

Abstract
Objectives
To determine whether percentages of CD4+CD25high T cells (a group of regulatory T cells, Treg) differ in patients with multiple sclerosis (MS) in relapse vs remission after glucocorticoid treatment and whether treatment for relapses changes Treg population and the expression of Foxp3, a key Treg-associated molecule.
Materials and methods
Peripheral blood mononuclear cells (PBMC) were obtained from 20 patients with MS during relapse, just before and 2 days after starting steroid treatment (i.v. methyl-prednisolone 1 g / day for 3 days) and then 6 weeks after treatment. CD4+CD25hi cells were analysed by using flow cytometry. Cytokines were measured by using an ELISA and Foxp3, CD3 and CD25 expression by using quantitative real-time PCR.
Results
The percentage of CD4+CD25hi cells, plasma IL-10 and Foxp3 / CD3 ratio increased 48 h after methylprednisolone initiation and returned to baseline values by 6 weeks post-treatment.
Conclusions
Results suggest that glucocorticoids increase Treg cell functional molecules and percentages. This may be a mechanism whereby steroids expedite recovery from MS relapses.
Keywords: CD25, cytokines, Foxp3, glucocorticoids, methylprednisolone, multiple sclerosis, regulatory T cells
Introduction
Many features of multiple sclerosis (MS) strongly suggest autoimmune mechanisms. In autoimmune diseases, there is a failure of mechanisms that normally maintain immunological tolerance and thus prevent expansion of autoreactive T cells (1, 2). Such mechanisms include a subpopulation of regulatory T cells (Tregs) that are naturally arising, are CD4+CD25high(hi), are produced in the thymus and are also present in the periphery, representing up to ~5–10% of circulating CD4+ T cells. Treg cells express CD4, CD25, CTLA4, GITR and, most relevantly, the transcription factor Foxp3 (2, 3). Foxp3 is crucial for the function of Treg cells. It suppresses other transcription factors important in inflammation, including NF-κB (4).
Tregs require IL-2 and IL-2 receptor for their function. They inhibit proliferation and cytokine production of activated T cells. The suppressive cytokines, IL-10 or TGF-β, are implicated in the function of Treg cells (5).
In patients with experimental autoimmune encephalomyelitis (EAE), an autoimmune model of MS, transfer of CD4+CD25+ or CD25hi T cells suppresses the disease, while their depletion worsens the disease and allows its reinduction (6). Glucocorticoids enhance the IL-2-dependent expansion of Treg cells in patients with EAE and their ability to suppress EAE (7).
In patients with MS, studies have not shown consistent differences in the percentage of CD4+CD25hi compared with that of controls; however, there are alterations in the suppressive effect of CD4+CD25hi on the proliferation and effect or function of CD4+CD25− T cells (effector cells, Teff) (8).
Whether the number or function of Treg cells changes during relapses is unclear. Because infection can trigger relapses and infectious products, for example toll-like receptor (TLR) ligands can inhibit Treg cell suppressive activity (via IL-6) (9, 10), we were interested in determining whether the percentage of CD4+CD25hi decreases in patients with MS relapse compared with remission, and whether relapses are associated with changes in the levels of IL-6. Our unpublished observations found no differences in the percentage of CD4+CD25hi cells between a group of patients in relapse and another matched group of patients in remission or between patients in relapse and healthy normal donors. We wanted, however, to determine whether there are differences in individual patients between relapse and remission. Moreover, because relapses in patients with MS are frequently treated with glucocorticoids (e.g. 1 g / day i.v. methylprednisolone, IVMP, for 3 days), we were interested to see the effect of glucocorticoids on the Treg cells and Foxp3. Karagiannidis et al. (11) demonstrated that glucocorticoids increase the number and function of Treg in patients with asthma. In this study, we aimed to confirm this in patients with MS and investigate potential mechanisms of Treg cell regulation by glucocorticoids. Glucocorticoids increase CD25 expression on T cells by upregulating CD25 transcription (12). In view of the important role of the IL-2 / IL-2 receptor (CD25) pathway in Treg function and the role of this pathway in the glucocorticoid enhancing of Treg function in EAE, we wanted to determine other potential mechanisms whereby glucocorticoids might enhance CD25, for example their effect on CD25 mRNA stability or on the shedding of CD25 from the cell membrane.
Materials and methods
Subjects
Twenty patients (15 women and five men, mean ± SD age: 44 years ± 11, range 34–78 years), 18 with relapsing–remitting MS in relapse and two with secondary progressive MS, who presented to the MS Centre at the peak of their MS relapse, were included in the study. Blood was collected at the peak of relapse immediately prior to initiation of steroid treatment, then again after 2 days of IVMP and 6 weeks after treatment when most were in clinical remission (see below). Subjects underwent standardized neurological examination including calculation of Expanded Disability Status Scale (EDSS) scores (13). Eight patients (six women and two men) were on disease-modifying treatment at the time of relapse (six on beta interferon and two on glatiramer acetate). Local Ethics Committee approved the study and written informed consent was obtained from all patients.
Surface staining of blood cells
One hundred microlitres of blood was surface stained with ECD-conjugated anti-CD4, PC5-conjugated CD25 and IgG2a Isotope (Beckman Coulter, High Wycombe, UK). Blood was incubated for 1 h at room temperature. One millilitre of FACS lysing solution (Becton Dickinson, Oxford, UK) was added. Blood was incubated for 10 min in the dark at room temperature. Samples were centrifuged at 200 g, washed with 2% foetal calf serum in RPMI (Sigma-Aldrich, Dorset, UK) and fixed with 0.5% formaldehyde. Data were collected by using a Beckman Coulter flow cytometer. Analysis was performed by using WINMDI software (La Jolla, CA, USA).
Surface and intracellular staining of PBMC for CD4, CD25 and IL-10 antibodies
Peripheral blood mononuclear cells (PBMC) were isolated by using gradient centrifugation with Histopaque 1077 (Sigma-Aldrich) and treated with 10 μg of brefeldin for 8 h (37°C; 5% CO2). Cells were centrifuged and supernatant decanted. One millilitre of PBA (phosphate-buffered saline, 0.5% bovine serum albumin, 1% sodium azide; Sigma-Aldrich) was added, and samples were centrifuged. Anti-CD4 (2.5 μl), anti-CD25 (5 μl) and isotype were added to appropriate FACS tubes. Cells were incubated on ice for 30 min. One millilitre of 2% formaldehyde was added; cells were incubated for 5 min at room temperature, then exposed to 1 ml of PBA, centrifuged and blotted. Two washes with saponin buffer (PBA + 0.1% saponin; Sigma-Aldrich) and two washes with 10% FCS in saponin buffer were performed. Five microlitres of PE-conjugated anti-IL-10 antibody and isotype control (Beckman Coulter) was added to appropriate tubes. Cells were incubated on ice for 2 h. After two washes with saponin buffer, cells were fixed with 0.5% formaldehyde. Analysis was performed on a Beckman Coulter flow cytometer.
Cytokine measurements of IL-6, IL-10 and soluble CD25 in plasma
Plasma samples were aliquoted and stored at −80°C for durations ranging between 2 weeks and 6 months. Prior studies in our laboratory showed that freezing at −80°C within this time frame does not significantly influence cytokine measurements. IL-10 and IL-6 were measured by using Biotrack ELISA. Soluble CD25 (sCD25) was measured by using an ELISA (R&D systems, Abingdon, UK). Measurements were performed according to the manufacturer’s instructions.
Quantitative real-time PCR
Total RNA was isolated from 2.5 ml of blood collected in PAXgene tubes (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. RNA quality was assessed using the Agilent Bioanalyzer (Santa Clara, CA, USA).
cDNA was synthesized in a 20-μL reaction from 0.5 ug of total RNA starting material using Oligo dT primers and Superscript II as previously described (14) with the addition of 1 μl of peptide nucleic acid mix (Affymetrix, Santa Clara, CA, USA) to reduce the synthesis of globin cDNA. Peptide nucleic acid mix was prepared with equal volumes of the following concentrations; PNA1: 2.88 μm, PNAs 2, 3, and 4: 4.8 μm.
Gene-specific primers (IDT Inc., San Jose, CA, USA) were designed with Light Cycler Probe Design Software, Version 1.0 (Idaho Technology Inc., Salt Lake City, UT, USA) or retrieved from the literature:
GAPDH (forward: 5′-GTG AAG GTC GGA GTC AAC G-3′; reverse: 5′-TGA GGT CAA TGA AGG GGT C-3′).
CD3gamma (forward: 5′-ATA GGA GGA GAA CAC CTG GAC-3′; reverse: 5′-TAC CCA CCG CCA TAC TAC CT-3′).
FoxP3 (forward: 5′-CAGCACATTCCCAGAGTTCCTC-3′; reverse: 5′-GCGTGTGAACCAGTGGTAGATC-3′) (15).
Twenty microlitres of cDNA synthesis reaction was diluted to 100 ul and 5 μl was used per PCR reaction. PCR was performed in a 20-μL reaction on the Opticon Chroma IV (MJ Research, Ramsey, MN, USA) with the following cycle parameters: 95°C–3 min, 40 cycles (95°C–10 s, 60°C–10 s, 72°C–30 s, plate read), melting curve 55–99°C. Sybr Green I fluorescence intensity was measured after each cycle. Product specificity was assessed by using melting curve analysis and selected samples were run on a 1.8% Agarose gel to determine amplicon size.
Relative values were calculated using Chroma IV software comparing unknown samples to a seven-point standard cDNA curve made from Human Stratagene Universal Donor Total RNA with FoxP3 PCR product spiked in. Spiked-in FoxP3 was generated using full-length gene primers in RT-PCR. FoxP3 and CD3gamma expression was first normalized by dividing relative values by GAPDH. Ratio of FoxP3 to CD3gamma is reported.
CD25 RNA stability assay
To determine effects of glucocorticoids on CD25 mRNA stability, PBMCs were isolated from healthy donors by standard gradient centrifugation with Histopaque 1077 (Sigma-Aldrich). One million PBMCs were plated out in a 24-well plate. Cells were untreated (control), treated with 100 ng / ml dexamethasone alone (Sigma-Aldrich), 5 ug actinomycin C (Biochemika, Fluka, Dorset, UK) alone or with dexamethasone for 0, 4 and 8 h. RNA was extracted using RNeasy miniprep kit (Qiagen) following the manufacturer’s instructions, then converted to cDNA. Real-time quantitative PCR (qPCR) was performed with CD25 primers (forward: 5′-GAGAAAGACCTCCGCTTCAC-3′; reverse: 5′-CGAGTGGCTAGAGTTTCCTG-3). Values were normalized for beta2-microglobulin (forward: 5′CTCCGTGGCCTTAGCTGTG-3′; reverse: 5′-TGGATGAAACCCAGACACATAG-3′). q PCR was performed on a MX4000 Multiplex qPCR system (Stratagene, La Jolla, CA, USA).
Method validation and statistical analysis
Methods were validated in previous experiments and experimental conditions (incubation times, etc.) selected for consistent, optimal results.
Wilcoxon and Friedman’s tests were used to analyse the data using prism and spsswin (San Diego, CA, USA). Friedman’s test was used to evaluate the percentage of CD4+CD25hi, IL-6 and IL-10 at the three time points.
Results
Percentage of CD4+CD25hi cells in patients with MS and effects of IVMP
There was a significant increase in the percentage of CD4+CD25hi T cells 48 h after the administration of IVMP (P = 0.002). Although a trend toward reduction in relapse was seen, there was no significant difference in the percentage of CD4+CD25hi between patients with RRMS in relapse compared to that of patients in the remission phase 6 weeks post-IVMP treatment (P = 0.19) (Fig. 1). There were no differences in the above parameters between patients who were and were not on disease-modifying treatment.
Figure 1.
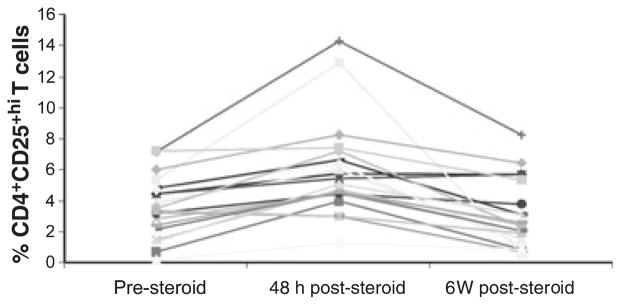
Percentage of CD4+CD25hi cells, 48 h before steroid treatment and 6 weeks after steroid treatment. Data from each patient are shown individually.
Cytokine ELISA data
Despite a trend toward reduction with treatment, there were no significant differences between IL-6 levels pre-IVMP treatment vs. after 48 h of IVMP treatment (P = 0.1) or pre-IVMP treatment vs. 6 weeks post-relapse (P = 0.81). Although there was no significant difference pre-IVMP treatment vs. 6 weeks post-relapse (P = 0.87), there was a significant difference between levels of IL-10 pre-IVMP treatment compared with that at 48 h post-IVMP (P = 0.02). The course of IL-10 expression paralleled that of the percentage of CD4+CD25hi T cells during IVMP treatment (Fig. 2). However, intracellular staining suggested that the source of the IL-10 determined by using an ELISA was not Treg cells. IVMP treatment did not consistently affect sCD25 and we did not find a correlation between surface and soluble CD25 before and after 48 h of IVMP treatment (data not shown).
Figure 2.

Parallel increase in IL-10 and in the percentage of CD4+CD25hi, pre-IVMP, 48 h post-IVMP and 6 weeks post-relapse.
qPCR for normalized CD3 and Foxp3 Ratio
Quantitative PCR detected an increase in Foxp3 / CD3 levels 48 h post-treatment (P = 0.02). However, no statistical difference in levels was observed pre-IVMP treatment vs 6 week post-relapse (Fig. 3).
Figure 3.
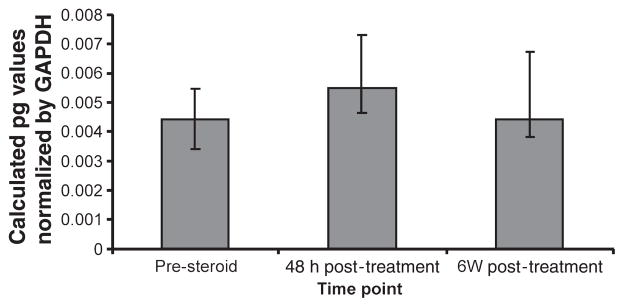
Ratio of Foxp3 / CD3 mRNA expression in steroid-treated patients with MS.
EDSS
All patients were clinically improved at 6 weeks of follow-up. The range of EDSS improvement from the peak of relapse to the 6-week time point was 2.0–0.5 steps. The only patient with only a 0.5-Kurtzke step improvement (7 to 6.5) had a minor reduction in CD4+CD25hi T cells at day 2 (3.26–3.04%).
CD25 mRNA stability assay
We wanted to determine whether glucocorticoids increase CD25 mRNA stability. In vitro treatment of normal PBMC with 100 ng of dexamethasone increased CD25 mRNA transcription, consistent with prior findings (12). CD25 mRNA levels decayed at similar rates after the addition of actinomycin C with or without dexamethasone (data not shown), indicating that the increase in CD25 mRNA under treatment with dexamethasone is due to increased transcription rate but not due to an increase in mRNA stability.
Discussion
We previously observed that the percentage of CD4+CD25hi cells in patients with RRMS in relapse was similar to that in healthy controls and a matched group of patients with RRMS in remission (M. Braitch and C.S. Constantinescu, unpublished data). In this study, we show that treatment with IVMP during relapses increases the percentage of CD4+CD25hi cells 48 h after the start of the treatment. A similar increase was observed in the Foxp3 / CD3 ratio and in plasma IL-10. Because treatment with steroids affects T-cell viability and can induce T-cell apoptosis (16), we chose to calculate the Foxp3-to-CD3 RNA ratio (both normalized to the GAPDH gene) rather than Foxp3 alone as this would best correct for T-cell numbers. We planned and began these studies just before Foxp3 antibodies for intracellular staining were available; thus, it was not possible to determine the percentages of Foxp3+ T cells.
We showed that glucocorticoids increase levels of anti-inflammatory, Treg-associated cytokine IL-10, a known effect of steroids (17), but IL-10 is not largely derived from regulatory T cells.
Foxp3 increased with treatment but decreased by 6 weeks after treatment to levels similar to those seen in relapse. CD4+ cell number did not change pretreatment and 48 h post-treatment or 6 weeks post-relapse, indicating that the transient increase in CD4+CD25hi T cells under IVMP treatment was not due to a general IVMP effect on the total population of T cells. Although IVMP is known to induce apoptosis (16), there was no indication of preferential apoptosis of the CD25− or CD25lo leading to a relative increase in CD25hi cells. There were also no significant differences in the forward scatter on flow cytometry analysis between these cell populations. We had considered the possibility of differential apoptosis, given that murine Treg cells are more resistant to glucocorticoid-induced apoptosis (18).
Several markers have been shown to be important for Treg cells. CD25 is one such marker; however, it is also transiently expressed on activated Teff. We think, nevertheless, that the increase in CD4+CD25hi T cells observed in our study was not due to some IVMP-induced general activation of T cells. Glucocorticoids have long been recognized to suppress T-cell activation, and our microarray analysis of gene expression before and after IVMP treatment in the patient population investigated in this study shows not an induction, but a downregulation of genes associated with T-cell activation. Similarly, although Foxp3 can be transiently upregulated during human T-cell activation (19), data from gene expression profiling do not point to other indicators of T-cell activation, thus making it an unlikely sole explanation for the rise observed in Foxp3 after steroid treatment. We did not perform in vitro suppression assays with CD25hi and CD25− cells from our patients, because the amount of blood we could obtain did not permit such assays. However, typically CD25hi cell percentage correlates well with both Foxp3 expression and Treg suppressive activity making it plausible that we witnessed a transient hike in these cells after steroid treatment. On the other hand, Pillai et al. (19) have shown that virtually all human CD4+CD25− T cells transiently upregulate Foxp3 and acquire transient suppressive properties on activation. It, therefore, cannot be excluded that this transient phenomenon occurred during steroid treatment and explains our findings. However, we can then argue that performing suppression assays would not have helped further, as suppressive ability could also, in principle, be attributed to either Treg or Teff activation. Our microarray results are in concordance with recent findings of the effects of glucocorticoid treatment for MS relapses, where Treg induction was associated with downregulation of T-cell activation after 5 days of treatment (20). A more detailed report of our microarray results, showing upregulation of pathways that inhibit T-cell activation, is the subject of a separate manuscript in preparation.
Infection can trigger MS relapses and for this reason we hypothesized that increasing levels of IL-6 before a relapse could diminish Treg suppression (9). However, patients with MS did not show significant changes in IL-6 levels pre- or post-relapse.
We demonstrated no differences in the plasma sCD25 levels pretreatment, 48 h after the start of IVMP or 6 weeks post-steroid treatment, suggesting that steroids did not decrease shedding of CD25 (as previously shown in a pediatric population) (21), thus prolonging expression of CD25. An increase in CD25 mRNA transcription confirmed previous results (12). Although glucocorticoids increase mRNA stability of several genes, including genes important in immune responses, such as MCP-1 (22), in our study in vitro dexamethasone did not delay CD25 mRNA degradation, arguing against increased mRNA stability as a mechanism of increasing CD25.
As mentioned above, results comparable with ours have recently been reported by another group (20). In that study, IVMP also led to an increase in the percentage of Treg cells, although, in contrast to our results, there was already a slight increase in Treg percentage in relapse, before IVMP. It is unlikely that our findings represent a similar upregulation of CD4+CD25hi cells, Foxp3 expression and IL-10 production due to the relapse per se, as the blood samples were obtained at the peak of the relapse and the changes we report were seen 2 days later. However, this possibility cannot be excluded, and future studies need to compare patients in relapse receiving and not receiving IVMP. Our results also corroborate well the data from the EAE model.
Glucocorticoids have pleiotropic effects in patients with MS and EAE (23). These include, in addition to apoptosis of leukocytes (16), down-regulation of cell adhesion molecules (24), regulation of innate immunity (25) and changes in the expression of chemokines (26) and chemokine receptors (27). Our study shows that an increase in the percentage of Tregs is possibly an additional, probably important, mechanism of action which explains the well known, transient benefits of glucocorticoids on the course of MS. Other MS treatments with longer lasting effects in MS such as IFN-β and glatiramer acetate are now known to enhance Treg function (28, 29). These treatments have been shown to have synergistic effects with glucocorticoids on a variety of immune functions (30) and combination treatments are being explored [for example the studies combining methylprednisolone with IFNβ-1a; see http://www.clinicaltrials.gov/ct2/show/NCT00168766 and Ref. (31)]. Such combinations may also result in synergistic Treg function enhancement.
In conclusion, glucocorticoids as used clinically in MS transiently enhance regulatory T cells and their expression of functional molecules. The clinical implications of these findings are that treatments that enhance Treg function contribute to the recovery from relapses, and that a more sustained enhancement of this function, possibly through combined immunomodulation, may benefit patients with MS in the longer term.
References
- 1.Sakaguchi S, Sakaguchi N. Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol. 2005;24:211–26. doi: 10.1080/08830180590934976. [DOI] [PubMed] [Google Scholar]
- 2.Curotto De Lafaille MA, Lafaille JJ. CD4(+) regulatory T cells in autoimmunity and allergy. Curr Opin Immunol. 2002;14:771–8. doi: 10.1016/s0952-7915(02)00408-9. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 4.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci USA. 2005;102:5138–43. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med. 2007;13:108–16. doi: 10.1016/j.molmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–6. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, Howard OM. Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur J Immunol. 2006;36:2139–49. doi: 10.1002/eji.200635873. [DOI] [PubMed] [Google Scholar]
- 8.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 10.Sutmuller RP, Morgan ME, Netea MG, Grauer O, Adema GJ. Toll-like receptors on regulatory T cells: expanding immune regulation. Trends Immunol. 2006;27:387–93. doi: 10.1016/j.it.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Karagiannidis C, Akdis M, Holopainen P, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol. 2004;114:1425–33. doi: 10.1016/j.jaci.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Lamas M, Sanz E, Martin-Parras L, et al. Glucocorticoid hormones upregulate interleukin 2 receptor alpha gene expression. Cell Immunol. 1993;151:437–50. doi: 10.1006/cimm.1993.1252. [DOI] [PubMed] [Google Scholar]
- 13.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–52. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 14.Kari L, Loboda A, Nebozhyn M, et al. Classification and prediction of survival in patients with the leukemic phase of cutaneous T cell lymphoma. J Exp Med. 2003;197:1477–88. doi: 10.1084/jem.20021726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yagi H, Nomura T, Nakamura K, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. 2004;16 :1643–56. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- 16.Leussink VI, Jung S, Merschdorf U, Toyka KV, Gold R. High-dose methylprednisolone therapy in multiple sclerosis induces apoptosis in peripheral blood leukocytes. Arch Neurol. 2001;58:91–7. doi: 10.1001/archneur.58.1.91. [DOI] [PubMed] [Google Scholar]
- 17.Gayo A, Mozo L, Suarez A, Tunon A, Lahoz C, Gutierrez C. Glucocorticoids increase IL-10 expression in multiple sclerosis patients with acute relapse. J Neuroimmunol. 1998;85:122–30. doi: 10.1016/s0165-5728(97)00262-2. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Murakami T, Oppenheim JJ, Howard OM. Differential response of murine CD4+CD25+ and CD4+CD25− T cells to dexamethasone-induced cell death. Eur J Immunol. 2004;34:859–69. doi: 10.1002/eji.200324506. [DOI] [PubMed] [Google Scholar]
- 19.Pillai V, Ortega SB, Wang CK, Karandikar NJ. Transient regulatory T-cells: a state attained by all activated human T-cells. Clin Immunol. 2007;123:18–29. doi: 10.1016/j.clim.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navarro J, Aristimuno C, Sanchez-Ramon S, et al. Circulating dendritic cells subsets and regulatory T-cells at multiple sclerosis relapse: differential short-term changes on corticosteroids therapy. J Neuroimmunol. 2006;176:153–61. doi: 10.1016/j.jneuroim.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 21.Sahid El-Radhi A, Hogg CL, Bungre JK, Bush A, Corrigan CJ. Effect of oral glucocorticoid treatment on serum inflammatory markers in acute asthma. Arch Dis Child. 2000;83:158–62. doi: 10.1136/adc.83.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhawan L, Liu B, Blaxall BC, Taubman MB. A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein-1 mRNA stability. J Biol Chem. 2007;282:10146–52. doi: 10.1074/jbc.M605925200. [DOI] [PubMed] [Google Scholar]
- 23.Sloka JS, Stefanelli M. The mechanism of action of methylprednisolone in the treatment of multiple sclerosis. Mult Scler. 2005;11:425–32. doi: 10.1191/1352458505ms1190oa. [DOI] [PubMed] [Google Scholar]
- 24.Wust S, Van Den Brandt J, Tischner D, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol. 2008;180:8434–43. doi: 10.4049/jimmunol.180.12.8434. [DOI] [PubMed] [Google Scholar]
- 25.Tsutsui S, Vergote D, Shariat N, Warren K, Ferguson SS, Power C. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells. FASEB J. 2008;22:786–96. doi: 10.1096/fj.07-9002com. [DOI] [PubMed] [Google Scholar]
- 26.Moreira MA, Tilbery CP, Monteiro LP, Teixeira MM, Teixeira AL. Effect of the treatment with methylprednisolone on the cerebrospinal fluid and serum levels of CCL2 and CXCL10 chemokines in patients with active multiple sclerosis. Acta Neurol Scand. 2006;114:109–13. doi: 10.1111/j.1600-0404.2006.00629.x. [DOI] [PubMed] [Google Scholar]
- 27.Elovaara I, Kuusisto H, Paalavuo R, et al. Effect of high-dose methylprednisolone treatment on CCR5 expression on blood cells in MS exacerbation. Acta Neurol Scand. 2006;113:163–6. doi: 10.1111/j.1600-0404.2005.00566.x. [DOI] [PubMed] [Google Scholar]
- 28.De Andres C, Aristimuno C, De Las Heras V, et al. Interferon beta-1a therapy enhances CD4+ regulatory T-cell function: an ex vivo and in vitro longitudinal study in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2007;182:204–11. doi: 10.1016/j.jneuroim.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 29.Weber MS, Hohlfeld R, Zamvil SS. Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics. 2007;4:647–53. doi: 10.1016/j.nurt.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fahey AJ, Robins RA, Kindle KB, Heery DM, Constantinescu CS. Effects of glucocorticoids on STAT4 activation in human T cells are stimulus-dependent. J Leukoc Biol. 2006;80:133–44. doi: 10.1189/jlb.0605296. [DOI] [PubMed] [Google Scholar]
- 31.Cohen JA, Calabresi PA, Chakraborty S, et al. Avonex Combination Trial in relapsing–remitting MS: rationale, design and baseline data. Mult Scler. 2008;14:370–82. doi: 10.1177/1352458507083189. [DOI] [PubMed] [Google Scholar]