

Abstract
Early primitive stem cells have long been viewed as the cancer cells of origin (tumor initiating target cells) due to their intrinsic features of self-renewal and longevity. However, emerging evidence suggests a surprising capacity for normal committed cells to function as reserve stem cells upon reprogramming as a consequence of tissue damage resulting in inflammation and wound healing. This results in an alternative concept positing that tumors may originate from differentiated cells that can re-acquire stem cell properties due to genetic or epigenetic reprogramming. It is likely that both models are correct, and that a continuum of potential cells of origin exists, ranging from early primitive stem cells to committed progenitor or even terminally differentiated cells. A combination of the nature of the target cell and the specific types of gene mutations introduced determine tumor cell lineage, as well as potential for malignant conversion. Evidence from mouse skin models of carcinogenesis suggests that initiated cells at different stages within a stem cell hierarchy have varying degrees of requirement for reprogramming (e.g. inflammation stimuli), depending on their degree of differentiation. This article will present evidence in favor of these concepts that has been developed from studies of several mouse models of skin carcinogenesis.
Keywords: Skin Cancer, Reprogramming, Stem cell hierarchy, Cell of origin, Malignant potential, Inflammation, p53
1. Introduction
One of the core questions in cancer biology relates to the identity and nature of the cancer “cell of origin,” the target cell in which the first oncogenic driver mutation occurs and leads to tumor initiation [1]. With the emerging concepts of “cancer-stem-cells”, i.e. that sub-population of cells with tumor-initiating capacity in serial transplantation assays, it has been hypothesized that the primitive early stem cells might be the cancer cells of origin, because of their intrinsic features of self-renewal and longevity. However, evidence for the plasticity of completely normal cells, demonstrating their ability to acquire stem cell characteristics [2] raised the possibility that more committed progenitor cells can also serve as target cells for initiation, as long as they maintain or can re-acquire stem cell-like features. Among the questions that arise in light of these new concepts, are the following: 1) do tumors arise from stem cells, or committed progenitor cells that require reprogramming through inflammation or biological stress? 2) How does the cell of origin (stem cell or committed progenitor cell) affect malignant potential? 3) Do cancers of different histological subtypes arise by reprogramming of the same target cells?
In general, cellular reprogramming refers to the concept of rewiring the epigenetic and transcriptional network of one cell type to that of a different cell type [3, 4]. In this review, we refer to “reprogramming” as those additional genetic, epigenetic, and micro-environmental alterations that are required for target cells to initiate and maintain/propagate a tumor.
2. Stem cell hierarchy and malignant potential
Increasing evidence suggests a surprising role for normal committed progenitor cells as a backup reservoir for adult stem cells after reprogramming in response to stress conditions. Using a method to trace the lineage of quiescent label-retaining cells (LRCs) in vivo in the intestinal crypt, it has been demonstrated that upon wounding, a population of LRCs that normally gives rise to Paneth cells can be reprogrammed to repopulate the stem cell niche and contribute to the regeneration of all intestinal cell lineages [5]. Similar results have been obtained by lineage tracing of Bmi1-positive quiescent stem cells exposed to radiation damage [6]. Another study, also using in vivo lineage tracing in mice, has demonstrated that upon depletion of airway stem cells, differentiated luminal secretory cells can be de-differentiated into basal stem cells [7]. This capacity of committed cells to function as reserve stem cells via reprogramming is very relevant to the tumor cell of origin question, as these studies support the idea that terminally differentiated cells can also become tumor-initiating cells. Similarly, it has been shown that inflammatory tumor promoting environments mediated by enhanced NF-κB activity and subsequent Wnt pathway activation induced intestinal epithelial non-stem cells to acquire a stem-cell like fate and function as tumor-initiating cells [8]. A mouse model of brain cancer also has shown that aggressive glioblastoma can originate from a range of different cell types including astrocytes and mature neurons in the nervous system via direct reprogramming [9].
These studies support the notion that tumors can originate from progenitor cells as well as stem cells. It is likely that there is a continuum of potential cells of origin ranging from early primitive stem cells to committed progenitor or even terminally differentiated cells, and the combination of the nature of the target cell and the specific types of gene mutations introduced determine their malignant potential [1]. For example, tumors that arise from early epidermal stem cells within the bulge region of the hair follicle might intrinsically possess more aggressive properties than tumors with the same genetic mutations but arising from a more differentiated, committed cell type in the epidermal lineage. Conversely, initiated cells within the more differentiated cell compartment would have the greatest requirement for reprogramming, in order to facilitate self renewal and malignant progression (Fig. 1). Evidence in favor of these concepts has come from studies of the skin model system, as discussed later in more detail [10, 11].
Figure 1. A continuum of potential cells of origin for skin cancer.
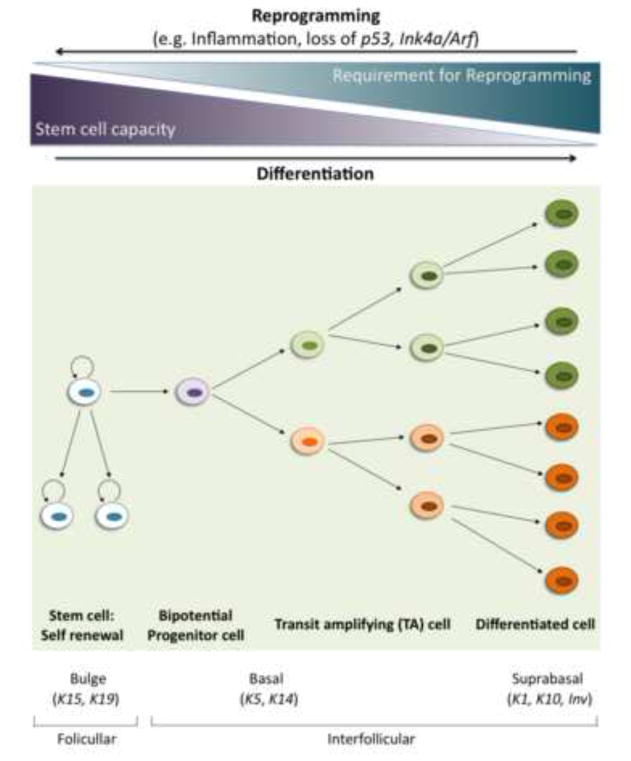
Stem cells with high self-renewal capacity produce, by asymmetrical division, a new stem cell and a more differentiated one (progenitor cell) with higher proliferative rate but low or absent self-renewal capability. Alternatively, stem cells can undergo a symmetrical division to generate two new stem cells or two more-differentiated ones. More differentiated cells (transit amplifying cells) are more numerous within the tissue and can progressively lead to different lineages by differentiating into specialized cells that maintain the tissue. In skin, cells within the bulge region of the hair follicle have a high stem-cell self-renewal capacity and high malignant potential, and therefore initiating mutations in the bulge stem cells can give rise to papillomas or carcinomas without extensive cellular reprogramming. Alternatively, papillomas might be derived from cells within the interfollicular epidermis, either the basal stem-cell population that still has some self-renewal capacity or from the transit amplifying or even suprabasal cells. However, target cells within the more differentiated cell compartment would have greater requirements for reprogramming such as inflammation, loss of p53, or loss of Ink4/Arf, in order to facilitate self renewal and malignant progression
3. Inflammation and multistage skin tumorigenesis
Inflammation is a highly complex process involving cellular and humoral components of the immune system that exhibit both pro- and anti-tumor properties during cancer development [12, 13]. Since the initial study in 1863 by Virchow [14], who observed the presence of a leukocytic infiltrate in tumor tissues and hypothesized a connection between the sites of inflammation and tumorigenesis, critical roles for inflammatory stimuli have been demonstrated in initiating, maintaining, and advancing tumors. These insights have led to various therapeutic strategies targeting inflammation for the prevention and treatment of cancer [12, 13, 15–18].
A critical role for inflammation in skin cancer is well characterized from studies using the classical two-stage DMBA/TPA chemical carcinogenesis model. This involves a single topical application of a mutagen, such as DMBA (7,12-dimethylbenz-[a]-anthracene) to introduce an initiating mutation, followed by prolonged exposure to a tumor promoter, such as TPA (12-O-tetradecanoyl phorbol-13-acetate) for 20 weeks. The predominant types of tumors that arise in this model are benign squamous papillomas, and a small portion of these tumors progress to malignant squamous cell carcinomas (SCCs). Some of these SCCs further undergo epithelial-mesenchymal transition (EMT) and become a more aggressive form of poorly differentiated carcinomas known as spindle cell carcinomas [19]. In the DMBA/TPA protocol, DMBA can form covalent adducts with the DNA of all epidermal target cells and causes tumor initiation commonly through a specific mutation in codon 61 of Hras (Q61L) [20–22]. However, the initiating Ras mutation by itself is not sufficient to form tumors in most experimental models, emphasizing the need for a tumor promoter in the second stage. The type of promoter as well as the duration and frequency of the tumor promoter treatment profoundly affect the incidence of papillomas in two-stage carcinogenesis experiments [23, 24].
The tumor-promoting role of TPA in the DMBA/TPA model is not simply limited to stimulation of cell proliferation. TPA regulates activation of protein kinase C (PKC) isoforms and induces a pleiotropic tissue inflammatory reaction by the production of a wide range of cytokines such as Tgfβ, Tnfα and Il-1α that are crucial mediators of wound inflammatory responses [25–29]. This wound inflammatory reaction is critical in tumor promotion in that 1) other promoters that are equally potent as TPA in enhancing cell proliferation but without inducing cutaneous inflammation do not cause papilloma development, and 2) TPA treatment can be replaced by repeated wounding as well as by the injection of wound growth factors such as TGF-β with resultant papilloma formation [25, 30–32].
Skin carcinogenesis induced in the DMBA/TPA model was presumed to proceed in a linear fashion through distinct stages including development of inflammation-dependent benign papillomas, conversion into malignant squamous cell carcinomas (SCCs), and progression of some of SCCs to undifferentiated spindle cell carcinomas via EMT [19]. There is however evidence that some aggressive spindle cell carcinomas arise by a separate route that is distinct from the classical pathway that is heavily dependent on inflammatory stimuli and Hras mutation [11]. Detailed analysis of carcinomas by gene expression profiling and histology revealed two distinct categories representing pure SCCs or those with only a minor spindle cell component (Class A), or essentially pure spindle cell carcinomas (Class B). Unlike the Class A carcinomas, the Class B carcinomas exhibit characteristics of EMT including 1) up-regulation of Snai1, Zeb1 and Vimentin, 2) down-regulation of E-cadherin, Krt5, and Krt14, and 3) “Claudin low signature,” which denotes stem cell-like features that have been shown in a subset of human breast cancer [33, 34].
Striking differences between the Class A and Class B carcinomas were seen by analysis of 1) the integrity and expression of the Ink4a/Arf locus, and 2) their dependency on inflammation and Hras signaling, suggesting that Class B tumors may arise via a separate route involving distinct genetic and molecular reprogramming mechanisms. The fact that the Class B tumors frequently undergo loss of the Ink4a/Arf locus is notable. Three genes, p16/Cdkn2a, p15/Cdkn2b, and p19/Arf, are encoded from the deleted regions in the Class B carcinomas, and p16 and p19 have been specifically implicated in hindering self-renewal of neural and hematopoietic stem cells and blocking direct reprogramming of somatic cells to iPSCs [35–37]. This is reminiscent of the mechanisms by which p53 modifies the stem-like state and malignant potential of target cells (discussed later).
Exposure of mice to different levels of inflammation by modulating the length of TPA treatment revealed a differential dependence of these two tumor classes on inflammatory responses. While reduced exposure to inflammation dramatically decreased overall yields of papillomas and Class A carcinomas, more Class B carcinomas with high frequency of Ink4/Arf genetic alterations were observed. Mice treated with an abbreviated duration of TPA treatment of 5 weeks, rather than the usual 20 weeks, showed the highest relative proportion of Class B to Class A carcinomas. This observation is reminiscent of the “high-risk papillomas” reported by Hennings et al. three decades ago, which gave rise to carcinomas with a rather high 20% conversion rate after an abbreviated TPA treatment of 5 weeks [38]. We conclude that most papillomas and Class A SCCs seem to have a greater requirement for reprogramming by exposure to inflammatory agents, while the Class B carcinomas, which express high levels of Cd34 and some other known bulge stem cell markers, have a reduced requirement for this reprogramming stimulus. Intriguingly, mutant Hras signaling was downregulated in Class B spindle carcinomas prompting a study of skin tumor development in mice lacking the Hras target gene. Despite the fact that very few papillomas developed in Hras-deficient mice, surprisingly, the majority of null mice developed Class B carcinomas, most of which harbored mutations in Kras and deletions of the Ink4/Arf locus. All these data suggest the Class B carcinomas do not arise from pre-existing Class A carcinomas but utilize a novel pathway leading to malignant progression that is less dependent on inflammation and Hras signaling. Two possible explanations are 1) the Class B tumors may undergo alternative genetic events possibly involving Ink4A at an early stage of carcinogenesis, or 2) the Class B tumors may arise from an early stem cell that does not require such extensive reprogramming by inflammatory and tumor-promoting stimuli (Fig. 2).
Figure 2. Models of the possible origins of Class A and Class B carcinomas.
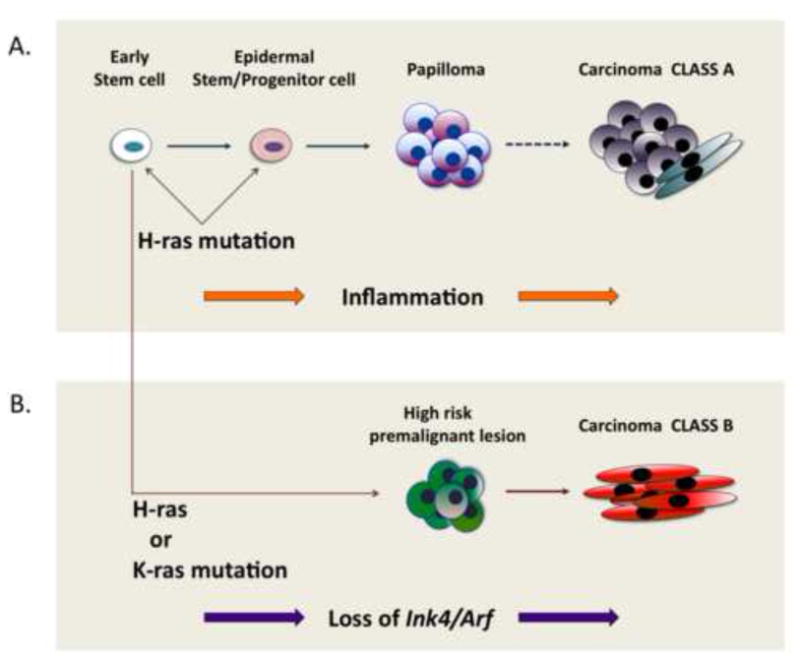
Class B carcinomas may not arise from pre-existing Class A carcinomas but utilize a novel pathway leading to malignant progression that is less dependent on inflammation and Hras signaling. Class A and Class B carcinomas could have the same cell of origin but diverge at an early stage (e.g. loss of Ink4/Arf), giving rise to different kinds of premalignant lesions (papilloma and high-risk premalignant lesion, respectively). Conversely, Class A and Class B carcinomas could have two completely different cells of origin in the skin and types of Ras mutations may also be different depending on the nature of target cells.
4. Inflammation and reprogramming in skin cancer
The data described above suggest a model by which Ras-driven tumors arising from early stem cells may require minimal inflammation-related reprogramming, whereas more differentiated target cells require extensive reprogramming by wounding and/or exogenous inflammatory stimuli (Fig. 1). Several groups have tried to model these events by targeting mutant Ras to different epidermal cell compartments. Overexpression of mutant Hras under the control of a truncated K5-promoter that targets the outer root sheath (ORS) of hair follicles, where stem cells are believed to reside, gave rise to malignant SCCs that are independent of wounding or extra TPA treatments [10]. On the other hand, expression of the same mutant gene in interfollicular differentiated cells resulted in the formation of terminally differentiating papillomas, but only at sites subject to inflammation by wounding, such as the base of the tail or the ear [10, 39, 40]. Moreover, these tumors were strongly stimulated by treatment with TPA [39, 40], but regressed once TPA was withdrawn, demonstrating their high dependency on chronic inflammation. These data are compatible with the concept that the requirement for inflammation-induced reprogramming is inversely correlated with the degree of differentiation of the target cells. Interestingly, the ability of committed lung cells to undergo reprogramming has also been reported to be inversely related to their stage of differentiation [7].
More recently, using an approach combining knock-in technology and tissue-specific expression of targeted genes using Cre-recombinase, several groups activated an oncogenic endogenous Kras allele (KrasG12D) in different epithelial cell populations in an attempt to trace tumor cells of origin [41–45] (summarized in Table 1). Apart from the Shh-Cre, which is expressed in transit-amplifying (TA) matrix cell populations, expression of KrasG12D in epithelial cells in the follicle or interfollicular region resulted in spontaneous development of cystic or proliferative lesions and some papillomas, but no malignant carcinomas. Direct comparison of these various studies is complicated by the use of different promoters to drive either mutant Hras or Kras in skin epithelial compartments. There may be intrinsic differences between oncogenic Kras and Hras in their capacity to confer stem-like features on target cells, or in their patterns of expression in subsets of epithelial cells. Only some of these studies addressed the role of wounding or inflammation by TPA treatment on development of benign or malignant skin tumors [10, 39, 40, 45], and much remains to be done in this area. An interesting possibility is that tumors developing from different compartments, or driven by different Ras genes, may show changes in dependence on exogenous inflammatory agents.
Table 1. Models of ras-driven skin carcinogenesis targeting different epidermal cell populations.
Tracing cells of origins for SCCs has been attempted using keratin and other gene promoters by targeting oncogenic Hras or Kras in different epithelial compartments. The role of wounding/inflammation has been addressed by some of these studies [10, 39, 40, 45], but not all. Only simultaneous activation of KrasG12D and loss of Trp53 by Cre-mediated recombination led to development of malignant SCCs from cell populations expressing K5, K14, K15, and K19 [42–44].
| Genes targeted |
Hras, Tg | LSL-Kras-G12D | ||||||
|---|---|---|---|---|---|---|---|---|
| Promoters used | K1, K10 | K5 | Lrig1 | INV | K14 | K5 | K15 | K19 |
|
Representative cell populations |
Suprabasal layer of IFE |
ORS of HF | Junctional zone |
Suprabasal layer of IFE, Basal layer (K5- like) |
Basal cells of HF, IFE, sebaceous glands |
Basal cells of IFE and ORS of HF |
Bulge | Bulge |
|
Wounding/TPA used |
Yes | Yes | Yes | No* | No* | Yes* | No* | No* |
|
Papilloma (dorsal) development |
Yes | Yes | Yes | Yes | Yes** | Yes | Yes | Yes |
|
Inflammation dependency |
Wounding/TPA -dependent |
Wounding/TPA -independent |
Wounding -dependent |
Wounding/TPA -independent ? |
Wounding/TPA -independent ? |
Wounding/TPA -independent ? |
Wounding/TPA -independent ? |
Wounding/TPA -independent ? |
|
Trp53 flox/flox used |
No | No | No | No | Yes | Yes | Yes | Yes |
|
Carcinoma development |
No, Not tested with Trp53 f/f |
Yes, Not tested with Trp53 f/f |
No, Not reported with Trp53 f/f |
No, Not reported with Trp53 f/f |
Yes, with Trp53 f/f |
Yes, with Trp53 f/f, with Trp53 R172H/f |
Yes, with Trp53 f/f |
Yes, with Trp53 f/f |
| References |
Bailleul et al. 1990 Greenhalgh et al. 1993 [39, 40] |
Brown et al., 1998 [10] |
Page et al. 2013 [45] |
Lapouge et al., 2011 [44] |
Lapouge et al., 2011 Lapouge et al., 2012 [44, 85] |
Caulin et al., 2007 [42] |
White et al., 2011 Lapouge et al., 2011 [43, 44] |
Lapouge et al., 2011 [44] |
Tg; Transgenic overexpression, LSL; Lox-Stop-Lox, INV; Involucrin, IFE; Interfollicullar Epidermis, HF; Hair Follicle, ORS; Outer Root Sheath, f/f; flox/flox
Sensitivity to additional wounding/inflammation has not been addressed.
The precise locations of these papillomas have not been described in detail.
Despite some discrepancies, the above data lead to a plausible hypothesis that different target cells within the epidermal lineage have varying degrees of requirement for reprogramming (shown as chronic inflammatory promotion here) in order to develop even benign papillomas. The specific characteristics of the target cells that are intrinsically capable of producing papillomas that progress to carcinomas have not been identified.
5. Stem cell hierarchy and tumor type
A related but distinct question is whether tumors of different histological subtypes, but arising within the same tissue, have the same or different cells of origin. Basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs), representing two different types of non-melanoma skin tumors, are the commonest forms of cancer in Caucasian populations, but whether they come from the same target cell through acquisition of separate genetic events, or from completely distinct cell compartments, has been a controversial issue. Transgenic mouse approaches that involve activation of Ras and Sonic Hedgehog pathways known to be drivers of SCC and BCC development, respectively, have provided confusing and sometimes contradictory results. The use of various experimental strategies have led to the conclusions that BCCs in the mouse can arise from the hair follicle bulge region [46] from the interfollicular epidermis and upper infundibulum [47] or from both of these target cell populations [48]. Direct comparison of these results is complicated by the fact that all used different genetic events to activate Shh signaling (loss of Ptch, activation of Smo, or expression of activated Gli2 respectively), and the locations of the resulting BCCs (dorsal, ear, or tail skin) showed substantial variation. Parallel questions regarding the origins of SCCs have been addressed using keratin and other gene promoters to drive activated forms of Hras or Kras in various epithelial compartments [41–45] (see Table 1). These studies have shown that proliferative squamous lesions and papillomas, often at sites of scratching or wounding, can arise as a consequence of Ras activation in several distinct cell compartments, ranging from regions of the bulge containing stem cells (marked by Krt15 or Krt19 expression) to fully differentiated cell compartments (expressing K1, K10, or involucrin) with low proliferative capacity.
One possible conclusion from all of these targeting studies is that virtually every cell is capable of becoming transformed, and that no specific target cell population exists for either BCCs or SCCs. However, the use of tissue-specific promoters to drive expression of potent activated oncogenes raises a number of possible caveats. Many of these promoters are infamously “leaky” and may be expressed in regions outside the expected cell compartment. Secondly, cancer initiation by chemical or physical carcinogens entails mutational events involving one or more driver genes in single cells. This is a different scenario from that resulting from oncogene activation simultaneously in many, possibly contiguous, cells within an epithelial compartment. In the latter case, initiated cells may not have to out-compete their neighbors – an important facet of “normal” initiation at the single cell level [49–51]. The exact locations of these single initiated cells, in particular those with the highest malignant potential, remain to be determined.
The observation that the hair follicle bulge as well as the interfollicular region contain putative cells of origin of both SCCs [10, 43, 44, 52] and BCCs [46, 48] raises the possibility that the stem cells in these regions may be capable of forming both tumor types. If this were the case, what are the main determinants of tumor cell fate? As is often the case, studies of normal epidermal lineage selection during mouse development provide some important clues. The roles of the Wnt-β-catenin signaling in promoting Hair Follicle (HF) lineage differentiation during development have been well characterized [53–55], and a study by Jahoda and colleagues has suggested a critical antagonistic function for EGFR/Ras signaling in specifying interfollicular epidermis (IFE) lineage at the expense of hair follicle development [56]. First, they showed that increased levels of EGFR and KGF ligands promoted epidermal differentiation while inhibiting hair follicle formation. Consistent with these findings, gene expression analysis of KGF and HBEFG-treated embryonic skin demonstrated that while genes involved in epidermal differentiation such as S100a18, S100a6, loricrin, and keratin 6A, were rapidly induced, Shh signaling factors including Shh, Gli1, Ptch2, which are implicated in hair follicle morphogenesis were downregulated [56]. This interesting observation illustrates an important concept in developmental biology: positive activators of one cell fate decision simultaneously inhibit the alternative pathway [57]. These data suggest a model in which the nature of the first genetic alteration in this stem cell at the pre-lineage selection stage might determine the fate of resulting tumors (Fig. 3). Activation of the Ras pathway mimics the effect of Egf/Hbegf in commitment to the epidermal squamous lineage, while at the same time inhibiting activation of the alternative cell fate by the Shh pathway. Experimental evidence in favor of this model came from analysis of the opposing effects of the patched gene (Ptch) in BCC and SCC formation. The Ptch gene is a well-known suppressor of the Shh pathway and loss of function of Ptch promotes BCC formation in a mouse model resembling human BCC development [58, 59]. Paradoxically, genetic mapping in mouse models of SCCs identified a variant of PtchFVB that promotes the development of SCCs arising from the epidermal lineage, synergizing with the chemically induced initiating Hras mutation [60, 61]. Mechanistically, over-expression of the PtchFVB variant appears to influence cell fate commitment at the initiation stage and reinforce the commitment to the epidermal lineage that is stimulated by activation of Ras/Egfr signaling. We conclude that genetic alterations in the Ras and Shh pathways in stem cells have critical and antagonistic roles in determining tumor lineage selection, which closely resembles the process of normal skin epithelial lineage selection during development.
Figure 3. The effect of specific genetic pathways on tumor-cell fate.
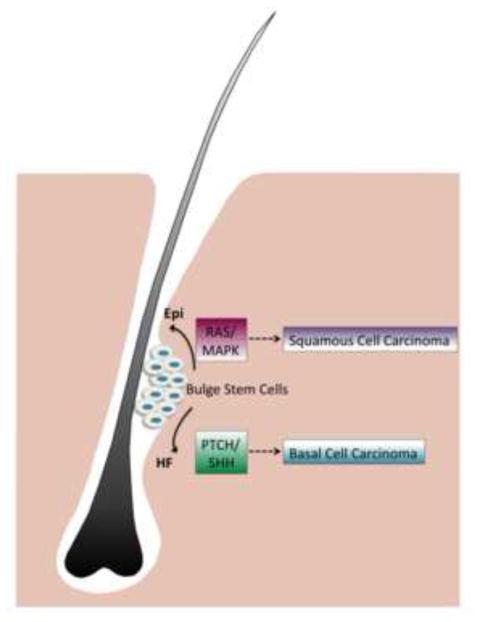
The nature of the first genetic alteration in a single target-cell population within the bulge stem-cell region at the pre-lineage selection stage might determine the fate of resulting tumors. Activation of each of the pathways (RAS/MAPK or PTCH/SHH signaling) in a bulge stem cell promotes either epidermal (Epi) differentiation or hair follicle (HF) formation, and may further induce the formation of particular tumor types (squamous-cell carcinomas or basal-cell carcinomas, respectively).
Oncogenic alterations have also been proposed to determine specific lineage phenotypes in other tumor types, possibly through epigenetic reprogramming. This has been particularly nicely demonstrated for leukemia mouse models, where the identity of the driver oncogene, when expressed in haematopoietic stem cells, determines the lineage of the resulting tumor cells [62].
6. Role of p53 in reprogramming target cells
p53 is an extensively characterized tumor-suppressor that plays a central role in multiple cellular pathways including apoptosis, senescence, cell cycle arrest, and DNA damage repair. Recently, an emerging role for p53 in regulating cell differentiation, stem cell reprogramming, and self-renewal has suggested the intriguing possibility that p53 also functions in reprogramming of cancer target cells, a role that goes beyond its classically known functions in preserving genome integrity.
In DMBA/TPA-mediated skin carcinogenesis, the loss or mutation of Trp53 is coupled with progression of benign papilliomas to malignant SCCs [63]. Kemp et al. showed that when Trp53−/− mice were subjected to the DMBA/TPA protocol, while the yield of papillomas was reduced, malignant carcinomas arose more rapidly and with an extremely high malignant conversion rate approaching 50% [64]. Usually, only 3 to 10% of benign papillomas progress to SCCs from most strains of wild-type mice. Thus, p53 functions in the stage of malignant progression. The studies of KrasG12D-driven skin tumorigenesis described above, surprisingly, reported no spontaneous progression to carcinomas in any of these models. However, simultaneous loss of Trp53 by Cre-mediated recombination led to development of malignant SCCs from different epithelial populations (derived from target cells expressing K5, K14, K15, and K19, see Table 1). This common effect of p53 loss on target cells at different stages of the epidermal lineage is surprising, and raises questions as to the mechanisms by which this conversion takes place.
One straightforward explanation stems from the well known tumor-suppressive roles of p53; when classical functions of p53 in apoptosis, senescence, and cell cycle arrest are all withdrawn, increased genomic instability may greatly accelerate malignant tumor progression. An alternative mechanism may reflect the emerging roles of p53 in stem cell regulation, cell differentiation, and epidermal lineage selection. It has been suggested that loss of p53 empowers a differentiated cell to restore stem-like features and also that absence of p53 can increase stem cell numbers in various tissue systems [65–69]. This occurs as p53 normally regulates asymmetric division to generate one daughter cell with a stem-cell fate (“self-renewal”) and another that differentiates, but loss of p53 results in symmetric stem cell division to give rise to two identical daughter stem cells [70]. Trp53−/− mice have higher numbers of mammary stem cells that can form mammospheres in vitro and in vivo, conceivably because p53 favors asymmetric divisions of stem cells, thereby limiting their numbers and promoting cell differentiation [66, 67]. Similarly, in epidermis, loss of p53 may also increase numbers of bulge stem cells or may allow committed progenitor cells to adopt a more stem-like state, leading to expansion of the population of target cells carrying high malignant potential that may give rise to SCCs.
In support of these concepts, several groups have demonstrated that p53 functions as a barrier to direct reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) via its effects on cell cycle arrest and apoptosis [35, 71–74]. Of particular interest is the role of a miRNA family activated by p53 that has been shown to modulate diverse cellular processes including stem cell reprogramming and development. For example, p53-mediated induction of miR-34s provides a blockade for somatic reprogramming [75–78]. Genetic ablation of miR-34s, including miR-34a and miR-34b/c, was shown to enhance reprogramming efficiency for iPSCs generation. The mechanisms by which miR-34s functions may not be entirely through cell cycle control, because suppression of reprogramming by miR-34a was also partially due to repression of a number of reprogramming transcription factors such as Nanog, Sox2, and N-myc [77]. Furthermore, p53-activated miR-34a targets β-catenin and suppresses canonical Wnt signaling [79]. Similarly, a negative impact of p53 on trans-activation of Wnt pathway genes has been previously reported, and p53 also induced SIAH, an E3 ligase that targets β-catenin for proteasome-mediated degradation [80–82].
This miR-34a-dependent and -independent suppression of Wnt-β-catenin signaling by p53 should affect the known activity of Wnt signaling in regulation of stem cell self-renewal in several tissue systems [83, 84], and as discussed earlier, the Wnt-β-catenin pathway is critical in making cell fate decisions at the early stage of epidermal lineage selection [53, 55]. These data overall suggest that p53 plays a role in promotion of the epidermal lineage during the stage of cell fate choice and that loss of p53 confers stem cell plasticity to this differentiating target cell population.
A comparable argument may be possible for BCC development in the context of p53 loss. It has been reported that BCC formation is much more efficient when Ptch1 is deleted in combination with loss of p53 using K14-Cre-ER (where target cells are mostly in the interfollicular epidermis) compared to K15-Cre-PR1 (target cells are the bulge stem cells) [46]. Given that K15-representing cells in the bulge area already possess stem cell features, the effects of loss of p53 on conferring stem-like characters to this population of target cells for BCC formation is relatively minimal compared to the expected effects on the K14-positive IFE target cells. Altering p53 functions may be amongst the most powerful means of reprogramming the malignant potential of target cells. The combination of two potent genetic events at an early stage of carcinogenesis, such as Ras mutation and loss of p53, may abrogate the need for additional reprogramming effects including inflammation.
7. Concluding remarks
Studies of multistage carcinogenesis in the skin have provided many of the concepts of initiation, promotion and progression that are operative in most forms of human cancer. These models have identified causal roles for specific genes at particular stages of carcinogenesis in ways that would not have been possible using human samples. Since mouse skin is also the most widely studied solid tissue model of normal stem cell organization, it seems likely that this model will continue to provide us with information that will be critical for understanding the origins of cancer stem cells from normal stem cells or reprogrammed differentiated progenitors in response to tissue damage.
Acknowledgments
The authors thank members of the Balmain lab for insightful discussion. Research in the Balmain lab is supported by U01 CA84244, U01 CA141455, and R01 CA111834-06A1 (A.B.) and T32-CA108462-10 (I.Y.S).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat Rev Cancer. 2003:434–43. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Egli D, Birkhoff G, Eggan K. Mediators of reprogramming: transcription factors and transitions through mitosis. Nat Rev Mol Cell Biol. 2008:505–16. doi: 10.1038/nrm2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanna J, Carey BW, Jaenisch R. Reprogramming of somatic cell identity. Cold Spring Harb Symp Quant Biol. 2008:147–55. doi: 10.1101/sqb.2008.73.025. [DOI] [PubMed] [Google Scholar]
- 5.Buczacki SJA, et al. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013:65–9. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 6.Yan KS, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA. 2012:466–71. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tata PR, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013:218–23. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwitalla S, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Friedmann-Morvinski D, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012:1080–4. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown K, et al. The malignant capacity of skin tumours induced by expression of a mutant H-ras transgene depends on the cell type targeted. Curr Biol. 1998:516–24. doi: 10.1016/s0960-9822(98)70203-9. [DOI] [PubMed] [Google Scholar]
- 11.Wong CE, et al. Inflammation and Hras signaling control epithelial-mesenchymal transition during skin tumor progression. Genes Dev. 2013:670–82. doi: 10.1101/gad.210427.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Colotta F, et al. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009:1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 14.Virchow R. An Address on the Value of Pathological Experiments. Br Med J. 1881:198–203. doi: 10.1136/bmj.2.1075.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elinav E, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nature Publishing Group. 2013:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 16.Balkwill FR, Mantovani A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin Cancer Biol. 2012:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Gonda TA, Tu S, Wang TC. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle. 2009:2005–13. doi: 10.4161/cc.8.13.8985. [DOI] [PubMed] [Google Scholar]
- 18.Mantovani A, et al. Cancer-related inflammation. Nature. 2008:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 19.Klein-Szanto AJ, et al. Multistage chemical carcinogenesis protocols produce spindle cell carcinomas of the mouse skin. Carcinogenesis. 1989:2169–72. doi: 10.1093/carcin/10.11.2169. [DOI] [PubMed] [Google Scholar]
- 20.Quintanilla M, et al. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 21.Balmain A, et al. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature. 1984:658–60. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- 22.Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983:72–4. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- 23.Viaje A, et al. Effects of antiinflammatory agents on mouse skin tumor promotion, epidermal DNA synthesis, phorbol ester-induced cellular proliferation, and production of plasminogen activator. Cancer Res. 1977:1530–6. [PubMed] [Google Scholar]
- 24.Verma AK, Boutwell RK. Effects of dose and duration of treatment with the tumor-promoting agent, 12-O-tetradecanoylphorbol-13-acetate on mouse skin carcinogenesis. Carcinogenesis. 1980:271–6. doi: 10.1093/carcin/1.3.271. [DOI] [PubMed] [Google Scholar]
- 25.Hensler S, Mueller MM. Inflammation and skin cancer: old pals telling new stories. Cancer J. 2013:517–24. doi: 10.1097/PPO.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 26.Hansen LA, Monteiro-Riviere NA, Smart RC. Differential down-regulation of epidermal protein kinase C by 12-O-tetradecanoylphorbol-13-acetate and diacylglycerol: association with epidermal hyperplasia and tumor promotion. Cancer Res. 1990:5740–5. [PubMed] [Google Scholar]
- 27.Fournier A, Murray AW. Application of phorbol ester to mouse skin causes a rapid and sustained loss of protein kinase C. Nature. 1987:767–9. doi: 10.1038/330767a0. [DOI] [PubMed] [Google Scholar]
- 28.Castagna M, et al. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982:7847–51. [PubMed] [Google Scholar]
- 29.Akhurst RJ, FEE F, Balmain A. Localized production of TGF-beta mRNA in tumour promoter-stimulated mouse epidermis. Nature. 1988:363–5. doi: 10.1038/331363a0. [DOI] [PubMed] [Google Scholar]
- 30.Fürstenberger G, et al. Stimulatory role of transforming growth factors in multistage skin carcinogenesis: possible explanation for the tumor-inducing effect of wounding in initiated NMRI mouse skin. Int J Cancer. 1989:915–21. doi: 10.1002/ijc.2910430531. [DOI] [PubMed] [Google Scholar]
- 31.Fürstenberger G, et al. Effects of the phorbol ester 4-O-methyl-12-O-tetradecanoylphorbol-13-acetate on mouse skin in vivo: evidence for its uselessness as a negative control compound in studies on the biological effects of phorbol ester tumor promoters. Cancer Res. 1982:342–8. [PubMed] [Google Scholar]
- 32.Yamasaki H, et al. Comparative effects of a complete tumor promoter, TPA, and a second-stage tumor promoter, RPA, on intercellular communication, cell differentiation and cell transformation. Carcinogenesis. 1985:1173–9. doi: 10.1093/carcin/6.8.1173. [DOI] [PubMed] [Google Scholar]
- 33.Prat A, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 35.Li H, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009:1136–9. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molofsky AV, et al. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003:962–7. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park I-K, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003:302–5. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 38.Hennings H, et al. Induction of papillomas with a high probability of conversion to malignancy. Carcinogenesis. 1985:1607–10. doi: 10.1093/carcin/6.11.1607. [DOI] [PubMed] [Google Scholar]
- 39.Bailleul B, et al. Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell. 1990:697–708. doi: 10.1016/0092-8674(90)90115-u. [DOI] [PubMed] [Google Scholar]
- 40.Greenhalgh DA, et al. Induction of epidermal hyperplasia, hyperkeratosis, and papillomas in transgenic mice by a targeted v-Ha-ras oncogene. Mol Carcinog. 1993:99–110. doi: 10.1002/mc.2940070208. [DOI] [PubMed] [Google Scholar]
- 41.Caulin C, et al. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004:5054–8. doi: 10.1158/0008-5472.CAN-04-1488. [DOI] [PubMed] [Google Scholar]
- 42.Caulin C, et al. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. The Journal of clinical investigation. 2007:1893–901. doi: 10.1172/JCI31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White AC, et al. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci USA. 2011:7425–30. doi: 10.1073/pnas.1012670108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lapouge G, et al. Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci USA. 2011:7431–6. doi: 10.1073/pnas.1012720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Page ME, et al. The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell. 2013:471–82. doi: 10.1016/j.stem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang GY, et al. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell. 2011:114–24. doi: 10.1016/j.ccr.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Youssef KK, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol. 2010:299–305. doi: 10.1038/ncb2031. [DOI] [PubMed] [Google Scholar]
- 48.Grachtchouk M, et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. The Journal of clinical investigation. 2011:1768–81. doi: 10.1172/JCI46307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dejosez M, et al. Safeguards for cell cooperation in mouse embryogenesis shown by genome-wide cheater screen. Science. 2013:1511–4. doi: 10.1126/science.1241628. [DOI] [PubMed] [Google Scholar]
- 50.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010:309–22. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vermeulen L, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013:995–8. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 52.Li S, et al. A keratin 15 containing stem cell population from the hair follicle contributes to squamous papilloma development in the mouse. Mol Carcinog. 2013:751–9. doi: 10.1002/mc.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andl T, et al. WNT signals are required for the initiation of hair follicle development. Dev Cell. 2002:643–53. doi: 10.1016/s1534-5807(02)00167-3. [DOI] [PubMed] [Google Scholar]
- 54.Watt FM, Jensen KB. Epidermal stem cell diversity and quiescence. EMBO Mol Med. 2009:260–7. doi: 10.1002/emmm.200900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, et al. Activation of beta-catenin signaling programs embryonic epidermis to hair follicle fate. Development. 2008:2161–72. doi: 10.1242/dev.017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson GD, et al. KGF and EGF signalling block hair follicle induction and promote interfollicular epidermal fate in developing mouse skin. Development. 2009:2153–2164. doi: 10.1242/dev.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davidson EH. Emerging properties of animal gene regulatory networks. Nature. 2010:911–20. doi: 10.1038/nature09645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008:2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 59.Nitzki F, et al. Patched knockout mouse models of Basal cell carcinoma. J Skin Cancer. 2012:907543. doi: 10.1155/2012/907543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wakabayashi Y, et al. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature. 2007:761–5. doi: 10.1038/nature05489. [DOI] [PubMed] [Google Scholar]
- 61.Kang HC, et al. Ptch1 Overexpression Drives Skin Carcinogenesis and Developmental Defects in K14Ptch(FVB) Mice. J Invest Dermatol. 2012 doi: 10.1038/jid.2012.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vicente-Dueñas C, et al. Function of oncogenes in cancer development: a changing paradigm. EMBO J. 2013:1502–13. doi: 10.1038/emboj.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bremner R, Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990:407–17. doi: 10.1016/0092-8674(90)90523-h. [DOI] [PubMed] [Google Scholar]
- 64.Kemp CJ, et al. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993:813–22. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- 65.Meletis K, et al. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006:363–9. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 66.Tao L, et al. Repression of mammary stem/progenitor cells by p53 is mediated by Notch and separable from apoptotic activity. Stem Cells. 2011:119–27. doi: 10.1002/stem.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cicalese A, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009:1083–95. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 68.Zhao Z, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010:1389–402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin T, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005:165–71. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 70.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006:1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 71.Kawamura T, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009:1140–4. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marión RM, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009:1149–53. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanabe K, et al. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc Natl Acad Sci USA. 2013:12172–9. doi: 10.1073/pnas.1310291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hong H, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009:1132–5. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hünten S, et al. The p53/microRNA network in cancer: experimental and bioinformatics approaches. Adv Exp Med Biol. 2013:77–101. doi: 10.1007/978-94-007-5590-1_5. [DOI] [PubMed] [Google Scholar]
- 76.Lin C-P, et al. The emerging functions of the p53-miRNA network in stem cell biology. Cell Cycle. 2012:2063–72. doi: 10.4161/cc.20207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi YJ, et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol. 2011:1353–60. doi: 10.1038/ncb2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chang T-C, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim NH, et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci Signal. 2011:ra71. doi: 10.1126/scisignal.2001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fiucci G, et al. Siah-1b is a direct transcriptional target of p53: identification of the functional p53 responsive element in the siah-1b promoter. Proc Natl Acad Sci USA. 2004:3510–5. doi: 10.1073/pnas.0400177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iwai A, et al. Siah-1L, a novel transcript variant belonging to the human Siah family of proteins, regulates beta-catenin activity in a p53-dependent manner. Oncogene. 2004:7593–600. doi: 10.1038/sj.onc.1208016. [DOI] [PubMed] [Google Scholar]
- 82.Lee K-H, et al. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci USA. 2010:69–74. doi: 10.1073/pnas.0909734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He XC, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004:1117–21. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- 84.Reya T, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 85.Lapouge G, et al. Skin squamous cell carcinoma propagating cells increase with tumour progression and invasiveness, in. EMBO J. 2012:4563–75. doi: 10.1038/emboj.2012.312. [DOI] [PMC free article] [PubMed] [Google Scholar]