

Abstract
Macrophages, owning tremendous phenotypic plasticity and diverse functions, were becoming the target cells in various inflammatory, metabolic and immune diseases. Cytochrome P450 epoxygenase 2J2 (CYP2J2) metabolizes arachidonic acid to form epoxyeicosatrienoic acids (EETs), which possess various beneficial effects on cardiovascular system. In the present study, we evaluated the effects of EETs treatment on macrophage polarization and recombinant adeno-associated virus (rAAV)-mediated CYP2J2 expression on lipopolysaccharide (LPS)-induced cardiac dysfunction, and sought to investigate the underlying mechanisms. In vitro studies showed that EETs (1μmol/L) significantly inhibited LPS-induced M1 macrophage polarization and diminished the proinflammatory cytokines at transcriptional and post-transcriptional level; meanwhile it preserved M2 macrophage related molecules expression and upregulated antiinflammatory cytokine IL-10. Furthermore, EETs down-regulated NF-κB activation and up-regulated peroxisome proliferator-activated receptors (PPARα/γ) and heme oxygenase 1 (HO-1) expression, which play important roles in regulating M1 and M2 polarization. In addition, LPS treatment in mice induced cardiac dysfunction, heart tissue damage and infiltration of M1 macrophages, as well as the increase of inflammatory cytokines in serum and heart tissue, but rAAV-mediated CYP2J2 expression increased EETs generation in heart and significantly attenuated the LPS-induced harmful effects, which mechanisms were similar as the in vitro study. Taken together, the results indicate that CYP2J2/EETs regulates macrophage polarization by attenuating NF-κB signaling pathway via PPARα/γ and HO-1 activation and its potential use in treatment of inflammatory diseases.
Inflammation is an adaptive response that protects body from endogenous and exogenous pathogens (Takeuchi and Akira, 2010). Whether or not the inflammation can be effectively controlled determines the outcome of diseases. Sepsis is a serious medical condition featured by dysregulated systemic inflammatory response to infection with profound effects on all organs and most notably the cardiovascular system. Sepsis-induced cardiovascular dysfunction is one of the major predictors of morbidity and mortality of sepsis (Coquerel et al., 2014). It remains as major challenge for both scientists and clinicians since there is no effective cure method. As regarded its pathogenesis, continued activation of neutrophils and monocytes/macrophages may attribute to accelerate the septic response (Stearns-Kurosawa et al., 2011).
Macrophages have long been considered to be important immune response effector cells and the functional diversity of macrophages can be attributed to their ability to respond to different microenvironmental cues by displaying equally diverse functional phenotypes (Gordon and Taylor, 2005). Macrophages can be categorized into at least two phenotypically and functionally distinct subsets, that is, classically activated M1 macrophages (“killer” macrophages) and alternatively activated M2 macrophages (“repair” macrophages) (Mantovani et al., 2004; Gordon and Taylor, 2005; Martinez et al., 2009). Priming with interferon γ (IFNγ), tumor necrosis factor-α (TNF-α), or sub-stimulatory concentrations of lipopolysaccharide (LPS) will program macrophages into the M1 phenotypic state, which is associated with tissue damage, inhibition of cell proliferation, and generation of pro-inflammatory cytokines. In contrast, exposure to interleukin-4 (IL-4) or IL-13 generates M2 macrophages, which is associated with tissue repair, cell proliferation, and production of anti-inflammatory factors such as IL-10 and TGF-β (Vats et al., 2006; Mosser and Edwards, 2008). Deregulation of macrophage polarization and the resulting pro-inflammatory consequences are pathologically related to various adverse cardiovascular events. The mechanisms through which control the balance of pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages are of particular importance since shifting the balance pharmacologically is an attractive target for attenuating the tissue damage observed in numerous inflammatory pathophysiological conditions.
Cytochrome P450 epoxygenases of the CYP2J and CYP2C subfamilies are abundantly expressed in the cardiovascular system and metabolize arachidonic acid (AA) to produce four biologically active epoxyeicosanoic acids (EETs): 5,6-, 8,9-, 11,12-, and 14,15-EETs. EETs can be rapidly hydrolyzed to biologically less active dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolase (sEH/Ephx2) (Xu et al., 2011). Inhibitors of sEH (sEHi) prevent the conversion of EETs to DHETs, resulting in stabilized EETs levels (Imig and Hammock, 2009). EETs possess diverse biological functions, and observations revealed that EETs exert protective effects on various cardiovascular diseases, including attenuation of heart injuries and anti-hypertension (Schmelzer et al., 2005; Wang et al., 2005; Wang et al., 2014). EETs also possess potent anti-inflammatory properties in the vasculature by suppressing NF-κB activation (Node et al., 1999; Cai et al., 2013). Moreover, CYP2J2 is expressed in primary human monocytes and monocytic cell lines (THP-1 and U937). In vitro differentiation of monocytes to macrophages results in increased CYP2J2 expression (Nakayama et al., 2008). CYP epoxygenase activity can also modulate the activation of macrophages (Bystrom et al., 2011). However, whether and how EETs regulate macrophages phenotypes remains unclear.
While a putative receptor for EETs has yet to be identified, EETs have been shown to activate peroxisome proliferator-activated receptors (PPARα/γ) (Liu et al., 2005; Wray et al., 2009). This activation and associated repression of NF-κB signaling are at least partially responsible for the anti-inflammatory effects of EETs (Plutzky, 2003; Cai et al., 2013). Similar to the physiological effects of EETs, increased heme oxygenase 1 (HO-1) activity has anti-apoptotic and anti-inflammatory properties in the cardiovascular system (Alcaraz et al., 2003). Recent studies have suggested that the vascular protective effects of EETs and HO-1 are functionally linked (Sacerdoti et al., 2007; Sodhi et al., 2012; Elmarakby et al., 2013). In the current study, we tested the hypothesis that EETs regulate macrophage polarization by attenuating NF-κB signaling pathway via PPARα/γ and HO-1 activation, and attenuate LPS-induced cardiac dysfunction.
Materials and Methods
Reagents
8,9-EET, 11,12-EET, 14,15-EET, and 14,15-EEZE were purchased from Cayman Chemical Company (Ann Arbor, MI). CD206 antibody was purchased from Biolegend (San Diego, CA). CYP2J2 and HO-1 antibodies were purchased from Abcam (Cambridge, UK). CD11c antibody was purchased from R&D Systems (Minneapolis, MN). Arginase-1, iNOS, CD68, F4/80, c-Rel, α-SMA, vimentin, desmin, Lamin B1 and β-actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). IκBα, and NF-κB p65 antibodies were purchased from Cell Signaling Technology (Danvers, MA). Zinc protoporphyrin IX (ZnPP) was purchased from Frontier Scientific (Logan, UT). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Life Technologies (Grand Island, NY). Horseradish peroxidase-conjugated secondary antibodies were purchased from Thermo Fisher Scientific (Rockford, IL). LPS (L2880, Escherichia coli 055:B5), Hemin, CORM (carbon monoxide-releasing molecule), bilirubin and all other chemicals were purchased from Sigma–Aldrich (St. Louis, MO) unless otherwise indicated.
Construction and preparation of rAAV vectors
The recombinant adeno-associated virus (rAAV) vectors (type 2) containing CYP2J2 or green fluorescent protein (GFP) were prepared by triple plasmids co-transfection in HEK293 cells as previously described (Cai et al., 2013). The vectors were purified, tittered and stored at −80°C before use.
Animals
All animal care and experimental procedures were approved by the Experimental Animal Research Committee of Tongji Medical College, Huazhong University of Science & Technology, and in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. Eight weeks old C57BL/6 mice were randomly assigned into four groups: rAAV-GFP +Saline, rAAV-CYP2J2 + Saline, rAAV-GFP+LPS, rAAV-CYP2J2 + LPS. One hundred microliter containing rAAV-GFP or rAAV-CYP2J2 (1 × 1011 p.f.u), respectively, were injected via tail veins 4 weeks before LPS injection. For the lethal endotoxin shock model, mice were intraperitoneally administered (i.p.) 20 mg/kg LPS. The survival rate of mice were monitored for 5 days (n = 15/group). Mice were subjected to hemodynamic measurements and echocardiography, blood samples and tissues were collected 6 h after 10 mg/kg LPS (i.p) challenge in several independent experiments (n = 8/group).
Hemodynamic measurements and echocardiography
Echocardiographic examination was performed with a 30-MHz high frequency scanhead (VisualSonicsVevo770, VisualSonics Inc. Toronto, Canada) as previously described (Ma et al., 2012). Left ventricle hemodynamic measurements were performed with a Millar Catheter System via the left carotid artery as described previously (Shioura et al., 2007).
Histological analysis
Heart tissues were harvested, fixed in 4% paraformaldehyde in PBS, and embedded in paraffin for histological analysis. Heart sections were stained with hematoxylin/eosin (HE). Infiltration of macrophages in the myocardium were examined by immunohistochemistry using antibody against F4/80 and CD11c. Immunohistochemical staining was performed according to the manufacture's description (Zsbio, Beijing, China). For all sections, 3,3′-diaminobenzidine was used as the indicator substrate.
Preparation of mouse peritoneal macrophages
Peritoneal macrophages from 6 to 8 weeks old C57BL/6 mice were elicited by intraperitoneal injection of 3 ml of 3% thioglycollate in distilled water. After 3 days, cells were collected by infusing their peritoneal cavity with ice-cold sterile PBS as previously described with minor modification (Ranganathan et al., 2013). After a soft abdominal massage for 30 sec, the peritoneal liquid was collected and centrifuged at 1,000 rpm for 10min, resuspended in DMEM media with 10% FBS, 2mM L-glutamine, 100 units/ml penicillin and 0.1 mg/ml streptomycin. After 2 h, non-adherent cells were removed by washing twice with PBS and the adherent monolayer cells were used for experiments. The viability of the adherent cells was assessed by trypan blue dye exclusion test and the proportion of macrophages was determined by immunofluorescence staining of macrophages for the surface marker CD68.
Cell culture and treatments
Peritoneal macrophages and RAW 264.7 cells (a mouse macrophage cell line) were maintained at 37°C in a 5% CO2 incubator. Experiments were conducted on cells at approximately 80–90% confluence. Macrophages were incubated in the presence of 1 μM 8,9-, 11,12-, or 14,15-EET with/without LPS (1 μg/ml) for 12h. As indicated, 1 μM 14,15-EEZE (an inhibitor of EET), 1 μM GW9662 (an antagonist of PPARγ), 1 μMGW6471 (an antagonist of PPARα), 10μM ZnPP (an inhibitor of HO-1), hemin (10 μM), CORM (20μM) or bilirubin (10 μM) were added 1 h before LPS treatment. At the end of the incubation for 12 h, culture supernatants were collected and stored at −80°C prior to analysis. For time course experiment, RAW 264.7 cells were treated with LPS (1 μg/ml) for various incubation times (0.5, 1, 3, 6, 12, and 24 h). For mRNA determination, cells were stimulated with LPS (1 μg/ml) for 3 h. To measure the stability of mRNA, 10 μg/ml actinomycin D (ActD) was added directly to cell cultures previously treated with LPS for 3 h without removal of the stimulant. Cells were incubated for the appropriate time after the addition of ActD, following by real-time RT-PCR analysis of mRNA.
Transfection of RAW 264.7 macrophages with siRNA were performed with Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Commercially available mouse HO-1 specific siRNA and negative control siRNA (RiboBio, Guangzhou, China) were used for transfection. The cells were transfected for 48 h and then subjected to treatment as described in figure legends, protein levels were further measured by western blot analysis.
Cell viability measurement
The CCK-8 detection kit was used to measure cell viability as previously described (Tang et al., 2013). Briefly, RAW 264.7 cells were seeded in a 96-well microplate at a density of 5,000 cells/well and then treated with different concentrations of 11,12-EET (250, 500, 750, 1000 nM) in the presence or absence of LPS (1 μg/ml) for 12 h. The control group was treated with 0.1% DMSO. The cell monolayer was washed three times with PBS, then a 1:10 diluted CCK8 solution in DMEM was added to the cells and incubated at 37°C for 2 h, viable cells were counted by absorbance measurements with amicroplate reader at a wavelength of450nm. The optical density value at 450 nm was reported as the percentage of cell viability in relation to the control group (set as 100%).
Detection of cytokine production
TNF-α, IL-1β, IL-6, MCP-1, and IL-10 levels in serum, heart extracts, and cell culture supernatants were measured using ELISA kits according to the manufacturer's instructions (Boster Bio, Pleasanton, CA). Protein levels were calculated from a cytokine standard curve.
Reverse transcription and quantitative real-time PCR
Total RNA extracts were isolated from mice hearts or peritoneal macrophages and RAW 264.7 cell lysates using Trizol reagent (Invitrogen, USA) following the manufacturer's instructions. The concentration and purity of the RNA were determined by using NanoDrop (Thermo Scientific). cDNA was synthesized using TransScript First-Stand cDNA Synthesis Kit(TransGen Biotech) in a total volume of 50 μl. After reverse transcription of the RNA, real-time quantitative PCR was performed using TransStart Eco Green Qpcr SuperMix (TransGen Biotech) on an Applied Biosystem 7900 Real-time PCR System. Primer pairs were listed in Table 1. The relative expression level of target genes normalized to GAPDH were calculated as 2−ΔΔCT. Samples were analyzed in triplicate and every experiment was performed at least three independent times.
Table 1. Primer sequences for quantitative real-time PCR.
| Gene | Forward | Reverse |
|---|---|---|
| CD11c | GCTGTCTCCAAGTTGCTCAGA | GAGCACACTGTGTCCGAACT |
| iNOS | CCAAGCCCTCACCTACTTCC | CTCTGAGGGCTGACACAAGG |
| IL-6 | GGACCAAGACCATCCAATTCATCTTGAAA | GACCACAGTGAGGAATGTCCACAAA |
| TNF-α | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| IL-1β | TACAAGGAGAAGAAAGTAATGACAA | AGCTTGTTATTGATTTCTATCTTGT |
| MCP-1 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| CD206 | CCCAAGGGCTCTTCTAAAGCA | CGCCGGCACCTATCACA |
| Arg-1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| IL-10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| GAPDH | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
Western blot analysis
Heart tissue homogenates and cell lysates were extracted according to the manufacturer's protocol (Boster Biological). Protein concentrations were determined using the BCA assay. Western blots were performed using various antibodies. Briefly, equal amounts of extracts were separated by 10% SDS–PAGE, transferred onto PVDF membranes. After blocking with 5% (w/v) bovine serum albumin (BSA), the membranes were incubated with appropriate primary antibodies at 4°C overnight, followed by incubation with peroxidase-conjugated secondary antibodies at room temperature for 2 h. The blots were visualized with enhanced chemiluminescence detection reagent. β-Actin and lamin B1 were used as internal controls for total/cytosolic protein and nuclear protein, respectively. Bands were quantified by densitometry using Quantity One software (Bio-Rad, Hercules, CA).
Macrophage migration determination
Macrophages migration was determined in Boyden chambers as described previously (Cai et al., 2013). RAW 264.7 macrophages were starved in serum-free medium for 24 h, resuspended in 200 μl serum-free medium, and then added to the upper chambers (5 × 105 cells per well), while the lower chambers were filled with 500 μl DMEM medium supplemented with 10% bovine serum. 1 μg/ml LPS was added to stimulate macrophage migration, 1 μM 11,12-EET, 1 μM 14,15-EEZE, 1 μM GW9662, 1 μM GW6471, or 10 μM ZnPP were added 1 h prior to the addition of LPS to the lower chambers. After incubating for 6 h at 37°C, unmigrated cells were wiped off the upper side of the filter with a cotton swab, while the migrated cells attached to the bottom were fixed with methanol and stained with crystal violet. Five random fields per sample were counted.
Statistical analysis
All results were expressed as the means ± SEM and each experiment was repeated at least three times. Statistical differences were evaluated by one-way ANOVA and Bonferroni post-test. Differences were considered to be significant at P < 0.05.
Results
11,12-EET shifts LPS-treated macrophages from pro-inflammatory M1 to anti-inflammatory M2 phenotype via inhibition of NF-κB signaling
In the time course experiment of LPS treatment on NF-κB activation and macrophage polarization, immunoblotting analysis were performed and revealed that LPS induced rapidly and strongly cytoplasmic IκBα degeneration, but gradually went back to basal line with the newly synthesized IκBα accumulated. NF-κB p65 and c-Rel nuclear translocation with a peak of activity at about 1 h after LPS treatment, the activity subsequently declined slightly, but remained significantly elevated above basal for at least 24 h (Fig. 1A, B). With the stimulus factors persist, the macrophage phenotype makers (CD11c and iNOS for M1, CD206 and arginase-1 for M2) were getting more and more significant different (Fig. 1C). Taken all of this changes into consideration, we selected 12 h for our intervention time in most experiments except otherwise indicated.
Fig 1.
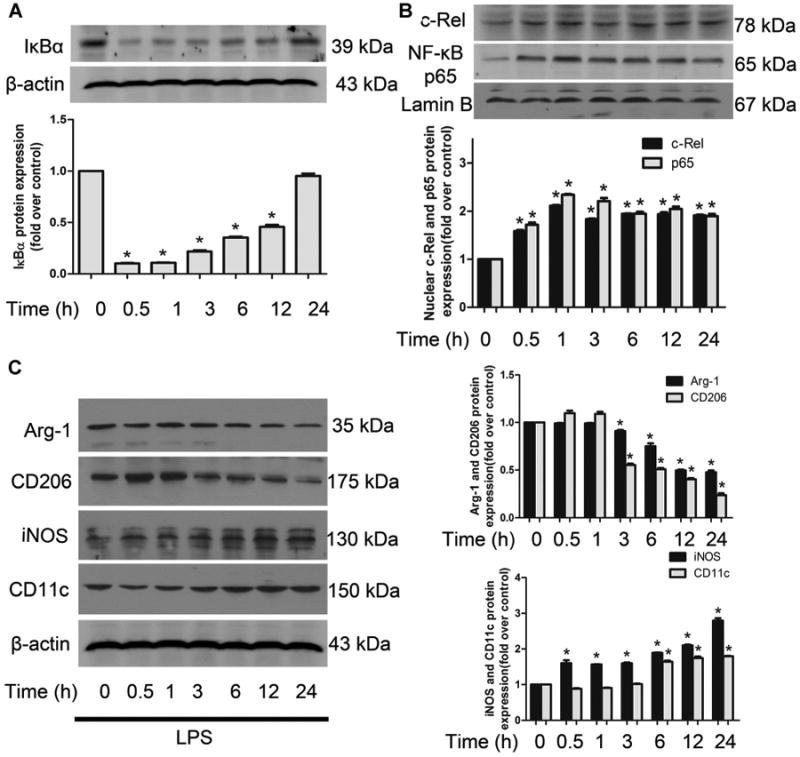
Time course for LPS-induced NF-κB activation and macrophage polarization. A–C: Representative immunoblots and quantitation for NF-κB activation (IκBα, NF-κB p65 and c-Rel) and macrophage phenotype markers (CD11c and iNOS for M1, CD206 and Arg-1 for M2) induced by LPS at different time points in RAW 264.7 cells. Data are shown as mean ± SEM from three independent experiments. *P < 0.05 versus control.
To elucidate the effects of EETs on macrophage inflammation, we treated LPS-activated RAW 264.7 macrophages with 8,9-, 11,12-, or 14,15-EET. Maximal inhibition of LPS-induced NF-κB p65 activation was achieved with 11,12-EET, followed by 14,15-EET and 8,9-EET, indicating differential bioactivity of the different EET regioisomers, while all kinds of EETs had no obvious effect on LPS-induced c-Rel activation (Fig. S1). We further performed cell viability test and found that 11,12-EET showed no significant pro-proliferation or toxicity effect in resting or LPS activated cells (Fig. 2A). Based on these pre-experiments and previous research, 1μM 11,12-EET were selected for in vitro experiments.
Fig 2.
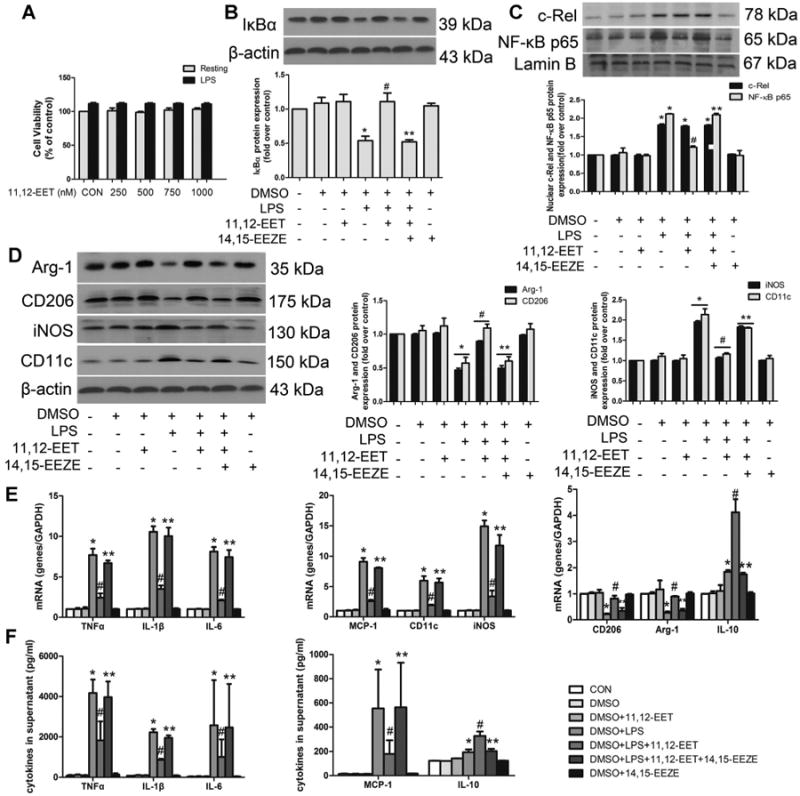
11,12-EET shifts LPS-induced M1 macrophage polarization to M2 phenotype via inhibition of NF-κB signaling in primary cells. Cells were incubated with various dose of 11,12-EET in the absence or presence of 1 μg/ml LPS for 12 h. Cell viability was detected by CCK8 assay (A). Representative immunoblots and quantitation of IκBα (B), NF-κB p65 and c-Rel nuclear translocation (C). D: Representative immunoblots and quantitation of M1 (CD11c and iNOS) and M2 (CD206 and Arg-1) markers. E: The mRNA levels of pro-inflammatory M1 markers (TNFα, IL-1 β, IL-6, MCP-1, CD11c, iNOS) and anti-inflammatory M2 markers (CD206, Arg-1, IL-10) were measured by quantitative real-time PCR after LPS stimulation for 3 h. The mRNA signal for each gene was normalized over GADPH signal. F: TNFα, IL-1β, IL-6, MCP-1, and IL-10 were assessed by ELISA after LPS stimulation for 12 h. Data are shown as mean ±SEM from three independent experiments. *P< 0.05 versus control; #P<0.05 versus DMSO + LPS; **P< 0.05 versus DMSO + LPS + 11,12-EET.
To determine directly whether 11,12-EET can induce macrophage M2 activation and suppress LPS-induced M1 activation, primary peritoneal macrophages and RAW 264.7 cells were subjected to different treatments. LPS-treatment activated NF-κB signaling pathway, allowing NF-κB to translocate to the nucleus, leading to M1 polarization. 11,12-EET significantly attenuated most of these effects and retained M2 related molecules, but showed no significant effect on c-Rel nuclear translocation. The protective effects observed with 11,12-EET were blocked by the putative EET receptor antagonist, 14,15-EEZE in primary macrophages (Fig. 2B–D) and RAW264.7 cells (Fig. S2A–C). The addition of 11,12-EET without LPS did not alter the macrophage phenotype, indicating that EETs are capable of skewing macrophages towards an M2 phenotype but do not influence the macrophages polarization under quiescent conditions.
11,12-EET regulates LPS-induced cytokines at transcription and post-transcription level
We assessed the effects of EET on LPS-induced cytokines mRNA levels and expression of related genes in macrophages (Figs. 2E, F and S2D, E). 11,12-EET pre-incubation attenuated the LPS-induced increase in the expression of CD11c, iNOS, IL-1β, TNF-α, IL-6, and MCP-1, and restored the expression levels of IL-10, CD206 and Arg-1. 14,15-EEZE significantly inhibited these effects of 11,12-EET. Transcription of mRNA is not the sole determinant of steady-state mRNA levels, but contributes to a balance of the relative rates of mRNA transcription and degradation; hence stability of mRNA represents an important factor in controlling mRNA abundance. The mRNA stability of IL-1β, TNF-α, IL-6, and MCP-1 in the presence of 11,12-EET were significantly reduced when compared to those treated with LPS alone, while IL-10 mRNA stability was maintained (Fig. S2F). These data suggested an important role of 11,12-EET on various cytokines mRNA stability and release.
11,12-EET mediated M1 to M2 phenotype polarization confers anti-inflammatory action via PPARα/γ activation
It has been previously documented that PPARα/γ are effectors of EET actions (Liu et al., 2004; Ng et al., 2007), therefore we investigated whether the macrophage polarization effect of EET was mediated by PPARα/γ. We found that 11,12-EET significantly up-regulated the expression of PPARα/γ and restored the protein levels decreased by LPS (Fig. 3A). Furthermore, pre-treatment with 11,12-EET markedly inhibited LPS-induced M1 macrophage activation and NF-κB activation, which could be significantly suppressed by addition of GW9662 (a selective PPARγ inhibitor) and GW6471 (a selective PPARα inhibitor) in primary cells (Fig. 3B–D) and RAW 264.7 cells (Fig. S3A–C).
Fig 3.
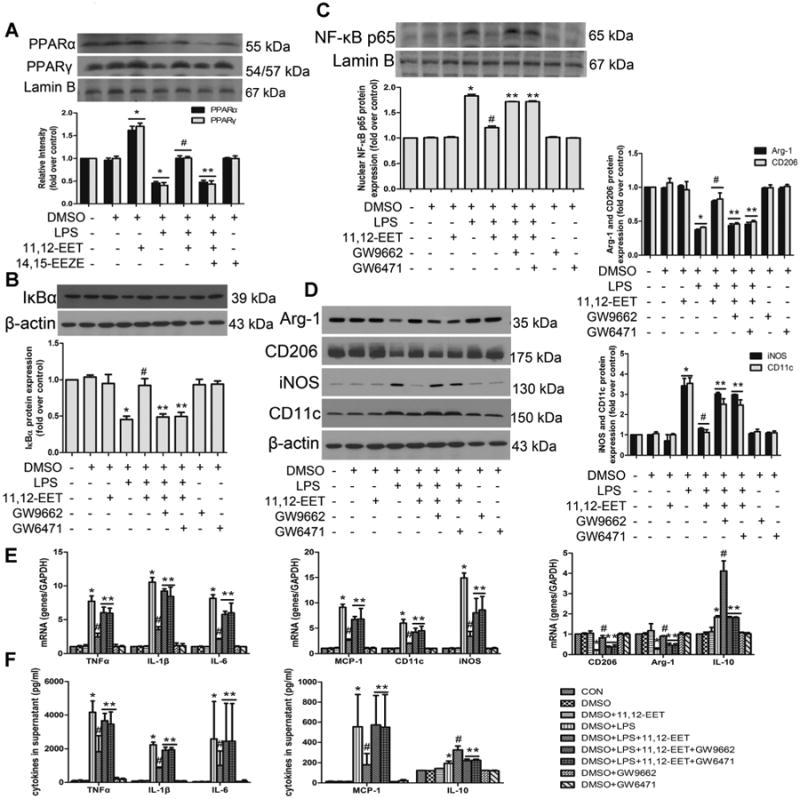
11,12-EET mediated M1 to M2 phenotype polarization and anti-inflammatory effects are mediated by PPARα/β activation in primary cells. A: Representative immunoblots and quantitation of peroxisome proliferator-activated receptor α and γ (PPARα and PPARγ) expression in nuclear following 11,12-EET treatment in the presence or absence of LPS in primary cells. Representative immunoblots and quantitation of NF-κB signaling pathway (B, C) and M1, M2 markers (D). Administration of PPAR antagonists (GW9662/GW6471) significantly abolished EET effects on LPS-induced M1 macrophage polarization. E: Relative mRNA expression of inflammatory genes (TNFα, IL-1β, IL-6, MCP-1, IL-10) or phenotype molecules (CD11c, iNOS, CD206, Arg-1) were determined after LPS stimulation for 3 h. F: TNFα, IL-1β, IL-6, MCP-1, and IL-10 were assessed by ELISA after LPS stimulation for 12h. Data are shown as mean ±SEM from three independent experiments. *P< 0.05 versus control; #P<0.05 versus DMSO + LPS; **P< 0.05 versus DMSO + LPS + 11,12-EET.
We further assessed the role of PPARα/γ on LPS-induced inflammatory cytokine expression in macrophages by quantitative real-time PCR and ELISA (Figs. 3E, F and S3D, E). 11,12-EET treatment reduced the expression of M1 marker CD11c, iNOS, and inflammatory cytokines IL-1β, TNF-α, IL-6 and MCP-1 in LPS-induced macrophages, and up-regulated M2 marker CD206, Arg-1, and the anti-inflammatory cytokine IL-10. Pre-incubation with either GW9662 or GW6471 significantly inhibited these effects.
11,12-EET mediated M1 to M2 phenotype shift is associated with HO-1 expression
We examined the effects of EETs on HO-1 expression in RAW 264.7 macrophages. HO-1 expression was increased in a dose-dependent manner by EET-treatment (Fig. S4A). In addition, EET-mediated rescue of HO-1 protein expression during LPS-treatment was significantly attenuated by GW9662 or GW6471 co-treatment (Fig. S4B). To further delineate the role of HO-1 in regulating macrophage polarization and inflammation, we downregulated HO-1 expression by HO-1 siRNA. All three commercially available siRNAs decreased HO-1 expression levels (Fig. S4C). HO-1 siRNA3 was the most effective in inhibiting HO-1 protein expression and was used for the silencing of HO-1. Both HO-1 siRNA and HO-1 inhibitor ZnPP significantly attenuated EET-induced M1 to M2 polarization and the anti-inflammatory effects (Fig. 4A–F). These results indicated that the macrophage polarization effect of EET was partially mediated by HO-1 activation. HO-1 activator hemin and its metabolic by-products, bilirubin or CORM significantly attenuated LPS-induced NF-κB signaling pathway, and hemin regulated macrophage polarization while bilirubin and CORM not (Fig. S4D, E), suggesting that different mechanisms may involve in the regulation of inflammation.
Fig 4.
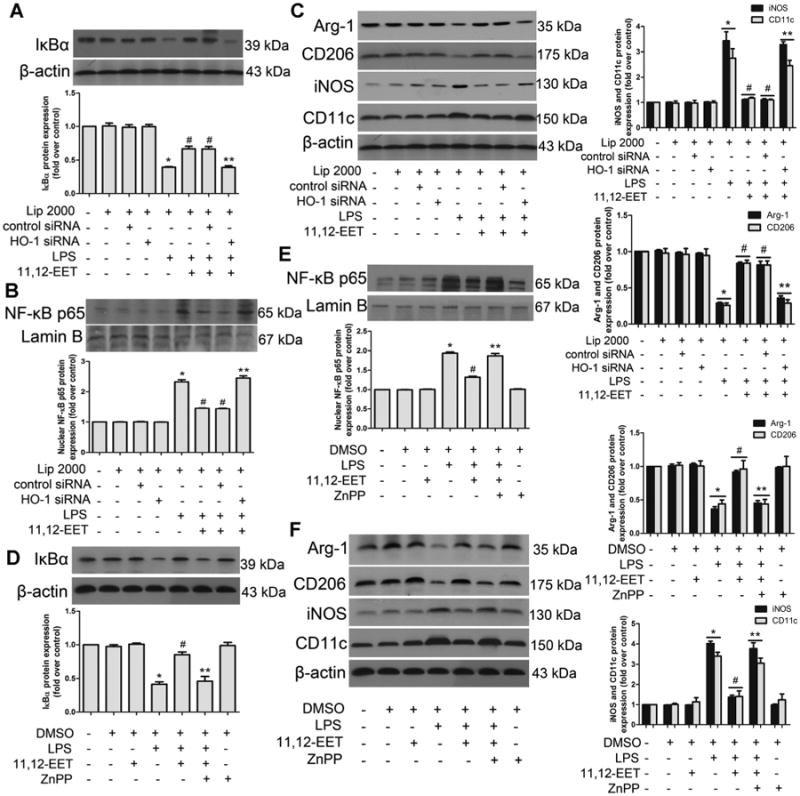
11,12-EET mediated M1 to M2 phenotype polarization and anti-inflammatory effects are mediated by HO-1 activation. A–F: Representative immunoblots and quantitation of IκBα, p65 nuclear translocation, M1 and M2 markers. The EET effects on LPS-induced M1 to M2 polarization were abolished by pre-treatment with heme oxygenase (HO-1) specific siRNA (A–C) and zinc protoporphyrin IX (ZnPP) (D–F). Data are shown as mean ± SEM from three independent experiments. *P <0.05 versus control; #P< 0.05 versus DMSO + LPS or Lip2000 + LPS, **P < 0.05 versus DMSO + LPS +11,12-EET or Lip2000 + LPS +11,12-EET.
11,12-EET inhibits macrophages migration via PPARα/γ and HO-1 activation
Tissue infiltration of macrophages is thought to be a major factor in many inflammatory diseases. Using a Transwell system, we assessed the effect of 11,12-EET on LPS-induced macrophage migration. LPS-treatment alone prompted RAW 264.7 cells to migrate, and 11,12-EET inhibited the chemotactic effect of LPS (Fig. 5). Pre-treated cells with 14,15-EEZE, GW9662, GW6471, or ZnPP significantly blocked the effect of 11,12-EET. In the absence of LPS-treatment, macrophage migration was not significantly altered by any of the compounds.
Fig 5.
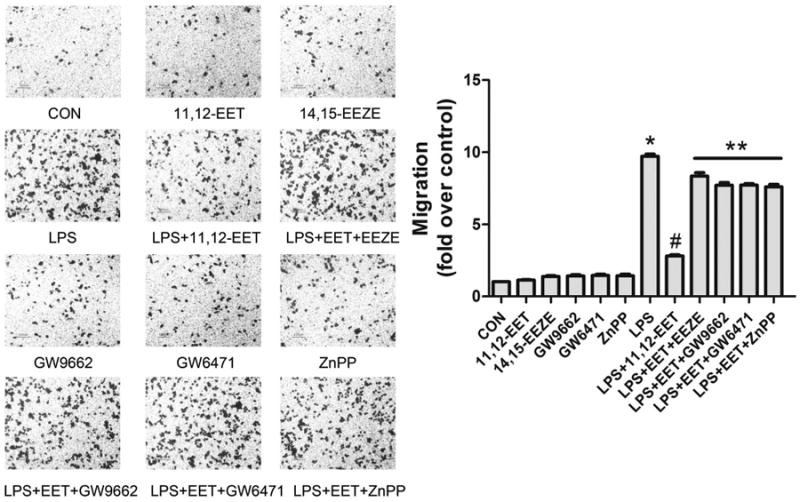
11,12-EET suppresses LPS-induced macrophage migration via PPARα/γ and HO-1 activation. Representative pictures of RAW 264.7 macrophages migration with indicated treatment and calculation of migrated cell number. Data are shown as mean ± SEM from three independent experiments. *P<0.05 versus control; #P <0.05 versus DMSO + LPS; **P< 0.01 versus DMSO + LPS+ 11,12-EET.
CYP2J2 protects mice from LPS-induced cardiac dysfunction via inhibition of inflammation and infiltration of M1 macrophages
Four weeks after rAAV-CYP2J2 virus injection, CYP2J2 expression in the heart tissue was abundant as evaluated by western blot (Fig. 6F). Our previous studies (Zhao et al., 2012b; Cai et al., 2013) indicated that rAAV-mediated CYP2J2 delivery led to abundant heart CYP2J2 expression and increased both cardiac and systemic EETs levels. Echocardiography and hemodynamic assessments showed that LPS administration markedly deteriorated myocardial contractile function evidenced by reduction of the percentage of left ventricular fractional shortening, ejection fraction, increased LVID(S) (left ventricular internal dimension in systole) and impaired LV hemodynamics as assessed via load-dependent (dp/dtmax, dp/dtmin). RAAV-mediated CYP2J2 expression showed significant improvement of cardiac function and parameters as showed in Table 2. In the LPS-induced mice sepsis, CYP2J2 significantly increased the survival rate of mice from 29.22% to 60.11% (Fig. 6A). CYP2J2 also dramatically suppressed the levels of pro-inflammatory cytokines TNFα, IL-1β, IL-6, MCP-1 and up-regulated the anti-inflammatory cytokine IL-10 in serum and heart tissue (Fig. 6B, C). LPS-induced mRNA levels TNFα, IL-1β, IL-6, MCP-1 of heart tissue were significantly attenuated in CYP2J2 expression mice, while IL-10 mRNA level was increased (Fig. 6D). Hematoxylin/eosin analysis showed that CYP2J2 ameliorated the heart lesion of sepsis mice with descent of tissue damage and reduced inflammatory infiltration. Immunohistochemical assay of F4/80 and CD11c revealed a decreasing of infiltrated M1 positive macrophages in heart sections (Fig. 6E). Western blots showed that CYP2J2 significantly increased the expression of PPARα/γ and HO-1 levels and reduced the NF-κB activation compared to LPS-induced cardiac dysfunction mice in the heart tissue (Fig. 6F–H). Further research found that conditioned medium (CM) from LPS-treated macrophages upregulated the expression of α-SMA in cardiac fibroblasts, but addition of 11,12-EET significantly reversed this effect. CYP2J2 overexpression also protected angiotensin II (Ang-II) induced mice from cardiac injury via PPARα/β and HO-1 activation (Fig. S5). These results indicated that CYP2J2/EETs play important role in inflammatory diseases.
Fig 6.
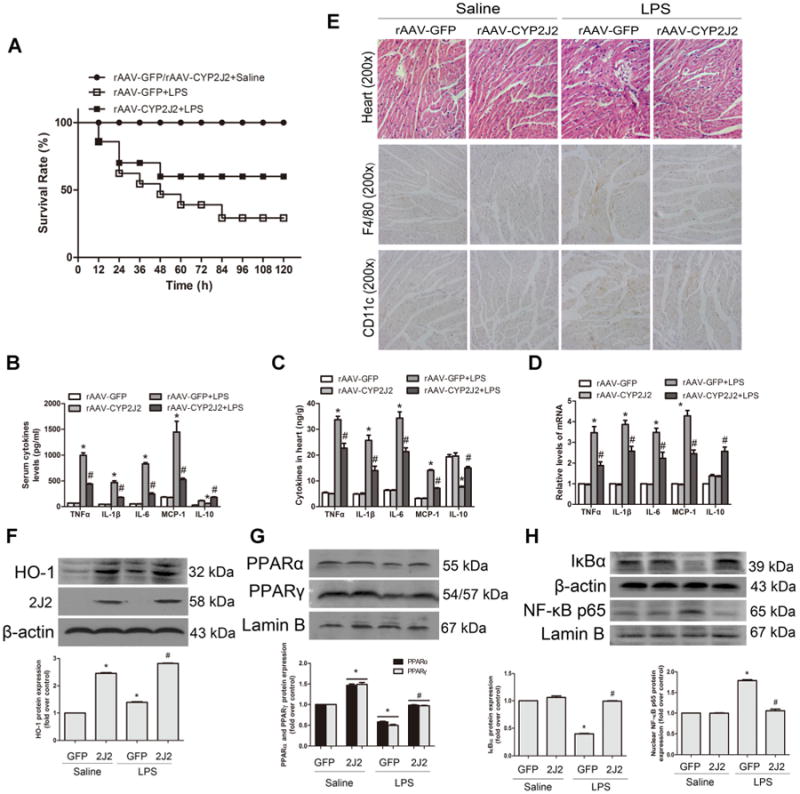
CYP2J2/EETs attenuate LPS-induced cardiac dysfunction via inhibition of inflammation and infiltration of macrophage through PPARα/γ and HO-1 activation. RAAV-CYP2J2 and rAAV-GFP virus were injected via tail veins 4 weeks before LPS challenge. (A) Survival rate was observed for 5 days and calculated after LPS (20 mg/kg) administration (n = 15/group). Mice treated as shown in other illustrations were 6 h after LPS (10 mg/kg) treatment (n = 8/group). Serum (B) and heart (C) cytokines levels of TNFα, IL-1 β, IL-6, MCP-1 and IL-10 were measured by ELISA. D: Total RNA was isolated from heart and assessed by quantitative real-time PCR. E: Hematoxylin/eosin and immunohistochemical staining of F4/80 and CD11c in heart sections were performed (original magnification, 200×). Representative pictures of immunohistochemical detection of positive cells are shown. F–H: Representative immunoblots and quantitation of total heart protein expression of HO-1, CYP2J2, IκBα, and nuclear protein levels of PPARα, PPARγ and NF-κB p65 were shown. Data are shown as mean ±SEM; *P<0.05 versus rAAV-GFP + Saline; #P<0.05 versus rAAV-GFP + LPS.
Table 2. Hemodynamic and echocardiography results.
| Saline | LPS | |||
|---|---|---|---|---|
|
|
|
|||
| rAAV-GFP | rAAV-CYP2J2 | rAAV-GFP | rAAV-CYP2J2 | |
| Hemodynamic | ||||
| HR (bmp) | 416 ± 27 | 421 ± 30 | 405 ± 29 | 411 ± 28 |
| dp/dt max (mmHg/sec) | 8347 ± 277 | 8470 ± 447 | 2467 ± 372* | 5437 ± 399# |
| dp/dt min (mmHg/sec) | 5398 ± 352 | 5527 ± 446 | 1949 ± 214* | 4081 ± 403# |
| Echocardiography | ||||
| Ejection fraction (%) | 78.6 ± 0.9 | 79.3 ± 0.5 | 46.6 ± 0.6* | 75.9 ± 1.0# |
| Fraction shortening (%) | 43.4 ± 0.8 | 44.4 ± 0.7 | 29.3 ± 0.6* | 38.3 ± 0.9# |
| LVID(d) (mm) | 3.69 ± 0.04 | 3.69 ± 0.02 | 3.61 ± 0.03 | 3.65 ± 0.01 |
| LVID(S) (mm) | 2.72 ± 0.05 | 2.68 ± 0.02 | 3.49 ± 0.06* | 2.83 ± 0.04# |
LPS, lipopolysaccharide; dp/dt max, maximal slope of systolic pressure increment; dp/dt min, minimal slope of diastolic pressure decrement; LVID(d), left ventricular internal dimension in diastole; LVID(s), left ventricular internal dimension in systole; LVEDP, left ventricular end diastolic pressure; HR, heart rate; rAAV-GFP, rAAV-CYP2J2, recombinant adeno-associated virus mediated GFP and CYP2J2 gene delivery mice. (Data are shown as mean ± SEM; n = 8,
P<0.05 vs. rAAV-GFP ± Saline,
P<0.05 vs. rAAV-GFP ± LPS).
Discussion
The current study investigated the effects of EETs treatment on macrophage polarization and rAAV-mediated CYP2J2 expression on LPS-induced cardiac dysfunction. Herein we showed that EETs significantly inhibited M1 macrophage polarization and diminished the proinflammatory cytokines at transcriptional and post-transcriptional level, meanwhile it retained M2 macrophage related molecules expression invitro. Moreover, in vivo study showed that CYP2J2 significantly protected mice from cardiac dysfunction induced by LPS administration, which were mediated by inhibition of inflammation and M1 macrophage infiltration in the heart. The mechanisms were attributed to EETs's down-regulation on NF-κB activation and up-regulation on PPARα/γ and HO-1, which can be the regulatory molecules for M1 and M2 polarization (Fig. 7).
Fig 7.
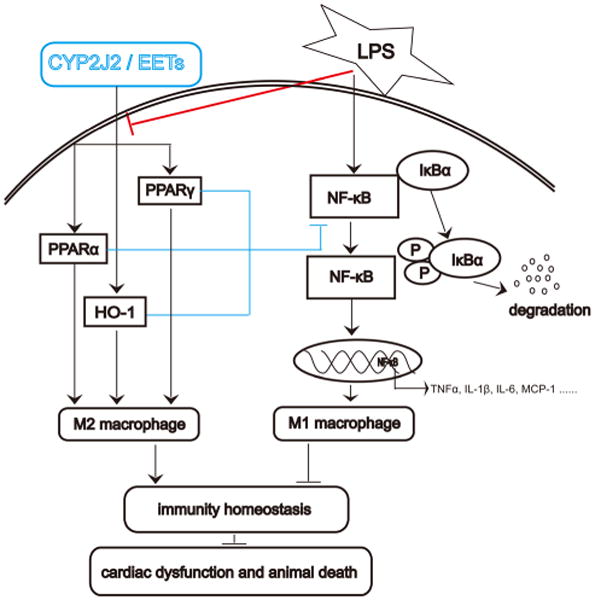
Schematic of mechanisms on CYP2J2/EETs mediated macrophage polarization in response to LPS-induced inflammation and cardiac dysfunction. A stop (┴) indicates inhibitory or blocking effects, and an arrow (↓) indicates increased or enhanced effects. LPS-treatment induced M1 macrophage polarization via NF-κB activation. Phosphorylation of IκB resulted in its dissociation from the transcription factor NF-κB, allowing NF-κB to translocate to the nucleus and participate in transcription of genes involved in inflammation. Ultimately, LPS-treatment induced cardiac dysfunction and animal death. CYP2J2 overexpression or CYP2J2 derived EETs regulates M2 macrophage polarization via up-regulation on PPARα/γ and HO-1, down-regulation on NF-κB signaling pathway, which maintains immunity homeostasis and improves cardiac dysfunction and survival in sepsis mice.
Several studies have shown that CYP2J2 in the cardiovascular system produces EETs that reduce inflammation both in vivo and in vitro (Deng et al., 2011; Sanders et al., 2012; Cai et al., 2013). Whether EETs regulate macrophages polarization and regulatory pathways that direct macrophages into distinct functional subsets need further study. Macrophages were mainly characterized by highly diversity and plasticity, and functional phenotypes of macrophages involved in various pathological processes. In chronic inflammation, macrophages undergo M1 polarization and contribute to tissue injury. Class A scavenger receptor (SRA) deficient mice have a reduction in M2 macrophages and an increase in M1 macrophages, which results in exacerbation of cardiac dysfunction and fibrosis following myocardial infarction (Deng et al., 2011). Similarly, roles for inflammatory macrophage recruitment have been proposed for hypertensive cardiac remodeling (Kai et al., 2009), atherosclerosis (Woollard and Geissmann, 2010), and several other myocarditis diseases model (Barin et al., 2012). Moreover, M1-polarized macrophage was one of the major cells causing the aberrant cytokines pattern in sepsis mice, the transfer of M2-BMDMs caused a prolonged survival of mice with sepsis (Feng et al., 2014). Accumulating studies suggested macrophage as a key mediator in sepsis development. Whereas, the LPS-induced mortality, systemic hypotension, and histologically evaluated tissue injury were substantially diminished by administration of urea-based, small-molecule inhibitors of sEH to C57BL/6 mice (Schmelzer et al., 2005), but further studies clarifying the precise molecular mechanisms are needed. Meanwhile, the interaction among immune cells, cardiomyocytes and cardiac fibroblasts indicate that all these cells would be receptive to reciprocal paracrine signaling, pointing to a potentially important role for macrophages in cardiac homeostasis (Fujiu and Nagai, 2013). Previous research and our previous data demonstrated that cardiomyocyte-specific overexpression of CYP2J2 or rAAV-mediated CYP2J2 delivery led to abundant heart CYP2J2 expression and increased both cardiac and systemic EETs levels, endogenous and exogenous EETs showed significant protective effects in cardiovascular and renal systems (Xiao et al., 2010; Zhao et al., 2012a; Cai et al., 2013; Chaudhary et al., 2013). In order to assess the bioactivity of EETs in vivo, we established LPS-induced cardiac dysfunction in mice. Overexpression of CYP2J2 significantly attenuated LPS-induced cardiac dysfunction, inhibited inflammation and reduced M1 macrophage infiltration in the heart. Another in vivo experiment showed an increase of M1 macrophages infiltrated in chronic inflammatory heart induced by Ang-II infusion; cardiac-specific CYP2J2 transgenic mice significantly alleviated these effects. Furthermore, 11,12-EET effectively inhibited macrophage migration induced by LPS in vitro. Both the LPS-induced acute and Ang-II-induced chronic inflammation were attenuated by EETs in vivo via PPARα/γ and HO-1 activation, which consistent with our in vitro experiments, indicating that EETs play an important role in macrophage polarization and macrophage-mediated cardiac inflammatory diseases.
A major locus of control for macrophage phenotype polarization occurs at the transcriptional level: STAT-1, IRF-5, and NF-κB regulate M1 polarization, whereas STAT-6, IRF-4, and PPARγ regulate M2 macrophage polarization (Tugal et al., 2013). Among these transcription factors, we focused on the PPAR nuclear receptors that are highly expressed in macrophages (Chawla, 2010). M1 macrophages produce pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, which are mediators of the development of inflammatory diseases. Additionally, the pro-inflammatory chemokine MCP-1 is a potent chemoattractant capable of promoting additional monocyte recruitment to a site of inflammation (Biswas et al., 2012). Our results demonstrated that 11,12-EET regulated inflammatory cytokines at transcriptional and post-transcriptional levels, and showed obvious anti-inflammatory effect, indicating its potential application prospect in preventing and delaying the progression of inflammatory diseases.
Previous studies have demonstrated that the anti-inflammatory properties of EETs requires either direct or indirect activation of PPARα/γ (Liu et al., 2004; Liu et al., 2005; Wray et al., 2009; Bystrom et al., 2011). A study by Liu et al. (2005) showed that PPARγ inhibition abolished the EET-mediated anti-inflammatory effect in endothelial cells subjected to laminar flow. Other studies have demonstrated that activation of PPARγ signaling has a protective role for tissue remodeling and wound healing by reducing oxidative stress and inflammation (Zhao et al., 2012a; Cai et al., 2013). Indeed, local administration of rosiglitazone increased the expression of M2-specific markers such as ARG1, resistin-like β, and IL-10 (Hasegawa-Moriyama et al., 2012). Numerous studies have demonstrated that PPARγ activation has anti-inflammatory effects, which can regulate macrophage polarization (Bouhlel et al., 2007; Hasegawa-Moriyama et al., 2012). In contrast, there is limited evidence linking PPARα and macrophage polarization. PPARα is expressed in the vascular endothelium, vascular smooth muscle cells, and monocyte/macrophages where, like EETs, it can limit NF-κB activation (Bystrom et al., 2011). PPARα-deficient monocyte/macrophages exist in inflammatory states producing large amounts of thrombospondin-1 and inhibiting tumor growth (Kaipainen et al., 2007). Bystrom et al. (2011) showed that an epoxygenase-EET-PPARα axis provides a critical negative feedback loop in classical M1 activation. Consistent with these findings, we found that 11,12-EET significantly prevented the loss of PPARα/γ expression following LPS treatment. Furthermore, both the PPARγ inhibitor GW9662 and PPARα inhibitor GW6471 significantly attenuated the 11,12-EET effects, indicating that the macrophage polarization effect of EET was partially mediated by of PPARα/γ activation.
HO-1 catalyzes the first and rate-limiting step in the metabolism of free heme into ferrous iron, carbon monoxide (CO), and biliverdin. Numerous studies have demonstrated the importance of HO-1 as a cytoprotective defense mechanism against oxidative insults through the antioxidant activity of biliverdin and its metabolite, bilirubin, as well as the anti-inflammatory actions of CO (Hu et al., 2004; Sacerdoti et al., 2007; Elmarakby et al., 2013). EETs and HO-1 share many anti-inflammatory properties and direct links between EETs and HO-1 have recently been investigated. In vivo, EET agonist reduced vascular dysfunction and inflammation via induction of HO-1 in high fat diet fed rats (Sodhi et al., 2012). A recent study also showed that HO-1 induction was involved in the protective effect of sEH inhibition against diabetic renal injury as HO-1 had antioxidant and anti-inflammatory properties, and EETs induced up-regulation of HO-1 via a HIF-1α dependent mechanism (Elmarakby et al., 2013). HO-1 induction mediated by multiple pathways can drive the phenotypic shift to M2 macrophages and suggests that HO-1 induction in macrophages is a potential therapeutic approach to immunomodulation in widely diverse diseases (Naito et al., 2014). In the current study, 11,12-EET increased HO-1 expression in a concentration-dependent manner in macrophages. Pretreatment of cells with ZnPP or HO-1 siRNA significantly abolished EET-mediated M1 to M2 macrophage polarization and anti-inflammatory effects. Hemin (HO-1 activator), CORM and bilirubin were used to compare the effect of EET on macrophage polarization. All of them significantly attenuated LPS-induced NF-κB signaling pathway, but had no significant effect on macrophage polarization except hemin, which consistent with previous studies, indicating that HO-1 and its metabolites may show protective effect through different mechanisms (Hu et al., 2004; Jadhav et al., 2013; Dong et al., 2014; Naito et al., 2014). Further studies will need to be completed to determine the exact mechanisms involved in this process.
In summary, our results demonstrate that EETs significantly diminished the M1 polarization and preserved M2 polarization. RAAV-mediated CYP2J2 expression significantly attenuated LPS-induced cardiac dysfunction via suppressing inflammation and the M1 macrophage infiltration. The macrophage polarization effect of CYP2J2 and its metabolites EETs are possibly mediated by PPARα/γ and HO-1 activation. Our data provide evidences that CYP2J2/EETs have potential for the treatment of macrophage-mediated inflammation, such as sepsis and cardiac remodeling via blocking M1-polarized macrophage and aiding M2-polarized macrophage.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 31130031, 81400369 and 81471021) and National Basic Research Program of China (Nos. 2012CB518004 and 2012CB517801) and supported by Program for Changjiang Scholars and Innovative Research Team in University IRT_14R20.
Abbreviations
- EETs
epoxyeicosatrienoic acids
- 14,15-EEZE
14,15-epoxyeicosa-5(Z)-enoic acid
- LPS
lipopolysaccharide
- Ang-II
angiotensin II
- M1
classically activated macrophage
- M2
alternatively activated macrophage
- rAAV
recombinant adeno-associated virus
- PPAR
peroxisome proliferator-activated receptor
- HO
heme oxygenase
- ZnPP
zinc protoporphyrin IX
- CORM
carbon monoxide-releasing molecule
Footnotes
Supporting Information: Additional supporting information may be found in the online version of this article at the publisher's web-site.
The authors have no financial conflicts of interest.
Literature Cited
- Alcaraz MJ, Fernandez P, Guillen MI. Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr Pharma Des. 2003;9:2541–2551. doi: 10.2174/1381612033453749. [DOI] [PubMed] [Google Scholar]
- Barin JG, Rose NR, Cihakova D. Macrophage diversity in cardiac inflammation: A review. Immunobiology. 2012;217:468–475. doi: 10.1016/j.imbio.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res. 2012;53:11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Bystrom J, Wray JA, Sugden MC, Holness MJ, Swales KE, Warner TD, Edin ML, Zeldin DC, Gilroy DW, Bishop-Bailey D. Endogenous epoxygenases are modulators of monocyte/macrophage activity. PLoS ONE. 2011;6:e26591. doi: 10.1371/journal.pone.0026591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zhao G, Yan J, Liu W, Feng W, Ma B, Yang L, Wang JA, Tu L, Wang DW. CYP2J2 overexpression increases EETs and protects against angiotensin II-induced abdominal aortic aneurysm in mice. J Lipid Res. 2013;54:1448–1456. doi: 10.1194/jlr.M036533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary KR, Zordoky BN, Edin ML, Alsaleh N, El-Kadi AO, Zeldin DC, Seubert JM. Differential effects of soluble epoxide hydrolase inhibition and CYP2J2 overexpression on postischemic cardiac functionin aged mice. Prostaglandins Other Lipid Mediat. 2013:104–105. 8–17. doi: 10.1016/j.prostaglandins.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559–1569. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquerel D, Neviere R, Delile E, Mulder P, Marechal X, Montaigne D, Renet S, Remy-Jouet I, Gomez E, Henry JP, do Rego JC, Richard V, Tamion F. Gene deletion of protein tyrosine phosphatase 1B protects against sepsis-induced cardiovascular dysfunction and mortality. Arterioscler Thromb Vasc Biol. 2014;34:1032–1044. doi: 10.1161/ATVBAHA.114.303450. [DOI] [PubMed] [Google Scholar]
- Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, DeGraff LM, Lih FB, Foley J, Bradbury JA, Graves JP, Tomer KB, Falck JR, Zeldin DC, Lee CR. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J. 2011;25:703–713. doi: 10.1096/fj.10-171488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Huang H, Yun X, Kim DS, Yue Y, Wu H, Sutter A, Chavin KD, Otterbein LE, Adams DB, Kim YB, Wang H. Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology. 2014;155:818–828. doi: 10.1210/en.2013-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmarakby AA, Faulkner J, Pye C, Rouch K, Alhashim A, Maddipati KR, Baban B. Role of haem oxygenase in the renoprotective effects of soluble epoxide hydrolase inhibition in diabetic spontaneously hypertensive rats. Clin Sci. 2013;125:349–359. doi: 10.1042/CS20130003. [DOI] [PubMed] [Google Scholar]
- Feng L, Song P, Zhou H, Li A, Ma Y, Zhang X, Liu H, Xu G, Zhou Y, Wu X, Shen Y, Sun Y, Wu X, Xu Q. Pentamethoxyflavanone regulates macrophage polarization and ameliorates sepsis in mice. Biochem Pharmacol. 2014;89:109–118. doi: 10.1016/j.bcp.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Fujiu K, Nagai R. Contributions of cardiomyocyte-cardiac fibroblast-immune cell interactions in heart failure development. Basic Res Cardiol. 2013;108:357. doi: 10.1007/s00395-013-0357-x. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Hasegawa-Moriyama M, Ohnou T, Godai K, Kurimoto T, Nakama M, Kanmura Y. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates postincisional pain by regulating macrophage polarization. Biochem Biophy Res Commun. 2012;426:76–82. doi: 10.1016/j.bbrc.2012.08.039. [DOI] [PubMed] [Google Scholar]
- Hu CM, Chen YH, Chiang MT, Chau LY. Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation. 2004;110:309–316. doi: 10.1161/01.CIR.0000135475.35758.23. [DOI] [PubMed] [Google Scholar]
- Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav A, Tiwari S, Lee P, Ndisang JF. The heme oxygenase system selectively enhances the anti-inflammatory macrophage-M2 phenotype, reduces pericardial adiposity, and ameliorated cardiac injury in diabetic cardiomyopathy in Zucker diabetic fatty rats. J Pharmacol Exp Ther. 2013;345:239–249. doi: 10.1124/jpet.112.200808. [DOI] [PubMed] [Google Scholar]
- Kai H, Kudo H, Takayama N, Yasuoka S, Kajimoto H, Imaizumi T. Large blood pressure variability and hypertensive cardiac remodeling – role of cardiac inflammation. Circ J. 2009;73:2198–2203. doi: 10.1253/circj.cj-09-0741. [DOI] [PubMed] [Google Scholar]
- Kaipainen A, Kieran MW, Huang S, Butterfield C, Bielenberg D, Mostoslavsky G, Mulligan R, Folkman J, Panigrahy D. PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS ONE. 2007;2:e260. doi: 10.1371/journal.pone.0000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: The role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102:16747–16752. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhu Y, Rannou F, Lee TS, Formentin K, Zeng L, Yuan X, Wang N, Chien S, Forman BM, Shyy JY. Laminar flow activates peroxisome proliferator-activated receptor-gamma in vascular endothelial cells. Circulation. 2004;110:1128–1133. doi: 10.1161/01.CIR.0000139850.08365.EC. [DOI] [PubMed] [Google Scholar]
- Ma H, Gong H, Chen Z, Liang Y, Yuan J, Zhang G, Wu J, Ye Y, Yang C, Nakai A, Komuro I, Ge J, Zou Y. Association of Stat3 with HSF1 plays a critical role in G-CSF-induced cardio-protection against ischemia/reperfusion injury. J Mol Cell Cardiol. 2012;52:1282–1290. doi: 10.1016/j.yjmcc.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: An immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. 2014;564C:83–88. doi: 10.1016/j.abb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nitto T, Inoue T, Node K. Expression of the cytochrome P450 epoxygenase CYP2J2 in human monocytic leukocytes. Life Sci. 2008;83:339–345. doi: 10.1016/j.lfs.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab Dispos. 2007;35:1126–1134. doi: 10.1124/dmd.106.013839. [DOI] [PubMed] [Google Scholar]
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plutzky J. Medicine. PPARs as therapeutic targets: Reverse cardiology? Science. 2003;302:406–407. doi: 10.1126/science.1091172. [DOI] [PubMed] [Google Scholar]
- Ranganathan PV, Jayakumar C, Ramesh G. Netrin-1-treated macrophages protect the kidney against ischemia–reperfusion injury and suppress inflammation by inducing M2 polarization. Am J Physiol Renal Physiol. 2013;304:F948–957. doi: 10.1152/ajprenal.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacerdoti D, Colombrita C, Di Pascoli M, Schwartzman ML, Bolognesi M, Falck JR, Gatta A, Abraham NG. 11,12-epoxyeicosatrienoic acid stimulates heme-oxygenase-1 in endothelial cells. Prostaglandins Other Lipid Mediat. 2007;82:155–161. doi: 10.1016/j.prostaglandins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Sanders WG, Morisseau C, Hammock BD, Cheung AK, Terry CM. Soluble epoxide hydrolase expression in a porcine model of arteriovenous graft stenosis and anti-inflammatory effects of a soluble epoxide hydrolase inhibitor. Am J Physiol Cell Physiol. 2012;303:C278–C290. doi: 10.1152/ajpcell.00386.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioura KM, Geenen DL, Goldspink PH. Assessment of cardiac function with the pressure–volume conductance system following myocardial infarction in mice. Am J Physiol Heart Circul Physiol. 2007;293:H2870–2877. doi: 10.1152/ajpheart.00585.2007. [DOI] [PubMed] [Google Scholar]
- Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG. EET agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in Bach 1 and an increase in HO-1 levels. Prostaglandins Other Lipid Mediat. 2012;98:133–142. doi: 10.1016/j.prostaglandins.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Tang L, Tan YX, Jiang BG, Pan YF, Li SX, Yang GZ, Wang M, Wang Q, Zhang J, Zhou WP, Dong LW, Wang HY. The prognostic significance and therapeutic potential of hedgehog signaling in intrahepatic cholangiocellular carcinoma. Clin Cancer Res. 2013;19:2014–2024. doi: 10.1158/1078-0432.CCR-12-0349. [DOI] [PubMed] [Google Scholar]
- Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. 2013;33:1135–1144. doi: 10.1161/ATVBAHA.113.301453. [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ni L, Yang L, Duan Q, Chen C, Edin ML, Zeldin DC, Wang DW. CYP2J2-derived epoxyeicosatrienoic acids suppress endoplasmic reticulum stress in heart failure. Mol Pharmacol. 2014;85:105–115. doi: 10.1124/mol.113.087122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther. 2005;314:522–532. doi: 10.1124/jpet.105.083477. [DOI] [PubMed] [Google Scholar]
- Woollard KJ, Geissmann F. Monocytes in atherosclerosis: Subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray JA, Sugden MC, Zeldin DC, Greenwood GK, Samsuddin S, Miller-Degraff L, Bradbury JA, Holness MJ, Warner TD, Bishop-Bailey D. The epoxygenases CYP2J2 activates the nuclear receptor PPARalpha in vitro and in vivo. PLoS ONE. 2009;4:e7421. doi: 10.1371/journal.pone.0007421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Li X, Yan J, Yu X, Yang G, Xiao X, Voltz JW, Zeldin DC, Wang DW. Overexpression of cytochrome P450 epoxygenases prevents development of hypertension in spontaneously hypertensive rats by enhancing atrial natriuretic peptide. J Pharmacol Exp Ther. 2010;334:784–794. doi: 10.1124/jpet.110.167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63:597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Zhao G, Tu L, Li X, Yang S, Chen C, Xu X, Wang P, Wang DW. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum Gene Ther. 2012a;23:688–699. doi: 10.1089/hum.2011.135. [DOI] [PubMed] [Google Scholar]
- Zhao G, Wang J, Xu X, Jing Y, Tu L, Li X, Chen C, Cianflone K, Wang P, Dackor RT, Zeldin DC, Wang DW. Epoxyeicosatrienoic acids protect rat hearts against tumor necrosis factor-alpha-induced injury. J Lipid Res. 2012b;53:456–466. doi: 10.1194/jlr.M017319. [DOI] [PMC free article] [PubMed] [Google Scholar]