

Summary
Rho family GTPases and their effector proteins regulate a wide range of cell signaling pathways. In normal physiological conditions their activity is tightly controlled and it is not surprising that their aberrant activation contributes to tumorigenesis or other diseases. For this reason, the identification of small, cell permeable molecules capable of inhibition of Rho GTPases can be extraordinarily useful, particularly if they are specific and act reversibly.
Herein we describe a flow cytometric assay, which allows us to measure the activity of six small GTPases simultaneously. GST-tagged small GTPases are bound to six glutathione bead sets each set having a different intensity of red fluorescence at a fixed wavelength. The coated bead sets were washed, combined, and dispensed into 384-well plates with test compounds, and fluorescent-GTP binding was used as the read-out. This multiplex bead-based assay was successfully used for to identify both general and selective inhibitors of Rho family GTPases.
Keywords: Rho GTPases, Cdc42, bead-based multiplex assay, fluorescent GTP binding, screen, flow cytometry
1. Introduction
1.1. Rho GTPase inhibitors
During the past few years, Rho GTPases and their effector proteins have been recognized as major regulators of a wide range of signaling pathways that control a number of biological processes such as cell-cycle progression and gene transcription. These molecules have also been implicated in cellular processes such as adhesion, migration, phagocytosis, cytokinesis, neurite extension and retraction, cellular morphogenesis and polarization, growth and cell survival (1–3).
The function of Rho-family GTPases in disease pathogenesis has been reviewed extensively (4–7). For these reasons, the identification of small, cell permeable molecules capable of regulating Rho GTPases activity can be extraordinarily useful, particularly if they are specific and act reversibly. There has been limited success in identifying inhibitors that specifically interact with individual small Rho GTPases. Most previous efforts have been focused on inhibiting post-translational GTPase modification by lipids, which are necessary for their membrane localization and activation. Unfortunately, these inhibitors and drugs are not specific to GTPases and affect other cell signaling pathways, which complicate the interpretation of results and create toxicity issues. Recently one small molecule inhibitor of Cdc42 activation (secramine) (8) and several low molecular weight inhibitors for Rac1 have been described (9, 10). Here we describe a bead-based multiplex flow cytometry high throughput screening (HTS) assay which allowed simultaneous screening of six GTPase targets against ~ 200,000 compounds in the Molecular Libraries Small Molecule Respository (MLSMR). This approach resulted in the publication of two probe reports, one describing the discovery of novel small molecule probes for a selective Cdc42 inhibitor, and the other describing a pan-activator family of probes for Ras-related GTPases (http://mli.nih.gov/mli/mlp-probes/) as well as a general inhibitor of Rho family GTPases (11).
1.2. Fluorescent based assay
Small GTPases exist in two interconvertible forms: GDP-bound inactive and GTP-bound active forms. GTP/GDP exchange studies frequently use guanine nucleotide analogues, which behave similarly to the native species and have been modified such that they can be sensitively detected. Radiolabeled GTP analogs such as [γ-32P] GTP and [γ-35S] GTPγS have been most commonly used. While these analogs are very sensitive and non-perturbing, their use has obvious drawbacks associated with radioactivity including the requirement for measurement at fixed time points. Recently developed BODIPY-labeled nucleotides are therefore increasingly being adopted for characterizing of GTPase nucleotide binding activities over time (12–14). The fluorescence emission of BODIPY-guanine nucleotides is directly affected by protein binding. Free BODIPY-nucleotides in solution exhibit quenched fluorescence, which is unquenched upon protein binding. The resulting 2–10-fold fluorescence enhancement allows real-time detection of protein-nucleotide interactions. Fluorescent polarization assay has also been used in these experiments, however flow cytometry measurements have several advantages over the polarization assay (15), and could be successfully used for HTS of chemical libraries for identification of Rho GTPase inhibitors.
Recently developed technology dramatically accelerates the speed of analysis. Whereas previous methods using flow cytometry only allowed the measurement of 10 samples per min, now HyperCyt technology allows analysis of up to 1 sample per second. Methods using flow cytometry as the read-out in screening assays and general principles for applying such systems in multiwell plates have been described previously (15, 16) Recently, assays using beads as a solid support matrix for molecular interactions, have become an industry standard of flow cytometry fluorescence-based measurements (15, 17). This approach has been used to develop a bead-based flow cytometric, fluorescent GTP-binding assay which is highly sensitive and allows real-time measurements (18). This bead-based format was subsequently extended to a multiplexed target assay and successfully utilized for HTS to identify regulators of small GTPases (11). The unique multiplexing capabilities of flow cytometry enabled the simultaneous quantitative analysis of the activation or inhibition of six or more GTPases immobilized on colorcoded beads. The high surface density of the fusion protein on the beads is believed to promote GST dimerization and to promote stable attachment of the fusion protein to the bead during the course of analysis. Since the beads were obtained in quantity at discount (~ 1 penny per color per well), the approach resulted in a low-cost, time saving, and highly efficient method for the simultaneous measurements of the activity for a subset of proteins in the family. Moreover, it simplifies the high-content analyses involved with comparison and detection of the specific inhibitors of individual GTPases.
1.3. High Throughput Screening
Identification of inhibitors by multiplex bead-based HTS is a multistep process. The first step usually includes determination of conditions that allow simultaneous measurment of several bead-attached enzymes. The second step of inhibitor discovery, so called primary screening, is performed with one fixed concentration of compound. This is often a fast, cost-efficient assay, to screen many compounds for identification of potential 'hits' (i.e., active compounds). The third step would be further analysis of potential hits (compounds initially identified in the primary screen) in a dose-response series. And finally, the fourth step would use molecular or cell based assays to validate the particular function of hit compounds.
Finding the right conditions for multiplex assay is a potentially difficult, and time consuming process requiring some preliminary data from pilot studies. Initially, optimal conditions are determined for individual enzyme activities in a single-plex format. Often, these conditions vary in accordance to individual GTPases. For example, BODIPY-GTPγNH is identified as a better substrate for some of the small GTPases such as RhoA wt, Rac1wt and Cdc42, while others (Rab proteins and Ras) prefer binding to BODIPY -FL-GTP. Mg2+ ions are crucial for the Rho enzyme activity, while Rab proteins and H-Ras more effectively bound fluorescent GTP in the presence of EDTA. The binding activity of H-Ras is completely inhibited in the presence of 0.01% dodecyl maltoside in the assay, while this detergent has negligible effect on GTP binding by other GTPases. Based on these preliminary results, we chose specific conditions that were not optimal for all GTPases, but allowed us to measure simultaneously GTP binding to several small GTPases. High affinity binding of GTP to GTPases in the presence of Mg2+ ions complicates screening of competitive inhibitors (Kd = 0.6 nM for Cdc42 binding BODIPY-FL-GTP). For this reason, during primary screening Mg2+ ions were excluded from the assay system, which significantly decreased nucleotide affinity to GTPases by adding 1mM EDTA (Kd~30nM). This approach was used for the multiplex analysis of GTPase activity and, therefore, the HTS of the MLSMR.
2. Materials
Prepare all solutions using deionized water and analytical grade reagents. Prepare and store all reagents at 4°C or on ice (unless indicated otherwise).
2.1. Reagents and buffers
Test compounds are provided in 384-well plates and screened using 10−3 M stocks in dimethylsulfoxide (DMSO). Obtained from MLSMR (http://mlsmr.glpg.com/MLSMR_HomePage/)
10% bovine serum albumin (BSA). Freeze in aliquots; do not use a microwave oven for thawing.
100 mM dithiothreitol (DTT). Freeze at −20°C in aliquots.
5mM BODIPY-FL-GTP stock. (G-12411, Invitrogen Molecular Probes). Store at −20°C.1 M HEPES pH 7.5: Weight 23.83 g of HEPES, add water to a volume of 90 mL. Mix and adjust pH with KOH. Make up to 100 mL. Store at 4°C.
5 M NaCl: Dissolve 292.2 g of NaCl in 1L of water.
1 M KCl: Dissolve 75 g of KCl in 1L of water.
1 M MgCl2: Dissolve 20.3 g of MgCl2 6H2O in 100 mL of water.
100 mM EDTA: Dissolve 3.7 g of EDTA in 80 mL of water. Adjust pH to 7.5 with NaOH. Then bring volume to 100 mL.
10% Nonidet P-40 (NP-40): Dilute 1 mL of NP40 in water. Bring volume up to 10 mL.
NP-HPS buffer: 30 mM HEPES pH 7.5, 100 mM KCl, 20 mM NaCl, 0.01% (vol/vol) NP-40. Mix 30 mL of 1M HEPES, 100 mL of 1M KCl, 4 mL of 5 mM NaCl, 1 mL of 10% NP-40 and bring volume up to 1 L with water. Store at 4°C.
NP-HSP buffer containing 1 mM EDTA (NP-HPSE): Add to 100 ml NP-HPS buffer 1 mL 100 mM EDTA.
NP-HSP buffer containing 1 mM MgCl2 (NP-HPSM): Add to 100 mL of NP-HPS buffer 100 µL of 1 M MgCl2.
200 nM BODIPY-FL-GTP in NP- HPSE buffer: Add in 50 mL of NP-HPSE 2 µL of 5 mM BODIPY-FL-GTP. Prepare fresh and keep on ice.
2 nM BODIPY-FL-GTP in NP-HPSM buffer for dose response experiments: Prepare a 2 µM solution of BODIPY-FL-GTP by adding 1 µL of the 5 mM stock in 2.5 mL of NP-HPSM. Use a 1:1000 dilution of this solution for preparation of 2 nM BODIPY-FL-GTP). Keep on ice.
GST–GTPases: wild type (wt) Cdc42, Rac1, RhoA, and H-Ras and constitutively active mutants Cdc42Q61L, Rac1Q61L, RhoAQ63L, and H-RasG12V are purchased from Cytoskeleton, Inc. Rab2 and Rab7 are purified from E. coli as described (18). Store as 1 mg/mL stocks at −80°C.
4 µm diameter glutathione-bead (GSH-beads) sets for multiplex assays, distinguished by seven different intensities of red fluorescence (representing several orders of magnitude variation of emission at 665 ± 10 nm with excitation at 635 nm) are obtained from Duke Scientific Corp.(but may now be ordered from Thermo Fisher). Each polystyrene bead set is supplied at 1.4 × 105 beads/µL with about 1.2 × 106 glutathione sites per bead as determined by using GST–green fluorescent protein (GFP).
Fluorescence standard beads (Bangs Laboratories, cat. No. 825B). This kit contains five sets of beads, with a measured green fluorescence for each set in the FITC, or fluorescein, channel, using a 488 nm laser for excitation and (in our instrument) a 530 nm +/− 40 nm emission filter. The fluorescence is given in mean equivalents of soluble fluorophores (MESF) ranging from 40,000 soluble fluorescein equivalents to 1,100,000 soluble fluorescein equivalents, and is used to calibrate the instrument response.
384-well assay plates (Greiner Bio-One), 30 µL maximum volume.
V-bottom 96-well PCR plates (ISC Bioexpress).
Sealing covers for plates (Gene Mate). A roller seals the cover onto the plate.
2.2. Equipment
Biomek FXP (Beckman-Coulter) multi-tip dispensing instrument, or robot, with a pin tool device (V&P Scientific).
Computer with Microsoft Windows 2000 or Windows XP, 512 MB or more RAM, 500 MB or more of free disk space, and a USB port.
HyperView™ program (IntelliCyt).
GraphPad Prism 4 or 5 software.
Flow cytometer (CyAn ADP Dako, now Beckman-Coulter) or LSRII (Becton-Dickinson) and an Accuri C6 (Accuri). For multiplex assay, both 488 and 635 nm lasers are required. The data acquisition software must include a time parameter capable of binning data at 100 ms intervals continuously for 15 min or more.
HyperCyt™ instrument (IntelliCyt). This instrument includes an autosampler, a peristaltic pump, 25G stainless steel tube inlet probes, and PVC tubing. HyperCyt is set up as described earlier (16). Briefly, the peristaltic pump speed is set to 15 r.p.m. to result in a flow rate of about 2 µL s−1. Faster or slower speed is typically suboptimal and can also result in increased particle carryover. Peristaltic pump clamping pressure: when adjusted properly, there should be uniform air bubbles on both sides of the pump. If the bubbles are broken up on the flow cytometer side of the pump, the tension on the tubing is too great and can be appropriately adjusted.
Peltier cooler for standard size plates (Inheco, TEC Control 96 and CPAC Ultra Flat). The cooling device is placed on the autosampler deck of the HyperCyt.
Software for HyperCyt™ (IntelliCyt). Includes two programs that are needed to run the HyperCyt™ platform: HyperCytSampler controls the autosampler, while HyperCytDataAnalysis is used to bin the time-resolved files stored in flow cytometry standard 2.0 or 3.0 formats.
3. Methods
3.1. Primary screening of 384-well plates
A set of color-coded glutathione-microspheres, having different intensities of red fluorescence, is coated with an individual low molecular weight GST-GTPase on each microsphere (Fig.1A). After washing, individual GTPase coupled beads are combined and 5 µL aliquots of the resulting suspension are added into each well of a 384-well plate. A green fluorescent-GTP is used as a binding ligand to look for molecules that could regulate the binding of GTP to small GTPases.
Fig.1. Experimental setup for primary screening and dose response analyses.

(A) Six GSH-bead sets of varying intensities of red fluorescence are individually coated with GST-Ras family GTPases, and the seventh set of blank beads serves as a scavenger. (B) Setup of 384-well plates for primary screening. The columns are marked by numbers 1–24, and the rows are marked by letters A–P. Wells with a symbol “b” have the multiplex (seven different bead sets) in each well. Wells with a symbol “c” have compounds in them to be screened, a total of 320 different compounds per plate. Wells in the first two columns have no compounds, and serve as positive controls. Wells with a “-” symbol in the last two columns have no beads or compounds, and are used to mark the end of each row when binning the data. (C) Setup of 384-well plates for dose response analyses. Wells in columns 14 and 24 are marked with a “-” symbol, do not have beads and compounds in them, they contain only BODIPY-FL-GTP in assay buffer and are used as markers for the end of the dose-response series for a given compound. (These wells wash the tube for traces of compound to reduce contamination). Each well in column 1 to 13 has the seven different kinds of beads. Wells in columns 1, 2, 12 and 13 do not have compounds in them, and serve as positive controls. Compound 1 is added to wells A3 through A11 at concentrations of 100 µM, 33µM ….15nM, and the other compounds are dosed in the same way (starting from B3, C3 and so on). The HyperCyt autosampler picks up ~2 µl of suspension from each well across each row in turn: A1 through A24, B1 through B24, ending at P24.
3.1.1. Coating of the bead sets with small GTPases and preparation of 384-well plates for primary screening
In primary screening, each well contains seven bead types in different intensities of red fluorescence, coupled with individual GST-GTPases. This mixture is incubated for 45 min with 200 nM fluorescent GTP in the presence of 10 µM compound, and the protein binding fluorescence is determined using a flow cytometer with the beads delivered by a HyperCyt system (11).
Mix the 4 µm glutathione bead slurries, and load 250 µL of each bead type into seven individual tubes (see Note 1)
Collect beads by centrifugation for 30 sec at maximum speed.
Aspirate supernatant and wash beads with NP-HPSE buffer.
Block non-specific binding in each bead set individually with 0.1% BSA in NP-HPSE buffer for 30 min at room temperature (see Note 2).
Collect bead sets (repeat step 2).
Re-suspend beads in 100 µL of NP-HPSE buffer.
Thaw aliquots of six recombinant GST-GTPases.
Add ~20 µg of each GTPase to the corresponding tube with the individual bead set. Pipet gently and incubate the separate suspensions on a rotator overnight at 4°C. One bead set consists of GSH-beads without any bound protein and serves as a “scavenger” for GST-proteins that might dissociate during the assay to minimize cross-contamination of protein-bound bead sets (see Notes 3).
To remove unbound GST-GTPase, collect beads by centrifugation (step 2). Wash individual GTPase-coupled beads twice with 100 µL of ice-cold NP-HPSE buffer supplemented with 0.1% BSA and 1 mM DTT and keep in separate tubes on ice.
To minimize the dissociation of protein from the beads, split equal volumes of protein–bead suspension into 4 tubes. This volume is enough for ten 384-well plates.
Re-suspend the contents of the first sets of tubes in 600 µL NP-HPSE/BSA/DTT buffer. Keep the protein–bead mixture in the remaining tubes on ice with a minimal amount of buffer (~10 µL) before use.
Load 60 µL aliquots of each GTPase-bead sets in a new tube, dilute up to 2100 µL with NP-HPSE/DTT/BSA buffer, and load 130 µL of this suspension in the first 2 columns of a 96-well plate for distribution to an assay plate by the Biomek FXP. Use 8 tip pod to deliver 5 µL of coated bead suspension from 96-well plate in wells of column 1–22 of a 384-well plate. Wells 23 and 24 in the last two columns have no beads, which results in a large temporal gap in data acquisition at the end of each row that allows row separation during data analysis (Fig.1B).
The Beckman Coulter Biomek FXP robot is programmed to deliver 0.1 µL of compound (wells #2–22 of each row) or control in DMSO (wells #1,2) to 384-well plates using the pin tool device (see Note 4).
Next, the robot adds 5 µL of 200 nM BODIPY-GTP to each well (using the 384 tip pod), and mixes them.
Cover sample-loaded 384-well plate with foil cover and rotate slowly to maintain suspension at 4°C for 45 – 60 min. We use a rotating mixer that rotates the plate at 10 rpm. The plate is hold on with rubber bands.
As a negative control, prepare a plate with the same GTPase-bead mixture with fluorescent GTP and 0.5 mM unlabeled GTP as a competitor. Measure separately.
3.1.2. Flow Cytometry measurement
Sample delivery from a multi-well plate to a flow cytometer occurs in a continuous stream with an autosampler, which is connected with flexible tubing and a peristaltic pump to a flow cytometer. The autosampler sips ~2 µL suspension from each of the wells of a multi-well plate, leaving an air gap between samples. Data acquisition is obtained in a single time-resolved data file (one file per 384-well plate).
If the lab is equipped with all the necessary equipment (robots and Flow cytometry) a single person could screen ~30–35 384-well plates per day. Screening of one plate takes ~15 min.
Use following conditions from the outset to measure fluorescence of the standard calibration beads with the CyAn flow cytometer: use ~550–650 V voltage setting of the photomultiplier tube for the fluorescein channel (488 nm excitation, 530 nm emission). Adjust the voltage/gain of the photomultiplier tube for the red channel (635 nm excitation, 665 nm emission) so that the bead sets are well separated (Fig.6 A and B) (see Note 5).
Fill sample tubing with 100 nM BODIPY-FL-GTP solution in NP-HPSE/DTT/BSA buffer, and allow it to coat the interior tubing wall for several seconds before running the assay plate (see Note 6).
Move the plate from the rotator (at 4°C) and place the plate-cooler on the HyperCyt™ autosampler deck for high throughput sampling and flow cytometric measurements.
Start the plate run. The autosampler will move from well to well, under the control of HyperCytSampler software, sampling for ~1.1 sec from each well and pausing 0.4 sec in the air before sampling the next well. The series of 384 bubble-separated samples are delivered to the flow cytometer and measured as a single data file (see Note 7).
Fig. 6. Multiplex dose-response analysis with CyAn.
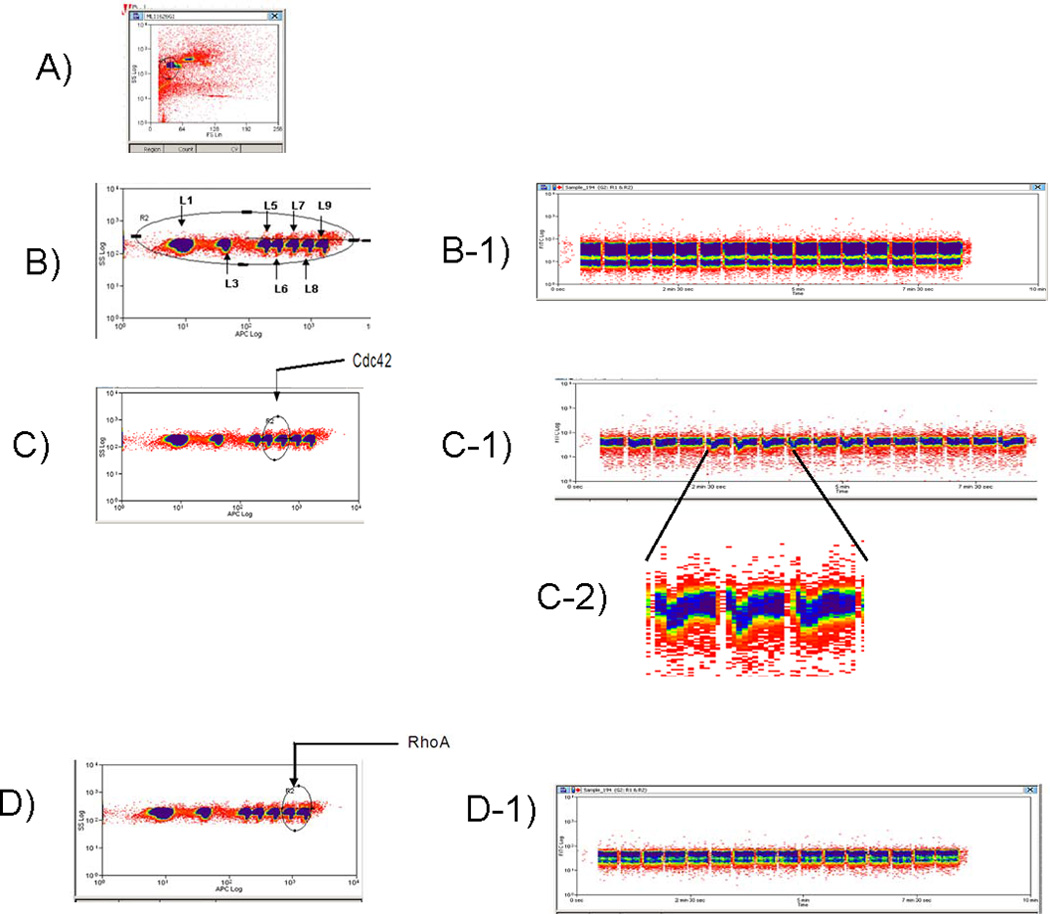
After incubation with fluorescent-GTP for ~45 min, the mixed bead set is analyzed as FCS vs. SSC (A), a gate is drawn around the singlet beads discriminated by SSC vs FL8 (uniformly sized bead populations discriminated by fluorescence intensity) (B). Each bead set is associated with a unique optical address. Electronic gates allow for separate analysis of each bead population. B1 shows green fluorescence (on the Y axis) versus time when all seven beads sets are gated (B). Gating Cdc42 containing beads (C) shows that test compounds affect Cdc42 binding to GTP in a dose dependent manner (C1). C2-shows an enlarged data fragment of C1; Gating RhoA wt and RhoA constitutively active mutant on coated beads (D) indicates no inhibition with test compounds, confirming that these compounds are specific to Cdc42 (D1).
3.1.3. Data analysis
Data analyses use HyperView™ software to merge the raw instrument flow cytometry standard (FCS) files and work lists associated with the compound library. Results from analyses of the compound activity in each well, corresponding to single compounds, are then processed through a Microsoft Excel spreadsheet template file segregating target specific data from each well and automatically calculating assay quality statistics (Z and Z’ analyses) (19, 20). Data is then normalized with respect to control wells and corrected for systematic error trends across plates (see Note 8). Data gating is performed as follows.
Open a data file with HyperView™ software.
Choose forward scatter (FCS linear) on the X-axis versus side scatter (SSC log) on the Y-axis. The beads on the screen will appear in a cluster based on particle size. As Fig. 2A shows, there may be a cluster of aggregated beads (increased FSC) in addition to singlet beads. Draw a gate around the singlet beads.
Select to display only the beads inside the gate, and in a two parameter plot of side scatter versus log red fluorescence intensity (FL8). The beads will be in seven clusters. Create a gate around each of the bead sets as shown in Fig. 2B.
These gates for the first plate become a template for subsequent plates measured that day under the same conditions. This is the benefit of using MESF beads to ensure the appropriate voltages for the entire library analysis.
Create a time versus number of events histogram to identify time bins. Time bins will be created in the HyperView™ program. Columns 23 and 24 of a 384-well plate have no beads; therefore there will be 352 bins of data to analyze.
These data are correlated with the compound list used for that plate and are exported to a Microsoft Excel spreadsheet. A decrease in the median fluorescence intensity (MFI) demonstrates that the compound in a particular well inhibits the binding of the GTPase to the fluorescent ligand.
Fig. 2. Data Analysis with HyperView™ software.

(A)In a plot of forward scatter versus side scatter, a gate is drawn around the singlet beads.
(B) Gates are drawn around each bead set using their red fluorescence.
(C) After gating of individual bead sets a time versus number of events histogram is created to identify time bins. A plot of events (beads per 0.1 s) versus time for a plate is displayed. There are no beads in columns 23 and 24, leaving 352 bins with beads. (D) An enlarged view of Row F. Software allows merging of the FCS files and work lists associated with the compound library. Results from analyses of the compound activity in each well, corresponding to single compounds, are then processed through a Microsoft Excel spreadsheet.
3.2. Dose-Response Measurements
As mentioned above (see Subheading 1.3) Mg2+ ions dramatically increase the affinity of GTP to GTPases. Equilibrium binding assay of single-plex Cdc42 binding to BODIPY-FL-GTP revealed that the Kd ~36 nM (in the presence of EDTA) drops up to 0.6 nM in the presence of Mg2+ (Fig. 3).
Fig. 3. Equilibrium binding assay.
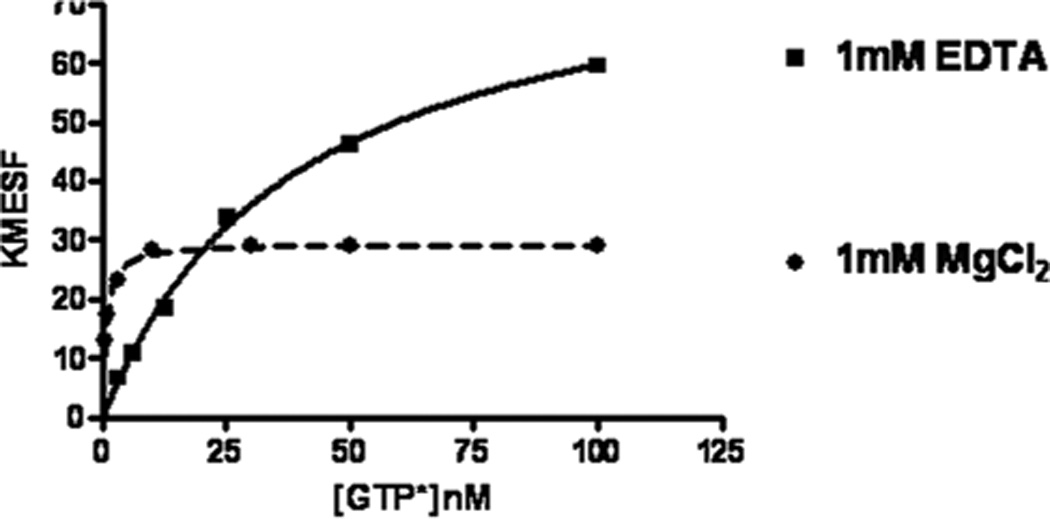
Cdc42 wt coupled on GSH-beads was depleted of nucleotide, and equilibrium binding of various concentration of fluorescent GTP in the presence of 1 mM EDTA or 1 mM MgCl2 was measured with a flow cytometer.
3.2.1 Equilibrium Binding Assay
Bind the wild-type GST-Cdc42 (4 µM) to GSH-beads overnight at 4°C. Any type of GSH-beads suitable for flow cytometry can be used. (see 3.1.1. steps 1 and 2).
Deplete Cdc42 on GSH-beads of nucleotide by incubating them with 10 mM EDTA containing NP-HPS buffer for 20 min at 30°C.
Wash twice with NP-HPS buffer, then re-suspended in the same buffer containing 1 mM EDTA/or 1 mM MgCl2, 1 mM DTT and 0.1% BSA.
Block Cdc42 binding to unbound sites by incubating the protein–bead complex for 15 min at room temperature.
Collect beads by centrifugation and re-suspend them in 30 µL of the same buffer and add 30 µL of appropriate dilutions (3 – 200 nM) of ice cold BODIPY-FL-GTP.
Incubate samples at 4°C for 45 min and measure the binding of fluorescent nucleotide to the enzyme using a flow cytometer (see Note 9).
Export raw data and plot using GraphPad Prism software.
3.2.2. Dose-Response Assay
Fig. 4A, panel 1 demonstrates a previously published example of a GTPase paninhibitor. The mean channel fluorescence of all individual GTPases is reduced. Panel 2 demonstrates an excerpt from the Excel file of the primary screen, in which one compound specifically inhibits the activity of only one GTPase (arrow) (11). In the next stage of evaluation, the inhibitory effect of active compounds obtained from the primary screening should be confirmed with dose response analyses in the presence of: a) 1 mM EDTA and b) 1 mM MgCl2. For assays with EDTA use primary screening conditions. As detected, with equilibrium binding Kd of Cdc42 to BODIPY-FL-GTP in the presence of Mg2+ ions changed, for this reason, in dose–response assays concentration of fluorescent GTP was decreased up to1 nM. Six Rho family GST-GTPases were assayed simultaneously in a single multiplex (Rac1 wt, Rac1Q61L, RhoA wt, RhoAL63, Cdc42 wt and Cdc42Q61L) and 1 nM BODIPY-FL-GTP binding was measured in the presence or absence of the serial drug dilution series. Both of these measurements confirmed that compound MLS000693334 (Fig. 3B) specifically inhibits GTP binding of Cdc42 (Fig. 5 A, B).
Fig.4. High-throughput screen identifies small-molecule inhibitors of small GTPases.
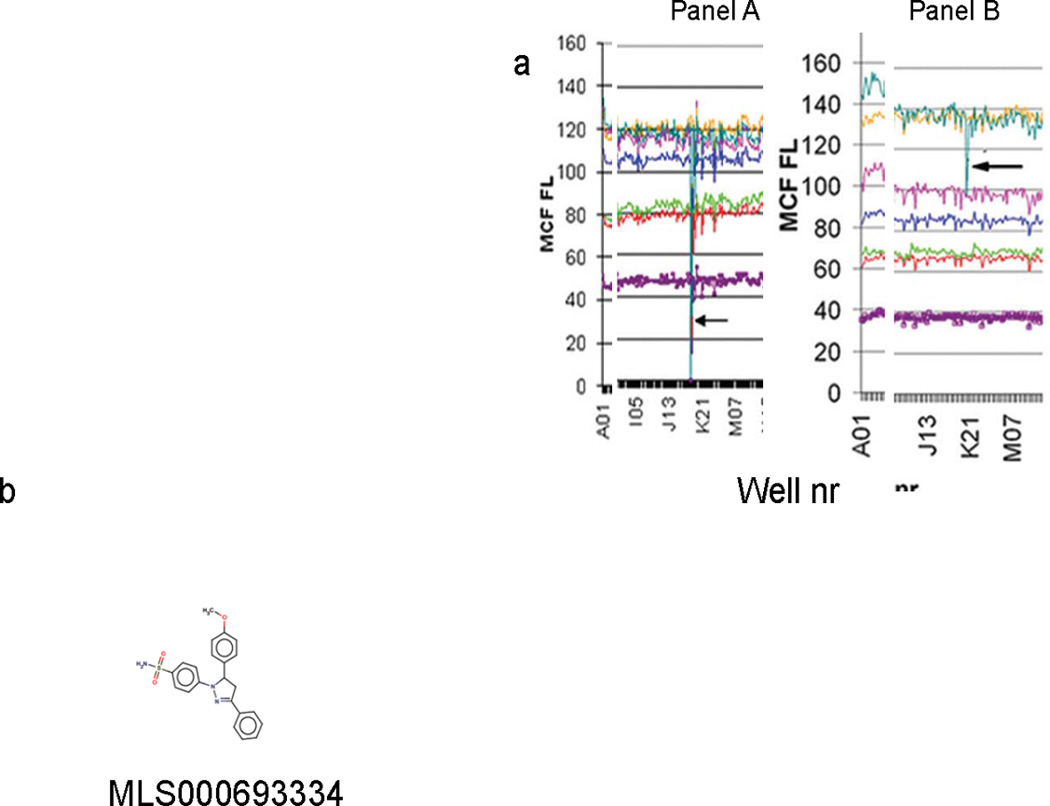
(A) The raw data were parsed in HyperView to produce annotated fluorescence summary data for each well. The parsed data were then processed through an Excel template file constructed specifically for the assay to segregate data for each target and the fluorescence scavenger in the multiplex. The Excel file shows one compound (arrow) that inhibits all GTPases (Panel A). Panel B shows a specific inhibitor of one GTPase. (B) Structure of Cdc42 specific inhibitor MLS000693334.
Fig. 5. Multiplex dose-response assay.
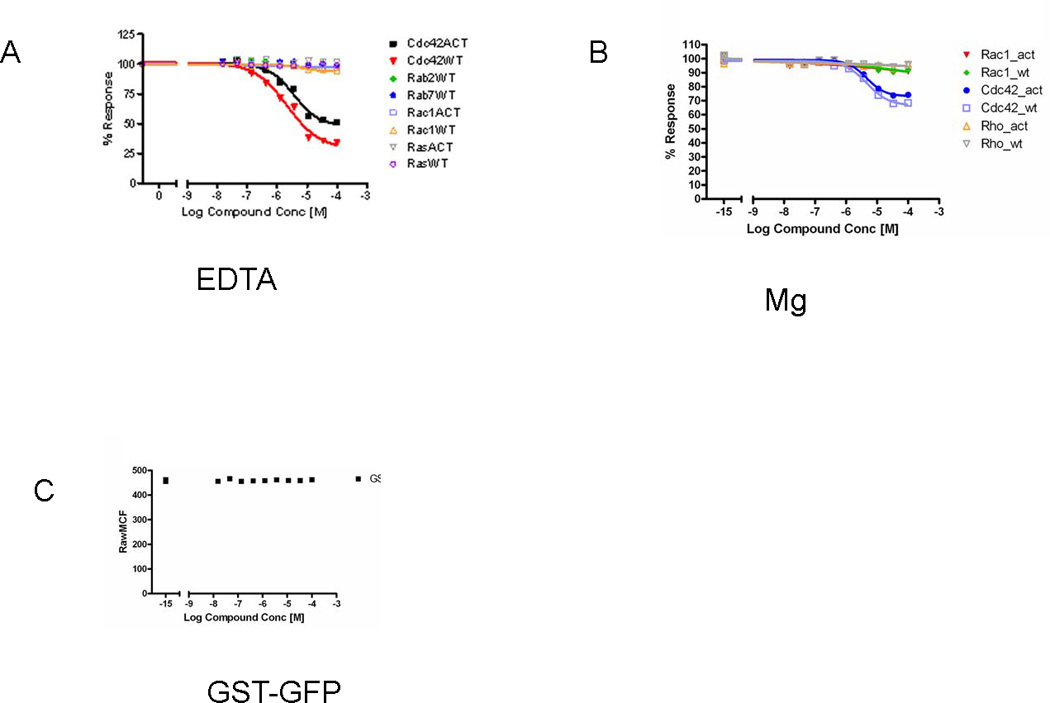
(A, B) Confirmation of active compounds identified in HTS multiplexed primary screening with dose response analysis of multiplex in the presence of EDTA (A) and Mg++ (B). Graphs show percent of activity versus compound concentration. (C) In order to evaluate the effect of compounds on the binding of GST fusion proteins to beads, bead sets were coated with GST-GFP and evaluated with the compounds of interest (active wells from HTS) in a dose response assay. The graph represents the raw mean channel fluorescence (MCF) value versus concentration of test compound for a nine level dose response. Analysis of the data demonstrates no interference between the compounds of interest and GST-GFP binding (see Note 11).
The inhibitory effect of the compound could be the result of dissociation of GST-tagged GTPase from the beads. To eliminate the possibility that the compound affects GST-protein binding to beads, additional single-plex analysis with GST-GFP was examined in the presence of EDTA and Mg2+ ions (Fig. 5C) (see Note 10).
For dose response assays compounds are diluted serially 1:3, a total of eight times from a starting concentration of 10 mM giving a 9-point dilution series in DMSO (Fig. 1C).
Load 10 µL of 100% DMSO in columns 1, 2 and 12, 13 of a 384-well plate. The wells in these columns do not have compounds in them, and serve as positive controls.
Dispense 9µL of 10mM compound in DMSO in column 3 and 6 µL of DMSO in columns 4–11.
Transfer 3µL of 10 mM compound (from column 3) in the adjacent well and mix with pipetting.
Continue sequentially for eight wells. The concentration of the final well will be 15 nM.
The Beckman Coulter Biomek FXP robot is programmed to deliver 5 µL of bead suspension into the wells of columns 1–13 of the 384-well plate.
Add 0.1 µL of compound (wells in columns 3–11) or control DMSO (wells in columns 1,2,12 and 13) using the pin tool device.
Add 5 µL of 200 nM BODIPY-FL-GTP (or 2 nM in assays with Mg2+) to each well, and mix them.
Cover the sample-loaded 384-well plate with a foil cover and rotate slowly to maintain suspension at 4°C for 45– 60 min.
Measure fluorescence as described above (see Subheading 3.1.2).
Acknowledgement
This work was supported by NIH grants U54 MH074425 and U54 MH084690
Footnotes
It is common to keep the beads suspended during coating with protein. We usually use overnight rotation at 4°C. Longer incubation times result in increased protein binding to beads. Occasionally, some fusion proteins lose about 50% of their binding activity while rotating. We let the proteins settle overnight, since all of them exhibited good binding in the process.
Blocking of beads with 0.1% BSA containing buffer can be done after coupling GST-GTPases with beads.
Most GST-GTPase fusion proteins will bind to the beads well, but some of these fusion proteins may have unstable binding to the glutathione beads. This binding stability depends on chosen conditions. For example, we found that GST-Rac1 dissociates from the beads in the presence of NP-HPSE, but not in the presence of Mg2+ ions. To minimize cross-contamination of beads with dissociated proteins we retain the “scavenger beads” (GSH-beads without any protein added) in the system. Do not use the particular GST-tagged protein that dissociates from the beads in multiplex sets during dose-response assays. Use a single-plex assay for such proteins. To minimize adsorption of beads to pipette tips and HyperCyt tube walls we use of 0.01% detergent in the assay buffer.
Using the robot (Biomek FXP) helps greatly in the preparation of 40 or more 384-well plates for large libraries. But if it is planned to investigate the effect of only a few compounds for GTPase ligand interaction, plates can easily be prepared manually.
Red laser alignment will help to solve problems with poor resolution of the separate bead sets.
Reduced fluorescent intensity at the start of plate screening could be due to the adsorption of fluorophore to the HyperCyt sample tubing walls. Running more fluorophore through the tubing to equilibrate fluorophore adsorption (just prior to sampling the plate) will solve this problem.
Sometimes the bead number in wells may be low. Usually increasing the sampling time from 0.9 sec to 1.3 sec doubles the number of beads sampled per well. But there may be other reasons, for example HyperCyt tubing or probe clogging, and replacing these parts will solve the problem.
The quality control Z' statistics were very good for each target. Beads are more homogeneous than cells, and therefore Z’ scores are better for bead-based assays than for cells. In particular, the average Z' was 0.87 ± 0.04 for Cdc42, 0.85 ± 0.04 for Rac1 wt, and 0.90 ± 0.03 for Rac1Q61L. The complete results from the multiplex screen are available on PubChem (http://pubchem.ncbi.nlm.nih.gov/, AID#: 757, 758, 759, 760, 761, 764, 1333–1337, 1339–1341).
For CyAn flow cytometer use 3.1.2 settings. New generation Accuri cytometers have pre-optimazed detector settings.
An observed inhibitory effect of a compound could be the result of inhibition of GSH-bead GST-protein interaction. In this case, the compounds would give the identical dose-response curve for all checked enzymes. To eliminate these false positive compounds, an additional assay should be done. Incubate 2 nM GSTGFP overnight with the beads, wash unbound GST-GFP and measure fluorescence with the addition of the suspect compounds. In our dose–response assays we have used GST-GFP screening for all compounds tested.
Some compounds may be fluorescent by themselves and will show higher fluorescence intensity than the positive control. For this reason, it is recommended to check all your interesting compounds in an auto-fluorescence assay. In an auto-fluorescence assay, use the same bead-protein sets, add compound, but instead of fluorescent GTP, add assay buffer (NP-HPSE, or NPHPSM), and then sample the plate. Some highly fluorescent compounds can carryover into the next wells. In this case, run the row or entire plate backwards (to designate the first highly fluorescent well as the last well sampled for that plate).
References
- 1.Chimini G, Chavrier P. Function of Rho family proteins in actin dynamics during phagocytosis and engulfment. Nature Cell Biol. 2000;2:E191–E196. doi: 10.1038/35036454. [DOI] [PubMed] [Google Scholar]
- 2.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 3.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Boettner B, Van Aelst L. The role of Rho GTPases in disease development. Gene. 2002;286:155–174. doi: 10.1016/s0378-1119(02)00426-2. [DOI] [PubMed] [Google Scholar]
- 5.Gomez del Pulgar T, Benitah SA, Valeron PF, Espina C, Lacal JC. Rho GTPase expression in tumourigenesis: evidence for a significant link. Bioessays. 2005;27:602–613. doi: 10.1002/bies.20238. [DOI] [PubMed] [Google Scholar]
- 6.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 7.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 8.Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, Macia E, Feng Y, Shair MD, Kirchhausen T. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nature Chem Biol. 2006;2:39–46. doi: 10.1038/nchembio751. [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 11.Surviladze Z, Waller A, Wu Y, Romero E, Edwards BS, Wandinger-Ness A, Sklar LA. Identification of a small GTPase inhibitor using a high-throughput flow cytometry bead-based multiplex assay. J Biomol Screen. 15:10–20. doi: 10.1177/1087057109352240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McEwen DP, Gee KR, Kang HC, Neubig RR. Fluorescent BODIPY-GTP analogs: real-time measurement of nucleotide binding to G proteins. Anal Biochem. 2001;291:109–117. doi: 10.1006/abio.2001.5011. [DOI] [PubMed] [Google Scholar]
- 13.Jameson EE, Roof RA, Whorton MR, Mosberg HI, Sunahara RK, Neubig RR, Kennedy RT. Real-time detection of basal and stimulated G protein GTPase activity using fluorescent GTP analogues. J Biol Chem. 2005;280:7712–7719. doi: 10.1074/jbc.M413810200. [DOI] [PubMed] [Google Scholar]
- 14.Evelyn CR, Ferng T, Rojas RJ, Larsen MJ, Sondek J, Neubig RR. High-throughput screening for small-molecule inhibitors of LARG-stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. J Biomol Screen. 2009;14:161–172. doi: 10.1177/1087057108328761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salas VM, Edwards BS, Sklar LA. Advances in multiple analyte profiling. Adv Clin Chem. 2008;45:47–74. doi: 10.1016/s0065-2423(07)00003-0. [DOI] [PubMed] [Google Scholar]
- 16.Edwards BS, Young SM, Oprea TI, Bologa CG, Prossnitz ER, Sklar LA. Biomolecular screening of formylpeptide receptor ligands with a sensitive, quantitative, high-throughput flow cytometry platform. Nature Protocols. 2006;1:59–66. doi: 10.1038/nprot.2006.9. [DOI] [PubMed] [Google Scholar]
- 17.Saunders MJ, Edwards BS, Zhu J, Sklar LA, Graves SW. Microsphere-based flow cytometry protease assays for use in protease activity 23 detection and high-throughput screening. Curr Protocols in Cytometry / editorial board, J. Paul Robinson, managing editor ... [et al Chapter 13. Unit 13(12):11–17. doi: 10.1002/0471142956.cy1312s54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz SL, Tessema M, Buranda T, Pylypenko O, Rak A, Simons PC, Surviladze Z, Sklar LA, Wandinger-Ness A. Flow cytometry for real-time measurement of guanine nucleotide binding and exchange by Raslike GTPases. Anal Biochem. 2008;381:258–266. doi: 10.1016/j.ab.2008.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 20.Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R. Statistical practice in high-throughput screening data analysis. Nature Biotech. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]