

Abstract
Benzoxazolone and benzothiazolone were used as template blocks to develop two series of dimers as anti-inflammatory and analgesic agents based on the concept of bivalent ligands. The first series (I) involved varying the carbon chain lengths extending from the piperazine core to the nitrogen atom of the dibenzo[d]oxazol-2(3H)-one or dibenzo[d]thiazol-2(3H)-one. The second series (II) was designed by changing the attachment point. All compounds were screened for their in vitro anti-inflammatory activity in terms of the inhibition of inducible nitric oxide synthase (iNOS) and nuclear factor kappaB (NF-κB). Seventeen compounds inhibited both targets. Eleven of them exhibited IC50 values below 3 μM while five compounds showed IC50 values of 1 μM or below. Most of the compounds were found to be devoid of cytotoxicity against mammalian kidney and solid tumors cell lines up to 25μg/mL. In vivo anti-inflammatory and antinociceptive studies revealed that compounds 3j, 5t and 8b have significant anti-inflammatory and analgesic activity comparable to that of indomethacin and ketorolac, respectively.
Keywords: Anti-inflammatory/analgesic, Nociception, iNOS, NF-κB, benzoxazolone, benzothiazolone, piperazine, dimers
1. Introduction
Inflammation is a multifactorial process caused by various stimuli including physical damage, irradiation, microbial infections and immune reactions.1 There are several molecular targets recognized from the inflammation cascades that could be antagonized to block the output of these cascades.2 The major anti-inflammatory targets include cyclooxygenases (COXs), lipoxygenases (LOXs), nitric oxide synthases (NOS) enzymes, nuclear factor kappa B (NF-κB),3 cytokines, cytokine receptors, tumor necrosis factor (TNF-α), chemokines, interferons (IFNs) and many other targets.4 Over the past few decades, three isoforms of nitric oxide synthase have been identified and found to catalyze the formation of nitric oxide (NO) from arginine.5 Of the three isoforms, inducible nitric oxide synthase (iNOS), which is responsible for the overproduction of NO, has been involved in a number of inflammatory diseases such as rheumatoid arthritis, asthma, inflammatory bowel disease and more recently, in neuropathic pain.6,7 Therefore, selective inhibition of iNOS would be a valid approach and prime target for the reduction of inflammation and alleviation of the associated pain. On the other hand, NF-κB represents a group of structurally related and evolutionarily conserved proteins that are responsible for the regulation of the expression of several genes involved in inflammatory response, including iNOS and dynorphins at the transcriptional level.8,9 In addition, it has been established that NF-kB signaling pathways promote the gene expression of many proinflammatory cytokines as well as tightly controlling COX-2 expression.10 Several studies have indicated the contribution of components of the NF-κB signaling pathways in the pathogenesis of chronic inflammatory diseases especially, rheumatic ones.11 Furthermore, there has been much evidence that a strong relationship exists between the pathological processes of inflammation and cancer.12 NF-kB was found to be the central molecular link between inflammation and carcinogenesis and its activation plays a pivotal role in growth and progression of inflammation-induced cancer.13 Moreover, many proinflammatory cytokines and chemokines in the tumor microenvironment which activate the IKKβ-dependent NF-κB signaling pathway have been demonstrated to be associated with cancer development and progression.14,15 Based on the critical role of the inflammatory microenvironment in progression of several cancer types, it was proposed that targeting the inflammatory components rather than the malignant cell may offer a better therapeutic approach that could prevent the quick mutation and acquired drug resistance.12,16,17 Numerous reports have demonstrated that NF-κB can stimulate cell proliferation, enhance angiogenesis, suppress apoptosis, and promote metastasis via inducing the expression of diverse target genes.18 Thus, it was suggested that the inhibition of NF-κB activation and its upstream signaling pathway may warrant a potential cancer therapy strategy.18 However, several challenges may counteract the using of NF-kB inhibitors in clinical practice. First, the inhibition of NF-kB may impair immune functions since NF-kB activation is important for maintaining both innate and acquired immunity.19 Also, the mechanisms that leads to the activation of IKK-β and NF-kB in both malignant and the inflammatory components of cancer need to be more clearly identified and fully understood.12
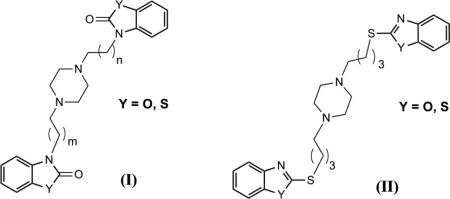
To date, Non-steroidal anti-inflammatory drugs (NSAIDs) continue to be the most commonly utilized medications for inflammation and pain.20 Concomitantly, NSAIDs are known to have several serious adverse effects such as gastric ulceration, renal injury and recently, cardiotoxicity.21 Therefore, there remains a substantial need for researchers to find new compounds devoid of these side-effects. In an attempt to overcome these associated liabilities, we herein, tried to introduce iNOS and NF-κB dual inhibitors as a potential strategy for the development of anti-inflammatory/analgesic agents without serious side effects. Our design of these agents was based on the previously reported compound (I), S-14080, that has anti-inflammatory and antinociceptive activities superior to acetylsalicylic acid and equivalent to glafenine. Compound (I) was found to inhibit the arachidonic acid inflammatory cascade.22 Moreover, scaffold (II) was found to have a significant analgesic activity.23 From the study of the structural features of the aforementioned compounds, benzoxazolonone and benzothiazolonone heterocycles in addition to the piperazine core are important for both anti-inflammatory and analgesic activities In this context, we decided to incorporate these moieties in one template structure (III) utilizing the fragment-based drug design concept (Fig. 1). Consequently, to investigate the effect of carbon linker length between the piperazine core and the heterocyclic moiety on the activity, we synthesized two series of dimers, homodimers and heterodimers, and varied the length of carbon chain from two to six carbons.
Figure 1.

Design strategy for the proposed dimers
2 Results and discussion
2.1. Chemistry
The synthesis of these compounds was facile and straightforward as outlined in Scheme 1. The first series was designed as homodimers with equal length carbon chains around piperazine ring. Commercially available 2(3H)-benzoxazolone and 2(3H)-benzothiazolone were N-alkylated with various dibromoalkanes to give intermediates 2a-j. Then, reaction of two equivalents of 2a-j with one equivalent of piperazine was performed to give the targeted compounds 3a-j that finally separated as hydrochloride salts for biological testing. While the second series was designed as heterodimers where the carbon chains around piperazine ring were different. The series 3a-j will aid in understanding the structure activity relationship for the inhibition of iNOS and NF-κB activities. The synthesis of these compounds is also outlined in Scheme 1. N-alkylation of 2(3H)-benzoxazolone or 2(3H)-benzothiazolone by various dibromoalkanes coupled with an equimolar amount of piperazine afforded intermediates 4a-h. These intermediates were then treated with bromoalkyl benzoxazolones or bromoalkyl benzthiazolones 2c-j to give the desired heterodimers 5a-t. Finally, dihydrochloride salts were formed for biological testing.
Scheme 1.

Reagents and conditions: (a) dibromoalkane, K2CO3, DMF, 60° C, 2 h; (b) piperazine, K2CO3, DMF, 60° C, 1.5 h; (c) piperazine, K2CO3, DMF, 60° C, 0.5 h; (d) K2CO3, DMF, 60° C, 2 h.
To explore the role of the heterocycle attachment position, a series of S-alkyltaed compounds were designed. Scheme 2 outlines the synthesis of the S-alkylated compounds wherein dibromobutane was reacted with the previously thionated 2(3H)-benzoxazolone or 2(3H)-benzothiazolone with Lawesson's reagent to give intermediates 7a-b. The dibromobutane was directed to the more reactive 2-thiocarbonyl. Subsequently, intermediates 7a-b were utilized to alkylate the piperazine ring resulting in compounds 8a-b.
Scheme 2.

Reagents and conditions: (a) Lawesson's reagent, toluene, reflux, 5 h; (b) 1,4 dibromobutane, K2CO3, DMF, 60° C, 2 h; (c) Piperazine, K2CO3, DMF, 60° C, 1.5 h.
2.2. Pharmacology
All the newly synthesized compounds, 3a-3j, 5a–5t and 8a-8b were screened through cellular assays for their anti-inflammatory/analgesic potential based on their inhibitory activities on two targets; iNOS and NF-κB. The results are expressed in terms of IC50 values (the concentration that caused a 50% inhibition) and presented in Table 1. In addition, the cytotoxicity towards a panel of mammalian cell lines was also determined. Based on the in vitro results, three compounds 3j, 5t and 8b were selected for in vivo efficacy study. The acetic acid induced-writhing test was used for testing the analgesic activity and the carrageenan-induced assay was used for assessment of the anti-inflammatory activity.
Table 1.
Inhibition of iNOS and NF-κB activity and cytotoxicity evaluation.
| Comp. | iNOS | NF-κB | Cytotoxicity (IC50 μM) | |||||
|---|---|---|---|---|---|---|---|---|
| IC50 | IC50 (MM) | LLC-PK11 | VERO | SK-OV-3 | BT-S49 | KB | SK-MEL | |
| 3a | NA | NA | NA | NA | NA | NA | NA | NA |
| 3b | NA | NA | NA | NA | NA | NA | NA | NA |
| 3c | NA | 4.91 | NA | NA | NA | NA | NA | NA |
| 3d | 4.62 | NA | 23.5 | NA | NA | NA | NA | NA |
| 3e | NA | NA | NA | NA | NA | NA | NA | NA |
| 3f | NA | 2.28 | NA | NA | NA | NA | NA | NA |
| 3g | 4.43 | 3.43 | NA | 19.4 | NA | NA | NA | NA |
| 3h | 0.41 | 0.43 | NA | NA | 25.0 | 19.3 | NA | 19.8 |
| 3i | 4.22 | NA | NA | NA | NA | 18.5 | NA | 21.3 |
| 3j | 0.28 | 0.41 | NA | NA | 24.3 | 24.3 | 20.8 | NA |
| 5a | NA | NA | NA | NA | NA | NA | NA | NA |
| 5b | NA | NA | 24.1 | NA | NA | NA | NA | NA |
| 5c | NA | NA | NA | NA | NA | NA | NA | NA |
| 5d | NA | NA | NA | NA | NA | NA | NA | NA |
| 5e | 4.98 | 4.78 | NA | NA | NA | NA | NA | NA |
| 5f | 4.51 | 1.71 | NA | 23.8 | NA | NA | NA | NA |
| 5g | 4.66 | 3.07 | NA | NA | NA | NA | NA | NA |
| 5h | 3.43 | 2.95 | NA | NA | NA | 25 | NA | 25 |
| 5i | NA | NA | NA | NA | NA | NA | NA | NA |
| 5j | 4.51 | 1.71 | 19.2 | NA | NA | NA | NA | NA |
| 5k | 4.86 | NA | NA | NA | NA | NA | NA | NA |
| 5l | 2.55 | 1.46 | NA | NA | NA | 14 | NA | 16.8 |
| 5m | 4.50 | NA | NA | NA | NA | NA | NA | NA |
| 5n | 2.57 | 2.40 | NA | NA | NA | 25 | NA | 12 |
| 5o | 4.54 | NA | NA | 17.6 | NA | NA | NA | NA |
| 5p | 1.44 | 1.18 | NA | NA | NA | 22.1 | NA | 17.5 |
| 5q | 1.77 | 1.24 | NA | NA | NA | NA | NA | NA |
| 5r | 0.51 | 0.83 | NA | NA | NA | 21.3 | 19.5 | 14.5 |
| 5s | 2.90 | 1.40 | NA | NA | NA | 22.5 | NA | 23 |
| 5t | 0.29 | 1.06 | NA | 25 | 25 | 19.6 | NA | NA |
| 8a | 1.12 | 1.40 | 23.5 | NA | NA | 22.5 | 25 | 25 |
| 8b | 0.41 | 0.34 | NA | NA | NA | NA | 25 | 17.5 |
| L-NMMA | 2.30 | - | - | - | - | - | - | - |
| Parthenolide | - | 2.57 | - | - | - | - | - | - |
| Doxorubicin | - | - | 0.79 | 0.92 | 0.20 | 0.17 | 0.22 | 0.14 |
Positive controls: L-NMMA for iNOS inhibition, Parthenolide for NF-κB inhibition and Doxorubicin for cytotoxicity. NA = no activity up to 25 μg/ml. IC50 = the test concentration that caused a 50% inhibition.
Four human cancer cell lines were used: SK-MEL (Melanoma), KB (epidermal carcinoma), BT-549 (breast carcinoma) and SK-OV-3 (ovarian carcinoma). Two non-cancerous kidney cell lines were also used: Vero (Monkey kidney fibroblast) and LLC-PK11 (Pig kidney epithelial cells).
2.2.1. iNOS enzyme inhibitory assay
The iNOS inhibitory assay was performed in LPS-induced mouse macrophages (RAW264.7) where the concentration of (NO) was determined by measuring the level of nitrite in the cell culture supernatant using Griess reagent.24 L-N-monomethyl Arginine (L-NMMA) was used as positive control. The homodimers 3h and 3j which contain benzothiazolone nucleus and the longest alkyl chains displayed the most potent inhibition of iNOS in this series with IC50 values of 0.41 and 0.28 μM, respectively (Table 1). On the other hand, the corresponding benzoxazolone dimers 3g and 3i were not as potent (IC50, 4.4 μM and 4.2 μM) although they have equal alkyl chains. The rest of the homodimer series, 3a-3f, with shorter alkyl chains did not show any activity with the exception of 3d (IC50, 4.6 μM). Subsequently, structure-activity relationship studies were performed to deduce how the variation of the alky chain length could affect the activity. In this regard, a series of heterodimers were synthesized and evaluated for their potential to inhibit iNOS activity. Out of the benzothiazolone heterodimers, two compounds 5r and 5t were the most active with IC50 values of 0.51 and 0.29 μM, respectively, while others (5h, 5l, 5n and 5p) with shorter alkyl chains were not as effective (IC50 values in the range of 1.44 - 3.43 μM). On the other hand, the benzoxazolone dimers 5q and 5s with the same alkyl chains length showed activity with IC50 values of 1.77 and 2.9 μM, respectively. However, other heterodimers were found to show weak activity (5e-5g, 5j-5k, 5m, 5o) or they were devoid of any activity (5a-5d and 5i). It was worth noting that the difference in attachment point of the alkyl chain to the heterocycle significantly affected the activity. Compounds 8a and 8b showed better iNOS inhibitory activities with IC50 values 1.12 and 0.41 μM, respectively. On the contrary, compounds 3e and 3f with the same alkyl chains length, attached to the nitrogen not to the sulfur atom, displayed no activity.
2.2.2. Inhibition of NF-κB transcriptional activity
The effect of compounds on the transcriptional activity of NF-κB was determined in PMA-induced human chondrosarcoma (SW1353) cells through a reporter gene assay.25 Parthenolide was used as a positive control. Interestingly, most compounds showing strong inhibition of iNOS also showed a strong inhibition of NF-κB activity. The benzothiazolone derivatives 3h and 3j exhibited the highest activity with IC50 values of 0.4 μM while, the corresponding benzoxazolone dimer 3g was not as effective with an IC50 value of 3.43 μM, and 3i was devoid of any activity (Table 1). Moreover, heterodimers 5p, 5r and 5t with long alkyl chains showed acceptable inhibitory activity with IC50 values of 1.18, 0.83 and 1.06 μM respectively. On the other hand, the benzoxazolone dimers 5q and 5s were equally active to the corresponding benzothiazolone dimers 5r and 5t with IC50 values 1.24 and 1.40 μM, respectively. Nevertheless, compounds 5h, 5j and 5n were moderately active while most of the corresponding benzoxazolone dimers were devoid of activity except compound 5g which displayed a weak activity with an IC50 value of 3.07 μM.
It should be mentioned that, changing the attachment point of the alkyl chains to the heterocycle increases the activity similar to its effect on the iNOS inhibition. Compounds 8a and 8b more potent than the corresponding dimers 3e and 3f where their alkyl chains attached directly to the nitrogen atom of the heterocycle rather than the sulfur atom. In addition, it was noted that the benzothiazolone dimer 8b has higher inhibitory activity than the benzoxazolone dimer 8a with IC50 values of 0.34 and 1.40 μM, respectively.
These findings indicate that the heterodimers were, in general, more active than homodimers. Furthermore, the benzothiazolone dimers that have long alkyl chains were more potent inhibitors of both iNOS and NF-κB than the corresponding benzoxazolone dimers. None of the compounds showed inhibition of Sp-1 activity which confirms that their activity towards NF-κB is specific and is not related to cytotoxicity.
2.2.3. Acetic acid-induced writhing test
Two compounds, 3j and 8b, which exhibited the highest inhibitory activities in vitro against NF-κB and iNOS targets were selected to be evaluated for antinociceptive potential in vivo using the well-known acetic acid-induced writhing assay. The effects of these ligands on the acetic acid abdominal writhing assay are summarized in Fig. 2. Indomethacin was used as a standard control for this assay. In vehicle treated mice, acetic acid produced robust writhing responses during the observation period. Mice that received indomethacin displayed fewer writhing responses. The test compounds 3j and 8b produced significant reduction in writhing responses. However, the results revealed that the effect of the test compounds on the number of writhing was markedly increased with the dose elevating from 3 mg/kg to 10 mg/kg.
Figure 2. Acetic acid-induced writhing test.

Writhing responses for 20 minutes were measured in mice pretreated with vehicle, Indomethacin (Indo) or test compounds after i.p. injection of 0.9% acetic acid (1 ml/ 0.1 kg). The observations are mean ± SD. * p <0.05, ** p <0.01 (n = 4-6), compared to control.
2.2.4. Carrageenan-induced rat paw edema assay
The anti-inflammatory activities of compounds 3j, 5t and 8b were tested using carrageenan-induced rat paw edema assay where ketorolac was used as the reference drug.26 Mean changes in paw edema thickness of animals pretreated with the test compounds were measured after 1, 2, 3, and 4 hours from induction of inflammation. Data was analyzed by one-way ANOVA, followed by Dunnett's multiple comparisons test (n = 5). All compounds showed significant anti-inflammatory activity comparable to that of ketorolac (Table 2). Results revealed that compound 8b was the most active dimer while compounds 3j and 5t showed moderate activities compared to ketorolac. These results verified that the attachment point of the alkyl chain significantly influences the activity in vivo as well as in vitro. Another observation is the decline in activity of tested compounds after 3 hours. This could be explained by the expected rapid degradation of test compounds via N-dealkylation or other metabolic pathways.
Table 2.
Edema thickness and inhibition percent of control, ketorolac and tested compounds.
| Compound | Edema thickness (×10−2 mm) ±SEM (Inhibition %) |
|||
|---|---|---|---|---|
| 1h | 2h | 3h | 4h | |
| Control | 140.8 ± 1.35 | 155.6 ± 2.80 | 167.4 ± 2.09 | 195.6 ± 3.02 |
| Ketorolac 30 mg/kg | 41.6 ± 2.02* (70.46) | 40.2 ± 2.82* (74.16) | 41.2 ± 2.06* (75.38) | 40.4 ± 1.52* (79.34) |
| 3j 30 mg/kg | 95.50 ± 1.02* (32.17) | 97.24 ± 2.35* (37.51) | 101.6 ± 1.68* (39.31) | 123.5 ± 1.13* (36.86) |
| 5t 30 mg/kg | 93.01 ± 1.93* (33.94) | 91.57 ± 2.63* (41.15) | 109.24 ± 1.36* (34.74) | 127.34 ± 1.87* (34.89) |
| 8b 30 mg/kg | 56.48 ± 2.14* (59.89) | 55.6 ± 2.09* (64.27) | 61.32 ± 1.67* (63.37) | 103.54 ± 1.77* (47.07) |
The observations are mean ± SD.
p <0.01 (n = 5), compared to control.
2.2.5. Cytotoxicity assay
All compounds were evaluated for cytotoxicity against a panel of four human solid tumor cell lines (SK-MEL: malignant-melanoma; KB: oral epidermal carcinoma; BT-549: breast ductal carcinoma, and SK-OV-3: ovary carcinoma) and two kidney cell lines (Vero: monkey kidney fibroblasts and LLC-PK1: pig kidney epithelial cells).27 Most of the compounds were not cytotoxic up to a concentration of 25 μg/mL and do not seem to have any significant anti-cell proliferative activity towards cancer cells as shown in Table1.
2.3. Docking study
In order to understand the structural basis for activity and the putative binding mode of our ligands in the active site of iNOS, the best compounds 3j, 5t and 8b were docked by employing LIGANDFIT embedded in Discovery Studio software.28 The X-ray structure of iNOS complexed with the selective inhibitor AR-C120011 (pdb code: 3E65) was selected for this approach. All compounds adopted a disposition and orientation similar to that of the selective inhibitor AR-C120011. It has been reported that the correlated side-chain rotations of Gln257, Arg260 and other residues expose a new specificity pocket extending from the active-site heme pocket.29 This new, larger pocket may explain the accommodation of the iNOS binding site for these bulky dimers and potentially their enhanced inhibitory activity. All docked compounds showed three critical features and interactions within the binding pocket of iNOS: 1) The heterocycle, benzoxazolone or benzothiazolone, made a stacking interaction with heme propionate; 2) The piperazine core was enclosed by a polar cavity consisting of Gln257, Val346, and Arg260 residues forming a network of hydrogen bonds between the piperazine nitrogen atoms and the former amino acids; and 3) an additional hydrogen bond tethered the sulfur or the carbonyl group of the extended inhibitor tails to Asn115 residue, as shown in Fig. 3. It could also be appreciated from Fig. 3(A) how compound 8b formed another hydrogen-bonding interaction with NH of Val346. In conclusion, the results indicate contributions from heme stacking, hydrogen bonding and hydrophobic interactions within the active site account for the binding mode of these inhibitors and potentially explain their activity.
Figure 3.

A) Docking and binding mode of compound 8b into iNOS active site (PDB code: 3E65), B) Docking and binding mode of compound 5t into iNOS active site (PDB code: 3E65),C) Docking and binding mode of compound 3j into iNOS active site (PDB code: 3E65), D) The superimposition of the docked poses 3j (blue), 5t (pink), 8b (green) and the crystallographic ligand, AR-C120011 (yellow) within active site of iNOS. Hydrogen bonds are represented by dashed green lines. All hydrogens are removed for the purposes of clarity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
3. Conclusion
Two series of benzoxazolone and benzothiazolone dimers were synthesized and evaluated in vitro and in vivo for their anti-inflammatory and analgesic activities. The first series was designed by varying the carbon chain length extending from the piperazine core to the nitrogen atom of the dibenzo[d]oxazol-2(3H)-one or dibenzo[d]thiazol-2(3H)-one. The second series was designed by changing the attachment point. Compounds 3j, 5t and 8b were the most potent in inhibiting iNOS activity with IC50 values of 0.28, 0.29 and 0.41 μM respectively. On the other hand, compounds 3h, 3j and 8b were found to be the most potent in inhibiting NF-κB activity with IC50 values 0.43, 0.41 and 0.34 μM respectively. Based on the overall results, we found that heterodimers with longer alkyl chains around the piperazine core were more active than homodimers. Moreover, the attachment point on the heterocycle is important and significantly affects the activity. Based on the potent in vitro activity towards both iNOS and NF-κB, three compounds were selected for assessing their in vivo anti-inflammatory/analgesic potential. Compound 8b exhibited promising in vivo activity which was comparable to indomethacin and ketorolac. The docking simulation was carried out for some active dimers to determine the probable binding modes and poses. The results indicated that these active compounds fit nicely into the iNOS binding site, resulting in a stable complex and acting as potential inhibitors of this enzyme.
4. Experimental protocols
4.1. Chemistry
Reagents and starting materials were obtained from commercial suppliers and were used without purification. Precoated silica gel GF Uniplates from Analtech were used for thin-layer chromatography (TLC). Column chromatography was done on silica gel 60 (Sorbent technologies). 1H and 13C NMR spectra were obtained on a Bruker APX400 at 400 and 100 MHz, respectively. The high resolution mass spectra (HRMS) were recorded on a Waters Micromass Q-Tof Micro mass spectrometer with a lock of spray source. The mass spectra (MS) were recorded on a Waters Acquity Ultra Performance LC with ZQ detector in ESI mode. Elemental analysis (C, H, N) were recorded on an elemental analyzer, Perkin-Elmer CHN/SO series II Analyzer. Chemical names were generated using ChemDraw Ultra (CambridgeSoft, version 12.0).
General Procedure A. Synthesis of bromoalkyl benzo[d]oxazol-2(3H)-one and bromoalkyl benzo[d] thiazol-2(3H)-one derivatives. 3-(4-bromobutyl)benzo[d]oxazol-2(3H)-one (2e)
K2CO3 (9.2 g, 66.6 mmol) and 1,4-dibromobutane (21.0 mL, 177.6 mmol) were added, under stirring, to a solution of benzo[d]oxazol-2(3H)-one (3.0 g, 22.2 mmol) in anhydrous DMF (30 mL). The reaction mixture was heated at 60 °C for 3 h. After cooling, the reaction mixture was poured into 100 mL water and extracted with ethyl acetate (3 × 70 mL). The combined organic layers were washed with saturated aqueous NaCl and dried over sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using hexane/ ethyl acetate (8: 2) as eluent to give 3.6 g (62%) of 2e as a white solid. 1H NMR (CDCl3): δ7.30 – 7.05 (m, 3H), 7.00 (d, J = 7.4, 1H), 3.86 (t, J = 5.1, 2H), 3.44 (t, J = 10.6, 2H), 1.95 (m, 4H). 13C NMR (CDCl3): δ154.54, 142.67, 130.94, 123.89, 122.49, 110.09, 108.25, 41.28, 32.74, 29.41, 26.31. MS (EI) m/z 292 (M++23).
3-(2-bromoethyl)benzo[d]oxazol-2(3H)-one (2a)
General Procedure A, yield 58%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.38 – 6.79 (m, 4H), 4.21 (t, J = 6.2, 2H), 3.67 (t, J = 6.1, 2H). 13C NMR (101 MHz, CDCl3) δ 154.20, 142.54, 130.83, 123.96, 122.75, 110.17, 108.54, 43.88, 27.73. MS (EI) m/z 242 (M++1).
3-(2-bromoethyl)benzo[d]thaizol-2(3H)-one (2b)
General Procedure A, yield 57%, white solid. 1H NMR (400 MHz, CDCl3) δ 7.64 – 6.99 (m, 4H), 4.37 (t, J = 6.2, 2H), 3.82 (t, J = 6.1, 2H). 13C NMR (101 MHz, CDCl3) δ 166.07, 154.40, 142.70, 135.83, 134.62, 122.03, 120.40, 55.74, 39.59. MS (EI) m/z 258 (M++1).
3-(3-bromopropyl)benzo[d]oxazol-2(3H)-one (2c)
General Procedure A, yield 59%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.41 – 6.87 (m, 4H), 4.00 (t, J = 6.8, 2H), 3.46 (t, J = 6.3, 2H), 2.94 – 1.89 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 154.45, 142.65, 131.13, 123.97, 122.59, 110.14, 108.27, 40.61, 30.75, 29.87. MS (EI) m/z 279 (M++23).
3-(3-bromopropyl)benzo[d]thaizol-2(3H)-one (2d)
General Procedure A, yield 61%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.54 – 6.95 (m, 4H), 4.11 (t, J = 7.5, 2H), 3.46 (t, J = 6.4, 2H), 2.35 – 2.29 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 156.76, 144.96, 133.45, 126.29, 124.91, 112.46, 110.59, 42.93, 33.06, 32.18. MS (EI) m/z 272 (M++1).
3-(4-bromobutyl)benzo[d]thaizol-2(3H)-one 2f
General Procedure A, yield 63%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.56 – 6.74 (m, 4H), 3.94 (t, J = 6.6, 2H), 3.41 (t, J = 6.0, 2H), 2.14 – 1.63 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 169.79, 136.85, 126.44, 123.15, 122.72, 122.66, 110.56, 41.67, 33.07, 29.58, 26.16. MS (EI) m/z 308 (M++23).
3-(5-bromopentyl)benzo[d]oxazol-2(3H)-one (2g)
General Procedure A, yield 65%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.25 – 6.95 (m, 4H), 3.84 (t, J = 7.2, 2H), 3.39 (t, J = 6.7, 2H), 1.97 – 1.87 (m, 2H), 1.85-1.78 (m, 2H), 1.63 – 1.45 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 154.54, 142.68, 131.08, 123.82, 122.39, 110.07, 108.21, 42.00, 33.21, 32.09, 26.96, 25.21. MS (EI) m/z 306 (M++23).
3-(5-bromopentyl)benzo[d]thaizol-2(3H)-one (2h)
General Procedure A, yield 64%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.50 – 6.86 (m, 4H), 3.89 (t, J = 7.3, 2H), 3.33 (t, J = 6.7, 2H), 1.88 – 1.81 (m, 2H), 1.74 – 1.67 (m, 2H), 1.52 – 1.44 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 169.67, 136.95, 126.37, 123.05, 122.68, 122.66, 110.55, 42.42, 33.48, 32.20, 26.76, 25.32. MS (EI) m/z 323 (M++23).
3-(6-bromohexyl)benzo[d]oxazol-2(3H)-one (2i)
General Procedure A, yield 65%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.39 – 6.73 (m, 4H), 3.82 (t, J = 7.2, 2H), 3.38 (t, J = 6.7, 2H), 1.95 – 1.66 (m, 4H), 1.66 – 1.23 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 154.56, 142.67, 131.14, 123.79, 122.33, 110.01, 108.25, 42.10, 33.65, 32.48, 27.66, 27.61, 25.82. MS (EI) m/z 298 (M++1).
3-(6-bromohexyl)benzo[d]thaizol-2(3H)-one (2j)
General Procedure A, yield 63%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.52 – 6.77 (m, 4H), 3.91 (t, J = 7.2, 2H), 3.36 (t, J = 6.7, 2H), 1.94 – 1.60 (m, 4H), 1.56 – 1.24 (m, 4H). 13C NMR (400 MHz, CDCl3) δ 206.96, 198.73, 196.03, 195.20, 195.13, 195.11, 192.07, 174.98, 172.76, 172.45, 171.25, 171.17, 170.79. MS (EI) m/z 314 (M++1).
General Procedure B. Synthesis of piperazine dibenzo[d]oxazol-2(3H)-one Homodimers 3,3′-(piperazine-1,4-diylbis(butane-4,1-diyl))bis(benzo[d]oxazol-2(3H)-one) (3e)
K2CO3 (0.76 g, 5.55 mmol) and piperazine (0.1 g, 0.92 mmol) were added, under stirring, to a solution of 2e (0.5 g, 1.85 mmol) in anhydrous DMF (10 mL). The reaction mixture was heated at 60 °C for 1.5 h. After cooling, the reaction mixture was poured into 50 mL water and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with saturated aqueous NaCl and dried over sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using dichlormethane/methanol (9.5: 0.5) as eluent to give 0.3 g (70%) of 3e as a white solid. 1H NMR (CDCl3): δ 7.26-6.96 (m, 8H), 3.82 (t, J = 7.0, 4H), 2.39 (s, 8H), 2.37 – 2.29 (m, 4H), 1.78 -1.75 (m, 4H), 1.57-1.52 (m, 4H). 13C NMR (CDCl3): δ 154.51, 142.65, 131.12, 123.73, 122.27, 109.97, 108.30, 57.66, 53.13, 42.08, 25.61, 23.88. HRMS calcd for C26H32N4O4 [M+H]+ 464.2424, found 465.2509. Anal. calcd for C26H34Cl2N4O4: C, 58.10; H, 6.38; N, 10.42. Found: C, 58.34; H, 6.87; N, 10.21.
3,3′-(piperazine-1,4-diylbis(ethane-2,1-diyl))bis(benzo[d]oxazol-2(3H)-one) (3a)
General Procedure B, yield 51%, white solid. 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.00 (m, 8H), 4.06 (t, J = 7.1, 4H), 2.72 – 2.62 (t, J = 7.2, 4H), 2.56 (s, 8H). 13C NMR (101 MHz, CDCl3) δ 169.87, 137.08, 126.27, 123.03, 122.75, 122.66, 110.57, 54.85, 53.27, 40.40. HRMS calcd for C22H24N4O4 [M+H]+ 408.1798, found 409.1891. Anal. calcd for C22H26Cl2N4O4: C, 54.89; H, 5.44; N, 11.64. Found: C, 55.14; H, 5.87; N, 11.28.
3,3′-(2,2′-(piperazine-1,4-diyl)bis(ethane-2,1-diyl))dibenzo[d]thaizol-2(3H)-one (3b)
General Procedure B, yield 52%, white solid. 1H NMR (400 MHz, DMSO) δ 7.85 – 6.92 (m, 8H), 4.32 (t, J = 6.7, 4H), 3.30 – 3.16 (m, 12H). 13C NMR (101 MHz, DMSO) δ 169.53, 136.88, 127.15, 123.82, 123.40, 122.06, 112.00, 52.59, 49.59, 37.54. HRMS calcd for C22H24N4O2S2 [M+H]+ 440.1431, found 441.1432. Anal. calcd for C22H26Cl2N4O2S2: C, 51.46; H, 5.10; N, 10.91. Found: C, 51.31; H, 5.47; N, 10.55.
3,3′-(piperazine-1,4-diylbis(propane-3,1-diyl))bis(benzo[d]oxazol-2(3H)-one) (3c)
General Procedure B, yield 53%, white solid. 1H NMR (400 MHz, D2O) δ 7.27 – 7.03 (m, 8H), 3.86 (t, J = 6.5, 4H), 3.19 (s, 8H), 3.00 (t, J = 6.8, 4H), 2.07 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 154.28, 142.41, 131.79, 124.19, 122.40, 109.97, 109.62, 55.25, 52.99, 24.24. HRMS calcd for C24H28N4O [M+H]+ 436.2111, found 437.2173. Anal. calcd for C24H30Cl2N4O4: C, 56.58; H, 5.94; N, 11.00. Found: C, 56.91; H, 5.43; N, 11.29.
3,3′-(piperazine-1,4-diylbis(propane-3,1-diyl))bis(benzo[d]thiazol-2(3H)-one) (3d)
General Procedure B, yield 50%, white solid. 1H NMR (400 MHz, DMSO) δ 7.81 – 7.02 (m, 8H), 4.03 (t, J = 6.8, 4H), 3.69 – 3.03 (m, 12H), 2.27 – 1.96 (m, 4H). 13C NMR (101 MHz, DMSO) δ 169.35, 137.18, 127.11, 123.67, 123.36, 122.06, 111.89, 48.47, 22.61, 17.88. HRMS calcd for C24H28N4O2S2 [M+H]+ 468.1735, found 469.1736. Anal. calcd for C24H30Cl2N4O2S2: C, 53.23; H, 5.58; N, 10.35. Found: C, 53.59; H, 5.12; N, 10.76.
3,3′-(piperazine-1,4-diylbis(butane-4,1-diyl))bis(benzo[d]thiazol-2(3H)-one) (3f)
General Procedure B, yield 51%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.70 – 6.46 (m, 8H), 3.92 (t, J = 6.9, 4H), 2.42 - 2.32 (m, 12H), 2.02 – 1.60 (m, 4H), 1.56 – 1.54 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 169.81, 137.05, 126.26, 122.97, 122.73, 122.61, 110.69, 57.64, 53.06, 42.48, 25.37, 23.81. HRMS calcd for C26H32N4O2S2 [M+H]+ 496.1967, found 497.2057. Anal. calcd for C26H34Cl2N4O2S2: C, 54.82; H, 6.02; N, 9.84. Found: C, 54.45; H, 6.51; N, 9.65.
3,3′-(piperazine-1,4-diylbis(pentane-5,1-diyl))bis(benzo[d]oxazol-2(3H)-one) (3g)
General Procedure B, yield 53%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.23 – 6.92 (m, 8H), 3.80 (t, J = 7.2, 4H), 2.42 (s, 8H), 2.29 (t, J = 7.3, 4H), 1.86 – 1.71 (m, 4H), 1.56 - 1.48 (m, 4H), 1.44 – 1.31 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 154.55, 142.67, 131.17, 123.73, 122.27, 110.01, 108.23, 58.30, 53.19, 42.21, 27.64, 26.38, 24.63. HRMS calcd for C28H36N4O4 [M+H]+ 492.2737, found 493.2814. Anal. calcd for C28H38Cl2N4O4: C, 59.47; H, 6.77; N, 9.91. Found: C, 59.34; H, 6.97; N, 10.33.
3,3′-(piperazine-1,4-diylbis(pentane-5,1-diyl))bis(benzo[d]thiazol-2(3H)-one) (3h)
General Procedure B, yield 52%, yellow oil. 1H NMR (400 MHz, D2O) δ 8.04 – 6.96 (m, 8H), 4.24 (t, J = 6.9, 4H), 3.83 (s, 8H), 3.52 – 3.36 (m, 4H), 2.14 – 1.91 (m, 8H), 1.65 – 1.58 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 155.45, 143.57, 132.07, 124.63, 123.17, 110.90, 109.13, 59.20, 54.09, 43.11, 28.54, 27.28, 25.53. HRMS calcd for C28H36N4O2S2 [M+H]+ 524.2280, found 525.2373. Anal. calcd for C28H38Cl2N4O2S2: C, 56.27; H, 6.41; N, 9.37. Found: C, 56.62; H, 6.81; N, 9.26.
3,3′-(piperazine-1,4-diylbis(hexane-6,1-diyl))bis(benzo[d]oxazol-2(3H)-one) (3i)
General Procedure B, yield 52%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.20 – 6.95 (m, 4H), 3.80 (t, J = 7.2, 2H), 2.45 (s, 4H), 2.30 (t, J = 7.3, 2H), 1.80 – 1.73 (m, 2H), 1.51 -1.44 (m, 2H), 1.42 – 1.34 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 154.56, 142.68, 131.20, 123.72, 122.26, 110.00, 108.24, 58.45, 53.07, 42.22, 27.68, 27.08, 26.58, 26.56. HRMS calcd for C30H40N4O4 [M+H]+ 520.3050, found 521.3132. Anal. calcd for C30H42Cl2N4O4: C, 60.70; H, 7.13; N, 9.44. Found: C, 60.43; H, 7.17; N, 9.55.
3,3′-(piperazine-1,4-diylbis(hexane-6,1-diyl))bis(benzo[d]thiaazol-2(3H)-one) (3j)
General Procedure B, yield 53%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.68 – 6.71 (m, 8H), 3.89 (t, J = 6.8, 4H), 2.84 – 1.99 (m, 12H), 1.70 (t, J = 6.2, 4H), 1.45 – 1.33 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 169.76, 137.09, 126.25, 122.93, 122.75, 122.62, 110.56, 58.44, 53.02, 42.70, 27.50, 27.14, 26.66, 26.57. HRMS calcd for C30H40N4O2S [M+H]+ 552.2593, found 553.2695. Anal. calcd for C30H42Cl2N4O2S2: C, 57.59; H, 6.77; N, 8.95. Found: C, 57.14; H, 6.28; N, 9.41.
General Procedure C. Piperazine benzo[d]oxazol-2(3H)-one derivatives. 3-(4-(piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one (4e)
K2CO3 (2.3 g, 16.71 mmol) and piperazine (2.4 g, 27.85 mmol) were added, under stirring, to a solution of 2e (1.5 g, 5.57 mmol) in anhydrous DMF (20 mL). The reaction mixture was heated at 60 °C for 0.5 h. After cooling, the reaction mixture was poured into 60 mL water and extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with saturated aqueous NaCl and dried over sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using dichloromethane/ methanol: NH4OH (9: 1) as eluent to give 1.2 g (78%) of 4e as a white solid. 1H NMR (D2O): δ 7.16 – 7.06 (m, 4H), 3.72 (t, J = 6.0, 2H), 3.62 (s, 8H), 3.27 (t, J = 6.3, 2H), 1.76 -1.68 (m, 4H). 13C NMR (101 MHz, D2O) δ 156.03, 142.10, 130.26, 124.44, 123.05, 110.02, 109.36, 56.63, 48.47, 41.27, 40.81, 24.17, 20.65. MS (EI) m/z 276 (M++1).
3-(2-(piperazin-1-yl)ethyl)benzo[d]oxazol-2(3H)-one (4a)
General Procedure C, yield 67%, white solid. 1H NMR (400 MHz, D2O) δ 7.31 – 7.13 (m, 4H), 4.30 (t, J = 5.9, 2H), 3.77 (s, 4H), 3.72 (t, J = 6.3, 2H), 3.64 (s, 4H). 13C NMR (101 MHz, D2O) δ 156.20, 142.47, 129.53, 124.69, 123.70, 110.46, 109.22, 54.25, 48.86, 40.72, 36.50. MS (EI) m/z 248 (M++1).
3-(2-(piperazin-1-yl)ethyl)benzo[d]thaizol-2(3H)-one (4b)
General Procedure C, yield 68%, white solid 1H NMR (400 MHz, D2O) δ 7.69 – 6.78 (m, 4H), 4.29 (t, J = 6.0, 2H), 3.68 (s, 4H), 3.64 – 3.40 (m, 6H). 13C NMR (101 MHz, D2O) δ 173.78, 135.44, 127.08, 124.30, 123.12, 122.16, 111.08, 54.24, 48.90, 40.69, 36.85. MS (EI) m/z 264 (M++1).
3-(3-(piperazin-1-yl)propyl)benzo[d]oxazol-2(3H)-one (4c)
General Procedure C, yield 66%, white solid 1H NMR (400 MHz, D2O) δ 7.27 – 7.01 (m, 4H), 3.88 (t, J = 6.7, 2H), 3.62 (s, 8H), 3.37 (t, J = 6.5, 2H), 2.35 – 2.13 (m, 2H). 13C NMR (101 MHz, D2O) δ 156.00, 142.21, 130.12, 124.54, 123.26, 110.19, 109.23, 54.50, 48.65, 40.84, 39.08, 22.26. MS (EI) m/z 262 (M++1).
3-(3-(piperazin-1-yl)propyl)benzo[d]thiazol-2(3H)-one (4d)
General Procedure C, yield 63%, white solid 1H NMR (400 MHz, D2O) δ 7.17 – 7.06 (m, 4H), 3.72 (t, J = 6.8, 2H), 3.62 (s, 8H), 3.29 (t, J = 6.5, 2H), 1.79 – 1.76 (m, 2H). 13C NMR (101 MHz, D2O) δ 156.03, 142.10, 130.26, 124.44, 123.05, 110.02, 109.36, 56.63, 48.47, 41.27, 40.81, 24.17. MS (EI) m/z 277 (M+).
3-(4-(piperazin-1-yl)butyl)benzo[d]thaizol-2(3H)-one (4f)
General Procedure C, yield 65%, yellow oil 1H NMR (400 MHz, D2O) δ 7.67 – 6.90 (m, 4H), 3.95 (t, J = 5.9, 2H), 3.46 – 3.38 (m, 8H), 3.20 – 2.86 (m, 2H), 1.87 – 1.58 (m, 2H), 1.22 – 1.17 (m, 2H). 13C NMR (101 MHz, D2O) δ 156.91, 142.99, 131.15, 125.33, 123.93, 110.91, 110.25, 57.52, 49.36, 42.16, 41.70, 25.06, 21.54. MS (EI) m/z 292 (M++1).
3-(5-(piperazin-1-yl)pentyl)benzo[d]oxazol-2(3H)-one (4g)
General Procedure C, yield 67%, white solid 1H NMR (400 MHz, D2O) δ 7.22 - 7.11 (m, 4H), 3.75 (t, J = 6.8, 2H), 3.61 (s, 8H), 3.22 (t, J = 6.5, 2H), 1.86 – 1.63 (m, 4H), 1.41 -1.33 (m, 2H). 13C NMR (101 MHz, D2O) δ 156.26, 142.21, 130.51, 124.40, 123.00, 110.05, 109.50, 57.07, 48.40, 41.70, 40.79, 26.45, 22.85, 22.81. MS (EI) m/z 290 (M++1).
3-(5-(piperazin-1-yl)pentyl)benzo[d]thaizol-2(3H)-one (4h)
General Procedure C, yield 66%, white solid 1H NMR (400 MHz, D2O) δ 7.89 – 6.53 (m, 4H), 3.79 – 3.31 (m, 10H), 3.13 (t, J = 7.3, 2H), 1.72 – 1.57 (m, 2H), 1.54 – 1.52 (m, 2H), 1.24 – 1.23 (m, 2H). 13C NMR (101 MHz, D2O) δ 136.36, 136.32, 126.81, 123.73, 122.64, 121.80, 111.64, 100.00, 56.98, 48.32, 42.16, 40.71, 26.29, 22.81. MS (EI) m/z 306 (M++1).
General Procedure D. Synthesis of piperazine dibenzo[d]oxazol-2(3H)-one Heterodimmers. 3-(5-(4-(4-(2-oxobenzo[d]oxazol-3(2H)-yl)butyl)piperazin-1-yl)pentyl)benzo[d]oxazol-2(3H)-one (5o)
K2CO3 (0.75 g, 5.45 mmol) and 4e (0.5 g, 1.82 mmol) were added, under stirring, to a solution of 2g (0.49 g, 1.82 mmol) in anhydrous DMF (20 mL). The reaction mixture was heated at 60 °C for 2 h. After cooling, the reaction mixture was poured into 60 mL water and extracted with ethyl acetate (3 × 40 mL). The combined organic layers were washed with saturated aqueous NaCl and dried over sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using dichlormethane/ methanol (9.5: 0.5) as eluent to give 0.58 g (73%) of 5o as a white solid. 1H NMR (400 MHz, D2O) δ 7.42 – 6.82 (m, 8H), 3.74 (t, J = 6.7, 2H), 3.69 (t, J = 6.9, 2H), 3.57 (s, 8H), 3.22 (t, J = 7.8, 2H), 3.19 – 3.12 (m, 2H), 1.84 – 1.57 (m, 8H), 1.34 – 1.27 (m, 2H). 13C NMR (101 MHz, D2O) δ 156.21, 156.20, 142.17, 142.13, 130.41, 130.28, 124.37, 124.32, 123.03, 122.92, 110.04, 109.97, 109.40, 109.30, 56.65, 56.20, 48.60, 48.56, 41.59, 41.14, 26.38, 24.07, 22.88, 22.71, 20.66. HRMS calcd for C27H34N4O4 [M+H]+ 478.2580, found 479.3266. Anal. calcd for C27H36Cl2N4O4: C, 58.80; H, 6.58; N, 10.16. Found: C, 58.47; H, 6.13; N, 10.39.
3-(3-(4-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)ethyl)piperazin-1-yl)propyl)benzo[d]oxazol-2(3H)-one (5a)
General Procedure D, yield 67%, white solid. 1H NMR (400 MHz, D2O) δ 7.36 – 7.10 (m, 8H), 4.25 (t, J = 5.9, 2H), 3.93 (t, J = 6.6, 2H), 3.67 – 3.46 (m, 10H), 3.37 – 3.26 (m, 2H), 2.29 – 2.11 (m, 2H). 13C NMR (101 MHz, D2O) δ 156.40, 156.28, 142.53, 130.27, 124.59, 124.50, 123.58, 123.28, 110.44, 110.30, 109.18, 109.16, 54.10, 53.91, 49.35, 49.09, 39.01, 22.27. HRMS calcd for C23H26N4O4 [M+H]+ 422.1954, found 423.2025. Anal. calcd for C23H28Cl2N4O4: C, 55.76; H, 5.70; N, 11.31. Found: C, 56.14; H, 5.24; N, 11.01.
3-(3-(4-(2-(2-oxobenzo[d]thiazol-3(2H)-yl)ethyl)piperazin-1-yl)propyl)benzo[d]thiazol-2(3H)-one (5b)
General Procedure D, yield 63%, white solid. 1H NMR (400 MHz, CDCl3) δ 7.24 – 6.87 (m, 8H), 3.97 – 3.79 (m, 4H), 2.66 (t, J = 6.5, 2H), 2.46 – 2.32 (m, 10 H), 1.95 – 1.82 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 154.55, 142.63, 142.60, 131.43, 131.17, 123.69, 123.67, 122.26, 122.17, 109.91, 109.86, 108.47, 108.44, 55.14, 54.93, 53.15, 52.95, 40.38, 39.81, 24.58. HRMS calcd for C23H26N4O2S2 [M+H]+ 454.1497, found 455.1564. Anal. calcd for C23H28Cl2N4O2S2: C, 52.37; H, 5.35; N, 10.62. Found: C, 52.59; H, 5.12; N, 10.11.
3-(2-(4-(4-(2-oxobenzo[d]oxazol-3(2H)-yl)butyl)piperazin-1-yl)ethyl)benzo[d]oxazol-2(3H)-one (5c)
General Procedure D, yield 64%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.28 – 6.79 (m, 8H), 3.91 (t, J = 6.4, 2H), 3.82 (t, J = 7.0, 2H), 2.68 (t, J = 6.4, 2H), 2.52 (s, 4H), 2.39 – 2.32 (m, 6H), 1.88 – 1.67 (m, 2H), 1.63 – 1.46 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 154.59, 154.54, 142.67, 131.20, 131.11, 123.74, 123.68, 122.30, 122.28, 110.00, 109.96, 108.44, 108.29, 57.55, 55.18, 53.20, 53.02, 42.08, 39.85, 25.60, 23.85. HRMS calcd for C24H28N4O4 [M+H]+ 436.2111, found 437.2196. Anal. calcd for C24H30Cl2N4O4: C, 56.58; H, 5.94; N, 11.00. Found: C, 56.90; H, 5.88; N, 10.91.
3-(2-(4-(4-(2-oxobenzo[d]thiazol-3(2H)-yl)butyl)piperazin-1-yl)ethyl)benzo[d]thiazol-2(3H)-one (5d)
General Procedure D, yield 61%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.54 – 6.98 (m, 8H), 4.06 (t, J = 7.2, 2H), 3.96 (t, J = 7.4, 2H), 2.71 – 2.63 (m, 2H), 2.57 (s, 4H), 2.45 (s, 4H), 2.41 – 2.34 (m, 2H), 1.80 -1.73 (m, 2H), 1.61 - 1.54 (m, 2H). 13C NMR (101 MHz, CDCl3) 169.84, 137.10, 137.09, 126.25, 123.01, 122.97, 122.82, 122.77, 122.65, 122.64, 110.66, 110.56, 57.58, 54.92, 53.31, 53.03, 42.51, 40.42, 29.68, 25.38, 23.82. HRMS calcd for C24H28N4O2S2 [M+H]+ 468.1654, found 469.1735. Anal. calcd for C24H30Cl2N4O2S2: C, 53.23; H, 5.58; N, 10.35. Found: C, 53.31; H, 5.17; N, 10.83.
3-(5-(4-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)ethyl)piperazin-1-yl)pentyl)benzo[d]oxazol-2(3H)-one (5e)
General Procedure D, yield 62%, yellow oil. 1H NMR (400 MHz, D2O) δ 7.57 – 7.43 (m, 8H), 4.53 (t, J = 6.0, 2H), 4.08 (t, J = 6.8, 2H), 3.91 – 3.75 (m, 10H), 3.51 – 3.36 (m, 2H), 2.15 – 1.87 (m, 4H), 1.66 – 1.59 (m, 2H). 13C NMR (101 MHz, D2O) δ 165.86, 142.69, 131.02, 130.10, 124.95, 124.70, 123.93, 123.31, 110.79, 110.46, 109.84, 109.52, 99.99, 56.99, 54.19, 49.35, 49.28, 41.98, 37.36, 34.96, 26.58, 23.04. HRMS calcd for C25H30N4O4 [M+H]+ 450.2267, found 451.2339. Anal. calcd for C25H32Cl2N4O4: C, 57.36; H, 6.16; N, 10.70. Found: C, 57.14; H, 5.94; N, 10.26.
3-(5-(4-(2-(2-oxobenzo[d]thiazol-3(2H)-yl)ethyl)piperazin-1-yl)pentyl)benzo[d]thiazol-2(3H)-one (5f)
General Procedure D, yield 59%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.60 – 6.89 (m, 8H), 4.04 (t, J = 7.2, 2H), 3.92 (t, J = 7.4, 2H), 2.64 (t, J = 7.1, 2H), 2.56 (s, 4H), 2.42 (s, 4H), 2.30 (t, J = 7.3, 2H), 1.77 – 1.70 (m, 2H), 1.55 – 1.48 (m, 2H), 1.45 – 1.32 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 169.80, 169.79, 137.09, 126.25, 122.99, 122.95, 122.80, 122.73, 122.64, 110.57, 110.54, 58.23, 54.92, 53.33, 53.12, 42.70, 40.42, 27.45, 26.40, 24.71. HRMS calcd for C25H30N4O2S2 [M+H]+ 482.1810, found 483.1908. Anal. calcd for C25H32Cl2N4O2S2: C, 54.05; H, 5.81; N, 10.08. Found: C, 54.39; H, 6.17; N, 9.78.
3-(6-(4-(2-(2-oxobenzo[d]oxazol-3(2H)-yl)ethyl)piperazin-1-yl)hexyl)benzo[d]oxazol-2(3H)-one 5g
General Procedure D, yield 57%, colorless oil. 1H NMR (400 MHz, D2O) δ 7.34 – 6.83 (m, 8H), 4.25 (t, J = 7.3, 2H), 3.82 – 3.42 (m, 12H), 3.27 – 3.06 (m, 2H), 1.70 - 1.64 (m, 4H), 1.45 – 1.20 (m, 4H). 13C NMR (101 MHz, D2O) δ 156.34, 156.33, 142.47, 142.22, 130.58, 129.57, 124.59, 124.32, 123.60, 122.91, 110.41, 110.02, 109.52, 109.14, 56.85, 56.75, 53.94, 48.99, 48.72, 45.46, 41.86, 36.77, 36.74, 31.44, 26.58, 23.11, 23.09. HRMS calcd for C26H32N4O4 [M+H]+ 464.2424, found 465.2507. Anal. calcd for C26H34Cl2N4O4: C, 58.10; H, 6.38; N, 10.42. Found: C, 57.88; H, 6.10; N, 10.11.
3-(6-(4-(2-(2-oxobenzo[d]thiazol-3(2H)-yl)ethyl)piperazin-1-yl)hexyl)benzo[d]thiazol-2(3H)-one 5h
General Procedure D, yield 55%, yellow oil. 1H NMR (400 MHz, DMSO) δ 7.57 – 7.06 (m, 8H), 4.33 – 4.17 (m, 2H), 3.81 (t, J = 7.0, 2H), 3.74 – 3.01 (m, 12H), 1.82 – 1.57 (m, 4H), 1.48 – 1.24 (m, 4H). 13C NMR (101 MHz, D2O) δ 153.69, 153.67, 139.82, 139.57, 127.92, 126.92, 121.94, 121.66, 120.95, 120.25, 107.75, 107.36, 106.87, 106.48, 54.09, 51.28, 46.34, 46.06, 42.80, 39.21, 34.11, 28.78, 23.93, 20.43.. HRMS calcd for C26H32N4O2S2 [M+H]+ 496.1967, found 497.2053. Anal. calcd for C26H34Cl2N4O2S2: C, 54.82; H, 6.02; N, 9.84. Found: C, 54.30; H, 5.77; N, 10.23.
3-(3-(4-(4-(2-oxobenzo[d]oxazol-3(2H)-yl)butyl)piperazin-1-yl)propyl)benzo[d]oxazol-2(3H)-one 5i
General Procedure D, yield 53%, colorless oil. 1H NMR (400 MHz, D2O) δ 7.44 - 7.34 (m, 8H), 4.12 (t, J = 6.7, 2H), 4.02 (t, J = 6.9, 2H), 3.88 (s, 8H), 3.66 – 3.56 (m, 2H), 3.52 (t, J = 7.5, 2H), 2.59 – 2.35 (m, 2H), 2.01-2.00 (m, 4H). 13C NMR (101 MHz, D2O) δ 142.56, 142.55, 124.81, 124.71, 123.54, 123.34, 122.83, 110.52, 110.50, 110.40, 110.37, 109.66, 109.64, 109.47, 56.58, 54.49, 49.01, 48.84, 41.49, 39.31, 24.33, 22.51, 20.91. HRMS calcd for C23H26N4O4 [M+H]+ 4 422.1954, found 423.2025. HRMS calcd for C25H30N4O4 [M+H]+ 450.2267, found 451.2359. Anal. calcd for C25H32Cl2N4O4: C, 57.36; H, 6.16; N, 10.70. Found: C, 57.01; H, 5.84; N, 10.31.
3-(3-(4-(4-(2-oxobenzo[d]thiazol-3(2H)-yl)butyl)piperazin-1-yl)propyl)benzo[d]thiazol-2(3H)-one 5j
General Procedure D, yield 52%, yellow oil. 1H NMR (400 MHz, D2O) δ 7.79 – 7.44 (m, 8H), 4.28 (t, J = 6.6, 2H), 4.20 (t, J = 6.2, 2H), 3.76 (s, 8H), 3.50 – 3.44 (m, 4H), 2.50 – 2.32 (m, 2H), 2.09 – 1.87 (m, 4H). 13C NMR (101 MHz, D2O) δ 173.23, 173.17, 136.39, 136.16, 126.98, 126.87, 124.03, 123.89, 122.97, 122.84, 122.06, 122.04, 111.64, 111.35, 56.19, 54.20, 48.74, 48.69, 41.58, 39.43, 23.83, 22.21, 20.58. HRMS calcd for C25H30N4O2S2 [M+H]+ 482.1810, found 483.1900. Anal. calcd for C25H32Cl2N4O4S2: C, 54.05; H, 5.81; N, 10.08. Found: C, 54.14; H, 6.12; N, 10.45.
3-(3-(4-(5-(2-oxobenzo[d]oxazol-3(2H)-yl)pentyl)piperazin-1-yl)propyl)benzo[d]oxazol-2(3H)-one 5k
General Procedure D, yield 53%, yellow oil. 1H NMR (400 MHz, DMSO) δ7.85 – 6.76 (m, 8H), 4.02 (t, J = 6.3, 2H), 3.93 (t, J = 6.6, 2H), 3.68 – 3.08 (m, 12H), 2.11 (m, 2H), 1.73 – 1.64 (m, 4H), 1.33 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 158.95, 158.87, 126.46, 126.21, 115.40, 115.33, 112.08, 112.04, 111.88, 111.79, 111.76, 111.65, 99.93, 99.68, 47.44, 44.22, 42.35, 42.19, 31.82, 30.09, 16.59, 15.55, 13.96, 13.86. HRMS calcd for C26H32N4O4 [M+H]+ 464.2424, found 465.2491. Anal. calcd for C26H34Cl2N4O4: C, 58.10; H, 6.38; N, 10.42. Found: C, 58.65; H, 6.05; N, 9.97.
3-(3-(4-(5-(2-oxobenzo[d]thiazol-3(2H)-yl)pentyl)piperazin-1-yl)propyl)benzo[d]thiazol-2(3H)-one 5l
General Procedure D, yield 51%, colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.57 – 6.82 (m, 8H), 3.95 (t, J = 6.9, 2H), 3.88 (t, J = 7.1, 2H), 2.72 – 2.13 (m, 12H), 1.90 - 1.82 (m, 2H), 1.74 – 1.67 (m, 2H), 1.62 – 1.44 (m, 2H), 1.39 – 1.32 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 169.80, 169.72, 137.31, 137.06, 126.25, 126.18, 122.93, 122.89, 122.73, 122.64, 122.61, 122.51, 110.78, 110.54, 58.29, 55.07, 53.20, 53.04, 42.67, 40.94, 27.44, 26.41, 24.81, 24.71. HRMS calcd for C26H32N4O2S2 [M+H]+ 496.1967, found 497.2055. Anal. calcd for C26H34Cl2N4O2S2: C, 54.82; H, 6.02; N, 9.84. Found: C, 55.19; H, 5.94; N, 10.11.
3-(3-(4-(6-(2-oxobenzo[d]oxazol-3(2H)-yl)hexyl)piperazin-1-yl)propyl)benzo[d]oxazol-2(3H)-one 5m
General Procedure D, yield 56%, yellow oil. 1H NMR (400 MHz, DMSO) δ 7.85 – 6.76 (m, 8H), 4.02 (t, J = 6.3, 2H), 3.93 (t, J = 6.6, 2H), 3.67 – 3.04 (m, 12H), 2.17 – 2.05 (m, 2H), 1.69 – 1.61 (m, 4H), 1.33 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 165.67, 165.59, 133.29, 133.06, 122.10, 122.03, 118.77, 118.75, 118.72, 118.64, 118.50, 118.40, 106.64, 106.40, 54.36, 50.99, 49.10, 49.04, 48.96, 38.62, 36.89, 23.41, 23.04, 22.56, 22.53, 20.72. HRMS calcd for C27H34N4O4 [M+H]+ 478.2580, found 479.2675. Anal. calcd for C27H36Cl2N4O4: C, 58.80; H, 6.58; N, 10.16. Found: C, 58.48; H, 6.13; N, 9.85.
3-(3-(4-(6-(2-oxobenzo[d]thiazol-3(2H)-yl)hexyl)piperazin-1-yl)propyl)benzo[d]thiazol-2(3H)-one 5n
General Procedure D, yield 49%, colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.54 – 6.94 (m, 8H), 3.98 (t, J = 7.0, 2H), 3.90 (t, J = 7.1, 2H), 2.66 – 2.13 (m, 12H), 1.92 – 1.85 (m, 2H), 1.76 – 1.68 (m, 2H), 1.62 – 1.27 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 169.78, 169.69, 137.39, 137.16, 126.20, 126.14, 122.88, 122.85, 122.83, 122.74, 122.66, 122.60, 122.50, 110.75, 110.51, 58.46, 55.10, 53.21, 53.06, 42.73, 40.99, 27.51, 27.15, 26.67, 26.64, 24.83. HRMS calcd for C27H34N4O2S2 [M+H]+ 510.2123, found 511.2226. Anal. calcd for C27H36Cl2N4O2S2: C, 55.56; H, 6.22; N, 9.60. Found: C, 55.19; H, 6.57; N, 10.03.
3-(5-(4-(4-(2-oxobenzo[d]thiazol-3(2H)-yl)butyl)piperazin-1-yl)pentyl)benzo[d]thiazol-2(3H)-one 5p
General Procedure D, yield 54%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.57 – 6.85 (m, 8H), 3.94 – 3.88 (m, 4H), 2.64 – 2.19 (m, 12H), 1.74 – 1.69 (m, 4H), 1.63 – 1.44 (m, 4H), 1.40 – 1.34 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 169.71, 169.68, 137.13, 126.21, 122.89, 122.80, 122.78, 122.60, 122.57, 110.66, 110.50, 58.29, 57.61, 53.20, 53.10, 42.70, 42.52, 27.42, 26.40, 25.38, 24.72, 23.85. HRMS calcd for C27H34N4O2S2 [M+H]+ 511.2123, found 511.2203. Anal. calcd for C27H36Cl2N4O2S2: C, 55.56; H, 6.22; N, 9.60. Found: C, 56.14; H, 6.17; N, 9.69.
3-(6-(4-(4-(2-oxobenzo[d]oxazol-3(2H)-yl)butyl)piperazin-1-yl)hexyl)benzo[d]oxazol-2(3H)-one 5q
General Procedure D, yield 48%, yellow oil. 1H NMR (400 MHz, D2O) δ 7.18 (m, 8H), 3.84 (t, J = 6.1, 2H), 3.76 (t, J = 6.7, 2H), 3.53 (s, 8H), 3.21 (t, J = 7.3, 2H), 3.15 (t, J = 7.5, 2H), 1.90 – 1.54 (m, 8H), 1.31 (m, 4H). 13C NMR (101 MHz, D2O) δ 156.44, 142.36, 142.31, 130.66, 130.46, 124.41, 124.34, 123.11, 122.94, 110.18, 110.07, 109.56, 109.38, 56.78, 56.76, 56.22, 48.71, 48.66, 41.88, 41.18, 26.59, 25.17, 25.09, 24.09, 23.17, 20.73. HRMS calcd for C28H36N4O4 [M+H]+ 492.2737, found 493.2828. Anal. calcd for C28H38Cl2N4O4: C, 59.47; H, 6.77; N, 9.91. Found: C, 59.14; H, 6.53; N, 10.26.
3-(6-(4-(4-(2-oxobenzo[d]thiazol-3(2H)-yl)butyl)piperazin-1-yl)hexyl)benzo[d]thiazol-2(3H)-one 5r
General Procedure D, yield 50%, colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.59 – 6.82 (m, 8H), 3.98 – 3.76 (m, 4H), 2.80 – 2.06 (m, 12H), 1.75 – 1.65 (m, 4H), 1.61 – 1.24 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 169.73, 137.08, 137.05, 126.24, 122.93, 122.91, 122.74, 122.72, 122.60, 122.58, 110.69, 110.54, 58.49, 57.61, 53.15, 53.03, 42.69, 42.48, 27.51, 27.14, 26.66, 26.62, 25.36, 23.81. HRMS calcd for C28H36N4O2S2 [M+H]+ 524.2280, found 525.2384. Anal. calcd for C28H38Cl2N4O2S2: C, 56.27; H, 6.41; N, 9.37. Found: C, 56.31; H, 6.66; N, 9.01.
3-(5-(4-(6-(2-oxobenzo[d]oxazol-3(2H)-yl)hexyl)piperazin-1-yl)pentyl)benzo[d]oxazol-2(3H)-one 5s
General Procedure D, yield 55%, colorless oil. 1H NMR (400 MHz, D2O) δ 7.22 – 7.22 (m, 8H), 3.87 – 3.67 (m, 4H), 3.55 (s, 8H), 3.18 -3.14 (m, 4H), 1.81 – 1.52 (m, 8H), 1.35 – 1.30 (m, 6H). 13C NMR (101 MHz, D2O) δ 156.49, 142.39, 142.37, 130.73, 130.67, 124.41, 124.39, 123.04, 122.99, 110.16, 110.13, 109.62, 109.55, 56.74, 48.59, 48.55, 41.93, 41.67, 34.61, 26.62, 26.40, 25.20, 25.12, 23.18, 22.93, 22.76. HRMS calcd for C29H38N4O4 [M+H]+ 506.2893, found 507.2983. Anal. calcd for C29H40Cl2N4O4: C, 60.10; H, 6.96; N, 9.67. Found: C, 59.84; H, 6.81; N, 9.29.
3-(5-(4-(6-(2-oxobenzo[d]thiazol-3(2H)-yl)hexyl)piperazin-1-yl)pentyl)benzo[d]thiazol-2(3H)-one 5t
General Procedure D, yield 56%, yellow oil. 1H NMR (400 MHz, DMSO) δ7.74 – 7.08 (m, 8H), 3.96 – 3.91 (m, 4H), 3.70 – 3.06 (m, 12H), 1.91 – 1.48 (m, 8H), 1.45 – 1.18 (m, 6H). 13C NMR (101 MHz, D2O) δ 156.45, 142.31, 142.30, 130.65, 130.58, 124.33, 124.32, 122.97, 122.92, 110.08, 110.05, 109.55, 109.48, 56.76, 48.52, 48.50, 41.85, 41.59, 34.51, 34.46, 26.57, 26.35, 25.14, 25.05, 23.12, 22.87, 22.68. HRMS calcd for C29H38N4O2S2 [M+H]+ 538.2436, found 539.2540. Anal. calcd for C29H40Cl2N4O2S2: C, 56.94; H, 6.59; N, 9.16. Found: C, 57.15; H, 6.09; N, 9.52.
General Procedure E. Synthesis of benzo[d]thiazole-2(3H)-thione 6b
Lawesson's reagent (3.56 g, 8.82 mmol) was added, under stirring, to a solution of benzo[d]thiazol-2(3H)-one (2 g, 13.24 mmol) in anhydrous toluene (80 mL). The reaction mixture was heated under reflux for 5 h. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using hexane/ethyl acetate (7: 3) as eluent to give 1.4 g (63%) of 6b as a white solid. 1H NMR (400 MHz, DMSO) 13.72 (s, SH, 1H), 7.76 – 7.06 (m, 4H). 13C NMR (101 MHz, DMSO) δ 190.28, 141.70, 129.80, 127.54, 124.60, 122.14, 112.86. MS (EI) m/z 168 (M++1).
Benzo[d]oxazole-2(3H)-thione 6a
General Procedure E, yield 65%, white solid. 1H NMR (400 MHz, DMSO) δ 13.16 (s, SH, 1H), 7.10 – 6.67 (m, 4H). 13C NMR (101 MHz, DMSO) δ 187.52, 138.94, 127.04, 124.78, 121.84, 119.39, 110.10. MS (EI) m/z 152 (M++1).
General Procedure F. Synthesis of 2-(4-bromobutylthio)benzo[d]thiazole 7b
K2CO3 (2.47 g, 17.94 mmol) and 1,4-dibromobutane (5.65 mL, 47.84 mmol) were added, under stirring, to a solution of 6b (1.0 g, 5.98 mmol) in anhydrous DMF (20 mL). The reaction mixture was heated at 60 °C for 2 h. After cooling, the reaction mixture was poured into 70 mL water and extracted with ethyl acetate (3 × 40 mL). The combined organic layers were washed with saturated aqueous NaCl and dried over sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using hexane/ ethyl acetate (8: 2) as eluent to give 1.2 g (67%) of 7b as yellow oil. 1H NMR (CDCl3): δ 7.95 – 7.24 (m, 4H), 3.47 (t, J = 5.3, 2H), 3.40 (t, J = 6.7, 2H), 2.16 – 1.90 (m, 4H). 13C NMR (CDCl3) δ 166.58, 153.23, 135.21, 126.06, 124.25, 121.51, 120.98, 32.83, 32.49, 31.52, 27.88. MS (EI) m/z 302 (M++1).
2-(4-bromobutylthio)benzo[d]oxazole 7a
General Procedure F, yield 69%, dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.43 – 6.80 (m, 4H), 3.89 (t, J = 6.4, 2H), 3.37 (t, J = 5.9, 2H), 1.85 – 1.81 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 169.71, 136.82, 126.45, 123.14, 122.70, 122.59, 110.59, 41.66, 33.16, 29.59, 26.17. MS (EI) m/z 286 (M++1).
General Procedure G. Synthesis of 1,4-bis(4-(benzo[d]thiazol-2-ylthio)butyl)piperazine 8b
K2CO3 (0.68 g, 4.98 mmol) and piperazine (0.07 g, 0.83 mmol) were added, under stirring, to a solution of 7b (0.5 g, 1.66 mmol) in anhydrous DMF (10 mL). The reaction mixture was heated at 60 °C for 1.5 h. After cooling, the reaction mixture was poured into 50 mL water and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with saturated aqueous NaCl and dried over sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (SiO2) using dichlormethane/ methanol (9.5: 0.5) as eluent to give 0.53 g (61%) of 8b as a yellow oil. 1H NMR (CDCl3): δ 7.26 (m, 8H), 3.29 (t, J = 7.2, 4H), 2.41 (s, 8H), 2.32 (t, J = 7.2, 4H), 1.82 (m, 4H), 1.62 (m, 4H). 13C NMR (CDCl3) δ 165.01, 151.75, 141.96, 124.18, 123.72, 118.30, 109.76, 57.80, 53.16, 32.17, 27.31, 25.81. HRMS calcd for C26H32N4S4 [M+H]+ 528.1567, found 529.1573. Anal. calcd for C26H34Cl2N4S4: C, 51.90; H, 5.70; N, 9.31. Found: C, 52.11; H, 6.17; N, 9.29.
1,4-bis(4-(benzo[d]oxazol-2-ylthio)butyl)piperazine 8a
General Procedure G, yield 63%, colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.05 – 7.01 (m, 8H), 3.33 (t, J = 7.2, 4H), 2.43 (s, 8H), 2.33 (t, J = 10.2 , 4H), 1.82 (m, 4H), 1.63 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.07, 153.32, 135.16, 125.98, 124.11, 121.44, 120.91, 57.86, 53.17, 33.48, 27.31, 25.94. HRMS calcd for C26H32N4O2S2 [M+H]+ 496.2032, found 497.2035. Anal. calcd for C26H34Cl2N4O2S2: C, 54.82; H, 6.02; N, 9.84. Found: C, 54.97; H, 6.52; N, 10.07.
4.2. Pharmacology
4.2.1. Assay for iNOS inhibition
The assay was performed in mouse macrophages (RAW264.7, obtained from ATCC)) cultured in phenol red free RPMI medium supplemented with 10% bovine calf serum and 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin at 37 °C in an atmosphere of 5% CO2 and 95% humidity. Cells were seeded in 96-well plates (50,000 cells/well) and incubated for 24 hrs for a confluency of 75% or more. Test samples diluted in serum free medium were added and after 30 minutes of incubation LPS (5 μg/mL) was added and cells were further incubated for 24 hrs. The concentration of nitric oxide (NO) was determined by measuring the level of nitrite in the cell culture supernatant by using Griess reagent.24 Percent inhibition of nitrite production by the test compound was calculated in comparison to the vehicle control. IC50 values were obtained from dose curves. L-NMMA was used as positive control.
4.2.2. Assay for NF-κB inhibition
The assay was performed in human chondrosarcoma (SW1353, obtained from ATCC) cells as described earlier.26 Cells were cultured in 1:1 mixture of DMEM/F12 supplemented with 10% FBS, 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin at 37 °C in an atmosphere of 5% CO2 and 95% humidity. Cells (1.2 × 107) were washed once in an antibiotic and FBS-free DMEM/F12, and then resuspended in 500 μL of antibiotic-free DMEM/F12 containing 2.5% FBS. NF-κB luciferase plasmid construct was added to the cell suspension at a concentration of 50 μg/mL and incubated for 5 min at room temperature. The cells were electroporated at 160 V and one 70-ms pulse using BTX disposable cuvettes model 640 (4-mm gap) in a BTX Electro Square Porator T 820 (BTX I, San Diego, CA). After electroporation, cells were plated to the wells of 96-well plates at a density of 1.25 × 105 cells per well. After 24 h, cells were treated with different concentrations of test compound for 30 min prior to the addition of PMA (70 ng/mL) and incubated for 8 h. Luciferase activity was measured using the Luciferase Assay kit (Promega). Light output was detected on a SpectraMax plate reader. Percent inhibition of luciferase activity was calculated as compared to vehicle control and IC50 values were obtained from dose curves. Parthenolide was used as positive control. Sp-1 was used as a control transcription factor which is unresponsive to inflammatory mediators (such as PMA). This is useful in detecting agents that nonspecifically inhibit luciferase expression due to cytotoxicity or inhibition of luciferase enzyme activity.
4.2.3. Assay for in vitro cytotoxicity
Cytotoxicity was determined against four human tumor cell lines [SK-MEL (malignant melanoma); KB (epidermal carcinoma, oral); BT-549 (ductal carcinoma, breast); SK-OV-3 (ovary carcinoma)] and two noncancerous kidney cell lines [Vero cells (African green monkey kidney fibroblasts) and LLC-PK11 (pig kidney epithelial cells)] as described earlier.18 All the cell lines were obtained from ATCC and cultured in RPMI-1640 medium supplemented with bovine calf serum (10%) and amikacin (60 mg/L), at 37°C, 95% humidity, 5% CO2. Cells were seeded at a density of 25,000 cells/well and grown for 24 h. Samples, diluted appropriately in serum free medium, were added to the cells and again incubated for 48 h. The number of viable cells was determined by Neutral Red assay.30 Percent decrease in cell viability was calculated in comparison to vehicle control. IC50 (the concentration of the test compound that caused a 50% decrease in cell viability) was calculated from the dose curves. Doxorubicin was used as positive control. All assays were performed in triplicate and the mean values are given in Table 1.
4.2.4. In vivo assays: (Acetic acid-induced writhing in mice)
Male Swiss mice (20-30g, Harlan, Indianapolis, IN, USA) were grouped housed (n=3) in polycarbonate cages (20×35×12 cm). Food (Purina 5001 Laboratory Rodent Chow, St Louis, MO, USA) and water were available ad libitum. Room temperature was maintained at 22 +/-1 °C and overhead illumination was maintained on a 12-h light-dark cycle.
Procedure: Acetic acid-induced writhing assay was performed as mentioned before.31 Briefly, mice received IP injections of test compounds or vehicle 15 min before nociceptive testing. The positive control 3 mg/kg indomethacin was administered IP 30 min before nociceptive testing. Subjects received 15 min of apparatus habituation in a 15 cm diameter acrylic observation enclosure immediately before i.p. injection of 0.9% acetic acid (1 ml/ 0.1 kg). The number of writhing responses was counted for 20 minutes. In this assay, it is typical for 10 – 20% of vehicle treated animals to be non-responders.32 In the present study 5 of the 36 subjects (i.e. 13.9%) did not respond to acetic acid injections and their data were omitted prior to statistical analyses; this yielded groups sizes of n = 4-6. Data were analyzed one-way ANOVA followed by Dunnett Multiple Comparisons Test. These procedures were conducted under the ethical standards of the International Association for the Study of Pain and in accordance with the principles of laboratory animal care as detailed in the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985) and were approved by the University of Mississippi IACUC (Protocol # 10-007).
4.2.5. Carrageenan-induced rat paw edema assay
Adult albino rats of both sexes weighing between 120–150 g were used. Rats were uniformly hydrated by giving 3 ml water/rat through gastric inoculation to reduce variability to edema response. Animals were divided into groups each of five animals. The control group was given saline containing few drops of Tween 80. Test compounds or standard anti-inflammatory compound, ketorolac tromethamine, were suspended in distilled water by the aid of few drops of Tween 80 and administrated intraperitoneally one hour before induction of inflammation. Induction of inflammation was performed by S.C. injection of 50 ml of 1% carrageenan-sodium gel (Sigma–Aldrich, USA), into the subplantar region of the right hind paw. The dorso-ventral diameter (thickness) of the right and left hind paws of each rat was measured using a pair of dial thickness gauge calipers accurate to 0.0001 cm 1 h, 2 h, 3 h and 4 h after induction of inflammation. The left hind paw diameter served as a control for the degree of inflammation in the right hind paw. The percentage of anti-inflammatory activity (% inhibition of inflammation) was calculated according to the following equation: % inhibition= (1 – Lt/Lc) × 100
Lt is the mean increase in paw thickness in rats treated with the tested compounds.
Lc is the mean increase in paw thickness in control group.
Data were analyzed and results were presented in Table 2.
5.2. Docking study
The binding site was generated from the co-crystallized ligand (AR-C120011) within iNOS protein (PDB code: 3E65). Selected active ligands were docked using the following docking configuration: (i) number of Monte Carlo search trials = 30000, search step for torsions with polar hydrogens = 30°. (ii) The Root Mean Square Difference (RMS) threshold for ligand-to-binding site shape match was set to 2.0 employing a maximum of 1.0 binding site partitions and 1.0 site partition seed. (iii) The interaction energies were assessed employing Consistent Force Field (CFF) force field with a non-bonded cutoff distance of 10.0 Å and distance-dependent dielectric. An energy grid extending 3.0 Å from the binding site was implemented. (iv) Rigid body ligand minimization parameters were: 10 iterations of steepest descend (SD) minimization followed by 20 Broyden–Fletcher–Goldfarb–Shanno (BFGS) iterations applied to every successful orientation of the docked ligand. (v) A maximum of 10 diverse docked conformations/poses of optimal interaction energies were saved. (vi) The saved conformers/poses were further energy-minimized within the binding site for a maximum of 1000 rigid-body iterations. (vii) Six scoring functions were used to score the high ranking docked poses/conformers: Jain,33-35 LigScore1, LigScore2,34,35 PLP1,36 PLP2 and PMF.37,38
Acknowledgments
This work was supported in part by NIGMS P20 GM104932 (CRM). The authors wish to thank Prof. Dr. Mutasem O. Taha, Jordan University for his help and valuable discussion in performing the docking study. Also, we deeply thank Mr. Paul Bates and Ms. Katherine Martin for excellent technical help in carrying out the in vitro bioassays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Geronikaki AA, Lagunin AA, Hadjipavlou-Litina DI, Eleftheriou PT, Filimonov DA, Poroikov VV, Alam I, Saxena AK. J. Med. Chem. 2008;51:1601. doi: 10.1021/jm701496h. [DOI] [PubMed] [Google Scholar]
- 2.Gautam R. a. J., S. M. Med. res. rev. 2009;29:767. doi: 10.1002/med.20156. [DOI] [PubMed] [Google Scholar]
- 3.Schmitz ML BS. Phytochem. rev. 2005;4:19. [Google Scholar]
- 4.Bremner P HM. Phytoch. rev. 2005;4:27. [Google Scholar]
- 5.Levy D, Zochodne DW. Pain Pract. 2004;4:11. doi: 10.1111/j.1533-2500.2004.04002.x. [DOI] [PubMed] [Google Scholar]
- 6.Alderton WK, Cooper CE, Knowles RG. Biochem. J. 2001;357:593. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne JE, Bonnefous C, Symons KT, Nguyen PM, Sablad M, Rozenkrants N, Zhang Y, Wang L, Yazdani N, Shiau AK, Noble SA, Rix P, Rao TS, Hassig CA, Smith ND. J. Med. Chem. 2010;53:7739. doi: 10.1021/jm100828n. [DOI] [PubMed] [Google Scholar]
- 8.Corea G, Fattorusso E, Lanzotti V, Di Meglio P, Maffia P, Grassia G, Ialenti A, Ianaro A. J. Med. Chem. 2005;48:7055. doi: 10.1021/jm050321r. [DOI] [PubMed] [Google Scholar]
- 9.Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, Park JS, Cho HJ. Eur. J. Neurosci. 2004;19:3375. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang M, Zhao M, Qiu H, Shi D, Wang J, Tian Y, Lin L, Deng W. PLoS One. 2014;9:109951. doi: 10.1371/journal.pone.0109951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roman-Blas JA, Jimenez SA. Osteoarthr. Cartil. 2006;14:839. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Karin M, Greten FR. Nat. Rev. Immunol. 2005;5:749. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 13.Karin M, Cao Y, Greten FR, Li ZW. Nat. Rev. Cancer. 2002;2:301. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 14.Balkwill F, Mantovani A. Lancet. 2001;357:539. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 15.Karin M. Nature. 2006;441:431. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 16.Baud V, Karin M. Nat. Rev. Drug Discov. 2009;8:33. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin W-W, Karin MJ. Clin. Invest. 2007;117:1175. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CH, Jeon YT, Kim SH, Song YS. Biofactors. 2007;29:19. doi: 10.1002/biof.5520290103. [DOI] [PubMed] [Google Scholar]
- 19.Inoue J, Gohda J, Akiyama T, Semba K. Cancer Sci. 2007;98:268. doi: 10.1111/j.1349-7006.2007.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salgin-Goksen U, Gokhan-Kelekci N, Goktas O, Koysal Y, Kilic E, Isik S, Aktay G, Ozalp M. Bioorg. Med. Chem. 2007;15:5738. doi: 10.1016/j.bmc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Wolfe MM, Lichtenstein DR, Singh GN. Engl. J. Med. 1999;340:1888. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 22.Yous S, Poupaert JH, Chavatte P, Espiard JG, Caignard DH, Lesieur D. Drug Des. Discov. 2001;17:331. [PubMed] [Google Scholar]
- 23.Viaud M-C, Jamoneau P, Flouzat C, Bizot-Espiard J-G, Pfeiffer B, Renard P, Caignard D-H, Adam G, Guillaumet GJ. Med. Chem. 1995;38:1278. doi: 10.1021/jm00008a006. [DOI] [PubMed] [Google Scholar]
- 24.Quang DN, Harinantenaina L, Nishizawa T, Hashimoto T, Kohchi C, Soma G, Asakawa Y. Biol. Pharm. Bull. 2006;29:34. doi: 10.1248/bpb.29.34. [DOI] [PubMed] [Google Scholar]
- 25.Ma G, Khan SI, Benavides G, Schuhly W, Fischer NH, Khan IA, Pasco DS. Cancer Chemother. Pharmacol. 2007;60:35. doi: 10.1007/s00280-006-0344-0. [DOI] [PubMed] [Google Scholar]
- 26.Winter CA, Risley EA, Nuss GW. Proc. Soc. Exp. Biol. Med. 1962;111:544. doi: 10.3181/00379727-111-27849. [DOI] [PubMed] [Google Scholar]
- 27.Mustafa J, Khan SI, Ma G, Walker LA, Khan IA. Lipids. 2004;39:167. doi: 10.1007/s11745-004-1215-5. [DOI] [PubMed] [Google Scholar]
- 28.Accelrys Software Inc. Discovery Studio Modeling Environment, Release 2.5. Accelrys Software Inc.; San Diego: 2007. [Google Scholar]
- 29.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Nat. Chem. Biol. 2008;4:700. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borenfreund E, Babich H, Martin-Alguacil N. In Vitro Cell Dev. Biol. 1990;26:1030. doi: 10.1007/BF02624436. [DOI] [PubMed] [Google Scholar]
- 31.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Proc. Natl. Acad. Sci. USA. 2003;100:9044. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeffrey M, Sonya W, You W. Assessing Nociception in Murine Subjects, Methods in Pain Research. CRC Press; 2001. [Google Scholar]
- 33.Jain AN. J. Comput. Aided Mol. Des. 1996;10:427. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 34.Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. J. Mol. Graph. Model. 2005;23:395. doi: 10.1016/j.jmgm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Venkatachalam CM, Jiang X, Oldfield T, Waldman M. J Mol Graph Model. 2003;21:289. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- 36.Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DB, Fogel LJ, Freer ST. Chem Biol. 1995;2:317. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 37.Muegge I. J. Comput. Chem. 2001;22:418. [Google Scholar]
- 38.Muegge I, Martin YC. J. Med. Chem. 1999;42:791. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]