

Abstract
Molecular imaging using optical techniques provides insight into disease at the cellular level. In this paper, we report on a novel dual-modality probe capable of performing molecular imaging by combining simultaneous three-dimensional optical coherence tomography (OCT) and two-dimensional fluorescence imaging in a hypodermic needle. The probe, referred to as a molecular imaging (MI) needle, may be inserted tens of millimeters into tissue. The MI needle utilizes double-clad fiber to carry both imaging modalities, and is interfaced to a 1310-nm OCT system and a fluorescence imaging subsystem using an asymmetrical double-clad fiber coupler customized to achieve high fluorescence collection efficiency. We present, to the best of our knowledge, the first dual-modality OCT and fluorescence needle probe with sufficient sensitivity to image fluorescently labeled antibodies. Such probes enable high-resolution molecular imaging deep within tissue.
OCIS codes: (170.0180) Microscopy, (170.4500) Optical coherence tomography, (170.2520) Fluorescence microscopy, (110.2350) Fiber optics imaging
1. Introduction
Molecular imaging [1,2] provides insight into cellular dysfunction by the targeting of specific molecular markers of disease. Modalities such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) have demonstrated highly disease-specific, low-spatial resolution imaging capabilities using radioisotope-based molecular imaging tracers, and are utilized in the diagnosis of a range of diseases, including cancer [3,4], Alzheimer’s disease [5] and Parkinson’s disease [6]. Optical molecular imaging modalities provide insight potentially at a much higher spatial resolution, typically at the cellular or subcellular level [7,8]. Fluorescence-based optical molecular imaging techniques [9–11] typically target specific cells or signaling pathways through the use of fluorescently labeled antibodies, which comprise a protein that binds to a specific antigen, and a fluorophore that enables imaging. Such labels have been developed for a wide range of disease-related targets [12,13].
However, the fluorescence excitation and emission signals are subject to scattering and absorption and thereby attenuate rapidly in tissue, especially in the visible wavelength range. Whole-body fluorescence tomography has been well studied in small animal models [14], but multiple scattering of the fluorescence signal rapidly degrades spatial resolution to a few millimeters. In contrast, intravital microscopy [15] uses miniaturized intravascular or endoscopic scanning probes to detect molecular targets with high resolution, but is limited to cells that are located on the surfaces of blood vessels, airways or other endoscopically accessible locations. Such surface imaging excludes from examination the tissue involved in many disease processes.
One avenue to access such tissue is to utilize a rigid fluorescence microendoscope, which has the potential to be inserted interstitially into tissue. A range of designs has been proposed [16,17] that differ in the focusing optics and the scheme used to scan the imaging beam. Several groups have proposed the use of graded-index (GRIN) lenses that may be positioned adjacent to, or within, the tissue and coupled to an external scanning mechanism [18,21]. A two-dimensional (2D) image is produced by raster scanning the light beam across the proximal end of the lens. Alternative designs have explored the use of fiber-bundles [16,22], resonant scanning fibers [23,24] and microelectromechanical systems (MEMS) [25]. Such designs have typically been restricted to a forward-viewing configuration, where the area of tissue being imaged is immediately in front of the endoscopic probe. In these designs, miniaturization of the probe restricts the complexity of the focusing optics, and the beam is typically focused at a fixed distance. Imaging at different depths in the tissue is achieved by physically inserting the probe further into the tissue. In such a forward-viewing configuration, the lateral field of view is highly restricted, limited by the diameter of the probe, and improvements in miniaturization result in further decreases in the area that can be imaged.
A side-viewing configuration [26], in which the light beam is emitted perpendicular to the probe, can offer significant improvements in the field of view, as rotating and translating the probe enables imaging of a 2D cylindrical image plane at a fixed distance from the probe, whereby out-of-focus light is rejected via confocal sectioning. Whilst the distance of the focal plane from the probe can be tuned over a certain range, the extension of this scheme to three-dimensional (3D) imaging via focus scanning over a depth range on the order of millimeters is problematic due to the extreme miniaturization, which limits the complexity of the focusing optics. Alternatively, one could abandon the confocal sectioning concept and utilize distal optics with a low numerical aperture (NA) and hence a large depth of focus. However, such probes lack any ability to depth section the detected fluorescence as the collected fluorescence represents a projection image, superimposing signal from multiple depths and complicating interpretation of the image.
One approach to mitigate the lack of depth sectioning in low-NA fluorescence imaging systems has been to augment the fluorescence signal with optical coherence tomography (OCT), which achieves depth sectioning through interferometry rather than through the focusing optics [27]. This dual-modality approach combines the molecular imaging capability of fluorescence with the 3D structural imaging capability of OCT [28,29]. Recently, miniaturized dual-modality imaging probes have been developed utilizing double-clad fiber (DCF) [30]. DCF offers a convenient way to deliver the OCT and fluorescence excitation beams through the core, whilst efficiently collecting emitted fluorescence through the multimode, higher NA inner cladding. DCF also provides a convenient means of separating the detection channels of the two imaging modalities using a double-clad fiber coupler (DCFC) [31,32]. In addition, the co-incidence of the OCT and fluorescent imaging beams means that the images are inherently laterally co-registered. Dual-modality probes based on this concept have been demonstrated in the form of endoscopes [33–35], intravascular probes [36,37] and needle probes [38].
OCT needle probes [39–45] represent a very high degree of miniaturization of rigid microendoscopes, with true 3D imaging capability and outer diameters down to 310 µm [45,46]. Our own work has recently extended the capabilities of such needle probes to dual-modality imaging [38] using a side-facing fiber probe based on DCF that was encased in a 23-gauge needle (640 μm outer diameter), which allowed imaging at a depth of tens of millimeters into tissue. However, this preliminary design lacked the fluorescence sensitivity required for cellular-level molecular imaging.
In this paper, we report the development of an all-fiber, dual-modality needle-based system capable of imaging individual cells marked with fluorescently labeled antibodies. It features an improved 24-gauge (outer diameter 570 µm) side-viewing needle probe, referred to as a molecular imaging needle (MI needle), which simultaneously acquires a co-registered 3D OCT data volume and a 2D fluorescence image. Advances over our earlier work [38] are achieved through an improvement in the sensitivity of the distal focusing optics and use of an asymmetric DCFC [32] to achieve better fluorescence collection efficiency. We present a thorough characterization of the needle and demonstrate its capability by imaging thin sections of human liver, validated against a benchtop wide-field fluorescence microscope. We also present the first use of an MI needle to image fluorescently labeled cells within an intact fresh mouse liver. This is the first demonstration of a dual-modality OCT and fluorescence needle capable of imaging fluorescently labeled antibodies deep within tissue.
2. Materials and methods
2.1 OCT system hardware
A schematic of the complete dual-modality MI needle system is shown in Fig. 1. A custom-built swept-source OCT (SS-OCT) system forms the OCT imaging component. It comprises a 50 kHz repetition rate, wavelength-swept laser source (Axsun Technologies Inc., USA) centered at 1310 nm with full sweep bandwidth of 100 nm and an average output power of 40 mW. A 90/10 coupler forms the interferometer with sample and reference arms as shown. Detection of the returning interferometric signal is achieved using a balanced photodetector (PDB420C-AC, Thorlabs, USA), with the balancing channel supplied by a portion of the source power split off using a 90/10 coupler and attenuated with a variable optical attenuator (VOA50-APC, Thorlabs, USA). The detected signal was acquired using a digitizer card (ATS9350, AlazarTech, Canada). As the light source does not have a Gaussian spectrum, the axial resolution was measured by applying a Tukey window with α = 0.1 to the sample spectrum obtained from an air/glass interface and compensated for second- and third-order dispersion to measure an axial resolution of 14 µm in air, corresponding to ~9 µm in tissue, assuming a refractive index, n, of 1.4. When used with a bulk-optic sample arm, the system achieves a measured sensitivity of 114 dB.
Fig. 1.
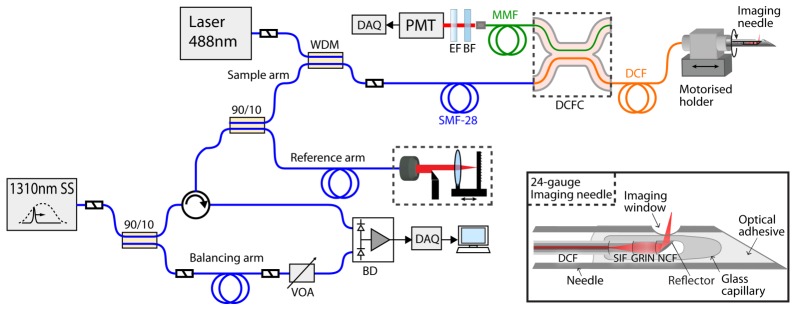
Schematic of the dual-modality OCT + fluorescence imaging system with asymmetric double-clad fiber coupler and MI needle (inset). SS, swept source; WDM, wavelength-division multiplexing coupler; SMF-28, single-mode fiber; MMF, multimode fiber; DCF, double-clad fiber; DCFC, double-clad fiber coupler; EF, long-pass emission filter; BF, blocking filter; PMT, photomultiplier tube; SIF, step-index fiber; GRIN, gradient-index fiber; NCF, no-core fiber; VOA, variable optical attenuator; BD, balanced photodetector; DAQ, data acquisition.
2.2 Fluorescence imaging hardware
For fluorescence excitation, 488-nm wavelength light from a frequency-doubled semiconductor laser (Sapphire SF 488, Coherent Inc., USA) is injected into the core of SMF28 fiber in the OCT sample arm using a wavelength-division multiplexing coupler (INT-MSI-1300, Thorlabs, USA). The sample arm fiber is connectorized to DCF (SM-9/105/125-20A, Nufern, USA) that delivers both OCT and fluorescence excitation wavelengths to the sample through the 9 µm core. The core is single mode over the 1300 nm wavelength band and multimodal at the fluorescence excitation wavelength of 488 nm (~20 modes). The 488-nm excitation power at the sample can be adjusted continuously between 90 µW and 8 mW.
For fluorescence detection, returning fluorescence is collected in the inner cladding of the DCF (0.2 NA). The inner cladding signal is coupled into the multimode output fiber of a custom-made double-clad fiber coupler before detection using a photo-multiplier tube (PMT) (9136B, Electron Tubes, United Kingdom). The asymmetric DCFC was fabricated using a custom computer-controlled fusion and tapering setup and a combination of DCF and custom highly multimode fiber. It is optimized for single-mode core transmission of more than 95% and highly efficient multimode inner-cladding collection with an achromatic multimodal coupling ratio of greater than 70% [32]. This is a more than two times improvement in coupling efficiency compared to our previous DCFC [31]. The unused arm was index matched to eliminate parasitic backreflections from the endface that could introduce noise and artifacts into the OCT image. Low-noise fluorescence detection is achieved using a 500-nm long-pass emission filter (FF01-500/LP-25, Semrock, USA) and a 488-nm blocking filter (NF02-488S-25, Semrock, USA) to reduce detection of back-coupled excitation light. Fluorescence data was acquired using a digitizer card (NI PCIe-6351, National Instruments Corporation, USA). Our system is designed for use with AlexaFluor 488 fluorescent dye, which has excitation and emission maxima similar to those of fluorescein (490 nm and 525 nm, respectively). However, the system may also be readily adapted for other fluorophores.
Radial fluorescence imaging can be performed simultaneously with 3D OCT imaging using the side-viewing needle by rotating the needle in the sample during insertion or pullback. Our system uses counter-rotation of the needle (alternating clockwise and anticlockwise rotation over 360 degrees) in order to avoid the need for a DCF rotary junction. Whilst laboratory prototypes of DCF rotary junctions based on free-space optics have been described in the literature [35], such devices are not yet commercially available. Alternatively, fast scanning for clinical use could be achieved using a configuration with high-frequency linear actuation and slow rotation of the needle inside a transparent sheath [47].
2.3 Needle optical design and fabrication
Several technological innovations were required to reach the sensitivity necessary to detect single-cell fluorescence. We utilize a low-loss optical design based on a capillary encapsulated all-fiber assembly, which achieves OCT imaging sensitivity and resolution comparable to conventional free-space OCT sample arms [42]. A schematic of the design is shown in the inset in Fig. 1. It consists of three short sections of different specialty fibers directly fusion-spliced to the sample arm DCF: a section of large-core step-index fiber (SIF) which serves as a beam-expanding spacer, a section of GRIN fiber, which functions as a lens, and an angle-polished section of no-core fiber (NCF) for high-efficiency sideways beam deflection by total internal reflection (TIR). TIR is maintained by encasing the fiber probe in a glass microcapillary that is collapsed and fused onto the fiber and which also provides an optical-quality low-loss imaging window. 1310-nm wavelength light for OCT is directed from the DCF core through the focusing optics to the sample, where it is elastically backscattered and collected via the return optical path to the DCF core. 488-nm wavelength light for fluorescence excitation is also directed from the DCF core through the focusing optics to the sample and emitted fluorescence from the sample is collected via the return optical path to the DCF inner cladding.
Figure 2 shows the fabricated angle-polished fiber probe (Fig. 2(a)), the capillary-encased probe (Fig. 2(b)), and the final imaging needle (Fig. 2(c)). All steps of probe construction, with the exception of polishing, were performed on a commercial fusion splicing workstation (LFS-4000, Vytran, USA). The fiber sections in the fabricated fiber probe consist of large-core SIF (DrakaElite Low OH Step-Index Multimode Optical Fiber 105/125 µm, Prysmian Group, The Netherlands), large-core GRIN fiber (DrakaElite Graded-Index Multimode Optical Fiber 100/125 µm, Prysmian Group, The Netherlands) and 125-µm diameter pure silica NCF (NCF125, Prime Optical Fiber Corporation, Taiwan). The NCF end section is polished to an angle of 48 degrees using a commercial fiber polisher (Ultrapol Fiber Lensing Machine, ULTRATEC Manufacturing Inc, USA). Our second-generation dual-modality needle uses GRIN fiber with a larger 100-µm diameter core compared to our previous design [38] which used a core diameter of 62.5 µm. This increases the fluorescence collection efficiency and also allows flexibility in the optical design so that a more tightly focused beam can be achieved with longer working distance. In addition, this design uses a SIF spacer instead of NCF. As in lensed fiber designs employing NCF spacers, the SIF behaves as a beam expanding section for the light exiting the core of the DCF, so that the full diameter of the GRIN fiber can be exploited for focusing. For returning fluorescence light, however, the SIF forms an extra guiding section, which ensures that more of the light captured by the GRIN fiber is transmitted to the DCF inner cladding. The entire assembly is glued into a needle with a side-viewing window that is electrochemically etched using a custom-built setup.
Fig. 2.
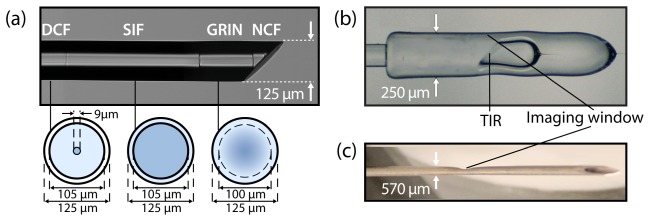
The fabricated MI needle. (a) Angle-polished all-fiber probe with cross-sectional schematics of each of the fibers used in the final design. (b) Capillary encased probe with total internal reflection (TIR) for side-viewing. (c) Final 24-gauge molecular imaging needle.
The lengths of each of the fiber sections determine the properties of the output beam for imaging. The beam properties for the OCT imaging wavelength (including spot size, working distance, and Rayleigh range) were computed given the lengths of each element using paraxial ray matrix transformations for Gaussian beams implemented in Matlab. At the 488-nm fluorescence excitation wavelength, the DCF core is multi-modal. For this reason, the exact characteristics of the output beam at 488 nm cannot be easily simulated, as they will depend on the relative power in the various modes. However, the focal spots at 488 nm and 1300 nm will be at the same axial location if the chromatic aberration of the GRIN fiber lens can be neglected (this expectation is confirmed by our measurements presented in Sections 3.1 and 3.2). Therefore, the beam at 1300 nm gives a reasonable approximation for the working distance and NA of the fluorescence excitation beam.
Since the OCT and fluorescence excitation beams share the same optical path, our final design lengths were a compromise between the somewhat different requirements of the two modalities. Fluorescence would ideally use a beam with a high excitation NA and short working distance in order to excite the sample with high intensity close to the needle (where fluorescence collection is most efficient); whereas, OCT normally uses lower NA and longer working distances in order to obtain a sufficient imaging range and depth-of-focus. The final lengths of SIF and GRIN sections for the fabricated needle (Fig. 2) were 424 µm and 179 µm, respectively, giving a simulated OCT working distance of 400 µm and a simulated focal spot size of 12 µm in water (n = 1.32 at 1300nm and n = 1.33 at 488nm) and, consequently, a fluorescence working distance of ~400 µm and excitation NA of 0.07. The detection NA is calculated to be 0.17, which is limited by the core diameter of the GRIN fiber. A detailed experimental characterization of the output beam properties is given in Section 3.
2.4 MI needle: OCT characterization
The OCT imaging beam from the fabricated MI needle was measured in air using a commercial CCD-based beam profiler (SP620U, Ophir-Spiricon, USA). The full-width at half-maximum (FWHM) output beam diameters were obtained via Gaussian fitting of the recorded 2D beam intensities over a series of distances from the needle up to 1.2 mm in air. The measured beam diameters were subsequently compared with the simulated diameters obtained using paraxial ray matrix transformations implemented in Matlab. When used in tissue, the imaging window will be surrounded by tissue fluid, which is assumed to have a refractive index close to that of water, n = 1.32 at a wavelength of 1300 nm. Therefore, a simulated beam profile in water was also determined as an indication of the beam properties that will be realized during imaging. The OCT lateral resolution, defined as the FWHM diameter of the Gaussian beam at its waist, was inferred from the simulated profile in water.
To determine the OCT sensitivity of the needle, the signal-to-noise ratio (SNR) was recorded for the reflection from a fused-silica/water interface, which was aligned at the focus of the water-immersed side-viewing needle. The summation of the measured SNR, the Fresnel reflectivity of ~27 dB for a fused-silica/water interface, and attenuation introduced into the system during the measurement gave the final value for the OCT sensitivity.
2.5 MI needle: fluorescence characterization
Both OCT and fluorescence imaging beams travel along the same optical path to the sample and, to a first approximation, the properties of the output beam at 488 nm can be expected to be similar to those at 1310 nm. As explained in Section 2.3, exact modeling of the 488-nm excitation beam is not trivial due to the multimodal nature of the incident light at this wavelength. In addition, the collection of emitted fluorescence is also not straightforward to simulate and would require extensive ray-tracing calculations. To address these complications in determining the fluorescence imaging characteristics of the needle, we imaged fluorescent beads in water and used the recorded images to estimate the fluorescence point-spread function (PSF) for different depths. From the estimated PSFs, we can determine the lateral resolution as well as the decay in fluorescence collection efficiency as a function of depth. We used 7.7 µm fluorescent beads (AlexaFluor® 488 Reference Standard (886), Bangs Laboratories Inc, USA) in water, which we dehydrated on a glass plate before covering with an almost cured silicone matrix, and finally curing completely in a warm oven. Using this method, the beads strongly adhered to the surface of the silicone upon removal from the glass plate and remained fixed during imaging under water immersion.
To measure the PSF, we used a two-axis raster scanning stage with the side-viewing needle positioned perpendicularly over the silicone phantom to record 2D images of a single fluorescent bead. Water was used between the needle and coverslip to minimize backreflections from the imaging window and from the coverslip. The PMT output voltage was acquired while raster scanning the phantom, with acquisition triggered in 4 µm intervals, to record a two-dimensional fluorescence image of the bead. Imaging was performed in this configuration at progressively increasing distances from the imaging window, up to 1.5 mm. The 488-nm excitation power was held constant at 90 µW for all distances. From the images, cross-sectional profiles in x and y were generated by averaging 10 line-scans at the center of the bead in each axis, and were normalized and fit to a Gaussian curve. A value for the fluorescence lateral resolution was determined from the FWHM of the fitted Gaussian curve by applying a simple deconvolution operation, assuming Gaussian profiles for the PSF and the bead fluorescence signal. The decay in fluorescence excitation/collection efficiency was also assessed during this experiment by measuring the amplitude of the cross-sectional profiles at each depth.
Finally, to characterize fluorescence detection sensitivity of the needle, we recorded the fluorescence signal from a range of fluorescent dye solutions that were prepared by diluting 1% fluorescein (Minims Fluorescein Sodium 1% w/v, Chauvin Pharmaceuticals Ltd, UK) into four concentrations from 100 nM to 5 nM, using saline at pH 9.5 to maintain optimal fluorescence efficiency. The fluorescence detection sensitivity was measured by raster scanning the needle across four thin glass tubes (inner/outer diameters of 0.4∕0.55 mm) filled with the fluorescein dilutions, and recording the averaged signal from each solution as well as the noise floor. The 488-nm excitation power for the sensitivity measurements was 90 µW.
2.6 3D OCT + fluorescence imaging phantom
To assess the dual-modality imaging performance of the MI needle, it was inserted into a tissue-mimicking phantom. The phantom consisted of sub-resolution TiO2 scatterers (average size 0.5 µm) dispersed evenly in a silicone matrix (n = 1.4) with a concentration of 0.9 mg/ml. Distinct features were incorporated into the phantom by embedding three nylon monofilament lines (0.3-mm diameter fishing lines), one of which was non-fluorescent (line 1 in Fig. 3) and two of which were fluorescent (lines 2 and 3 in Fig. 3). The geometrical arrangement of the three nylon monofilament lines within the scattering matrix was such that the two fluorescent lines (lines 2 and 3) were located at different radial distances from the needle axis, in order to gauge the difference in the measured fluorescence signals from targets located at different distances from the probe.
Fig. 3.
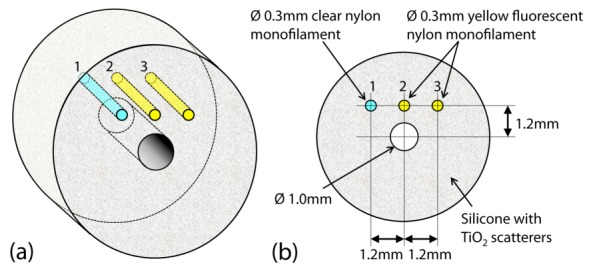
Schematic of the tissue-mimicking, scattering and fluorescent phantom. (a) 3D view. (b) Cross-sectional radial view showing the dimensions and positions of the three nylon monofilament lines relative to the needle hole in the center. Lines 2 and 3 are fluorescent; whereas, line 1 is non-fluorescent.
A 3D OCT + 2D radial fluorescence data set was obtained by inserting the needle into the 1-mm diameter hole in the centre of the phantom whilst counter-rotating the needle over ~360° at 1 Hz and translating over a distance of 2 mm in 4 µm steps. The 488-nm excitation power was set to 200 µW for this measurement. These images were then combined to yield a 3D cylindrical volume. During imaging, the central hole was filled with water in order to ensure fluid-immersion of the needle, similar to the conditions encountered when inserting into biological tissue, and to reduce the backreflection from the silicone surface.
2.7 Liver tissue preparation, labeling and imaging
The capability of the MI needle to image fluorescently labeled antibodies bound in tissue was assessed using two sets of ex vivo experiments. The first experiment involved imaging of fixed, thin sections of human liver, allowing validation against standard wide-field fluorescence microscopy. The second experiment assessed the ability of MI needles to perform imaging in an intact antibody-perfused whole mouse liver.
The human liver study was approved by the Human Research Ethics Committees of the University of Western Australia, Fremantle Hospital and Sir Charles Gairdner Hospital (Western Australia) and informed consent was obtained from patients. A specimen of normal adult liver was fixed in formalin and paraffin-embedded, before being sectioned at a thickness of 15-20 µm and mounted on glass microscope slides. Labeling of the sections was performed using standard two-step indirect immunofluorescence protocols. In brief, liver sections were incubated overnight with a mouse-anti-human GCTM-5 primary antibody (obtained from Prof. Martin Pera, University of Melbourne). In human liver, the GCTM-5 epitope is expressed on the surface of biliary epithelial cells and a subset of liver progenitor cells (LPCs) and, therefore, the GCTM-5 antibody will bind and specifically mark these cells [48]. After incubating with the primary antibody, sections were incubated with a rabbit-anti-mouse secondary antibody, conjugated with an Alexa Fluor 488 dye (Alexa Fluor® 488 Rabbit Anti-Mouse IgG, Merck Millipore, USA) to fluorescently label the bound GCTM-5 antibody. The fluorescence of the Alexa Fluor 488 dye is much brighter than that of fluorescein and, unlike fluorescein, is insensitive to changes in pH between 4 and 10. Therefore, the pH of the tissue was not controlled in these experiments. Labeled sections were covered with a standard No. 1 coverslip (0.13-0.16 µm thick) to preserve the tissue and facilitate imaging.
Sections on microscope slides were mounted on a sample holder and dual-modality imaging was performed by holding the needle adjacent to the sample and raster scanning the sample beneath the needle using a two-axis scanning stage (Fig. 4). Imaging was performed in this configuration with sections located approximately at the focus of the fluorescence excitation and OCT imaging beams. The fluorescence excitation power was 90 µW for imaging of the liver tissue samples. OCT and fluorescence signals were acquired synchronously, such that each OCT A-scan is associated with a corresponding fluorescence measurement, representing the integrated signal from that position. Data acquisition while raster scanning was triggered at 4 µm intervals. At every raster location, the PMT output voltage as well as four OCT A-scans were acquired (the four OCT A-scans were averaged in post-processing to reduce noise). The distance scanned in each dimension was adjusted according to the size of the sample. Acquisition time for one line-scan of 5-mm length, for example, was 1 second. Acquired OCT and fluorescence images were intrinsically co-registered (in two dimensions) due to the common optics and trigger signal. Images obtained using the MI needle were benchmarked against images obtained using a standard conventional wide-field fluorescence microscope (IX2-ILL100 Inverted Microscope, Olympus Corporation, USA) with 4 × objective (UPlanFLN 4 × /0.13, Olympus Corporation, USA).
Fig. 4.

Side-viewing needle scanning configuration for imaging prepared liver sections using x-y raster scanning stage (water immersion not shown).
In the second experiment, where imaging was performed on a fresh (not fixed) whole mouse liver, tissue was obtained under institutional tissue sharing protocols. The liver was freshly dissected from a mouse model (Balb/C x C57BL/6 x FVB/N background) and cleared of blood by portal vein perfusion with phosphate buffered saline (PBS) containing 1% Heparin. 1.5 mL of primary EpCAM antibody conjugated to Alexa Fluor 488 was injected into the common bile duct and incubated for 20 minutes. Unbound antibody was removed by flushing 5 mL of PBS through catheters inserted into the common bile duct, hepatic arteries and portal veins.
The MI needle was inserted into the whole liver and retracted whilst counter-rotating over ~360° at 1 Hz, allowing acquisition of 2D radial OCT images and co-registered fluorescence projection images. The 488-nm excitation power was set to 200 µW. Data acquisition was triggered to give 1600 radial scans per rotation of the needle. The rotational scanning speed of this system is comparable to that previously published for ex vivo measurements [40–42], although more rapid needle scanning is feasible [43].
3. Results
3.1 MI needle OCT characterization
The beam profile for the fabricated needle, measured in air, is shown in Fig. 5(a). The FWHM beam diameters measured along both the x- (squares) and y- (circles) axes, along with the Matlab simulated beam profiles for each axis (solid and dashed lines, respectively) are shown. The x- and y-axes are in the directions orthogonal and parallel to the needle axis, respectively. The measured Gaussian fit diameters agree well with the simulated diameters. The output beam profile for the water-immersed probe, simulated based on the measured profile in air (in Fig. 5(a)), is given in Fig. 5(b).
Fig. 5.
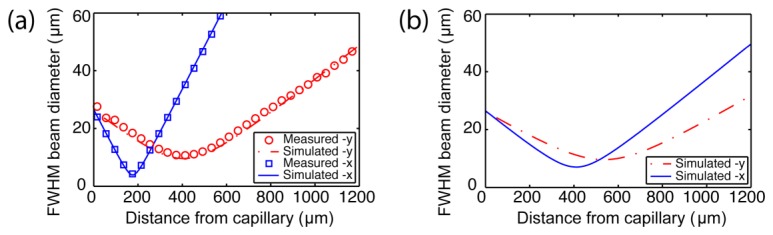
Output beam profiles (using FWHM beam diameters) at 1310 nm wavelength. (a) Measured output beam diameters for the fabricated side-viewing MI needle in air (squares and circles) with corresponding simulated diameters obtained from ray-matrix transformations (Solid and dashed lines). (b) Simulated beam diameters calculated from (a) for the side-viewing MI needle in water.
Astigmatism resulting from the curved interface of the capillary is evident in the measured beam profile in air (Fig. 5(a)). However, the astigmatism is greatly reduced when the needle output window is immersed in fluid with a refractive index close to that of water (Fig. 5(b)), which is the case when imaging inside biological tissue. The distance to the closest focal plane (x-axis) in water is considered to be the working distance of the needle and, from the simulated profile, this is 410 µm from the capillary window. The FWHM beam diameters determine the OCT lateral resolution at this location and are 7 µm and 9.6 µm in the x- and y-directions, respectively. The ellipticity (ratio of the diameters in x and y) of the beam in water is less than 1.65 over a range of 1 mm from the imaging window.
The OCT sensitivity of the fabricated side-viewing MI needle was measured to be 100 dB using the attenuated specular reflection from a glass/water interface. This is on par with typical benchtop OCT systems.
3.2 MI needle fluorescence characterization
To characterize the fluorescence lateral resolution, as well as the decay in fluorescence excitation/collection efficiency of the MI needle as a function of depth, we measured its 2D fluorescence PSF using a fluorescent bead in water as described in Section 2.5. Images of the fluorescent bead at distances of 300 µm and 1050 µm from the needle are shown in Fig. 6(a) and Fig. 6(c), respectively. The bead image cross sections at 300 µm and 1050 µm from the needle, extracted from the averaged fluorescence signal along each axis, are shown in Fig. 6(b) and 6(d), respectively. Gaussian curves were fitted to the cross sections in order to quantify the FWHM and amplitude of the PSF as a function of distance from the needle, resulting in the plots in Fig. 6(e) and Fig. 6(f), respectively.
Fig. 6.

Fluorescence PSF measured using a fluorescent bead in water. (a) and (c) 2D fluorescence image (raster scanned) of a bead at 300 µm and 1050 µm from the needle, respectively. (b) and (d) Corresponding fluorescence cross-sectional profiles (blue lines) and Gaussian curve fitting (red dashed lines) along x-axis (top panel) and y-axis (bottom panel) for a bead at 300 µm and 1050 µm from the needle, respectively. (e) FWHM diameters of the fluorescence PSF in x- and y-axes as a function of depth through water, obtained from Gaussian fitting. (f) Normalized amplitude of the fluorescence PSF as a function of depth in water. (g) Log (dB) normalized amplitude of the fluorescence PSF as a function of depth in water, with line of best fit (red line) showing rate of decay of fluorescence efficiency.
The resulting best lateral resolutions in the x- and y- dimensions, are 12 µm and 13 µm, respectively, obtained in a plane ~400 µm from the needle. The decay in the PSF amplitude is shown in Fig. 6(f), and on a dB scale in Fig. 6(g). A line of best fit in Fig. 6(g) indicates that fluorescence efficiency drops off rapidly at a rate of 22.7 dB/mm. Thus, the majority of the fluorescence signal is obtained within 1 mm of the needle.
Finally, the sensitivity of fluorescence detection was measured using dilutions of fluorescein from 100 nM to 5 nM at pH 9.5, as shown in Fig. 7(a). For these measurements the glass tubes were positioned such that the top of each tube was ~300 µm from the imaging window of the needle. The fluorescence signal from the tubes was used to calculate a line-of-best-fit (Fig. 7(b)) and its intersection with the noise floor gave a minimum detectable concentration of fluorescein of 1.5 nM. This is a substantial improvement of over 130 times compared to our previous dual-modality needle design [38]. We observed that the noise floor of our system (with needle probe connected but without a sample present) was proportional to the excitation power level, and that increasing the excitation power level to values higher than the minimum setting of 90 µW did not improve the SNR in these measurements. We believe that the origin of this excitation-power-dependent noise floor is autofluorescence of the system components. Both the MI needle and the DCF coupler were identified as sources of autofluorescence. In the needle, the optical adhesive used to mount the fiber probe is the most likely source of autofluorescence when it interacts with stray or backscattered 488-nm wavelength excitation light. In the DCF coupler, the optical adhesive used to mount the coupler in its packaging was determined to be the source of the autofluorescence. Whilst this autofluorescence determines the ultimate sensitivity limit of the system, our tissue imaging results in Section 3.4 show that it is negligible compared to the autofluorescence of the tissue. The system autofluorescence is, therefore, not a limiting factor for the practical imaging performance in biological tissue.
Fig. 7.
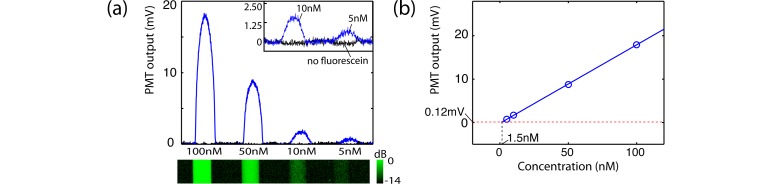
Fluorescence sensitivity measured using fluorescein at pH 9.5. (a) Measured fluorescence signal (upper panel) with log-scaled fluorescence image (lower panel) of four glass capillary tubes filled with decreasing concentrations of fluorescein. Inset: magnified plot of signal from 10 nM and 5 nM fluorescein filled tubes (blue line) compared to the same tubes with no fluorescein (black line). In the fluorescence colourbar, dB is measured relative to maximum measured fluorescence signal. (b) Plot of maximum fluorescence signal from each tube (blue circles) with line of best fit (blue line) compared to the RMS noise floor (red dashed line).
3.3 3D OCT + fluorescence imaging performance
The dual-modality image obtained for the 3D scattering and fluorescent tissue phantom is presented in Fig. 8. The detected fluorescence at each radial position is depicted by a projection of the fluorescence signal onto the outer circumference of the radial OCT image. The resulting 3D image (Fig. 8(a)) shows the intrinsic co-registration of the two modalities as seen by the overlap of the fluorescence signal with the two fluorescent monofilament lines, which appear as low scattering inclusions in the OCT image. As expected, the non-fluorescent line produces no signal in the fluorescence channel, whilst appearing identical to the two fluorescent lines in the OCT image. Notably, the radial image in Fig. 8(b) shows that the fluorescent line closer to the needle (~880 µm from the imaging window) produces a signal that is significantly higher (approximately 9.9 dB) than the line further away from the needle (~1.33 mm from the imaging window). This is consistent with our characterization of the fluorescence collection efficiency decay with depth presented in Fig. 6, from which the difference in measured fluorescence for these two lines is expected to be approximately 10 dB. Importantly, this experiment also highlights the utility of the depth information provided by the structural OCT images, as it is the understanding of the 3D structural features that enables interpretation of the 2D fluorescence signal.
Fig. 8.

Dual-modality images obtained for the scattering and fluorescent tissue-mimicking phantom using the side-viewing needle with rotation and translational scanning. (a) A cutaway view of the 3D OCT image with the measured fluorescence signal for each radial location projected onto the surface of the cutaway plane. (b) A cross-sectional radial view showing the image of the three lines with the measured fluorescence signal for each radial position projected onto the circumference of the radial image. In the fluorescent colourbar, dB is measured relative to maximum measured fluorescence signal.
3.4 Molecular imaging needle performance
In order to evaluate the fluorescence imaging performance of our dual-modality system we first used the MI needle to image a 15-µm section of human liver fluorescently labeled at the GCTM-5 epitope as described in Section 2.7. The liver section was obtained from a normal healthy donor liver containing positive staining ductal epithelial cells and liver progenitor cells. Proof-of-principle imaging was performed by comparing the MI needle images with corresponding benchtop wide-field fluorescence microscope images of the section.
Figure 9 shows the resulting needle images compared with the conventional fluorescence microscope image of the section. The needle OCT image in Fig. 9(a) is taken from a plane through the center of the thin section. Figure 9(b) shows the corresponding needle fluorescence image. Focal areas of fluorescence from labeled cells and a background of autofluorescence from the tissue are observable. Note that while the fluorescence signal detected by the PMT was measured over a range of 80mV, the distinction between labeled cells and background autofluorescence has been more clearly visualized by setting the saturation of the fluorescence colorbar to 27mV. A region of interest, indicated by the dashed boxes in Figs. 9(a) and 9(b), is shown magnified in Figs. 9(c) and 9(d), respectively. Figure 9(d) depicts focal areas of fluorescently labeled epithelial cells from bile ducts. The magnified OCT image (Fig. 9(c)) indicates that these ducts lie within a surrounding cord of lower scattering connective tissue. The ducts are readily distinguishable within the connective tissue in the combined OCT plus fluorescence image in Fig. 9(e). Other fluorescently labeled cells that do not form ducts are also visible in the liver parenchyma and are highlighted in the inset in Fig. 9(e). From the corresponding wide-field fluorescence image of this section (Fig. 9(f)) these labeled cells are identified as liver progenitor cells (LPCs), as confirmed by the high magnification inset shown (top right of Fig. 9(f)), which was taken using a 10 × microscope objective. Liver progenitor cells are implicated in occurrence and recurrence of liver tumor and are of particular interest for understanding the mechanism of tumor recurrence after therapy [48]. Detection of these cells using a side-viewing MI needle provides new opportunities to study their distribution in situ.
Fig. 9.

Side-viewing MI needle images obtained for a section of normal human liver, showing labeled biliary ducts and liver progenitor cells. (a) En face OCT needle image obtained after averaging 4 co-located OCT A-scans at each radial position. (b) Corresponding fluorescence needle image. (c) Magnified OCT image of selected region indicated by dashed box in a. (d) Corresponding magnified fluorescence needle image of selected region indicated by dashed box in b. (e) OCT + fluorescence magnified image of selected region. Inset: Magnified view of region indicated by small pink box. (f) Corresponding standard wide-field fluorescence microscopy image for selected region. Inset: High magnification (10 × objective) image of region indicated by pink box confirming liver progenitor cells (LPCs).
Excellent correspondence is observed between the needle images and the wide-field fluorescence microscope images. While the microscope images exhibit higher resolution, as expected from the larger numerical aperture (0.13 compared to 0.07 for the needle), these images show that the MI needle is capable of detecting fluorescence from labeled cells.
Figure 10 shows results obtained using the MI needle deep within an antibody-labeled whole mouse liver. Results acquired at two distinct pullback locations within the mouse liver are presented in Figs. 10(a) and 10(b). Adopting the graphical representation described in Fig. 8, the detected radial fluorescence is projected onto the outer circumference of the 2D radial OCT image. The radial fluorescence data from each image indicates ducts that show high levels of fluorescence as a result of fluorescently labeled EpCAM that has bound specifically to epithelial cells. In a control study with an unlabeled control liver (results not shown), no fluorescence was detected, confirming that the observed fluorescence is due to specific labeling of the tissue. The OCT images at each location clearly depict cross-sectional views of duct-like structures with low scattering lumens, corresponding to the detected fluorescence. The structures visible in the OCT aid in interpretation of the detected fluorescence.
Fig. 10.
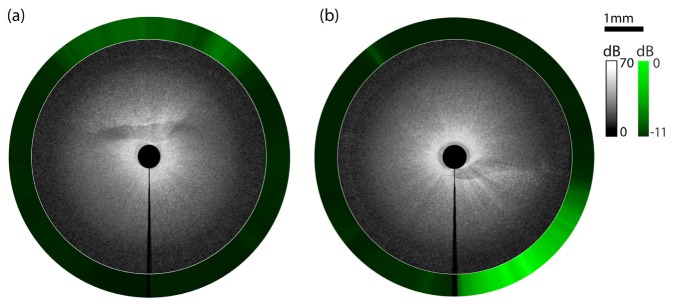
Radial images obtained at two pullback locations using the MI needle deep within a whole mouse liver. The fluorescence signal for each radial position is projected onto the circumference. Fluorescently labeled epithelial cells are visible in each image. OCT images show corresponding ductal structures surrounding low backscattering lumen. The vertical black line at the 6 o’clock position marks the start and end location of the needle rotation. Fluorescent signal (in dB) is measured relative to maximum measured fluorescence signal.
4. Discussion
These results demonstrate that MI needles are capable of imaging with sensitivity comparable to benchtop fluorescence microscopes whilst enabling imaging deep within solid tissue. The high sensitivity to fluorescence presented here can be attributed to the efficient fluorescence collection as well as low optical losses in all parts of the system. It is to be expected that, in tissue, the cellular-level imaging resolution and sensitivity that was achieved in scans of thin liver sections can only be realized very close to the needle due to scattering. However, results acquired within an intact organ in Fig. 10 demonstrate that the detection of fluorescently labeled aggregations of cells or tissue regions is feasible over much greater depth ranges.
A dominant feature in our imaging experiments is that tissue autofluorescence is a limiting factor when using MI needles with 488 nm excitation. This makes the use of longer-wavelength or near-infrared (NIR) fluorophores highly desirable in deep-tissue needle imaging. This can be seen in Fig. 9, where the autofluorescence from the surrounding tissue limits contrast of fluorescently labeled cells. Our choice of fluorescence excitation wavelength for this work was motivated by the availability of well-established fluorophore-labeling protocols for liver cells (GCTM-5(human) and EpCAM (mouse) antibodies, both with Alexa Fluor 488 dye). By transitioning to longer-wavelength excitation in the red or NIR, the autofluorescence of the tissue will be substantially reduced or completely eliminated [49]. Furthermore, the reduced scattering at longer wavelengths will reduce the aforementioned degradation of resolution and sensitivity with distance from the needle. Finally, the noise floor of the system can be expected to decrease in line with a reduction in autofluorescence within the system itself. By changing the wavelength-sensitive components such as the wavelength-division multiplexing coupler, the PMT and the filters, we believe this system can readily be adapted for use with NIR fluorescent dyes [36,37,50] or upconverting nanoparticles [51,52].
5. Conclusions
In this paper, we have developed a dual-modality 24-gauge (outer diameter 570 µm) optical imaging needle capable of molecular imaging with high sensitivity. The design augments a 2D fluorescence projection image with the depth-sectioning capabilities of co-registered 3D-OCT. Using DCF, and incorporating a high-efficiency DCFC together with a low-loss optical probe design, our needle achieves OCT and fluorescence imaging sensitivities that are on par with benchtop systems. Our results show the first demonstration of an OCT and fluorescence needle to detect fluorescently labeled antibodies deep within a fresh intact organ, indicating the potential of MI needle probes to enable high-resolution molecular imaging deep in tissue.
Acknowledgments
The authors would like to thank Prof. Martin Pera from The University of Melbourne for supply of the GCTM-5 antibody. This work was supported by Cancer Council WA, Breast Cancer Research Centre WA, National Health and Medical Research Council (Australia) and the Australian Research Council. Canadian authors acknowledge support from the National Science and Engineering Research Council.
References and links
- 1.Massoud T. F., Gambhir S. S., “Molecular imaging in living subjects: Seeing fundamental biological processes in a new light,” Genes Dev. 17(5), 545–580 (2003). 10.1101/gad.1047403 [DOI] [PubMed] [Google Scholar]
- 2.Shah K., Jacobs A., Breakefield X. O., Weissleder R., “Molecular imaging of gene therapy for cancer,” Gene Ther. 11(15), 1175–1187 (2004). 10.1038/sj.gt.3302278 [DOI] [PubMed] [Google Scholar]
- 3.Gambhir S. S., “Molecular imaging of cancer with positron emission tomography,” Nat. Rev. Cancer 2(9), 683–693 (2002). 10.1038/nrc882 [DOI] [PubMed] [Google Scholar]
- 4.Baum R. P., Prasad V., Müller D., Schuchardt C., Orlova A., Wennborg A., Tolmachev V., Feldwisch J., “Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules,” J. Nucl. Med. 51(6), 892–897 (2010). 10.2967/jnumed.109.073239 [DOI] [PubMed] [Google Scholar]
- 5.Nordberg A., Rinne J. O., Kadir A., Långström B., “The use of PET in Alzheimer disease,” Nat. Rev. Neurol. 6(2), 78–87 (2010). 10.1038/nrneurol.2009.217 [DOI] [PubMed] [Google Scholar]
- 6.Nandhagopal R., Kuramoto L., Schulzer M., Mak E., Cragg J., Lee C. S., McKenzie J., McCormick S., Samii A., Troiano A., Ruth T. J., Sossi V., de la Fuente-Fernandez R., Calne D. B., Stoessl A. J., “Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study,” Brain 132(11), 2970–2979 (2009). 10.1093/brain/awp209 [DOI] [PubMed] [Google Scholar]
- 7.Weissleder R., Pittet M. J., “Imaging in the era of molecular oncology,” Nature 452(7187), 580–589 (2008). 10.1038/nature06917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hellebust A., Richards-Kortum R., “Advances in molecular imaging: Targeted optical contrast agents for cancer diagnostics,” Nanomedicine (Lond) 7(3), 429–445 (2012). 10.2217/nnm.12.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ntziachristos V., Bremer C., Weissleder R., “Fluorescence imaging with near-infrared light: New technological advances that enable in vivo molecular imaging,” Eur. Radiol. 13(1), 195–208 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi H., Choyke P. L., “Target-cancer-cell-specific activatable fluorescence imaging probes: Rational design and in vivo applications,” Acc. Chem. Res. 44(2), 83–90 (2011). 10.1021/ar1000633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett T., Koyama Y., Hama Y., Ravizzini G., Shin I. S., Jang B. S., Paik C. H., Urano Y., Choyke P. L., Kobayashi H., “In vivo diagnosis of epidermal growth factor receptor expression using molecular imaging with a cocktail of optically labeled monoclonal antibodies,” Clin. Cancer Res. 13(22), 6639–6648 (2007). 10.1158/1078-0432.CCR-07-1119 [DOI] [PubMed] [Google Scholar]
- 12.Daly C. J., McGrath J. C., “Fluorescent ligands, antibodies, and proteins for the study of receptors,” Pharmacol. Ther. 100(2), 101–118 (2003). 10.1016/j.pharmthera.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 13.te Velde E. A., Veerman T., Subramaniam V., Ruers T., “The use of fluorescent dyes and probes in surgical oncology,” Eur. J. Surg. Oncol. 36(1), 6–15 (2010). 10.1016/j.ejso.2009.10.014 [DOI] [PubMed] [Google Scholar]
- 14.Leblond F., Davis S. C., Valdes P. A., Pogue B. W., “Pre-clinical whole-body fluorescence imaging: Review of instruments, methods and applications,” J. Photochem. Photobiol. B-Biol. 98(1), 77–94 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuebler W. M., Parthasarathi K., Lindert J., Bhattacharya J., “Real-time lung microscopy,” J. Appl. Physiol. 102(3), 1255–1264 (2006). 10.1152/japplphysiol.00786.2006 [DOI] [PubMed] [Google Scholar]
- 16.Flusberg B. A., Cocker E. D., Piyawattanametha W., Jung J. C., Cheung E. L. M., Schnitzer M. J., “Fiber-optic fluorescence imaging,” Nat. Methods 2(12), 941–950 (2005). 10.1038/nmeth820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh G., Chung E., Yun S. H., “Optical fibers for high-resolution in vivo microendoscopic fluorescence imaging,” Opt. Fiber Technol. 19(6), 760–771 (2013). 10.1016/j.yofte.2013.07.008 [DOI] [Google Scholar]
- 18.Jung J. C., Mehta A. D., Aksay E., Stepnoski R., Schnitzer M. J., “In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy,” J. Neurophysiol. 92(5), 3121–3133 (2004). 10.1152/jn.00234.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levene M. J., Dombeck D. A., Kasischke K. A., Molloy R. P., Webb W. W., “In vivo multiphoton microscopy of deep brain tissue,” J. Neurophysiol. 91(4), 1908–1912 (2004). 10.1152/jn.01007.2003 [DOI] [PubMed] [Google Scholar]
- 20.Llewellyn M. E., Barretto R. P. J., Delp S. L., Schnitzer M. J., “Minimally invasive high-speed imaging of sarcomere contractile dynamics in mice and humans,” Nature 454(7205), 784–788 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillai R. S., Lorenser D., Sampson D. D., “Deep-tissue access with confocal fluorescence microendoscopy through hypodermic needles,” Opt. Express 19(8), 7213–7221 (2011). 10.1364/OE.19.007213 [DOI] [PubMed] [Google Scholar]
- 22.Muldoon T. J., Pierce M. C., Nida D. L., Williams M. D., Gillenwater A., Richards-Kortum R., “Subcellular-resolution molecular imaging within living tissue by fiber microendoscopy,” Opt. Express 15(25), 16413–16423 (2007). 10.1364/OE.15.016413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engelbrecht C. J., Johnston R. S., Seibel E. J., Helmchen F., “Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo,” Opt. Express 16(8), 5556–5564 (2008). 10.1364/OE.16.005556 [DOI] [PubMed] [Google Scholar]
- 24.Lee C. M., Engelbrecht C. J., Soper T. D., Helmchen F., Seibel E. J., “Scanning fiber endoscopy with highly flexible, 1 mm catheterscopes for wide-field, full-color imaging,” J. Biophotonics 3(5-6), 385–407 (2010). 10.1002/jbio.200900087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piyawattanametha W., Ra H., Qiu Z., Friedland S., Liu J. T. C., Loewke K., Kino G. S., Solgaard O., Wang T. D., Mandella M. J., Contag C. H., “In vivo near-infrared dual-axis confocal microendoscopy in the human lower gastrointestinal tract,” J. Biomed. Opt. 17(2), 021102 (2012). 10.1117/1.JBO.17.2.021102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J. K., Lee W. M., Kim P., Choi M., Jung K., Kim S., Yun S. H., “Fabrication and operation of GRIN probes for in vivo fluorescence cellular imaging of internal organs in small animals,” Nat. Protoc. 7(8), 1456–1469 (2012). 10.1038/nprot.2012.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang D., Swanson E. A., Lin C. P., Schuman J. S., Stinson W. G., Chang W., Hee M. R., Flotte T., Gregory K., Puliafito C. A., Fujimoto J. G., “Optical coherence tomography,” Science 254(5035), 1178–1181 (1991). 10.1126/science.1957169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan Y. T., Xie T. Q., Du C. W., Bastacky S., Meyers S., Zeidel M. L., “Enhancing early bladder cancer detection with fluorescence-guided endoscopic optical coherence tomography,” Opt. Lett. 28(24), 2485–2487 (2003). 10.1364/OL.28.002485 [DOI] [PubMed] [Google Scholar]
- 29.Yuan S., Roney C. A., Wierwille J., Chen C. W., Xu B., Griffiths G., Jiang J., Ma H., Cable A., Summers R. M., Chen Y., “Co-registered optical coherence tomography and fluorescence molecular imaging for simultaneous morphological and molecular imaging,” Phys. Med. Biol. 55(1), 191–206 (2010). 10.1088/0031-9155/55/1/011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu S. Y., Choi H. Y., Na J., Choi E. S., Lee B. H., “Combined system of optical coherence tomography and fluorescence spectroscopy based on double-cladding fiber,” Opt. Lett. 33(20), 2347–2349 (2008). 10.1364/OL.33.002347 [DOI] [PubMed] [Google Scholar]
- 31.Lemire-Renaud S., Rivard M., Strupler M., Morneau D., Verpillat F., Daxhelet X., Godbout N., Boudoux C., “Double-clad fiber coupler for endoscopy,” Opt. Express 18(10), 9755–9764 (2010). 10.1364/OE.18.009755 [DOI] [PubMed] [Google Scholar]
- 32.Madore W. J., De Montigny E., Ouellette O., Lemire-Renaud S., Leduc M., Daxhelet X., Godbout N., Boudoux C., “Asymmetric double-clad fiber couplers for endoscopy,” Opt. Lett. 38(21), 4514–4517 (2013). 10.1364/OL.38.004514 [DOI] [PubMed] [Google Scholar]
- 33.Mavadia J., Xi J., Chen Y., Li X., “An all-fiber-optic endoscopy platform for simultaneous OCT and fluorescence imaging,” Biomed. Opt. Express 3(11), 2851–2859 (2012). 10.1364/BOE.3.002851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xi J., Chen Y., Zhang Y., Murari K., Li M. J., Li X., “Integrated multimodal endomicroscopy platform for simultaneous en face optical coherence and two-photon fluorescence imaging,” Opt. Lett. 37(3), 362–364 (2012). 10.1364/OL.37.000362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pahlevaninezhad H., Lee A. M. D., Shaipanich T., Raizada R., Cahill L., Hohert G., Yang V. X. D., Lam S., MacAulay C., Lane P., “A high-efficiency fiber-based imaging system for co-registered autofluorescence and optical coherence tomography,” Biomed. Opt. Express 5(9), 2978–2987 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoo H., Kim J. W., Shishkov M., Namati E., Morse T., Shubochkin R., McCarthy J. R., Ntziachristos V., Bouma B. E., Jaffer F. A., Tearney G. J., “Intra-arterial catheter for simultaneous microstructural and molecular imaging in vivo,” Nat. Med. 17(12), 1680–1684 (2011). 10.1038/nm.2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H., Gardecki J. A., Ughi G. J., Jacques P. V., Hamidi E., Tearney G. J., “Ex vivo catheter-based imaging of coronary atherosclerosis using multimodality OCT and NIRAF excited at 633 nm,” Biomed. Opt. Express 6(4), 1363–1375 (2015). 10.1364/BOE.6.001363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lorenser D., Quirk B. C., Auger M., Madore W. J., Kirk R. W., Godbout N., Sampson D. D., Boudoux C., McLaughlin R. A., “Dual-modality needle probe for combined fluorescence imaging and three-dimensional optical coherence tomography,” Opt. Lett. 38(3), 266–268 (2013). 10.1364/OL.38.000266 [DOI] [PubMed] [Google Scholar]
- 39.Li X., Chudoba C., Ko T., Pitris C., Fujimoto J. G., “Imaging needle for optical coherence tomography,” Opt. Lett. 25(20), 1520–1522 (2000). 10.1364/OL.25.001520 [DOI] [PubMed] [Google Scholar]
- 40.Wu Y. C., Xi J. F., Huo L., Padvorac J., Shin E. J., Giday S. A., Lennon A. M., Canto M. I. F., Hwang J. H., Li X. D., “Robust high-resolution fine OCT needle for side-viewing interstitial tissue imaging,” IEEE J. Sel. Top. Quantum Electron. 16(4), 863–869 (2010). 10.1109/JSTQE.2009.2035362 [DOI] [Google Scholar]
- 41.Quirk B. C., McLaughlin R. A., Curatolo A., Kirk R. W., Noble P. B., Sampson D. D., “In situ imaging of lung alveoli with an optical coherence tomography needle probe,” J. Biomed. Opt. 16(3), 036009 (2011). 10.1117/1.3556719 [DOI] [PubMed] [Google Scholar]
- 42.Scolaro L., Lorenser D., McLaughlin R. A., Quirk B. C., Kirk R. W., Sampson D. D., “High-sensitivity anastigmatic imaging needle for optical coherence tomography,” Opt. Lett. 37(24), 5247–5249 (2012). 10.1364/OL.37.005247 [DOI] [PubMed] [Google Scholar]
- 43.Tan K. M., Shishkov M., Chee A., Applegate M. B., Bouma B. E., Suter M. J., “Flexible transbronchial optical frequency domain imaging smart needle for biopsy guidance,” Biomed. Opt. Express 3(8), 1947–1954 (2012). 10.1364/BOE.3.001947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLaughlin R. A., Quirk B. C., Curatolo A., Kirk R. W., Scolaro L., Lorenser D., Robbins P. D., Wood B. A., Saunders C. M., Sampson D. D., “Imaging of breast cancer with optical coherence tomography needle probes: Feasibility and initial results,” IEEE J. Sel. Top. Quantum Electron. 18(3), 1184–1191 (2012). 10.1109/JSTQE.2011.2166757 [DOI] [Google Scholar]
- 45.Lorenser D., Yang X., Kirk R. W., Quirk B. C., McLaughlin R. A., Sampson D. D., “Ultrathin side-viewing needle probe for optical coherence tomography,” Opt. Lett. 36(19), 3894–3896 (2011). 10.1364/OL.36.003894 [DOI] [PubMed] [Google Scholar]
- 46.Yang X., Lorenser D., McLaughlin R. A., Kirk R. W., Edmond M., Simpson M. C., Grounds M. D., Sampson D. D., “Imaging deep skeletal muscle structure using a high-sensitivity ultrathin side-viewing optical coherence tomography needle probe,” Biomed. Opt. Express 5(1), 136–148 (2014). 10.1364/BOE.5.000136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLaughlin R. A., Yang X., Quirk B. C., Lorenser D., Kirk R. W., Noble P. B., Sampson D. D., “Static and dynamic imaging of alveoli using optical coherence tomography needle probes,” J. Appl. Physiol. 113(6), 967–974 (2012). 10.1152/japplphysiol.00051.2012 [DOI] [PubMed] [Google Scholar]
- 48.Stamp L. A., Braxton D. R., Wu J., Akopian V., Hasegawa K., Chandrasoma P. T., Hawes S. M., McLean C., Petrovic L. M., Wang K., Pera M. F., “The GCTM-5 epitope associated with the mucin-like glycoprotein FCGBP marks progenitor cells in tissues of endodermal origin,” Stem Cells 30(9), 1999–2009 (2012). 10.1002/stem.1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frangioni J. V., “In vivo near-infrared fluorescence imaging,” Curr. Opin. Chem. Biol. 7(5), 626–634 (2003). 10.1016/j.cbpa.2003.08.007 [DOI] [PubMed] [Google Scholar]
- 50.Rao J., Dragulescu-Andrasi A., Yao H., “Fluorescence imaging in vivo: Recent advances,” Curr. Opin. Biotechnol. 18(1), 17–25 (2007). 10.1016/j.copbio.2007.01.003 [DOI] [PubMed] [Google Scholar]
- 51.Hilderbrand S. A., Shao F., Salthouse C., Mahmood U., Weissleder R., “Upconverting luminescent nanomaterials: Application to in vivo bioimaging,” Chem. Commun. (Camb.) (28), 4188–4190 (2009). 10.1039/b905927j [DOI] [PMC free article] [PubMed]
- 52.Haase M., Schäfer H., “Upconverting nanoparticles,” Angew. Chem. Int. Ed. Engl. 50(26), 5808–5829 (2011). 10.1002/anie.201005159 [DOI] [PubMed] [Google Scholar]