX, oxaliplatin.
Reduction of cell autophagy and increase of cell apoptosis in tumor tissues by combinational treatment with OX and 3-MA
To investigate the potential antitumor mechanism of combined OX plus 3-MA treatment in vivo, three sacrificed mice from each group were selected on day 27 after tumor cell inoculation, and tumor tissue from each mouse was collected, cut into ultrathin slices and used for detecting cell autophagy and cell apoptosis. As indicated in Fig. 5A, autophagosomes with a double membrane structure accompanied by typical swollen organelles were observed in the tumor tissues of mice treated with OX and OX plus 3-MA. However, the frequency of autophagosomes in the tumors of mice treated with OX alone were significantly higher compared with the tumors of mice treated with OX plus 3-MA (Fig. 5B; P<0.05), indicating a reduction of autophagy in tumor tissues by the cotreatment of mice with OX plus 3-MA. In contrast to tumor tissues obtained from the OX and OX plus 3-MA group mice, transmission electron microscopy of the tumor tissues obtained from the control and 3-MA groups showed little ultrastructure change and few autophagosomes (Fig. 5A and B). Consistent with these ultrastructure changes, the protein expression levels of LC3-II and ATG5 were increased in the tumor tissues of the OX group, and reduced in the OX plus 3-MA cotreatment group (Fig. 5C).
Figure 5.

Combination treatment with 3-MA and OX in vivo blocks OX-induced cell autophagy in tumor tissues. (A) Autophagosome formation in tumor tissues from each group was detected using electron microscopy. (B) Statistical data of the total number of autophagosomes counted in 100 randomly-selected fields from each group. A non-parametric Kruskal-Wallis test was used to analyze the differences between the groups. *P<0.05; **P<0.001. (C) Cell lysates from tumor tissues administered with different treatment regimens were used for western blot analysis to detect the protein expression levels of LC3 and ATG5. β-actin was used as the loading control. 3-MA, 3-methyladenine; OX, oxaliplatin; Bax, Bcl-2-associated X protein; LC3, microtubule-associated protein light chain 3; ATG5, autophagy related 5.
TUNEL staining of tumor tissues from mice that underwent different treatment regimens revealed that 3-MA treatment resulted in a low proportion of apoptotic cells while OX treatment markedly increased the proportion of apoptotic cells, compared with the control group. Furthermore, combined treatment with OX and 3-MA significantly enhanced tumor cell apoptosis, compared with OX treatment alone (Fig. 6A and B; P<0.05). The increased level of tumor cell apoptosis observed in mice treated with OX plus 3-MA supported the aforementioned observations of the current study, including the significant inhibition of tumor growth and the prolonged of survival time of mice receiving combinational treatment.
Figure 6.

Combination treatment with 3-MA and OX in vivo enhances OX-induced cell apoptosis in tumor tissues. (A) TUNEL staining was performed to determine the proportion of apoptotic cells in tumor tissues administered with different treatment regimens. (B) The statistical data showed the percentage of apoptotic cells detected in the tumor tissues by TUNEL staining. *P<0.05. 3-MA, 3-methyladenine; OX, oxaliplatin; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling.
Discussion
The present study demonstrated that OX treatment induces cancer cell autophagy as well as cell apoptosis, and that the induction of autophagy by OX compromised its apoptotic effects on colon cancer cells. Furthermore, inhibition of autophagy by 3-MA markedly augmented the OX-induced apoptosis of colon cancer cells in vitro and consequently enhanced OX-suppressed tumor growth in vivo.
Apoptosis and autophagy are two major cell death pathways in healthy cells and in tumor cells. It has previously been reported that the activation of autophagy in cancer cells inhibits therapy-induced apoptosis (17,18). In the current study, OX was observed to induce apoptosis as well as autophagy in CT26 cells. The observed increase in apoptosis was supported by the upregulated expression of apoptosis-related proteins, such as Bax, active caspase-3 and p53. Furthermore, OX administration was associated with the increased expression levels of a number of autophagy marker proteins, such as LC3-II, Beclin1 and ATG5. Consistent with these results, activation of apoptosis by OX was determined by conducting flow cytometric analysis and a TUNEL assay, while activation of autophagy was corroborated by immunofluorescence staining, AO staining, MDC staining and electronic microscopy. Notably, inhibition of the autophagy signaling pathway by 3-MA significantly enhanced OX-induced cell apoptosis in vitro and tumor suppression in vivo, indicating that the activation of autophagy by OX may reduce OX cytotoxicity. The results of the current study are consistent with previous findings (17,18) and a recent report (14), in which cisplatin-induced autophagy reduced its apoptotic effect by downregulating the ER stress-mediated apoptosis pathway and the mitochondrial apoptosis pathway in HeLa cells. Considering the results of the present study, it is plausible that OX-activated autophagy may be responsible for the observed clinical inefficiency of OX in specific patients with colon cancer. Although OX has proven beneficial in the treatment of advanced colon cancer, previous studies reported that ~50% of patients did not respond to a combination of OX and 5-FU treatment (19), and OX failed to eradicate micrometastatic disease in ~30% of patients who received combination therapy of OX, 5-FU and leucovorin (20,21). Therefore, incorporating autophagy inhibitors into the current regimens for advanced colon cancer may increase their therapeutic efficiency and improve the clinical outcomes of patients with advanced colon cancer.
The findings of the present study are in line with a recent report in which OX was demonstrated to activate autophagy in colon cancer cells via the ER stress and ROS pathways, and inhibition of this autophagy increased cell death upon OX treatment in vitro (13). However, the current study performed an additional step to demonstrate the potential therapeutic efficiency and feasibility in vivo, using a combinational treatment strategy of an autophagy inhibitor and OX in a mouse model. In addition, a potential mechanism for the suppression of OX-induced cytotoxicity in colon cancer cells was proposed; i.e., that OX-induced autophagy inhibits its own apoptotic effect in colon cancer cells. Similar to OX, a previous study identified that cisplatin induces autophagy, in turn antagonizing cisplatin-induced cytotoxicity in HeLa cells (14).
It appears to be a common phenomenon in colon cancer that systemic therapies, such as chemotherapy and radiotherapy, activate autophagy when they are intended to induce apoptosis. For example, 5-FU, an alternative standard chemotherapeutic reagent in colon cancer, has been demonstrated in vitro to induce autophagy, which compromised the cytotoxic effects of 5-FU. Furthermore, the administration of an autophagy inhibitor appeared to increase the sensitivity of the colon cancer cells to 5-FU (11,12). Similarly, radiotherapy was able to induce autophagy while inhibition of autophagy enhanced radiation-induced apoptosis in colon cancer cells in vitro (22). These studies support the clinical importance of inhibiting autophagy in colon cancer therapy. Notably, high expression levels of autophagy-related proteins, such as Beclin1 and LC3, have been detected in human tumor samples. This upregulated expression appears to significantly correlate with aggressive features and poor prognosis in colon cancer (23–25), indicating that autophagy is highly active in advanced colon cancer. Thus, the therapeutic effects of OX and 5-FU are compromised in patients exhibiting cancer with high expression levels of autophagy marker proteins, due to active autophagy in these tumors. This may be an alternative explanation for the failure of adjuvant therapeutic strategies containing OX and 5-FU in specific patients with advanced colon cancer. Therefore, in agreement with previous reports, the present study highlights the need to combine autophagy inhibitors and OX for the effective treatment of advanced colon cancer.
In conclusion, the present study identified that OX treatment induces autophagy as well as apoptosis in colon cancer cells, and that the autophagy induced by OX compromises its apoptotic effects. However, blockage of the autophagy signaling pathway using a combination of autophagy inhibitors and OX appears to significantly increase the cytotoxicity of OX and, therefore, enhance OX-induced tumor growth suppression in vivo. The current study indicates that the addition of an autophagy inhibitor to current regimens with OX may enhance the therapeutic effects and, thus, benefit patients with advanced colon cancer.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015 Feb 4; doi: 10.3322/caac.21262. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 3.Dahan L, Sadok A, Formento JL, Seitz JF, Kovacic H. Modulation of cellular redox state underlies antagonism between oxaliplatin and cetuximab in human colorectal cancer cell lines. Br J Pharmacol. 2009;158:610–620. doi: 10.1111/j.1476-5381.2009.00341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bleiberg H. Oxaliplatin (L-OHP): a new reality in colorectal cancer. Brit J Cancer. 1998:1–3. doi: 10.1038/bjc.1998.427. (Suppl 4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol. 2011;18:18–25. doi: 10.3747/co.v18i1.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howells LM, Sale S, Sriramareddy SN, et al. Curcumin ameliorates oxaliplatin-induced chemoresistance in HCT116 colorectal cancer cells in vitro and in vivo. Int J Cancer. 2011;129:476–486. doi: 10.1002/ijc.25670. [DOI] [PubMed] [Google Scholar]
- 7.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta. 2010;1806:220–229. doi: 10.1016/j.bbcan.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer. 2010;126:2457–2468. doi: 10.1002/ijc.24943. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol. 2009;16:761–771. doi: 10.1245/s10434-008-0260-0. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Tang B, Yu PW, et al. Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in Caco-2 cells. PloS One. 2012;7:e51076. doi: 10.1371/journal.pone.0051076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Yu H, Qin H, et al. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012;314:232–243. doi: 10.1016/j.canlet.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki K, Tsuno NH, Sunami E, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370. doi: 10.1186/1471-2407-10-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi W, Tang Q, Chen X, et al. Antitumor and antimetastatic activities of vesicular stomatitis virus matrix protein in a murine model of breast cancer. J Mol Med (Berl) 2009;87:493–506. doi: 10.1007/s00109-009-0444-5. [DOI] [PubMed] [Google Scholar]
- 17.Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romano S, D'Angelillo A, Pacelli R, et al. Role of FK506-binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ. 2010;17:145–157. doi: 10.1038/cdd.2009.115. [DOI] [PubMed] [Google Scholar]
- 19.de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–2947. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 20.André T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27:3109–3116. doi: 10.1200/JCO.2008.20.6771. [DOI] [PubMed] [Google Scholar]
- 21.Kuebler JP, Wieand HS, O'Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol. 2007;25:2198–2204. doi: 10.1200/JCO.2006.08.2974. [DOI] [PubMed] [Google Scholar]
- 22.He G, Wang Y, Pang X, Zhang B. Inhibition of autophagy induced by TSA sensitizes colon cancer cell to radiation. Tumour Biol. 2014;35:1003–1011. doi: 10.1007/s13277-013-1134-z. [DOI] [PubMed] [Google Scholar]
- 23.Giatromanolaki A, Koukourakis MI, Harris AL, Polychronidis A, Gatter KC, Sivridis E. Prognostic relevance of light chain 3 (LC3A) autophagy patterns in colorectal adenocarcinomas. J Clin Pathol. 2010;63:867–872. doi: 10.1136/jcp.2010.079525. [DOI] [PubMed] [Google Scholar]
- 24.Giatromanolaki A, Koukourakis MI, Koutsopoulos AV, Harris AL, Gatter KC, Sivridis E. Autophagy and hypoxia in colonic adenomas related to aggressive features. Colorectal Dis. 2013;15:e223–e230. doi: 10.1111/codi.12147. [DOI] [PubMed] [Google Scholar]
- 25.Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, Harris AL. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Brit J Cancer. 2010;103:1209–1214. doi: 10.1038/sj.bjc.6605904. [DOI] [PMC free article] [PubMed] [Google Scholar]