

Abstract
A high tricuspid regurgitant jet velocity (TRV) signifying risk for or established pulmonary hypertension (PH) is a serious complication in thalassemia patients. The underlying pathophysiology in thalassemia sub-groups and potential biomarkers for early detection and monitoring are not well defined, in particular as they relate to spleen removal. To better understand some of these unresolved aspects, we examined 76 thalassemia patients (35 non-transfused), 25 splenectomized non-thalassemia patients and 12 healthy controls. An elevated TRV (>2.5m/sec) was found in 25/76 (33%) of the patients, confined to non-transfused or those with a late start of transfusions, including patients with hemoglobin H-constant spring, a finding not previously described. These non, or late-transfused patients (76% splenectomized) had significantly increased platelet activation (sCD40L), high platelet count, endothelial activation (endothelin-1) and hemolysis (LDH, plasma free-Hb), while hypercoaguable and inflammatory markers were not significantly increased. The same markers were increased in the 7 patients with confirmed PH on cardiac catheterization, suggesting their possible role for screening patients at risk for PH. A combination of hemolysis and absence of spleen is necessary for developing a high TRV, as neither chronic hemolysis in the non-splenectomized thalassemia patients, nor splenectomy without hemolysis, in the non-thalassemia patients, resulted in an increase in TRV.
Keywords: Thalassemia, splenectomy, hemolysis, platelet activation, TRV, pulmonary hypertension
Introduction
The surgical removal of spleen aimed to ameliorate the clinical course and transfusion needs in thalassemia, was noted to predispose thalassemia patients, mostly thalassemia intermedia (TI), to increased pressure in the pulmonary arterial circulation, which can progress to clinically significant pulmonary hypertension (PH)[1–4]. Although recent advances in understanding some risk factors leading to increase in TRV, a proxy for PH, were made [5,6], the specific pathophysiology of PH in thalassemia, in particular as it relates to prior spelenectomy, has not been extensively studied. The pathophysiology has been generally attributed to the same processes causing PH in sickle cell disease (SCD). Though overlapping mechanisms of hemolysis, hypercoaguable state and oxidative stress are considered to contribute to the development of the condition in both disorders, these are distinct diseases and the relative contribution of each underlying mechanism is likely different. While hemolysis induced vascular endothelial dysfunction is a central concept in PH in SCD, a multifactorial pathogenesis, related to a hypercoaguable state and abnormal circulating red blood cells (RBCs) has been proposed for thalassemia. Considerable data suggests ongoing prothrombotic activity in TI, especially in those splenectomized, who are at a higher risk for primarily venous thrombotic events [7,5,8–11]. Whether PH, is another manifestation of the thrombotic process, caused by a common underlying pathogenesis, is less well established.
The frequency of an elevated TRV, in the various clinical types of thalassemia; transfusion dependent, non-transfused or partially transfused, splenectomized or not, differs in the available studies: Aessopos et al noted PH to be very rare among transfused TM patients [12]; while others have shown a rate of 10–37% among the transfused patients. However, these reports were limited by inconsistency in the selection of patients who were screened for their studies: Some had a low pre-transfusion Hb [13,14], in others the age of start of transfusions was unavailable, and variable TRV thresholds were used for screening [14,15,2,16–18]. Moreover, the existence of high TRV in severe forms of α-thalassemia, in particular Hb H-constant spring (H-CS) has not been examined.
In the present study we aimed to determine the frequency of an elevated TRV and explore the causative factors among thalassemia patients’ sub-groups in comparison to other pertinent patient groups, concentrating on the role of absence of spleen. Such characterizations of significant risk factors and biomarkers could broaden the ability to develop better clinical guidelines for screening thalassemia patients at-risk for PH as well as advance further study for therapeutic strategies.
Patients and Methods
Study population and TRV assessment
The study was approved by the institutional Review Board. Patients, ≥10 years old, consisted of those with a diagnosis of β-thalassemia major (TM), β-thalassemia intermedia and α thalassemia hemoglobin H-Constant Spring (H-CS) as well as subjects with hereditary spherocytosis (HS), and subjects without a red blood cell disorder who were splenectomized for other indications (e.g. trauma and immune thrombocytopenia). Healthy controls (age 20–35 years old) were enrolled for comparisons of biomarkers only. All subjects underwent a standard M-mode measurement, 2-dimensional and Doppler echocardiogram (Sonos 5500; Hewlett Packard, Palo Alto, CA). When a TRV was present, the peak systolic right ventricular to right atrial pressure gradient and estimate pulmonary artery (PA) pressure was calculated. A TRV > 2.5 m/s (corresponding to a pulmonary artery systolic pressure >35 mmHg) was used as a proxy for patients at risk for PH, with a sensitivity and specificity of 83% and 72%, respectively [19]. A TRV that equals 2.5m/sec, was considered normal, since it could represent variability or the upper limit of normal[20].
Platelet activation, coagulation, hemolytic and vascular markers
Serum P-selectin, soluble CD40 Ligand (sCD40L), thrombin-anti thrombin TAT, F1.2, and D-dimers were measured using commercial ELISA Kits: sP-selectin (R&D Systems; Minneapolis, MN); sCD40L (BenderMed Systems; Vienna, Austria) TAT, F1+2 (Siemens-Dade Behring; Munich, Germany), and D-Dimers (American Diagnostica; Stamford, CT, USA). Cell-free hemoglobin, measured with an improved cyano-hemoglobin method (BioAssay Systems, Hayward, CA). Nitric oxide (NOx) was purged from a solution and detected by chemiluminescence (NOA 280, Sievers Intruments Inc. Boulder CO). Red cell membrane expression of phosphatidylserine (PS) was determined by annexin V labeling [21].
Brain natriuretic peptide (BNP) was assayed using fluorescence immunoassay (Biosite Diagnostic, San Diego, CA). Endothelin-1 (ET-1) was detected as immunoreactive ET-1 with antibodies from Peninsula Labs (Belmont, CA). Serum platelet endothelial cell adhesion molecule-1 (sPECAM-1), IL-2, 4, 6, 8 and 10 and INF-γ and the adhesion molecules vascular endothelial growth factor (VEGF), soluble intercellular adhesion molecule-1 and -3 (sICAM1, sICAM 3), soluble vascular cell adhesion molecule-1 (sVCAM-1), and sE-selectin were determined by flowcytomix, Bender MedSystems (Vienna, Austria).
Statistical analysis
Statistical analysis was performed using SAS version 9.3 (SAS Institute Inc., Cary NC). To determine the association of biomarkers with TRV levels, frequency distributions for categorical data, and means and standard deviations for continuous data, were calculated for each subgroup being compared: splenectomized or not, transfused or not, and TRV > or < 2.5m/sec. Pearson correlations were used to explore the relationship of individual biomarkers with TRV within the different subgroups. Bivariate analyses compared the independent variables between the TRV groups (≤ 2.5 vs > 2.5). ANOVA models, with Tukey’s adjustment for significant group effect, were constructed to compare each of the biomarkers between the patients’ subgroups. Logistic regression models were used to examine the associations of each of the biomarkers with the binary outcome of occurrence of abnormal TRV. Based on the bivariate comparisons, we considered thebiomarkers which were significant at the 0.15 level for entry into the multivariate model. We examined the effect of splenectomy on TRV after adjusting for age in the models. A significance level of 0.05 was used for all statistical tests.
Results
Subject Characteristics and Doppler Echocardiography (TRV) results
After informed consent was obtained, 101 patients and 12 healthy controls were enrolled: Seventy six patients with thalassemia, 15 with hereditary spherocytosis and 10 splenectomized subjects without a red cell disorder. An elevated TRV (>2.5m/sec) was found in 25/76 (32.8%) thalassemia patients (mean, 2.91±0.4; range 2.6–3.25 m/sec). Of the 25 patients, 16 (64%) had TI and were not regularly transfused, 4 had Hb H-CS and 5 had TI at a younger age but transitioned to receive regular transfusions later in life, in 2 of them due to the diagnosis of PH (Table 1). TRV was normal or undetected in the 36 TM patients who have been on regular transfusions since early childhood, and in the HS and healthy splenectomized patients. Seven patients underwent cardiac catheterization which confirmed PH in all of them (mean TRV 3.5±0.4, mean pulmonary artery pressure 60±14mmHg).
Table 1.
Patients’ characteristics
| * TM (n=41) | TI (n= 27) | Hb H-CS (n=8) | HS (n=15) | Splenectomy (healthy) (n=10) | Total | |
|---|---|---|---|---|---|---|
| Sex, # (%) | ||||||
| Female | 20 (48.8) | 12 (44.4) | 4 (50) | 8 (53.3) | 5 (50) | 49 (48.5) |
| Male | 21 (51.2) | 15 (55.6) | 4 (50) | 7 (46.7) | 5 (50) | 52 (51.5) |
|
| ||||||
| Median Age, yrs/(range) | 27 (11–45) | 27 (12–56) | 25 (15–28) | 41 (17–54) | 18 (10–49) | 27 (11–55) |
|
| ||||||
| β thalassemia | 27 (66) | 12 (44) | - | - | - | 39 (57) |
|
| ||||||
| E/β0 thalassemia | 14 (34) | 15 | - | - | - | 29 (43) |
|
| ||||||
| Regular Transfusion, # (%) | 41 (100) | 8 (29) | 2 (25) | - | - | 51 (50) |
|
| ||||||
| Spleen removed, # (%) | 25 (59) | 17(62) | 5 (62) | 15 (100) | 10 (100) | 72 (71) |
|
| ||||||
| Years since splenectomy (SD) | 19 (9) | 17(9) | 14(5) | 26(12) | 12(12) | 19.5 (11.5) |
|
| ||||||
| TRV>2.5 m/sec (%) | 5 (12) | 16 (59) | 4 (50) | 0 | 0 | 25 (25) |
|
| ||||||
| TRV >2.5 m/sec and prior splenectomy (%) | 5 (100) | 11 (69) | 3 (75) | 0 | 0 | 19 (76) |
|
| ||||||
| Mean TRV m/sec (SD) | 2.33 (0.31) | 2.93 (0.4) | 3.23 (0.6) | <2.5 | <2.5 | - |
|
| ||||||
| LVEF, % (SD) | 0.67 (0.06) | 0.72 (0.06) | 0.75 (0.05) | 0.72(0.04) | 0.71(0.06) | - |
TM: Thalassemia major; includes patients with β0 thalassemia and E/β0 thalassemia, transfused every 3 weeks with mean pre transfusion Hb 10.3g/dL.
Ten of these patients have been diagnosed with TI in childhood, and transitioned to regular transfusions in second or third decade of life.
TI: includes patients with β thalassemia intermedia and H-CS: Hemoglobin H Constant-Spring who are non-transfused.
Splenectomy: Healthy individuals with history of splenectomy for non-RBC causes (trauma or chronic ITP).
Three of the TI patients with an elevated TRV had a prior thrombotic event, consisting of a post-splenectomy portal vein thrombosis, DVT and a cardiac thrombus. Five thalassemia patients (2 with TM, 3 with TI) expired during the study period. Three of these 5 patients had an increased TRV of > 2.6m/sec.
Hematologic markers for platelets, coagulation and hemolysis
Platelet count was elevated in TI and Hb H-CS patients, while normal in those with TM and only mildly elevated in patients with HS. Platelet activation marker, sCD40L, was elevated in the same thalassemia patient-groups while only mildly elevated in TM and HS patients and normal in the healthy splenectomized and normal control groups. P-selectin levels did not significantly differ between the 4 groups of patients (TM, TI, Hb H-CS, HS); the 2 markers correlated with each other (p=0.03; r = 0.4). P-selectin levels were lower in healthy splenectomized patients and in the normal controls (Table 2). TAT levels were higher in patients with TI, Hb H-CS compared to the other groups. PS expression on RBCs was overall low but higher among patients with TI and H-CS. There was no correlation of PS surface exposure, known to exert procoagulant effects, with the coagulation markers TAT, F1.2 and d-dimers. F1.2 and d-dimers were determined in a total of 60 patients (13 with an increased TRV). Results were mostly within the normal range (mean 133±73pmol/L and 175 ±130 ng/ml, respectively) and therefore not performed for the rest of the patients.
Table 2.
Hematologic and vascular biomarkers
| Marker (S.D.) | TM | TI | Hb H-CS | HS | Splenectomy | Controls | p |
|---|---|---|---|---|---|---|---|
| Coagulation and platelets | |||||||
| TAT, μg/mL | 3.2 (2.5) | 5.8 (10) | 6.7(8) | 2.8 (2.5) | 3.0 (0.9) | 2.1 (0.4) | ns |
| Plt count, ×109/L | 390 (107) | 559 (206) | 458 (184) | 406 (77) | 354 (214) | nd | 0.007 |
| P selectin, ng/ml | 35(13) | 30 (12) | 33(7) | 32 (12) | 21(7) | 25(5) | 0.06 |
| sCD40 L, ng/ml | 2.1(1.6) | 2.55(2.0) | 4.37(4.9) | 1.6 (1.4) | 0.93(0.3) | 0.95(0.2) | 0.04 |
| RBC and hemolysis | |||||||
| Hb, mg/dl | 10.3* (1.8) | 9.0 (2.1) | 8.5 (1.7) | 12.4 (2.6) | 12 (2.7) | nd | 0.002 |
| Free Hb (PFH), mg/dl | 68 (32) | 99 (41) | 90 (36) | 38 (15) | 27 (8) | 34 (27) | 0.001 |
| NOx, μmol/L | 9.1 (12) | 4.9 (2.5) | 4.0 (1.9) | 4.3 (3.7) | 4.0 (2.9) | 7.2 (2) | ns |
| LDH, U/L | 262 (198) | 589 (216) | 353 (248) | 300 (203) | 276 (187) | nd | 0.0001 |
| Bilirubin, mg/dl | 1.9 (0.9) | 2.9 (1.5) | 2.3 (1.6) | 1.5 (1) | nd | nd | 0.01 |
| §PS % | 0.6(0.7) | 1.2(1.5) | 1.0(1.1) | 0.1 (0.1) | 0.5(0.5) | 0.0 | ns |
| Endothelial markers/others | |||||||
| BNP, pg/ml | 29 (7) | 31 (37) | 16 (5) | 14 (14) | 15 (11) | 11 (8) | ns |
| sVCAM-1, ng/ml | 1043 (840) | 692 (600) | 680 (422) | 370 (140) | 320 (88) | 476 (120) | 0.003 |
| PECAM, ng/ml | 290 (170) | 704 (680) | 233(160) | 520(430) | 206(64) | 188(45) | 0.06 |
| ET-1, pg/ml | 38 (29) | 65 (45) | 73 (49) | 36 (19) | 39 (29) | 8 (1.8) | 0.0001 |
Mean value and SD (in parenthesis) for each patient-group. ANOVA models were constructed to compare each of the biomarkers between the patients’ subgroups.
pre transfusion Hb.
% phosphatidyl serine (PS) detected expression on RBCs
Increased hemolysis and a more severe anemia was noted in subjects with TI, HbH-CS as indicated by increased plasma free Hb (PFH), bilirubin and LDH levels and by low plasma NOx concentration. PFH correlated with LDH (r=0.4; p=0.05) and bilirubin inversely correlated with total Hb level (r= −0.7; p= 0.001). (Table 2 and Figure 1).
Fig. 1.


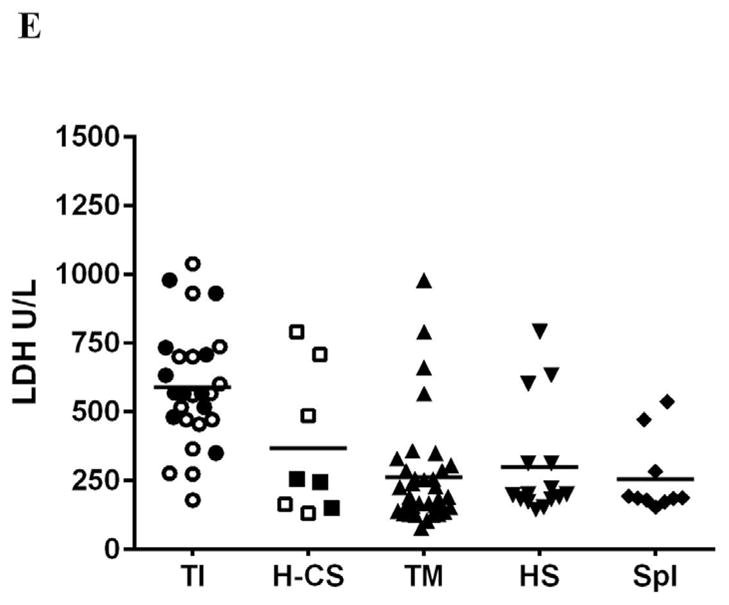
Platelet properties and hemolysis vary by patients’ subgroup. Platelets were higher in non-transfused thalassemia patients (TI and Hb H-CS) in comparison to other subgroups (A, p=0.007) and showed increase in platelet activation marker sCD40L(B, P=0.04) but not P-selectin (C, 0.06). Hemolysis was increased mostly in non-transfused thalassemia patients (D, C P <0.01; <0.01). Hollow symbols in TI and Hb H-CS groups represent splenectomized patients; bars represent the average
Endothelial activation, adhesion markers and BNP expression
ET-1 was significantly higher in TI and HbH-CS patients compared to TM patients and the other sub-groups and controls. Though highly variable, plasma soluble adhesion molecule sPECAM-1, but not sVCAM-1, had a trend towards a higher concentration in patients with TI. The other plasma cytokines and adhesion molecules were highly variable, had no association to TRV and were not included in this analysis. BNP was within the normal range, but higher levels in thalassemia patients compared to the other sub-groups were noted (Table 2).
Hematologic and vascular markers association with an elevated TRV
We investigated the relationship of the above markers with TRV. Platelet count was significantly higher in those with a TRV >2.5 compared to patients with a TRV ≤ 2.5 (p=0.02). Though, P-selectin and sCD40L correlated with each other, only sCD40L showed a correlation with the presence of increased TRV (p=0.05). There was no correlation of these two platelet activation markers with platelet count. Coagulation markers TAT or F1.2 and RBC PS expression were not associated with an increased TRV in our patient cohort. sPECAM-1 was significantly higher in patients with an increase in TRV (Table 3), possibly representing its expression on platelets, and platelets-endothelial interaction. Those with an increase in TRV had significantly higher markers of hemolysis than patients with a normal TRV; lower Hb and high PFH and LDH with an inverse correlation between PFH and NOx (r = −0.67; p=0.001. These markers were increased to the same range in 7 patients with a TRV >2.9m/sec who had confirmed PH on cardiac catheterization (Table 3).
Table 3.
Biomarkers in non-transfused patients (TI, H-CS and HS) by TRV
| Marker (S.D.) | TRV m/sec | p value | |
|---|---|---|---|
| Coagulation and Platelets | <2.5 | >2.5 | |
| TAT, μg/mL | 3.8 (5) | 7.8 (15) *3.6 (1.4) |
ns |
| Plt count, ×109/L | 402 (102) | 570 (156) *485(100) |
0.001 |
| P-selectin, ng/ml | 31 (13) | 31 (9) *30(8.0) |
ns |
| sCD40 L, ng/ml | 1.6(1.5) | 3.4 (3.0) *3.1(1.7) |
0.03 |
| RBC and Hemolysis | |||
| Hb, mg/dl | 12.2 (2.5) | 8.3 (1.3) *8.8(0.9) |
0.0001 |
| Free Hb (PFH), mg/dl | 61 (42) | 91(41) *89(27) |
0.04 |
| NOx, μmol/L | 4.1 (2.6) | 5.7 (2) *5.2(1.4) |
0.08 |
| LDH, U/L | 345 (213) | 591(250) *680(240) |
0.002 |
| Bilirubin, mg/dl | 2.0 (1.4) | 2.4 (1.3) *2.6(1.3) |
ns |
| PS, % | 1.0 (1.5) | 0.6 (0.8) * na |
ns |
| Vascular markers and other | |||
| BNP, pg/ml | 13.5 (15) | 42 (44) *65(44) |
0.01 |
| sVCAM-1, ng/ml | 520 (377) | 666 (496) *584(240) |
ns |
| sPECAM, ng/ml | 185 (107) | 683(880) *495(500) |
0.01 |
| ET-1, pg/ml | 43 (31) | 55 (39) *67(45) |
ns |
| Ferritin, ng/ml | 1872 (2534) | 486(355) | ns |
Mean value and SD (in parenthesis) for each TRV category. ns, not significant.
Indicates results in 7 patients (mean age 43±18 years) with a TRV >2.9 (mean 3.5±0.4). These patients underwent cardiac catheterization confirming PH. 6/7 of these patients were splenectomized for an average of 33.8±19 years.
When all potential variables were included in multivariate analysis, LDH (p<0.01) and platelet count (p<0.02) remained independently associated with an elevated TRV, when applied to the non-transfused patients.
Splenectomy, regular transfusions and association to TRV
TRV was significantly different within the non-transfused TI group when grouped by spleen status; higher in 15 splenectomized compared to the 10 not-splenectomized: 2.99 vs 2.40m/sec (p= 0.01). However, as noted before, TRV among transfused thalassemia patients, splenectomized or not, did not differ; and was within a normal range. The time interval since splenectomy was longer in the ones with an elevated TRV: 24.7±4 vs 11.4±3 years (p=0.03); Moreover, within the patient-group with an elevated TRV, those with a higher TRV >2.9m/sec had longer time periods without a spleen, 33.8 ±19 years, compared to patients with a TRV of 2.6–2.9 m/sec; 18.6±8 years (p=0.02). There was no significant interaction between patients’ age and time interval since splenectomy, as it affects TRV, implying that the effects of prolonged absence of spleen on TRV is independent from age. Logistic regression models applied to all the thalassemia study patients, showed that a patient who underwent a splenectomy was significantly more likely to have elevated TRV (OR 4.22; 95% CI 1.26–14.14). The non-transfused patient likelihood to have an elevated TRV is lower (OR 0.225; 95% CI 0.075–0.68). However, taken together, the non-transfused splenectomized thalassemia patients are more likely to get an elevated TRV by 4.44.
The association of splenectomy and the various biomarkers was evaluated in the non-transfused thalassemia patients (TI and Hb H-CS): Platelet count, markers of platelet activation (primarily sCD40L) and bilirubin were significantly higher in the splenectomized subjects. In contrast to previous data, RBC PS exposure was lower in the splenectomized patients (Table 4a). We examined the distinct role of platelet activation in the presence of hemolysis in the setting of absence of spleen; Platelet activation markers were higher in the splenectomized thalassemia group which had increased hemolysis, compared to the other splenectomized groups, HS and healthy patients, who had minimal or no hemolysis: sCD40L 4.7±1.2 in thalassemia patients and 1.6± 0.4 and 0.8± 0.1 in the other subgroups, respectively (p=0.003). P-selectin had a similar trend in the 3 groups, though not statistically significant: 34.1± 3.1; 31.8±3.3 and 23.4±3.4 (p=0.08). There was a significant difference in the frequency of an elevated TRV and increase in adhesion biomarkers in the thalassemia group (Table 4b).
Table 4.
Effect of splenectomy on biomarkers in non-transfused thalassemia patients (TI and H-CS) and in Non-thalassemia splenectomized patients (HS and healthy individuals)
| Marker/Finding | TI and H-CS patients | p value1 | p value2 | ||
|---|---|---|---|---|---|
| Splenectomized (n=15) | Non-splenectomized (n=10) | HS and healthy splenectomized (n=25) | |||
| TRV | 2.99 (0.6) | 2.4 (0.3) | 0.01 | 2.15 (0.25) | 0.0001 |
| TAT | 9 (16) | 3.3 (1.1) | ns | 2.9 (2.2) | ns |
| Plt count | 565 (177) | 413 (156) | 0.03 | 387 (142) | 0.001 |
| P-selectin | 34 (11) | 28 (9) | ns | 28 (12) | ns |
| sCD40-L | 4.7 (4) | 1.1 (0.9) | 0.01 | 1.3 (1.1) | 0.001 |
| Hb | 9.5 (2.1) | 9.6 (2.4) | ns | 12.2 (2.6) | 0.002 |
| Free Hb (PFH) | 88 (47) | 105 (34) | ns | 33 (14) | 0.0001 |
| NOx | 4.0 (2.2) | 5.6 (3) | ns | 4.1 (3.3) | ns |
| LDH | 494 (249) | 548 (219) | ns | 290 (192) | 0.003 |
| Bilirubin | 1.9 (1.2) | 3.2 (1.3) | 0.01 | 1.5 (1.0) | ns |
| PS (%) | 0.4 (0.5) | 1.1 (1.7) | ns | 0.4 (0.5) | ns |
| sVCAM-1 | 576 (322) | 768 (630) | ns | 348 (122) | 0.01 |
| ET 1 | 62 (38) | 69 (48) | ns | 37 (23) | 0.02 |
ns, not significant
sVCAM-1, soluble vascular cell adhesion molecule-1. ET-1, endothelin-1.
p value1: Splenectomized vs. non-splenectomized thalassemia patients.
p value2: Biomarkers in patients with chronic hemolysis (Splenectomized thalassemia; n=15) compared to patients with minimal or no hemolysis (splenectomized HS and healthy individuals; n=25).
Beyond the expected effect of regular transfusions on the anemia, it lowered hemolytic markers (PFH, LDH and NOx) while the effect on platelet count and platelet activation markers was less significant, though sCD40L levels trended down. Transfused patients had significantly higher serum ferritin levels and lower left ventricular ejection fraction (LVEF) (Table 5).
Table 5.
Transfusions effect on biomarkers in thalassemia patients
| Marker/finding | Transfused (n=51)* | Non-transfused (n=25) | p value |
|---|---|---|---|
| TRV | 2.3 (0.3) | 2.7 (0.6) | 0.0006 |
| EF | 0.68 (0.06) | 0.72 (0.06) | 0.004 |
| TAT | 3.5 (2.7) | 6.5(12) | ns |
| Platelet count | 433 (230) | 501 (180) | ns |
| P-selectin | 34 (12) | 31 (10) | ns |
| sCD40L | 2.1 (1.6) | 4.0 (5.1) | 0.05 |
| Free Hb (PFH) | 75 (33) | 95 (42) | 0.03 |
| NOx | 8.3 (11) | 4.8 (2.7) | ns |
| LDH | 329 (249) | 517 (234) | 0.002 |
| Bilirubin | 2.2 (1.4) | 2.5 (1.4) | ns |
| BNP | 28 (35) | 30 (36) | ns |
| ET-1 | 43 (36) | 66 (42) | 0.02 |
| Ferritin | 2447 (2680) | 847(1280) | 0.01 |
Regularly transfused patients consists of TM (n=41) and patients with TI and H-CS (n=10) transitioned to frequent or regular t transfusions (≥ 8times/year). TAT, thrombin-anti thrombin. BNP, brain natriuretic protein. ET-1, endothelin-1.
ns, not significant.
Discussion
Our data more clearly define patients at-risk for developing an increase in TRV, highlighting potential pathophysiologic mechanisms. Though increase in TRV and Doppler defined PH has some limitations in the context of hematologic diseases[20], there is strong predictive value and a general good correlation to right heart catheterization [19,18]. A third (25/76) of thalassemia patients had an elevated TRV, with a higher prevalence in non-transfused patients (TI and Hb H-CS), noted in 20/35 (57%), a similar rate to a previous report [6]. In 7 out of the 25 (28%) patients with an elevated TRV, PH was confirmed by cardiac catheterization. We have identified splenectomized patients with Hb H-CS as a group with a high risk for developing PH, 50% of them had an elevated TRV (mean TRV 3.23m/sec), a finding that to the best of our knowledge has not been previously recognized. Splenectomy is sometimes performed in order to treat the anemia in these patients [22], and a special attention to early screening with echocardiogram should be thought.
Our data showed a clear beneficial effect of transfusions; there were no cases of increase in TRV in the regularly transfused TM patients and an overall prevalence of 12 % when patients who started transfusions in second or third decade of life were included. This contradicts previous studies of a higher rate among transfused patients, however the transfusion schedule and Hb threshold varied in these reports [14,15,2,16,17].
The commencement of scheduled transfusions resulted in normalization of TRV in 2 adult patients as also observed in a previous case report [23]. However, in other 3 patients, transfusions have not reversed the increased TRV, suggesting the development of irreversible vasculature changes in the lung tissue [24], when instituted at later stages of evolving PH. The frequency and level of Hb required to prevent the pathophysiological changes resulting in an increase of TRV in TI patients who are not regularly transfused has not been studied. Prior splenectomy does not abate the beneficial effect of transfusions; 55% of our cohort of regularly transfused patients was splenectomized. The beneficial effect of transfusions seems mostly through reducing hemolysis and to a lesser extent minimizing platelet activation. Transfusions reduce effective and ineffective hematopoiesis, consequently lowering platelets and abnormal RBCs circulation. This possibly results in decreased platelet adhesion which was shown to be induced by both thalassemia platelets and by thalassemia RBC [25]. Despite adequate blood transfusion, as demonstrated by a mean pre-transfusion Hb of 10.3gr/dL, transfusions did not totally normalize the biomarkers to the levels observed in the normal control group (Tables 2 and 5).
Our data does not implicate a strong association of elevated TRV with a hypercoaguable state. Clinically, only 3 individuals (12%) of our patient population with an increase in TRV had prior thrombotic events, a lower rate than previously reported [4,8]. Laboratory tests showed only a modest increase in thrombin generation as indicated by TAT in splenectomized TI patients, and no increase F1.2 and D-dimers. Abnormal expression of negatively charged phospholipids, mostly phosphatidylserine (PS) triggering a prothrombotic state, is thought to be a main cause of the hypercoaguable state in thalassemia intermedia[5] but an association with PH was not analyzed. We did not find an association of RBC PS expression with TAT or with TRV. Still, PS RBC membrane extarnilization was higher in non-transfused thalassemia patients (62% splenectomized) than in the other subgroups. Other methods such as thromboelastometry on whole blood may yield more accurate measurements of thrombin generation in this patient population[11], and a correlation with TRV could be determined. Similarly, determination of RBC microparticles, found to be elevated in TI[26], may show an association with thrombotic markers and possibly with an increase in TRV. These methods were not utilized in our study.
Platelet count was overall higher in non-transfused splenectomized thalassemia patients, and associated with an increase in TRV, as opposed to the non-thalassemia splenectomized ones. A threshold risk of platelet count over 500K 109 L−1 was noted as also recently suggested[8]. Beyond the thrombocytosis, we have shown that increased platelet activation is present in this thalassemia patients and it is associated with the severity of TRV. Heightened platelet activation has been described in patients with thalassemia [27,5,28], though the mechanism is not fully understood. It was attributed to the accelerated thrombin generation through abnormal RBCs PS expression which in turn, triggers platelet activation[29,30]. However, there is limited data demonstrating such a correlation of abnormal RBCs with platelet activation; one study examined the association of P selectin with PS exposure, however it was examined on RBCs of transfused patients[30]. Additional presumed mechanisms, include increased platelet adhesion in association with a high platelet count, shown in splenectomized thalassemia patients [31]and platelets exposure to oxidative stress which causes their activation[31,32]. Our data found sPECAM-1, which has a role in platelet activation and adhesion signaling, to be higher among the TI patients [33,34], suggesting increased platelet adhesion having a role in the development of pulmonary vasculopathy. Platelet microparticles, elevated in thalassemia patients[35] have a significantly higher procoagulant activity than platelets [36] but were not analyzed in the context of PH or thrombotic events. Further study on the mechanisms of post-splenectomy platelet effect on the development of increased pulmonary vasculature pressure and RBCs-platelets interaction is warranted.
We have demonstrated increased hemolysis in thalassemia patients with a high TRV as indicated by an increased plasma free Hb and LDH, the latter also correlated with the extent of TRV (Table 3). Increased hemolysis resulted in nitric oxide scavenging by free hemoglobin, as seen by the inverse correlation between PFH and NOx in patients with a TRV>2.5m/sec (r= −0.67; p=0.001). However, NOx was not significantly different among TI patients with or without an increase in TRV. NO consumption, reduced bioavailability and consequent impaired blood flow may play a lesser role in the pathophysiology of pulmonary vasculopathy in thalassemia compared to SCD, where PH is thought to be largely derived from such mechanism, though somewhat controversial [37,38]. Alternatively, plasma free hemoglobin may act through alternative mechanisms such as a pro-oxidant and a trigger of pulmonary vasculature endothelial cell injury and in the initiation of platelet activation and aggregation [39–41], possibly contributing to the pulmonary vascular changes in splenectomized TI patients.
In contrast to SCD, our data did not show vascular injury in patients with a higher TRV as marked by endothelial cell adhesion molecules; sVCAM-1, sICAM, sE-selectin or inflammatory cytokins. ET-1, a potent vasoconstrictor produced by vascular endothelial and smooth muscle cells, was higher in the non-transfused thalassemia patients and is known to have a role in the development of pulmonary vascular dysfunction and PH [42,43] and found to. Further study of its value in treating and monitoring TRV in thalassemia is needed, in particular since treatment with ET-1 antagonists has shown benefit in patients with PH and an improvement was noted in a reported case of a thalassemia patient with PH[44]. BNP, a sensitive marker for PH and high right ventricle strain, though showing only a modest increase, was the highest in non-transfused TI patients. It may prove beneficial in guiding clinical decision and gauge response to treatment if validated in larger studies.
Taken together, we explored the selective role of absence of spleen in clinical settings with or without chronic hemolysis as it affects the above causative factors for an elevated TRV: Hemolysis in non-transfused thalassemia patients that have an intact spleen does not result in an increase in TRV, despite similar or higher markers of hemolysis than those measured in splenectomized TI patients. It is the combination of both hemolysis and increase in platelet number and activation, in the setting of absence of spleen, which seems to lead to an increase in TRV and overt PH. Absence of spleen without these 2 elements, as seen in HS and in healthy splenectomized individuals does not lead to an increase in TRV. HS patients had a longer time period of absence of spleen (mean 26±12 vs 17±9 years in TI patients) and none has developed an increase in TRV, in agreement with the findings of Crary et al [45]; Moreover, the risk for developing a high TRV increases over time after splenectomy in TI patients. We suggest a major role to thrombocytosis, coupled with platelet activation and platelet-endothelial adhesion in the development of a pulmonary vasculopathy in splenectomized TI patients. Therefore measuring sCD40L, P selectin and sPECAM-1 may prove to be useful in assessing the risk of developing PH in these patients. Plasma sCD40L, in particular, was shown to correlate with platelet activation and have a role in the interaction of platelets and endothelial cells in causing PH in other conditions[46–48].
In conclusion, a pathogenetic model could be proposed, in which ongoing hemolysis is not sufficient for triggering an increase in TRV unless a “second hit” of spleen removal with a consequent further thrombocytosis and platelet activation occurs. The interactions of platelets, circulating damaged RBCs, free hemoglobin and blood cells-derived microparticles subsequent to spelnectomy in the pathophysiology of PH, has not been adequately resolved. Thrombocytosis (>500 ×109/L), elevated sCD40L and increased LDH and possibly sPECAM-1 and ET-1 are markers suggestive of an increased risk for developing PH while activation of the coagulation system and inflammatory changes do not seem to play a significant role in our findings. These same markers proved to be elevated in the group with established PH suggesting their validity as indicators for risk for PH. Treatment with an anti-platelet agent or reducing platelet count with hydroxyurea are reasonable approaches in all splenectomized non-transfused TI patients [49]. However, more interventional study could result in multimodal treatment, tailored to the processes identified to be the most contributory in a given individual.
Acknowledgments
This work was supported by a grant from the National Institute of Health (NIH), National Heart, Lung, Blood Institute (NHLBI) Career Development award K23 HL077409-02. This study was also supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH, through UCSF-CTSI Grant Number UL1 RR024131.
Footnotes
Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Conflict-of Interest: The authors declare that they have no conflict of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study.
References
- 1.Morris CR, Kim HY, Trachtenberg F, Wood J, Quinn CT, Sweeters N, Kwiatkowski JL, Thompson AA, Giardina PJ, Boudreaux J, Olivieri NF, Porter JB, Neufeld EJ, Vichinsky EP. Risk factors and mortality associated with an elevated tricuspid regurgitant jet velocity measured by Doppler-echocardiography in thalassemia: a Thalassemia Clinical Research Network report. Blood. 2011;118 (14):3794–3802. doi: 10.1182/blood-2010-11-319152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, Saned MS, El-Chafic AH, Fasulo MR, Cappellini MD. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115 (10):1886–1892. doi: 10.1182/blood-2009-09-243154. [DOI] [PubMed] [Google Scholar]
- 3.Aessopos A, Stamatelos G, Skoumas V, Vassilopoulos G, Mantzourani M, Loukopoulos D. Pulmonary hypertension and right heart failure in patients with beta-thalassemia intermedia [see comments] Chest. 1995;107 (1):50–53. doi: 10.1378/chest.107.1.50. [DOI] [PubMed] [Google Scholar]
- 4.Karimi M, Musallam KM, Cappellini MD, Daar S, El-Beshlawy A, Belhoul K, Saned MS, Temraz S, Koussa S, Taher AT. Risk factors for pulmonary hypertension in patients with beta thalassemia intermedia. Eur J Intern Med. 2011;22 (6):607–610. doi: 10.1016/j.ejim.2011.05.013. S0953-6205(11)00105-1 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. British journal of haematology. 2000;111 (2):467–473. doi: 10.1046/j.1365-2141.2000.02376.x. [DOI] [PubMed] [Google Scholar]
- 6.Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, Joussef J, Rombos J, Loukopoulos D. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97 (11):3411–3416. doi: 10.1182/blood.v97.11.3411. [DOI] [PubMed] [Google Scholar]
- 7.Atichartakarn V, Angchaisuksiri P, Aryurachai K, Onpun S, Chuncharunee S, Thakkinstian A, Atamasirikul K. Relationship between hypercoagulable state and erythrocyte phosphatidylserine exposure in splenectomized haemoglobin E/beta-thalassaemic patients. British journal of haematology. 2002;118 (3):893–898. doi: 10.1046/j.1365-2141.2002.03711.x. [DOI] [PubMed] [Google Scholar]
- 8.Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, Saned M, Cesaretti C, Cappellini MD. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8 (10):2152–2158. doi: 10.1111/j.1538-7836.2010.03940.x. [DOI] [PubMed] [Google Scholar]
- 9.Taher A, Isma’eel H, Mehio G, Bignamini D, Kattamis A, Rachmilewitz EA, Cappellini MD. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thrombosis and haemostasis. 2006;96 (4):488–491. [PubMed] [Google Scholar]
- 10.Angchaisuksiri P, Atichartakarn V, Aryurachai K, Archararit N, Chuncharunee S, Tiraganjana A, Rattanasiri S. Hemostatic and thrombotic markers in patients with hemoglobin E/beta-thalassemia disease. Am J Hematol. 2007;82 (11):1001–1004. doi: 10.1002/ajh.20945. [DOI] [PubMed] [Google Scholar]
- 11.Tripodi A, Cappellini MD, Chantarangkul V, Padovan L, Fasulo MR, Marcon A, Mannucci PM. Hypercoagulability in splenectomized thalassemic patients detected by whole-blood thromboelastometry, but not by thrombin generation in platelet-poor plasma. Haematologica. 2009;94 (11):1520–1527. doi: 10.3324/haematol.2009.010546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aessopos A, Farmakis D, Deftereos S, Tsironi M, Tassiopoulos S, Moyssakis I, Karagiorga M. Thalassemia heart disease: a comparative evaluation of thalassemia major and thalassemia intermedia. Chest. 2005;127 (5):1523–1530. doi: 10.1378/chest.127.5.1523. [DOI] [PubMed] [Google Scholar]
- 13.Du ZD, Roguin N, Milgram E, Saab K, Koren A. Pulmonary hypertension in patients with thalassemia major. American heart journal. 1997;134 (3):532–537. doi: 10.1016/s0002-8703(97)70091-7. [DOI] [PubMed] [Google Scholar]
- 14.El-Beshlawy A, Youssry I, El-Saidi S, El Accaoui R, Mansi Y, Makhlouf A, Taher A. Pulmonary hypertension in beta-thalassemia major and the role of L-carnitine therapy. Pediatric hematology and oncology. 2008;25 (8):734–743. doi: 10.1080/08880010802244035. [DOI] [PubMed] [Google Scholar]
- 15.Kiter G, Balci YI, Ates A, Hacioglu S, Sari I. Frequency of pulmonary hypertension in asymptomatic beta-thalassemia major patients and the role of physiological parameters in evaluation. Pediatric hematology and oncology. 2010;27 (8):597–607. doi: 10.3109/08880018.2010.503338. [DOI] [PubMed] [Google Scholar]
- 16.Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation. 2011;123 (11):1227–1232. doi: 10.1161/circulationaha.110.988089. [DOI] [PubMed] [Google Scholar]
- 17.Morris CR, Kuypers FA, Kato GJ, Lavrisha L, Larkin S, Singer T, Vichinsky EP. Hemolysis-associated pulmonary hypertension in thalassemia. Annals of the New York Academy of Sciences. 2005;1054:481–485. doi: 10.1196/annals.1345.058. 1054/1/481 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Derchi G, Galanello R, Bina P, Cappellini MD, Piga A, Lai ME, Quarta A, Casu G, Perrotta S, Pinto V, Musallam KM, Forni GL on behalf of the Webthal Pulmonary Arterial Hypertension G. Prevalence and Risk Factors for Pulmonary Arterial Hypertension in a Large Group of beta-Thalassemia Patients Using Right Heart Catheterization: A Webthal(R) Study. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.002124. [DOI] [PubMed] [Google Scholar]
- 19.Janda S, Shahidi N, Gin K, Swiston J. Diagnostic accuracy of echocardiography for pulmonary hypertension: a systematic review and meta-analysis. Heart (British Cardiac Society) 2011;97 (8):612–622. doi: 10.1136/hrt.2010.212084. [DOI] [PubMed] [Google Scholar]
- 20.Nathan DG. Guilt by association. Blood. 2011;118 (14):3758–3759. doi: 10.1182/blood-2011-08-370338. [DOI] [PubMed] [Google Scholar]
- 21.Kuypers FA, Larkin SK, Emeis JJ, Allison AC. Interaction of an annexin V homodimer (Diannexin) with phosphatidylserine on cell surfaces and consequent antithrombotic activity. Thrombosis and haemostasis. 2007;97 (3):478–486. [PubMed] [Google Scholar]
- 22.Lal A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP. Heterogeneity of hemoglobin H disease in childhood. The New England journal of medicine. 2011;364 (8):710–718. doi: 10.1056/NEJMoa1010174. [DOI] [PubMed] [Google Scholar]
- 23.Atichartakarn V, Chuncharunee S, Chandanamattha P, Likittanasombat K, Aryurachai K. Correction of hypercoagulability and amelioration of pulmonary arterial hypertension by chronic blood transfusion in an asplenic hemoglobin E/beta-thalassemia patient. Blood. 2004;103 (7):2844–2846. doi: 10.1182/blood-2003-09-3094. [DOI] [PubMed] [Google Scholar]
- 24.Hassell KL. Pulmonary hypertension, tricuspid regurgitant velocity screening, and the nitric oxide pathway. Hematology Am Soc Hematol Educ Program. 2011;2011:419–426. doi: 10.1182/asheducation-2011.1.419. [DOI] [PubMed] [Google Scholar]
- 25.Goldfarb A, Grisaru D, Gimmon Z, Okon E, Lebensart P, Rachmilewitz EA. High incidence of cholelithiasis in older patients with homozygous beta-thalassemia. Acta Haematol. 1990;83 (3):120–122. doi: 10.1159/000205186. [DOI] [PubMed] [Google Scholar]
- 26.Habib A, Kunzelmann C, Shamseddeen W, Zobairi F, Freyssinet JM, Taher A. Elevated levels of circulating procoagulant microparticles in patients with beta-thalassemia intermedia. Haematologica. 2008;93 (6):941–942. doi: 10.3324/haematol.12460. [DOI] [PubMed] [Google Scholar]
- 27.Atichartakarn V, Angchaisuksiri P, Aryurachai K, Chuncharunee S, Thakkinstian A. In vivo platelet activation and hyperaggregation in hemoglobin E/beta-thalassemia: a consequence of splenectomy. Int J Hematol. 2003;77 (3):299–303. doi: 10.1007/BF02983790. [DOI] [PubMed] [Google Scholar]
- 28.Eldor A, Lellouche F, Goldfarb A, Rachmilewitz EA, Maclouf J. In vivo platelet activation in beta-thalassemia major reflected by increased platelet-thromboxane urinary metabolites. Blood. 1991;77 (8):1749–1753. [PubMed] [Google Scholar]
- 29.Borenstain-Ben Yashar V, Barenholz Y, Hy-Am E, Rachmilewitz EA, Eldor A. Phosphatidylserine in the outer leaflet of red blood cells from beta-thalassemia patients may explain the chronic hypercoagulable state and thrombotic episodes. Am J Hematol. 1993;44 (1):63–65. doi: 10.1002/ajh.2830440114. [DOI] [PubMed] [Google Scholar]
- 30.Ruf A, Pick M, Deutsch V, Patscheke H, Goldfarb A, Rachmilewitz EA, Guillin MC, Eldor A. In-vivo platelet activation correlates with red cell anionic phospholipid exposure in patients with beta-thalassaemia major. British journal of haematology. 1997;98 (1):51–56. doi: 10.1046/j.1365-2141.1997.1502965.x. [DOI] [PubMed] [Google Scholar]
- 31.Goldschmidt N, Spectre G, Brill A, Zelig O, Goldfarb A, Rachmilewitz E, Varon D. Increased platelet adhesion under flow conditions is induced by both thalassemic platelets and red blood cells. Thrombosis and haemostasis. 2008;100 (5):864–870. [PubMed] [Google Scholar]
- 32.Amer J, Fibach E. Oxidative status of platelets in normal and thalassemic blood. Thrombosis and haemostasis. 2004;92 (5):1052–1059. doi: 10.1267/thro04051052. [DOI] [PubMed] [Google Scholar]
- 33.Newman PJ, Newman DK. Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arteriosclerosis, thrombosis, and vascular biology. 2003;23 (6):953–964. doi: 10.1161/01.atv.0000071347.69358.d9. [DOI] [PubMed] [Google Scholar]
- 34.Varon D, Jackson DE, Shenkman B, Dardik R, Tamarin I, Savion N, Newman PJ. Platelet/endothelial cell adhesion molecule-1 serves as a costimulatory agonist receptor that modulates integrin-dependent adhesion and aggregation of human platelets. Blood. 1998;91 (2):500–507. [PubMed] [Google Scholar]
- 35.Chaichompoo P, Kumya P, Khowawisetsut L, Chiangjong W, Chaiyarit S, Pongsakul N, Sirithanaratanakul N, Fucharoen S, Thongboonkerd V, Pattanapanyasat K. Characterizations and proteome analysis of platelet-free plasma-derived microparticles in beta-thalassemia/hemoglobin E patients. Journal of proteomics. 2012;76(Spec No):239–250. doi: 10.1016/j.jprot.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Sinauridze EI, Kireev DA, Popenko NY, Pichugin AV, Panteleev MA, Krymskaya OV, Ataullakhanov FI. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thrombosis and haemostasis. 2007;97 (3):425–434. [PubMed] [Google Scholar]
- 37.Gladwin MT, Barst RJ, Castro OL, Gordeuk VR, Hillery CA, Kato GJ, Kim-Shapiro DB, Machado R, Morris CR, Steinberg MH, Vichinsky EP. Pulmonary hypertension and NO in sickle cell. Blood. 2010;116 (5):852–854. doi: 10.1182/blood-2010-04-282095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bunn HF, Nathan DG, Dover GJ, Hebbel RP, Platt OS, Rosse WF, Ware RE. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;116 (5):687–692. doi: 10.1182/blood-2010-02-268193. [DOI] [PubMed] [Google Scholar]
- 39.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA: the journal of the American Medical Association. 2005;293 (13):1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 40.Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Eaton JW, Balla G. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Molecular nutrition & food research. 2005;49 (11):1030–1043. doi: 10.1002/mnfr.200500076. [DOI] [PubMed] [Google Scholar]
- 41.Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100 (3):879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 42.Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovascular research. 2007;76 (1):8–18. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Vizza CD, Letizia C, Petramala L, Badagliacca R, Poscia R, Zepponi E, Crescenzi E, Nona A, Benedetti G, Ferrante F, Sciomer S, Fedele F. Venous endotelin-1 (ET-1) and brain natriuretic peptide (BNP) plasma levels during 6-month bosentan treatment for pulmonary arterial hypertension. Regulatory peptides. 2008;151 (1–3):48–53. doi: 10.1016/j.regpep.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Anthi A, Tsangaris I, Hamodraka ES, Lekakis J, Armaganidis A, Orfanos SE. Treatment with bosentan in a patient with thalassemia intermedia and pulmonary arterial hypertension. Blood. 2012;120 (7):1531–1532. doi: 10.1182/blood-2012-04-422568. [DOI] [PubMed] [Google Scholar]
- 45.Crary SE, Ramaciotti C, Buchanan GR. Prevalence of pulmonary hypertension in hereditary spherocytosis. Am J Hematol. 2011;86 (12):E73–76. doi: 10.1002/ajh.22182. [DOI] [PubMed] [Google Scholar]
- 46.Riondino S, Martini F, La Farina F, Spila A, Guadagni F, Ferroni P. Increased plasma levels of soluble CD40 ligand correlate with platelet activation markers and underline the need for standardized pre-analytical conditions. Clinical biochemistry. 2010;43 (7–8):666–670. doi: 10.1016/j.clinbiochem.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 47.Damas JK, Otterdal K, Yndestad A, Aass H, Solum NO, Froland SS, Simonsen S, Aukrust P, Andreassen AK. Soluble CD40 ligand in pulmonary arterial hypertension: possible pathogenic role of the interaction between platelets and endothelial cells. Circulation. 2004;110 (8):999–1005. doi: 10.1161/01.cir.0000139859.68513.fc. [DOI] [PubMed] [Google Scholar]
- 48.Allanore Y, Borderie D, Meune C, Lemarechal H, Weber S, Ekindjian OG, Kahan A. Increased plasma soluble CD40 ligand concentrations in systemic sclerosis and association with pulmonary arterial hypertension and digital ulcers. Annals of the rheumatic diseases. 2005;64 (3):481–483. doi: 10.1136/ard.2003.020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mannucci PM. Red cells playing as activated platelets in thalassemia intermedia. J Thromb Haemost. 2010;8 (10):2149–2151. doi: 10.1111/j.1538-7836.2010.04030.x. [DOI] [PubMed] [Google Scholar]