

Abstract
Mutations within the Aβ (amyloid β) peptide, especially those clustered at residues 21–23, are linked to early-onset AD (Alzheimer’s disease) and primarily associated with cerebral amyloid angiopathy. The Iowa variant, a substitution of an aspartic acid residue for asparagine at position 23 (D23N), associates with widespread vascular amyloid and abundant diffuse pre-amyloid lesions significantly exceeding the incidence of mature plaques. Brain Iowa deposits consist primarily of a mixture of mutated and non-mutated Aβ species exhibiting partial aspartate isomerization at positions 1, 7 and 23. The present study analysed the contribution of the post-translational modification and the D23N mutation to the aggregation/fibrillization and cell toxicity properties of Aβ providing insight into the elicited cell death mechanisms. The induction of apoptosis by the different Aβ species correlated with their oligomerization/fibrillization propensity and β-sheet content. Although cell toxicity was primarily driven by the D23N mutation, all Aβ isoforms tested were capable, albeit at different time frames, of eliciting comparable apoptotic pathways with mitochondrial engagement and cytochrome c release to the cytoplasm in both neuronal and microvascular endothelial cells. Methazolamide, a cytochrome c release inhibitor, exerted a protective effect in both cell types, suggesting that pharmacological targeting of mitochondria may constitute a viable therapeutic avenue.
Keywords: cerebral amyloid angiopathy, familial Alzheimer’s disease, methazolamide, mitochondrial dysfunction, post-translational modification
INTRODUCTION
Aβ (amyloid β) peptide is the major constituent of the fibrils deposited in parenchymal plaques and cerebral blood vessels of patients with AD (Alzheimer’s disease) and Down’s syndrome. It is an internal processing product of a larger transmembrane precursor molecule known as APP (amyloid precursor protein) encoded by a gene located on chromosome 21 [1,2]. The presence of mutations in the APP gene has been linked to familial forms of the disease that typically associate with early onset phenotypes [3,4]. However, the clinical manifestations differ with the type of amino acid substitution and the localization of the mutated residue within APP. Substitutions flanking the coding region for Aβ affect the processing of APP with either overproduction of Aβ or predominant generation of Aβ42, and are clinically associated with AD phenotypes. In contrast, mutations located within the Aβ sequence, predominantly those clustered at positions 21–23, are primarily linked to the development of CAA (cerebral amyloid angiopathy), although, depending on the genetic variant, they may manifest with either cerebral haemorrhage or dementia [1,5].
The Iowa variant, an autosomal dominant substitution of an aspartate residue for asparagine occurring at position 23 of Aβ (D23N), associates with cognitive impairment. Data from affected members showed onset of progressive, AD-like dementia in the sixth to seventh decade of life with cerebral atrophy, widespread neurofibrillary tangles, leukoencephalopathy and occipital lesions constituted by calcified amyloid-laden meningeal vessels. Vascular amyloid deposits together with abundant diffuse pre-amyloid lesions are predominant neuropathological features of the disease, significantly exceeding the incidence of neuritic plaques [6]. Sections of the cerebral cortex and white matter show severe amyloid angiopathy with the majority of meningeal and cortical vessels exhibiting thickened walls and reduced lumina, and many small blood vessels appearing entirely occluded. Although micro-haemorrhages could be identified by MRI and post-mortem examination, clinically manifested intracerebral haemorrhages have not been reported in this kindred. In contrast, a second family from Spain carrying the same mutation presented symptomatic cerebral haemorrhage in most of the affected members [7], suggesting that the presence of the mutation is not in itself sufficient for the induction of a specific clinical phenotype, and that other still undefined factors likely contribute to the diverse clinical presentation.
Biochemical analyses after sequential tissue extraction revealed a complex composition of the brain Iowa deposits. Amyloid lesions primarily consisted of a mixture of mutated and non-mutated Aβ molecules, presenting various degrees of solubility and partial aspartate isomerization at positions 1, 7 and 23 [8], all elements with the potential to play a significant role in disease pathogenesis. In general terms, the presence of intra-Aβ mutations has been shown to correlate in vivo with a decrease in the age of onset of the disease and in vitro with accelerated aggregation kinetics [9–11]. The formation of isoD (isoaspartate), a post-translational change resulting either from isomerization of aspartate or deamidation of asparagine residue, both chemically spontaneous non-enzymatic reactions, occurs during aging. IsoD has been reported in Aβ deposits in sporadic AD, in which isomerized Aβ peptides are found in senile plaques and amyloid-bearing vessels [12], as well as in diffuse plaques in Down’s syndrome cases [13]. The presence of isoD introduces an additional methylene group in the peptide backbone, with potential to alter structure and function influencing substrate recognition and turnover by proteases. In the present study, we analysed the influence of the D23N mutation and the presence of isoD residues on the aggregation properties of Aβ, assessing the similarities/differences in oligomerization/fibrillization kinetics among the different isoforms, and the cell death mechanisms elicited in neuronal cells and cerebral microvascular ECs (endothelial cells). The protective effect exerted by methazolamide, a CytC (cytochrome c) release inhibitor, highlights the potential of mitochondria pharmacological targeting as a viable therapeutic avenue.
MATERIALS AND METHODS
Peptide synthesis
Synthetic homologues of WT (wild-type) Aβ40, Aβ(isoD) (Aβ40 with isoD modifications at residues 1, 7 and 23), Aβ-Iowa (Aβ40 containing the D23N genetic variant) and Aβ-Iowa(isoD) (Aβ-Iowa bearing isoD modifications at residues 1 and 7) were synthesized using N-t-butyloxycarbonyl chemistry by James I. Elliott at Yale University (New Haven, CT, U.S.A.) and purified by reverse-phase HPLC on a Vydac C4 column (Western Analytical). Molecular masses were corroborated by MALDI–TOF-MS and concentrations were assessed by amino acid analysis as described previously [14].
Peptide aggregation
Synthetic Aβ homologues were dissolved to 1 mM in HFIP (hexafluoroisopropanol; Sigma), a pre-treatment that breaks down β-sheet structures and disrupts hydrophobic forces leading to monodisperse Aβ preparations [15]. After overnight incubation and lyophilization to remove HFIP, peptides were dissolved to 1.5 mM in 0.1%ammonium hydroxide followed by the addition of deionized water and 2-fold concentrated PBS (pH 7.4) to a final concentration of 1 mg/ml in PBS. Reconstituted peptides were incubated at 37°C for up to 3 days for the aggregation studies. Structural properties of the Aβ synthetic homologues at different time points were assessed by WB (Western blot) analysis under non-denaturing conditions, CD spectroscopy, Thioflavin T binding and TEM (transmission electron microscopy) as described below. For cell culture experiments, peptides were dissolved to 2 mM in 0.1% ammonium hydroxide followed by the addition of deionized water to 1 mM, and diluted into the pertinent culture medium at the required concentration.
CD spectroscopy
Changes in the secondary structure of the different Aβ peptides were estimated by CD spectroscopy as described previously [14]. Spectra in the far-UV light (wavelength range 190–260 nm; bandwidth 1 nm; intervals 1 nm; scan rate 60 nm/min) yielded by the different peptides at various time points of aggregation were recorded at 24°C with a Jasco J-720 spectropolarimeter, using a 0.2 mm path quartz cell and a peptide concentration of 1 mg/ml. For each sample, 15 consecutive spectra were obtained, averaged and baseline subtracted. Results are expressed in terms of molar ellipticity (°C · cm2 · dmol− 1).
Thioflavin T binding assay
Binding of the different Aβ peptides to Thioflavin T was monitored by fluorescence evaluation as described previously [14,16]. Briefly, 6 μl aliquots from each of the peptide aggregation time points were added to 184 μl of 50 mM Tris/HCl buffer, pH 8.5, and 10 μl of 0.1 mM Thioflavin T (Sigma). Fluorescence was recorded for 300 s in a LS-50B luminescence spectrometer (PerkinElmer) with excitation and emission wavelengths of 435 and 490 nm (slit width 10 nm) respectively as described previously [17].
TEM
Aliquots (3 μl) of the 3 h and 1 day time-point samples, aggregated at a concentration of 50 μM in EBM-2 (endothelial basal medium-2), were placed on to carbon-coated 400-mesh Cu/Rh grids (Ted Pella) and stained with 1% uranyl acetate in distilled water (Polysciences). Stained grids were examined in a Philips CM-12 TEM and images acquired with a Gatan (4k×4k) digital camera at the Microscopy Core Facility of NYU Langone Medical Center (New York, NY, U.S.A.) as described previously [14].
Native gel electrophoresis and WB analysis
Electrophoretic analysis for assessment of peptide aggregation was performed under native conditions using 5–30% gradient polyacrylamide gels, in the absence of SDS, using 25 mM Tris/glycine, pH 8.8, as running buffer, and molecular mass markers consisting of proteins with acidic pI (human albumin, ovalbumin, soybean trypsin inhibitor, lactoglobulin and insulin). Once the proteins reach their pore limit at the end of the run, the log of their molecular mass is proportional to the log of the protein’s relative mobility normalized to the dye front (RF). Estimation of the molecular mass of the different Aβ species was assessed by interpolation of the respective log RF values into the calibration curve generated from the standard proteins with the aid of GraphPad Prism software (GraphPad) as described previously [18]. Aβ oligomerization patterns were visualized by subsequent WB analysis. Briefly, after electrophoretic separation, proteins were electrotransferred on to nitrocellulose membranes (0.45 μM pore size; Hybond-ECL, GE Healthcare Life Sciences) at 400 mA for 2.5 h, using 10 mM Caps [3-(cyclohexylamino)propane-1-sulfonic acid; Sigma] buffer, pH 11.0, containing 10% (v/v) methanol. After blocking with 5% non-fat dried skimmed milk powder in TBST (TBS containing 0.1% Tween 20), membranes were immunoreacted with rabbit polyclonal antibodies specific against the C-terminus of Aβ40 (1:1000 dilution; Invitrogen), followed by incubation with HRP (horseradish peroxidase)-conjugated F(ab′)2 anti-(rabbit IgG) (1:5000 dilution; GE Healthcare) [14]. Fluorograms were developed by ECL with ECL WB detection reagent (GE Healthcare).
Dot-blot analysis
Oligomer formation during the peptide aggregation experiments was assessed by Dot-blot using rabbit anti-oligomer polyclonal (A11) antibody (Invitrogen) [19], as described previously [14]. Briefly, 800 ng aliquots of each of the aggregation data point samples were loaded on to a nitrocellulose membrane assembled into a Bio-Dot Microfiltration Apparatus (Bio-Rad Laboratories) and allowed to diffuse passively for 30 min before vacuum application. The membrane was then blocked in situ for 1 h with 1% non-fat dried skimmed milk powder in TBST, followed by vacuum application and two subsequent washes with TBST. After removal from the dot-blot apparatus and further blocking with 5% non-fat dried skimmed milk powder in TBST [1 h at room temperature (21°C)], the membrane was incubated overnight with A11 antibody (1:1000 dilution) followed by HRP-conjugated anti-rabbit secondary antibody. Immunoreactivity was assessed by ECL as above.
Cell cultures
Immortalized human brain microvascular ECs (HCMEC/D3, abbreviated as ECs) were obtained from Babette Weksler (Division of Hematology and Medical Oncology, Weill Medical College of Cornell University, NY, U.S.A.) [20] and maintained in complete EBM-2 (Lonza) with added growth supplements and 5% FBS. This cell line retains the morphological characteristics of primary brain ECs and expresses specific brain endothelial markers and cell-surface adhesion molecules. Human neuroblastoma cells (SH-SY5Y) were obtained from the A.T.C.C. (Manassas, VA, USA) and maintained in DMEM (Dulbecco’s modified Eagle’s medium) (Mediatech) with 10% FBS.
Cell death ELISA
The extent of apoptosis caused by the different Aβ peptides was assessed by quantification of histone–DNA complex fragments with Cell Death Detection ELISAplus (Roche) as described previously [14,17]. Cells (2×104/well) were seeded on to 24-well plates and allowed to attach for 1 day before treatment with the different Aβ peptides. WT Aβ40, Aβ(isoD), Aβ-Iowa and Aβ-Iowa(isoD), previously pre-treated in HFIP and solubilized as above, were diluted to a final concentration of 50 μM in EBM-2/1% FBS medium for EC challenge and DMEM without FBS for SH-SY5Y treatment. Following 1–3-day incubation with the various peptide homologues, the plates were centrifuged (for 10 min at 150 g; Beckman J-6B, Beckman Instruments) to collect the detached cells. After cell lysis, fragmented DNA–histone complexes (mono- and oligonucleosomes) were quantified by Cell Death Detection ELISAplus following the manufacturer’s specifications. Briefly, nucleosome-containing cell lysates were placed into streptavidin-coated microplate wells and added to a mixture of biotin-labelled anti-histone and HRP-conjugated anti-DNA antibodies. Following incubation, anti-histone antibodies immunoreact with the histone component of the nucleosomes capturing the complex on to the streptavidin-coated wells. Following binding of the anti-DNA antibodies to this complex, peroxidase activity, which is proportional to the amount of nucleosomes present in the cell lysates, was quantified photometrically with ABTS [2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)] substrate. For 3-day peptide treatments, exhibiting high apoptotic levels, a double volume of lysis buffer was used to avoid subsequent saturation of the colorimetric system.
Immunocytochemical evaluation of mitochondrial CytC release
Both EC and SH-SY5Y cells were plated on to glass chamber slides (Thermo Fisher Scientific), pre-coated with either collagen-I or poly-D-lysine (for ECs and SH-SY5Y cells respectively). After seeding, cells were allowed to attach for 1 day before treatment with the different peptides for 1–3 days, as above. Cells were washed with ice-cold PBS, fixed with 4%paraformaldehyde (10 min at room temperature) and blocked for 1 h with 20 mg/ml BSA in PBST (PBS containing 0.3%Triton X-100). Slides were further incubated with mouse anti-CytC monoclonal antibody (BD Biosciences; 1:200 dilution in PBST containing 5 mg/ml BSA; 2 h at room temperature) followed by Alexa Fluor® 488-conjugated anti-(mouse IgG) antibody (Invitrogen;1:200 in PBST with 5 mg/ml BSA; 1 h at room temperature). Fluorescence signals were visualized in a Nikon Eclipse E 800 deconvolution microscope using NIS Elements software (Nikon Instruments) for image acquisition and processing, and AutoQuant (Media Cybernetics) for 3D deconvolution.
Inhibition of CytC release by methazolamide
The effect of methazolamide in preventing the Aβ-induced release of CytC from the mitochondria into the cytoplasm was evaluated in EC and SH-SY5Y cultures. After peptide incubation in the presence and absence of methazolamide (Sigma), release of CytC was visualized by immunofluorescence microscopy and WB analysis, and corroborated by confocal assessment of CytC in conjunction with the mitochondrial marker MitoTracker®.
Immunofluorescence deconvolution microscopy
Detection of CytC after 1 day incubation with the most toxic of the Aβ peptides studied in the present paper, Aβ-Iowa(isoD), in the presence of methazolamide was performed exactly as described above.
WB analysis
Subcellular distribution of CytC in amyloid-challenged EC and SH-SY5Y cells was determined using mitochondrial protein extracts, prepared essentially as described previously [21]. Briefly, cells were collected in homogenization buffer [75 mM sucrose, 225 mM mannitol, 5 mM Tris/HCl, pH 7.4, containing 1 mM PMSF and CompleteTM protease inhibitor cocktail (Roche)] and disrupted with the aid of a Dounce glass homogenizer. Cell homogenates were centrifuged to remove unbroken cells and nuclei (600 g, 5 min, 4°C) and supernatants further centrifuged at 10300 g (5 min, 4°C) to subfractionate the mitochondria. The pellet, containing the mitochondrial fraction, was resuspended in homogenization buffer and further sonicated (Heat Systems Sonicator W-380, Ultrasonics; three 5 s cycles, power setting 8). For WB analysis, typically 10 μg of mitochondrial total proteins, assessed with BCA protein assay (Thermo Scientific), were separated on 16.5% polyacrylamide gels and electrotransferred to PVDF membranes (Immobilon, Millipore; 0.45 μm pore; 400 mA, 1.5 h) using Caps buffer, as above. Membranes were blocked with 5% non-fat dried skimmed milk powder in TBST, and subsequently immunoreacted with mouse anti-CytC monoclonal antibody (1:1000 dilution in 5% non-fat dried skimmed milk powder in TBST, overnight, 4°C) followed by HRP-labelled anti-(mouse IgG) antibody (1:10000 dilution; GE Healthcare), and ECL detection, as above. Densitometric assessment of band intensities was performed using ImageJ (NIH) software.
Confocal analysis
EC and SH-SY5Y cells (6×104), plated on to chamber slides coated with attachment factor for glass (Cell Systems), were treated with Aβ-Iowa (50 μM; 16 h) in the presence and absence of methazolamide (100 and 300 μM). After 30 min incubation with MitoTracker® Red CM-H2XRos (Life Technologies; 1.5 μM), cells were washed with warm PBS, fixed with 4% paraformaldehyde, and subjected to CytC immunocytochemistry as in immunofluorescence deconvolution microscopy. Confocal analysis was performed using a Zeiss LSM 510 microscope.
Prevention of Aβ-mediated apoptosis by inhibition of mitochondrial CytC release
Confirmation of CytC involvement in the Aβ-mediated activation of downstream cell-death pathways was achieved through evaluation of the protective effect of methazolamide by Cell Death Detection ELISAplus. Aβ peptides were incubated with EC and SH-SY5Y cultures under the same conditions listed above, in the presence and absence of 300 μM methazolamide, a concentration known to cause maximal inhibition of CytC release in EC treated with a different Aβ genetic variant [14]. The length of incubation with the different peptides was selected to yield maximal apoptosis in the absence of inhibitor and therefore varied depending on the inherent pro-apoptotic capabilities of the different peptides for the respective cell types. The most aggressive species, Aβ-Iowa(isoD), required shorter challenge (1 day for both EC and SH-SY5Y cells), whereas for the less aggressive peptides in all cases longer incubation times were needed [Aβ-Iowa: 2 days for ECs and 3 days for SH-SY5Y; Aβ(isoD): 3 days for both EC and SH-SY5Y). Induction of apoptosis was evaluated by Cell Death Detection ELISAplus, carried out as above.
Statistical analysis
ANOVA for comparison of multiple groups with Bonferroni or Tukey post-hoc tests was performed using GraphPad InStat (GraphPad). Values of P ≤ 0.05 were considered significant.
RESULTS
Structural analysis of Aβ variant homologues
Changes in the structural conformation and fibrillization propensity of Aβ40 induced by the presence of isoD modifications at positions 1, 7 and 23, and by the D23N mutation were studied by non-denaturing WB, CD spectroscopy, Thioflavin T binding, dot-blot and TEM, following peptide pre-treatment and aggregation as described in the Materials and methods section. As illustrated in Figure 1, WB analysis revealed an enhanced aggregation propensity of Aβ40 when bearing isoD-modified residues, the Iowa mutation at position 23, or a combination thereof. Both Aβ40 and Aβ40(isoD) showed mainly monomeric, dimeric and trimeric components up to 3 h incubation, with HMM (high-molecular-mass) species becoming evident after 6 h and steadily increasing during the 3-day duration of the experiment. However, the HMM signal was more intense for the Aβ(isoD) than for the WT counterpart. In the case of Iowa variant peptides, with and without the isoD modification, HMM aggregates were clearly noticeable after only 1 h incubation and became more relevant towards the end point of the experiment (Figure 1A). Although both Iowa variant homologues followed a comparable aggregation profile in WB, the signal intensity was more pronounced for the isoD-modified peptide, indicative of a higher content of oligomeric components.
Figure 1. Influence of aspartate residue isomerization and the D23N mutation on the structural properties of A β40.

(A) Native WB analysis. Samples collected at different aggregation time points (0–3 days) were separated on non-denaturing 5–30 % gradient gels, electrotransferred on to nitrocellulose, and membranes were probed with an antibody recognizing the C-terminus of Aβ40. Electrophoretic mobilities of monomers (m), dimers (d), trimers (t) and HMM species are indicated. (B) Secondary structure analysis by CD spectroscopy. CD studies were performed after incubation of the different synthetic homologues at 37 °C for up to 3 days. Data represent means of 15 scans after subtraction of background readings of buffer blanks. HFIP consistently induced α-helical structure formation in all peptides tested (exemplified for Aβ40). (C) Oligomerization/fibrillization analysis via Thioflavin T binding. Fluorescence evaluation (excitation 435 nm/emission 490 nm) of Thioflavin T binding at different time points during peptide aggregation was performed as described in the Materials and methods section. Upper panel, fluorescence kinetic measurements within the first 24 h. Bottom panel, Thioflavin T binding data after 3-day aggregation. In both cases, results are expressed in a.u. Data are presented as means ± S.D. for duplicate independent experiments.
Changes in secondary structure at the different time points were analysed by CD spectroscopy. After pre-treatment with HFIP, all peptides adopted the same typical α-helical conformation, exhibiting the classic scan with 2 minima at 208 and 222 nm (shown as a dotted line in Figure 1B for WT Aβ40). Following solubilization in a buffer containing a physiological salt concentration, Aβ40 adopted a typical unordered conformation (minimum at 198 nm) that remained unchanged after 1 day (Figure 1B). β-Sheet components (minimum at 218 nm) became evident after 3 days, although the presence of remaining negative ellipticity values below 200 nm were indicative of the co-existence of some residual random structures at this time point. As expected, and corroborating the WB data in Figure 1(A), Aβ40(isoD) showed a faster shift to β-sheet conformations compared with the unmodified counterpart, with β-structures becoming evident after 1 day; by day 3, residual random structures were still present. The aggressive Aβ-Iowa genetic variant, shown previously to exhibit higher aggregation tendency than WT Aβ40 [8,22], as well as the Aβ-Iowa(isoD), adopted predominantly β-sheet conformations immediately on solubilization, increasing their content in β-sheet components after 1 day of incubation and exhibiting positive ellipticity values below 210 nm. Both Iowa peptides began losing solubility after 3-day incubation, as illustrated by the more flattened spectra at this time point, indicating a lower protein concentration in solution. The effect was more pronounced in Aβ-Iowa(isoD) than in the peptide containing only the D23N substitution, suggesting that the mutation and the post-translational modifications had additive effects on the peptide conformation.
The fibrillization kinetics of the different Aβ species was evaluated with Thioflavin T binding, which displays fluorescence following binding to fibrillar and protofibrillar amyloid aggregates [14,16]. In accordance with the WB/CD data and its known poor fibrillogenic propensity, WT Aβ40 showed very low binding to Thioflavin T (Figure 1C), reaching only approximately 30 a.u. (arbitrary units) after 1-day aggregation and approximately 90 a.u. after 3 days (Figure 1C, bottom panel). As expected, the fibrillization propensity of WT Aβ40 increased with the presence of isoD, although the D23N mutation had a more profound structural effect. Thioflavin T binding values of Aβ(isoD) remained consistently lower than those obtained for Aβ-Iowa and Aβ-Iowa(isoD) throughout the duration of the experiment, with binding levels for the last two peptides reaching a plateau with similar fluorescence (~400 a.u.) after only ~15 h aggregation, a clear indication that the fibrillization mechanism is mainly influenced by the mutation.
Further analysis of the structures present at the different aggregation time points was assessed by TEM and by immunoreactivity with anti-oligomer antibody in dot-blot assays. As illustrated in Figure 2(A), TEM analysis indicated that WT Aβ40 formed only scarce small globular oligomeric structures, which typically precede the formation of protofibrils [23] up to 1-day aggregation. These globular assemblies coexisted with a small number of short protofibrils, structures shorter than 200 nm [16], in samples analysed after 3 days of incubation (results not shown). Aβ(isoD) showed, after 3 h aggregation, the presence of a few protofibrillar elements which increased in length and coexisted with a few slightly longer fibrillar elements (>200 nm) after 1 day. In line with the structural data described above and confirming their faster aggregation kinetics, Aβ-Iowa and Aβ-Iowa(isoD) showed already at the 3 h time point abundant protofibrillar structures coinciding with predominant fibrillar elements after 1-day aggregation. The formation of oligomeric assemblies during the aggregation experiments was evaluated by dot-blot using A11 anti-oligomer antibody as described previously [14]. This conformational antibody is known to recognize soluble oligomeric intermediates while failing to immunoreact with both low-molecular-mass oligomers as well as with Aβ fibrillar species [19]. As illustrated in Figure 2(B) and in agreement with the data shown above, Aβ-Iowa(isoD) aggregated aggressively, with strong oligomer formation after only 1 h, whereas Aβ-Iowa presented signals for A11-positive intermediate-size oligomers slightly later, after 6 h and 1 day. In agreement with TEM images illustrating an increase in fibrillar components with time, both Aβ-Iowa and Aβ-Iowa(isoD) A11 signals decreased at 3 days. Consistent with the slower oligomerization/fibrillization kinetics shown in Figure 1 and Figure 2(A), Aβ(isoD) exhibited A11 immunoreactivity at 1 and 3 days, without reaching the intensity of the Iowa peptide signals, whereas WT Aβ40, in line with its slower aggregation, presented oligomeric species detectable by A11 only after 3-day aggregation.
Figure 2. Structure of the different Aβ variants assessed by TEM and A11-immunoreactivity in dot-blot.
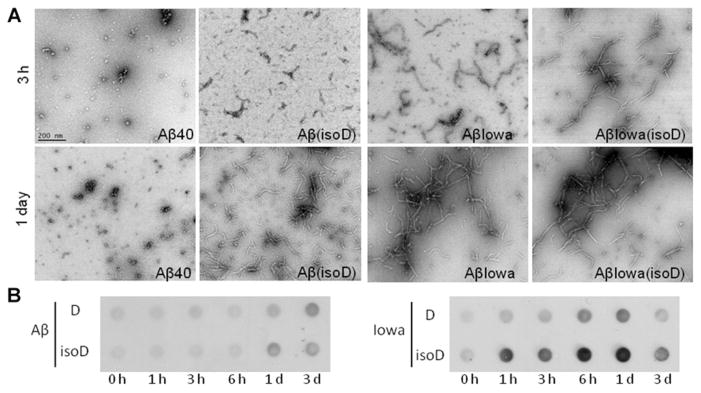
(A) TEM. Analysis of the structure of the different Aβ homologues was performed after aggregation for 3 and 24 h as described in the Materials and methods section. Images were acquired following negative staining with 1 % uranyl acetate, using a Philips CM-12 microscope equipped with a Gatan (4k×4k) digital camera. Magnification 88 000×; scale bar is 200 nm in all images. (B) Dot-blot. Nitrocellulose membranes, loaded with WT Aβ, Aβ(isoD), Aβ-Iowa and Aβ-Iowa(isoD) pre-aggregated 0–3 days, were probed with anti-oligomer (A11) antibody. Images illustrate oligomer immunoreactivity at selected aggregation time points for each variant tested. Left-hand panel, ECL signals of WT Aβ40 (D), and Aβ(isoD); right-hand panel, immunoreactivity of Aβ-Iowa (D) and Aβ-Iowa (isoD).
Aβ-mediated induction of apoptosis and mitochondrial CytC release in neuronal cells and microvascular ECs
The ability of the different Aβ variants studied in the present paper to induce DNA fragmentation, an event indicative of apoptosis, in SH-SY5Y and cerebral microvascular ECs was assessed at peptide concentrations typically used in in vitro assays measuring induction of cell death mechanisms by Aβ [24–27], with some studies testing concentrations even higher [9,28]. The actual levels of Aβ present in vivo in parenchymal and CAA lesions is very difficult to assess, but is by far much higher than the physiological concentration of soluble Aβ in biological fluids. In addition to the anticipated discrepancies on the basis of different areas of affected brains, individual and genetic differences among patients, and years of disease progression, many differences in the extraction procedures, biochemical and structural characterization of the different Aβ species, and methods used for evaluation of extracted materials contribute to obscuring the problem. Among the few quantitative data available, the amount of Aβ retrieved from brain deposits could reach an impressive ratio of 140 μg per gram of tissue depending on the brain area selected for analysis [29]. To increase the complexity of the problem, the actual in vivo ratio among the broad spectrum of Aβ oligomer species identified to date and their relevance to human disease [30] is also poorly defined. Findings aiming to categorize and quantify the different types of in vivo assemblies are scant, albeit recent reports indicate for a single type of oligomeric aggregate a concentration of 10 μg/ml in the Alzheimer’s brain [31]. All of these values are within the comparable micromolar range of our experimental dose.
As shown in Figure 3, the time frame for induction of DNA fragmentation in both cells, assessed by Cell Death Detection ELISAplus, was consistent with the aggregation propensity of the peptides and suggested that the initiation of the apoptotic pathway occurred in the presence of HMM aggregation species (large oligomers and protofibrils). As illustrated in Figure 3(A), SH-SY5Y cells challenged with WT Aβ40 for up to 3 days showed no increase in DNA fragmentation compared with control cells, consistent with the poor aggregation/fibrillization properties of the peptide. A 3-day treatment with Aβ(isoD) generated a clear apoptotic phenotype, consistent with the appearance of oligomers before the 3-day time point in the post-translationally modified variant. Results were more dramatic when Aβ peptides carrying the D23N Iowa mutation, alone or in combination with isomerized residues at positions 1, 7 and 23, were tested. The aggressive AβD23N Iowa mutant induced comparable levels of apoptosis after 2-day challenge, whereas the combination of D23N mutation plus isoDs, in line with the higher rate of oligomerization illustrated by the dot-blot and WB data, caused enhanced DNA fragmentation in only 1 day. Similar results were obtained for ECs (Figure 3B).
Figure 3. Apoptosis induction by the different Aβ species in neuronal cells and cerebral microvascular ECs.
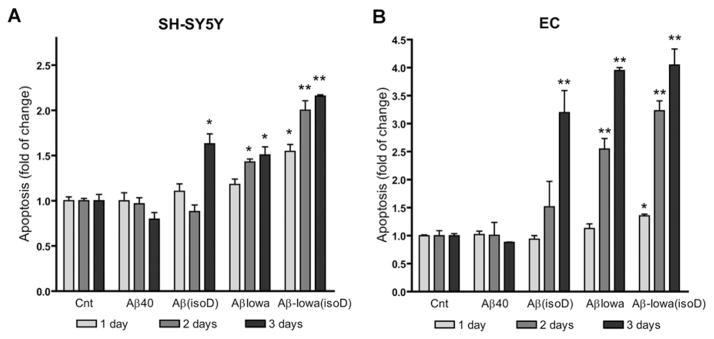
Cultures were challenged with 50 μM Aβ40, Aβ(isoD), Aβ-Iowa and Aβ-Iowa(isoD), and apoptosis was evaluated after 1–3 days by Cell Death Detection ELISAplus. (A) SH-SY5Y cells; (B) microvascular cerebral ECs. Results are expressed as fold change of nucleosome formation compared with no-peptide controls (Cnt) at the respective time points. Data are representative of at least three independent experiments performed in duplicate. Bars represent means ± S.E.M. *P < 0.05, **P < 0.01.
One of the main events driving the execution of apoptosis is the release of CytC from the mitochondria into the cytoplasm [14]; thus, assessment of CytC localization by immunofluorescence deconvolution microscopy was employed to corroborate the Cell Death Detection ELISAplus data. Figures 4 and 5 illustrate the shift from a punctate mitochondrial localization to a diffuse cytoplasmic CytC staining observed in both SH-SY5Y and ECs after 1 day of treatment with Aβ-Iowa and Aβ-Iowa(isoD) respectively. Whereas after a more prolonged treatment (3 days) with these aggressive peptides, cells detached from the plates and showed evidence of advanced stages of apoptosis (results not shown), 3-day challenge with Aβ(isoD) (Figure 6) induced in both cell types a release of CytC analogous to the 1-day treatment with Aβ-Iowa and Aβ-Iowa(isoD); CytC release was not evident in cells treated with WT Aβ40 under identical experimental conditions. Interestingly, the pattern of CytC release, as well as the time course of DNA fragmentation, was very similar in both ECs and neuronal cells, suggesting that the Aβ peptides in the present study induce similar cell death pathways in diverse cell types within time frames consistent with the aggregation/oligomerization properties of the different peptides.
Figure 4. Mitochondrial CytC release in SH-SY5Y cells after 1 day of peptide challenge.

Immunocytochemical evaluation of CytC in SH-SY5Y following 1 day of treatment with 50 μM Aβ40, Aβ(isoD), Aβ-Iowa or Aβ-Iowa(isoD). Green fluorescence highlights CytC localization; punctate staining represents mitochondrial localization, whereas diffuse green staining indicates release into the cytoplasm. Blue fluorescence represents nuclear DNA counterstained with DAPI. Magnification 40 ×. Middle panel illustrates enlarged cell images highlighting CytC subcellular localization. Cnt, control.
Figure 5. Mitochondrial CytC release in ECs after 1 day of Aβ challenge.

Immunocytochemical evaluation of CytC following 1 day of EC incubation with 50 μM Aβ40, Aβ(isoD), Aβ-Iowa or Aβ-Iowa(isoD). Green fluorescence illustrates CytC immunostaining; blue fluorescence represents nuclear DNA counterstained with DAPI. Magnification 40×. Middle panel depicts enlarged cell images illustrating the CytC intracellular staining pattern. Cnt, control.
Figure 6. Release of mitochondrial CytC in SH-SY5Y and ECs after 3 days of peptide challenge.

Immunocytochemical evaluation of CytC localization in SH-SY5Y and ECs treated for 3 days with the less aggressive Aβ40 and Aβ(isoD) peptides (50 μM). Green fluorescence depicts CytC immunostaining, as in Figures 4 and 5; punctate staining represents mitochondrial localization, and diffuse green staining indicates cytoplasmic release. Blue fluorescence signal depicts nuclear DNA counterstained with DAPI. Magnification 40×. Cnt, control.
Protection from apoptosis induced by Aβ-Iowa and isoD-modified Aβ peptide by treatment with the CytC release inhibitor methazolamide
Methazolamide, a pharmacological compound known to inhibit CytC release in isolated mitochondria [32,33], was shown in our previous study to exert a protective effect on the pro-apoptotic events elicited by specific vasculotropic Aβ variants in cerebrovascular cells [14]. As indicated above and in Figure 4, incubation of Aβ-Iowa(isoD), the most effective of the peptides in the present study, with neuronal and EC cultures resulted in a dramatic release of CytC from the mitochondria after 1 day. Co-incubation of the peptide with methazolamide completely prevented CytC release restoring the mitochondrial localization of the protein and rendering comparable immunofluorescence images with those of untreated control cells (Figure 7A). Quantitative evaluation of the number of cells exhibiting diffuse cytoplasmic CytC staining showed that the 4–5-fold increase induced by Aβ-Iowa(isoD) in both cells was abrogated by co-incubation with methazolamide, restoring levels close to the no-peptide controls (Figure 7B). Inhibition of CytC release by methazolamide was confirmed via WB experiments in enriched mitochondrial preparations (Figure 7C). Exposure of both cell types to Aβ-Iowa(isoD) resulted in a decrease in mitochondrial CytC after 1 day of incubation, an effect that was reversed by methazolamide. Interestingly, a reduction in the mitochondrial marker VDAC (voltage-dependent anion channel) was also observed on incubation with the peptide, suggesting that this treatment may have affected either the number of structurally intact mitochondria or the mitochondrial ratio within the subcellular fraction preparation, issues that are currently under investigation.
Figure 7. Protective effect of methazolamide on Aβ-mediated mitochondrial CytC release.

SH-SY5Y and ECs were treated with 50 μM Aβ-Iowa(isoD) in the presence (300 μM) and absence of methazolamide, followed by CytC visualization by deconvolution microscopy and WB after separation of mitochondrial fractions. (A) Immunofluorescence evaluation. Green signal illustrates CytC staining as above. Magnification 100×. (B) Quantification of cells depicting cytoplasmic CytC localization. Histograms represent the number of cells exhibiting cytoplasmic CytC diffuse staining by immunofluorescence, expressed as a percentage of total cells. Results illustrate cell counts in at least three 40× magnification fields. Bars represent means ± S.E.M. (C) WB analysis of mitochondrial fractions. CytC immunoreactivity in SH-SY5Y and ECs treated with Aβ-Iowa(isoD) in the presence and absence of methazolamide was assessed in mitochondrial fractions [(+) for VDAC; (−) for the cytoplasmic protein HSP90 (heat-shock protein 90)], prepared as described above. Histograms on the right-hand panels of each WB represent the densitometric quantification of the CytC band intensity normalized to the respective VDAC bands. Data are presented as means ± S.E.M. for at least three independent experiments. In (C) Aβ indicates Aβ-Iowa(isoD), M represents methazolamide (300 μM) and Cnt represents control.
Mitochondrial changes induced by Aβ were further visualized by confocal analysis of CytC and the mitochondrial marker MitoTracker®. As illustrated in Figures 8 and 9 for neuronal cells and ECs respectively, control untreated cells exhibited the characteristic punctate CytC and MitoTracker® signals which significantly overlapped in the merged images. Treatment with Aβ-Iowa resulted not only in release of CytC, but also in a less intense more diffuse MitoTracker® staining indicative of the poor localization of the oxidized dye to the organelles, which is dependent on mitochondrial membrane potential. In this line, it is noteworthy that Aβ treatment also appeared to affect levels of VDAC (Figure 7), a mitochondrial protein in which the function is also sensitive to changes in the organelle membrane potential. Co-incubation with methazolamide not only preserved the subcellular localization of CytC, but restored the mitochondrial membrane potential as visualized by the localization of MitoTracker® to the organelles, a feature that also occurs at a lower methazolamide concentration (100 μM).
Figure 8. Confocal microscopy evaluation of the effect of methazolamide in preventing CytC release and restoring mitochondrial membrane potential in SH-SY5Y.

After treatment with Aβ-Iowa (50 μM, 16 h) in the presence (100 and 300 μM) and absence of methazolamide, SH-SY5Y cells were incubated with MitoTracker® Red CM-H2XRos, followed by CytC immunocytochemistry, as above. Top panel, green fluorescence highlights CytC; middle panel, red fluorescence depicts the signal of oxidized MitoTracker®, the localization of which to the mitochondria is dependent on maintenance of the organelles’ membrane potential; bottom panel, merged images. Cnt, control; M, methazolamide. Magnification 40×.
Figure 9. Confocal microscopy assessment of the effect of methazolamide in preventing Aβ-mediated CytC release and restoring mitochondrial membrane potential in ECs.

ECs, challenged with Aβ-Iowa (50 μM, 16 h) in the presence and absence of methazolamide, were incubated with MitoTracker® Red and further immunostained for CytC, as above. Top panel, green fluorescence depicts CytC; middle panel, red fluorescence illustrates the signal of oxidized MitoTracker®, the localization of which to the mitochondria is dependent on the organelles’ membrane potential; bottom panel, merged images. Cnt, control; M, methazolamide. Magnification 40×.
Inhibition of CytC release into the cytoplasm was sufficient to protect neuronal cells and ECs from apoptosis, as evaluated by Cell Death Detection ELISAplus (Figure 10). Notably, 300 μM methazolamide, a concentration that did not cause toxicity in either cell line, reduced the amount of fragmented DNA to the levels of untreated controls for all the peptides studied in both neuronal cells and ECs. Whether therapeutic doses in humans will allow reaching a methazolamide brain concentration capable of preventing/ameliorating the effect of Aβ under disease conditions remains to be elucidated. Nevertheless, it should be noted that methazolamide long-term treatment in mouse models of Huntington disease and stroke, using significantly lower concentrations, were sufficient to exert a neuroprotective effect in vivo [33].
Figure 10. Protective effect of methazolamide on Aβ-mediated apoptosis induction.
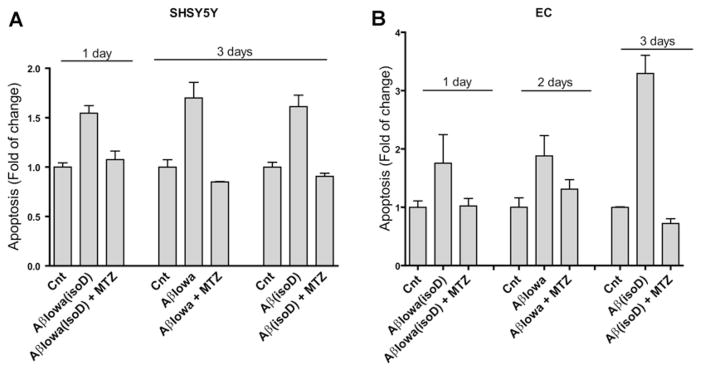
Cell cultures were treated with the different Aβ peptides at a concentration of 50 μM, in the presence (300 μM) and absence of methazolamide, and apoptosis induction was evaluated by Cell Death Detection ELISAplus. The length of incubation with the different peptides was selected to yield maximal apoptosis in the absence of inhibitor and therefore varied depending on the inherent pro-apoptotic capabilities of the different peptides, as described in the Materials and methods section. (A) SH-SY5Y cells. (B) Cerebral microvascular ECs. Results are expressed as fold change compared with no-peptide controls in the absence of inhibitor (Cnt) and are representative of three independent experiments performed in duplicate. Data are presented as means ± S.E.M. for duplicate experiments.
DISCUSSION
The mechanisms leading to amyloid deposition in AD are highly complex and interlink an array of molecular pathways, ultimately resulting in cell toxicity and death. Histopathological, genetic, biochemical and physicochemical studies, together with information obtained from transgenic animal models, strongly support the notion that abnormal aggregation/fibrillization and subsequent Aβ tissue accumulation are key players in the disease pathogenesis. The aggregation process is usually initiated by partially or completely unfolded forms of the peptides. Along this line, it is known that mutations affecting the mean hydrophobicity of proteins, the propensity to generate β-structures or those reducing the net charge of the molecule favour peptide aggregation from unfolded states which exist in dynamic equilibrium with folded structures [34]. In the case of the Iowa mutation in the present study, the D23N substitution greatly enhanced the oligomerization/fibrillization propensity of the molecule, as demonstrated by WB, Thioflavin T binding, TEM and dot-blot studies. The amino acid change occurring in Iowa kindred is similar to that introduced by the Dutch mutant; in both cases there is a loss of a negatively charged residue occurring at adjacent locations in the molecule (D23N compared with E22Q). Probably as a result of these similar changes, synthetic homologues of the D23N variant display comparable high content of β-sheet secondary structures and rapidly assemble in solution to form typical amyloid fibrils, as described previously for the E22Q substitution [22]. Supporting these findings, experimental and MD simulations with a set of Aβ21–30 decapeptides containing known mutations involved in familial AD have shown that the turn in the Val24–Lys28 region is destabilized by the presence of both D23N and E22Q substitutions. These conformational changes result in intramolecular interactions between the Aβ21–30 region and the rest of the full-length peptide that facilitate oligomerization and fibril formation [35]. This decapeptide region also forms a variety of loop structures and at least three types of metastable β-hairpins, motifs typically occurring as part of hydrogen-bonded strands comprising β-sheet structures. Notably, both the Iowa and Dutch mutations, in part as the result of the hydrophobic packing of side chains, generate β-hairpin structures with longer lifetimes, suggesting that these metastable structures may enhance pathogenicity of the peptides in vivo [36]. The contribution of the D23N mutation to the formation of intermediate metastable structures is also highlighted by recent magnetic resonance studies [37], demonstrating that, in contrast with full-length WT Aβ fibrils that mainly exhibit a supramolecular organization of parallel β-sheet structures, peptides bearing the Aβ-Iowa mutation are capable of forming both parallel and antiparallel β-sheet architectures [37]. Of interest for the present work, the antiparallel Aβ-Iowa components, which are thermodynamically metastable and transition to the more stable parallel structures, are also highly neurotoxic for SH-SY5Y cells, likely representing additional toxic intermediates in the aggregation process to the oligomeric assemblies capable of eliciting the cell death mechanisms described.
Contributing to the complexity of Iowa amyloid lesions, a considerable proportion of the deposited Aβ molecules exhibit, in addition to the mutation, partial isomerization of aspartic acid residues at positions 1, 7 and 23 [8]. This post-translational modification, fostered by aging, is also found in sporadic AD, in which it is known to modify both tau protein in PHFs (paired helical filaments), as well as deposited Aβ molecules [38]. In general terms, protein isomerization, together with post-translational modifications induced by oxygen radicals, protein truncations and formation of pyroglutamate have all been speculated as enhancers of aggregation likely to participate in disease pathogenesis. In the case of isoD formation, some conflicting data have been reported regarding the effect of amino acid isomerization on fibril formation, as well as its final role in the induction of neurotoxicity [39,40]. The results presented herein illustrate the structural effects introduced by the presence of isoD in WT Aβ40, demonstrating that the in vitro aggregation kinetics is clearly influenced by the post-translational modification resulting in a higher propensity for oligomerization/fibrillization compared with the unmodified WT counterpart, consistent with some of the previously reported findings [38]. Notably, in the case of the D23N mutated peptides, the additional presence of isoD modestly added to the exacerbated conformational changes induced by the mutation in itself, suggesting that the in vivo pathogenesis, although primarily driven by the presence of the genetic mutation, may also be partially influenced by the degree of aspartic acid residue isomerization.
Typically, the presence of accelerated oligomerization/ fibrillization correlates with enhanced amyloid-mediated cell toxicity. In the case of the D23N Iowa variant, previous studies in vascular smooth muscle cells demonstrated a significant loss of cell viability and proteolytic breakdown of vascular smooth muscle cell actin [22], similar to the effects caused by the E22Q substitution [27]. However, both mutations associate in vivo with very different clinical phenotypes. Whereas the Dutch variant associates with massive cerebral haemorrhagic episodes, the Iowa kindred presents with an AD-like dementia phenotype with dys-trophic neurites and neurofibrillary tangles coexisting with CAA and microhaemorrhages [6]. Thus it was relevant to analyse the effect of these peptides on endothelial and neuronal cells, for which very limited information is available. Although no studies have been published to our knowledge on EC, the reports on neurons are limited to the analysis of rat PC12 cells viability after challenge with Aβ42 D23N [9], a minor component of the brain Iowa deposits [8]. In the present study, we have used the more relevant Aβ40 D23N, more than 20-fold more abundant than the 42-residue-long isoform [41], and tested the additional effect of isoD modifications on microvascular endothelial and neuronal cells. In line with our aggregation studies, the presence of the D23N mutation significantly increased the peptide toxicity for both neuronal and ECs, whereas the isoD modification rendered both WT- and Iowa-Aβ more toxic than their unmodified counterparts. To date, the precise chronology for the deposition of the various Aβ species remains poorly defined. However, the presence of isoD has been reported in Congo Red (−) diffuse deposits in the Iowa kindred [41], as well as in Down’s syndrome [13], an indication that isomerization occurs before fibrillization. These results suggest that the presence of isoD-modified peptides in the Iowa peptidome may be an important, albeit unrecognized pathological factor contributing to the pathological features of the Iowa cases likely to enhance the aggregation properties of more soluble species through seeding mechanisms [42]. Once in the deposits, the isomerization contributes further to the stability of the deposits increasing their resistance to proteolytic degradation, a feature that limits the removal of the peptide by normal clearance mechanisms [43–45] promoting the accumulation of pathogenic species in affected brains and contributing to the disease pathogenesis.
Increasing evidence suggests that apoptotic biochemical cascades play pivotal roles in the cellular dysfunction and death observed in AD and related neurodegenerative disorders. The major form of apoptosis in mammalian cells proceeds through the mitochondrial pathway and is typically modulated by the Bcl-2 family of proteins involving mitochondrial outer membrane permeabilization and the release of proteins, including CytC, to the cytoplasm. These events facilitate downstream cell death cascades leading to sequential caspase activation, DNA fragmentation and formation of apoptotic bodies. Our data clearly demonstrate that comparable pre-fibrillar assemblies elicited by the presence of the Aβ D23N mutation and specific isoD post-translational modifications in the Aβ40 molecule are strong inducers of endothelial and neuronal cell apoptosis as indicated by the presence of cytosolic CytC and nuclear DNA fragmentation, the latter indicative of the execution phase of apoptosis. The fact that, in our studies, the induction of CytC release and the initial phases of toxicity closely followed the appearance of HMM oligomers and protofibrils for each variant, similar to our previous findings for Dutch and Piedmont mutants [14,46], suggested that these HMM species are also responsible for cell toxicity in the Iowa cases. Whether the induction of mitochondrial apoptotic pathways results from the primary involvement of death receptors, as demonstrated previously for the Aβ Dutch and Piedmont variants in microvascular endothelial and smooth muscle cells [14,46], or whether the mitochondria engagement is initiated through intracellular events, an intrinsic pathway, independently of death receptor signals, remains to be elucidated. Consistent with our findings in cell culture models, transgenic mice expressing the Iowa mutation in combination with the Dutch substitution (TgSwDI) present with robust and progressive accumulation of microvascular Aβ and exhibit apoptotic vascular cells in combination with cerebral vascular cell loss, features that highlight the in vivo relevance of these mutations in the induction of cell death pathways [47]. Although apoptosis indicators have not been investigated in familial cases exhibiting the D23N mutation, it is noteworthy that studies in AD patients have demonstrated alterations in the expression of apoptosis-related genes, many of them active participants in the mitochondrial cascade [48,49], suggesting that this process is also relevant in vivo.
Targeting mitochondrial dysfunction and preventing leakage of CytC into the cytoplasm with its detrimental downstream effects and critical contribution to cell death programmes are potentially effective therapeutic strategies. A number of drugs are currently available with the capacity to inhibit the release of CytC, one being methazolamide. This compound is capable of crossing the blood–brain barrier [32] and is currently FDA (Food and Drug Administration)-approved for use in the treatment of glaucoma owing to its capability to reduce aqueous fluid production and intraocular pressure. Confirming the crucial role of CytC release in amyloid-induced apoptosis, our results clearly demonstrated that specific inhibition of the process by methazolamide exerts a protective effect ameliorating endothelial and neuronal cell toxicity induced by pre-fibrillar assemblies of Iowa and isoD-modified Aβ, as we reported previously for the E22Q Dutch mutant [14,46]. Whether the agent will also display beneficial effects in animal models exhibiting Aβ deposition is currently under investigation. Nevertheless, it should be noted that methazolamide has been successfully employed previously in a transgenic mouse model of Huntington’s disease, in which intraperitoneal inoculation of the drug resulted in a significant dose-dependent delay of disease onset and mortality [32], highlighting the importance of this therapeutic strategy for neurodegenerative disorders.
Overall, our data highlight the contribution of HMM oligomeric/pre-fibrillar Aβ elements to the initiation of neuronal and cerebral microvascular EC apoptosis, demonstrating that genetic variants and/or post-translational modifications capable of accelerating the formation of intermediate-aggregation-state components are able to elicit comparable cell death responses. Through the induction of cellular dysfunction, Aβ likely plays a key role in altering the functionality of the neurovascular unit, a dynamic entity that regulates CNS development, modulates cerebral blood flow and influences the permeability properties of the blood–brain-barrier. The beneficial effect of methazolamide in preventing Aβ-mediated mitochondrial dysfunction will certainly contribute to preserving the cross-talk among the various cellular components (ECs, astrocytes, pericytes and neurons), indispensable for the maintenance of brain homoeostasis and the functional integrity of the neurovascular unit.
Acknowledgments
FUNDING
This work was supported by National Institute of Health [grant numbers NS051715 and AG030539] and the Alzheimer’s Association.
Abbreviations used
- a.u
arbitrary unit
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Aβ(isoD)
Aβ40 with isoD modifications at residues 1, 7 and 23
- Aβ
amyloid β
- Aβ-Iowa(isoD)
Aβ-Iowa bearing isoD modifications at residues 1 and 7
- Aβ-Iowa
Aβ40 containing the D23N genetic variant
- CAA
cerebral amyloid angiopathy
- Caps
3-(cyclohexylamino)propane-1-sulfonic acid
- CytC
cytochrome c
- DMEM
Dulbecco’s modified Eagle’s medium
- EBM-2
endothelial basal medium-2
- EC
endothelial cell
- HFIP
hexafluoroisopropanol
- HMM
high-molecular-mass
- HRP
horseradish peroxidase
- isoD
isoaspartate
- PBST
PBS containing 0.3 % Triton X-100
- TBST
TBS containing 0.1 % Tween 20
- TEM
transmission electron microscopy
- VDAC
voltage-dependent anion channel
- WB
Western blot
- WT
wild-type
Footnotes
AUTHOR CONTRIBUTION
Agueda Rostagno and Jorge Ghiso designed the experimental approach and directed the project. Silvia Fossati, Agueda Rostagno and Jorge Ghiso analysed the data and wrote the paper. Silvia Fossati, Krysti Todd and Krystal Sotolongo performed the experimental work. All authors read and approved the paper.
References
- 1.Rostagno A, Holton JL, Lashley T, Revesz T, Ghiso J. Cerebral amyloidosis: amyloid subunits, mutants and phenotypes. Cell Mol Life Sci. 2010;67:581–600. doi: 10.1007/s00018-009-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Zhang-Nunes SX, Maat-Schieman M, van Duinen S, Roos R, Frosch MP, Greenberg SM. The cerebral β-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu L, Rosa-Neto P, Hsiung GY, Sadovnick AD, Masellis M, Black SE, Jia J, Gauthier S. Early-onset familial Alzheimer’s disease. Can J Neurol Sci. 2012;39:436–445. doi: 10.1017/s0317167100013949. [DOI] [PubMed] [Google Scholar]
- 5.Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol. 2011;7:1–9. doi: 10.3988/jcn.2011.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. A novel APP mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg SM, Shin Y, Grabowski TJ, Cooper GE, Rebeck GW, Iglesias S, Chapon F, Tournier-Lasserve E, Baron JC. Hemorrhagic stroke associated with the Iowa amyloid precursor protein mutation. Neurology. 2003;60:1020–1022. doi: 10.1212/01.wnl.0000050140.10044.a8. [DOI] [PubMed] [Google Scholar]
- 8.Tomidokoro Y, Rostagno A, Neubert TA, Lu Y, Rebeck GW, Frangione B, Greenberg SM, Ghiso J. Iowa variant of familial Alzheimer’s disease: accumulation of posttranslationally modified AβD23N in parenchymal and cerebrovascular amyloid deposits. Am J Pathol. 2010;176:1841–1854. doi: 10.2353/ajpath.2010.090636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, Shirasawa T. Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. J Biol Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 10.Krone MG, Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea JE. Effects of familial Alzheimer’s disease mutations on the folding nucleation of the amyloid β-protein. J Mol Biol. 2008;381:221–228. doi: 10.1016/j.jmb.2008.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demeester N, Mertens C, Caster H, Goethals M, Vandekerckhove J, Rosseneu M, Labeur C. Comparison of the aggregation properties, secondary structure and apoptotic effects of wild-type, Flemish and Dutch N-terminally truncated amyloid β peptides. Eur J Neurosci. 2001;13:2015–2024. doi: 10.1046/j.0953-816x.2001.01579.x. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu T, Matsuoka Y, Shirasawa T. Biological significance of isoaspartate and its repair system. Biol Pharm Bull. 2005;28:1590–1596. doi: 10.1248/bpb.28.1590. [DOI] [PubMed] [Google Scholar]
- 13.Iwatsubo T, Saido T, Mann DM, Lee V, Trojanowski JQ. Full-length amyloid-β 1–42(43) and amino-terminally modified and truncated amyloid-β 1–42(43) deposit in diffuse plaques. Am J Pathol. 1996;149:1823–1830. [PMC free article] [PubMed] [Google Scholar]
- 14.Fossati S, Cam J, Meyerson J, Mezhericher E, Romero IA, Couraud P-O, Weksler B, Ghiso J, Rostagno A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. FASEB J. 2010;24:229–241. doi: 10.1096/fj.09-139584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stine WBJ, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 16.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe D, Teplow D. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofribrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 17.Viana RJ, Nunes AF, Castro RE, Ramalho RM, Meyerson J, Fossati S, Ghiso J, Rostagno A, Rodrigues CM. Tauroursodeoxycholic acid prevents E22Q Alzheimer’s Aβ toxicity in human cerebral endothelial cells. Cell Mol Life Sci. 2009;66:1094–1104. doi: 10.1007/s00018-009-8746-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rostagno A, Lashley T, Ng D, Meyerson J, Braendgaard H, Plant G, Bojsen-Moller M, Holton J, Frangione B, Revesz T, Ghiso J. Preferential association of serum amyloid P component with fibrillar deposits in familial British and Danish dementias: similarities with Alzheimer’s disease. J Neurol Sci. 2007;257:88–96. doi: 10.1016/j.jns.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 19.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 20.Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A, Bourdoulous S, Turowski P, et al. Blood–brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 21.Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, Chiarugi A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol. 2011;80:1136–1146. doi: 10.1124/mol.111.073916. [DOI] [PubMed] [Google Scholar]
- 22.van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid β-protein. J Biol Chem. 2001;276:32860–32866. doi: 10.1074/jbc.M104135200. [DOI] [PubMed] [Google Scholar]
- 23.Harper JD, Wong CW, Lieber CM, Lansbury PT. Assembly of Aβ amyloid protofibrils: an in vitro model for a possible early event in Alzheimer’s disease. Biochemistry. 1999;38:8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson AM, Wold LA, Walsh DM, Ferreira A. β-Amyloid carrying the Dutch mutation has diverse effects on calpain-mediated toxicity in hippocampal neurons. Mol Med. 2012;18:178–185. doi: 10.2119/molmed.2011.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25–35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology. 2011;217:397–410. doi: 10.1007/s00213-011-2291-7. [DOI] [PubMed] [Google Scholar]
- 26.Hoppe JB, Frozza RL, Horn AP, Comiran RA, Bernardi A, Campos MM, Battastini AM, Salbego C. Amyloid-β neurotoxicity in organotypic culture is attenuated by melatonin: involvement of GSK-3β, tau and neuroinflammation. J Pineal Res. 2010;48:230–238. doi: 10.1111/j.1600-079X.2010.00747.x. [DOI] [PubMed] [Google Scholar]
- 27.Davis JB, van Nostrand WE. Enhanced pathologic properties of Dutch-type mutant amyloid β-protein. Proc Natl Acad Sci USA. 1996;93:2996–3000. doi: 10.1073/pnas.93.7.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisenhauer PB, Johnson RJ, Wells JM, Davies T, Fine RE. Toxicity of various amyloid β peptide species in cultured human blood–brain barrier endothelial cells: increased toxicity of Dutch-type mutant. J Neurosci Res. 2000;60:804–810. doi: 10.1002/1097-4547(20000615)60:6<804::AID-JNR13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 29.Lue L-F, Kuo Y-M, Roher A, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel R, Rogers J. Soluble amyloid β concentration is a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 31.Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–358. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, et al. Inhibitors of cytochrome c release with therapeutic potential for Huntington’s disease. J Neurosci. 2008;28:9473–9485. doi: 10.1523/JNEUROSCI.1867-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Sirianni AC, Zhu S, Day AL, Kristal BS, Friedlander RM. Methazolamide and melatonin inhibit mitochondrial cytochrome c release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40:1877–1885. doi: 10.1161/STROKEAHA.108.540765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1042/BJ20130652. Received 9 May 2013/4 September 2013; accepted 13 September 2013 Published as BJ Immediate Publication 13 September 2013. [DOI] [PubMed] [Google Scholar]
- 35.Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, Rigby AC, Teplow DB. Familial Alzheimer’s disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc Natl Acad Sci USA. 2007;104:16522–16527. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruz L, Srinivasa Rao J, Teplow DB, Urbanc B. Dynamics of metastable β-hairpin structures in the folding nucleus of amyloid β-protein. J Phys Chem. 2012;116:6311–6325. doi: 10.1021/jp301619v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiang W, Yau W-M, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc Natl Acad Sci USA. 2012;109:4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu T, Watanabe A, Ogawara M, Mori H, Shirasawa T. Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch Biochem Biophys. 2000;381:225–234. doi: 10.1006/abbi.2000.1955. [DOI] [PubMed] [Google Scholar]
- 39.Murakami K, Uno M, Masuda Y, Shimizu T, Shirasawa T, Irie K. Isomerization and/or racemization at Asp23 of Aβ42 do not increase its aggregative ability, neurotoxicity, and radical productivity in vitro. Biochem Biophys Res Commun. 2008;366:745–751. doi: 10.1016/j.bbrc.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Shimizu T, Fukuda H, Murayama S, Izumiyama N, Shirasawa T. Isoaspartate formation at position 23 of amyloid β peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J Neurosci Res. 2002;70:451–461. doi: 10.1002/jnr.10350. [DOI] [PubMed] [Google Scholar]
- 41.Shin Y, Cho HS, Fukumoto H, Shimizu T, Shirasawa T, Greenberg SM, Rebeck GW. Aβ species, including IsoAsp23 Aβ, in Iowa-type familial cerebral amyloid angiopathy. Acta Neuropathol. 2003;105:252–258. doi: 10.1007/s00401-002-0639-0. [DOI] [PubMed] [Google Scholar]
- 42.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser PS, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 43.LeVine H., 3rd The amyloid hypothesis and the clearance and degradation of Alzheimer’s β-peptide. J Alzheimers Dis. 2004;6:303–314. doi: 10.3233/jad-2004-6311. [DOI] [PubMed] [Google Scholar]
- 44.Moro ML, Collins MJ, Cappellini E. Alzheimer’s disease and amyloid β-peptide deposition in the brain: a matter of ‘aging’? Biochem Soc Trans. 2010;38:539–544. doi: 10.1042/BST0380539. [DOI] [PubMed] [Google Scholar]
- 45.Kuo YM, Webster S, Emmerling MR, De Lima N, Roher AE. Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of Aβ peptides of Alzheimer’s disease. Biochim. Biophys. Acta. 1998;1406:291–298. doi: 10.1016/s0925-4439(98)00014-3. [DOI] [PubMed] [Google Scholar]
- 46.Fossati S, Ghiso J, Rostagno A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Aβ. Cell Death Dis. 2012;3:e321. doi: 10.1038/cddis.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid β protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 49.Sajan FD, Martiniuk F, Marcus DL, Frey WH, Hite R, Bordayo EZ, Freedman ML. Apoptotic gene expression in Alzheimer’s disease hippocampal tissue. Am J Alzheimers Dis Other Dement. 2007;22:319–328. doi: 10.1177/1533317507302447. [DOI] [PMC free article] [PubMed] [Google Scholar]